

Abstract
Background
Cardiac fibrosis following myocardial infarction (MI) results in heart failure. Caveolin-1, the main structural protein of caveolae, regulates signal transduction pathways controlling cell proliferation and apoptosis. Meanwhile, low phosphatase and tensin homolog (PTEN) activity enhances the PI3K/Akt signal pathway to induce cell proliferation. But whether caveolin-1 and PTEN activation regulates cardiac fibroblast proliferation and contributes to cardiac fibrosis from ischemic injury is incompletely understood. This study investigates whether hypoxia inducing cardiac fibroblast proliferation and phenotypic switch is caveolin-dependent.
Methods
We used in vitro and in vivo models of ischemic injury, immunohistochemical staining, and cell proliferation assays to address this hypothesis.
Results
We found that MI induced collagen deposition and cardiac dysfunction. After MI, mice displayed reduced caveolin-1 and PTEN expression and increased α-smooth muscle actin (α-SMA) expression in the infarct zone. Qualitative and quantitative analyses indicated that caveolin-1 expression was lowest at 7 days after MI, accompanied by increased collagen deposition and attenuated cardiac function. We cultured cardiac fibroblasts of mice were in hypoxia or normoxia conditions for 12, 24 and 48 hours. At all the time points, caveolin-1 and PTEN expression were gradually reduced, whereas, α-SMA was gradually increased. We also observed that cell viability was increased at 12 and 24 h after hypoxia then lightly decreased at 48 h. Additionally, disruption of caveolae with methyl-β-cyclodextrin (MβCD) enhanced p-Akt and α-SMA expression and fibroblast proliferation and phenotypic switch.
Conclusions
These findings suggest a key role for caveolae, perhaps through the caveolin-1/PTEN signaling pathway, in cardiac fibroblast proliferation and phenotypic switch under hypoxia.
Keywords: Caveolin-1, phosphatase and tensin homolog (PTEN), cardiac fibroblast, hypoxia
Introduction
Coronary artery disease and ischemic cardiomyopathy represent the leading cause of heart failure and continue to grow at exponential rates (1). After myocardial infarction (MI), myocardial cells die, cardiac fibroblasts proliferate, and results in pathologic fibrosis. Cardiac fibroblasts constitute more than 90% of the non-myocytes and play an essential role in the physiology of the heart. Cardiac fibroblasts produce extracellular matrix proteins and synthesize angiogenic and cardioprotective factors. Although cardiac fibroblasts are known to be resistant to apoptosis and to remain metabolically active in situations compromising cell survival, the underlying mechanisms are unknown (2). Cardiac fibrosis is an important contributor to the development of cardiac dysfunction in diverse pathological conditions, such as ischemia, and is typically characterized by uncontrolled proliferation of fibroblasts and excessive deposition of extracellular matrix proteins in the myocardium (3-5).
Caveolae are plasmalemmal invaginations enriched in cholesterol, glycosphingolipids, and lipid-anchored proteins relative to the bulk of the plasma membrane, and caveolae biogenesis and function depend on two distinct caveolar components: caveolins and cavins (6-9). The caveolin gene family consists of three distinct genes: namely, Caveolin-1, Caveolin-2, and Caveolin-3 (10-14). Caveolin-1 is a cholesterol-binding and integral membrane protein that regulates a variety of cellular processes, including integrin turnover and signal transduction pathways controlling cell proliferation and apoptosis (14-17). Caveolin-3 appears to be muscle specific and is expressed in cardiac, skeletal, and smooth muscle cells, whereas caveolin-1 and caveolin-2 are usually coexpressed and are particularly abundant in endothelial cells, fibroblasts, smooth muscle cells, adipocytes, and epithelial cells (12-14,18,19). Caveolin-1 has recently been found to be involved in the pathogenesis of ischemic injury (14,20-22). Cavins are also structural components of caveolae and has four isoforms.
Phosphatase and tensin homolog (PTEN) is a dual lipid/protein phosphatase that negatively regulates proliferation by repressing the integrin/PI3K/Akt pathway (15,23-30). In the cytoplasm, PTEN inhibits PI3K signaling by transforming PIP3 into PIP2 (31). Activated Akt positively regulates cell growth or activity, but negatively regulates cell autophagy and apoptosis. At low levels of PTEN in cytoplasm, PIP3 accumulates, and both Akt and phosphoinositide-dependent kinase-1 (PDK1), which contain a PH domain, binds to membrane-bound PIP3. Once bound to the membrane, PDK1 and the mammalian target of rapamycin1 (mTOR1) activate Akt through phosphorylation at various sites (32). Amino acid sequence analysis of PTEN indicates that PTEN contains the caveolin-1 consensus binding sequence OXOXXXXO corresponding to amino acids 271-278 (FHFWVNTF), where O represents the aromatic amino acid phenylalanine (F) (15,33). This suggests a relationship between caveolin-1 expression and PTEN function. However, it is unknown that whether caveolin-1 and PTEN, together, modulate cardiac fibroblast proliferation and phenotypic switch under hypoxia.
Materials and methods
Animal Model of acute MI and left ventricular function assay
Male C57BL/6 mice (Nanjing medical university laboratory animal center, Nanjing, China) were subjected to permanent ligation of left anterior descending coronary artery (LAD) to induce MI (n=10) (34). Sham-operated mice were used as controls. Sham-operated group (n=6). Mice underwent echocardiography at 7, 14 and 28 days, after surgery. Trans-thoracic echocardiography was performed with a 14 MHz il3L linear probe (Vivid 7 Ultrasound Machine, GE Medical). Data of heart function was measured according to modified recommendations by the American Society of Echocardiography. A mean value of 3 measurements was examined at day 7, 14 and 28. Hearts were also harvested at day 7, 14 and 28 for Masson trichrome staining, immunohistochemical staining and western blot analysis (n=6-10/group).
Cell culture
We obtained neonatal cardiac fibroblasts from the heart of 1-3-day-old C57BL/6 mice (Nanjing medical university laboratory animal center, Nanjing, China). After digestion of the hearts with Type-2 Collagenase (Gibco, New York, USA), cells were pelleted and seeded in 10-cm FALCON polystyrene dishes (Corning, NY, USA), and incubated for 120 min in media containing High glucose Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, Inc., USA), with 10% fetal calf serum (PAA, Dartmouth, MA), and antibiotics at 37 °C and 5% CO2. After 2 hours, the medium was removed to eliminate cardiomyocytes that did not attach to the non-coated plates, and replaced with fresh medium. Cardiac fibroblasts were allowed to grow until confluence, then trypsinized, and passaged twice before use. For induction of cellular hypoxia, cells were replaced by DMEM without glucose (GIBCO, Inc., USA) and incubated at 37 °C with 5% CO2, 1% O2 and 94% N2 in a GENbag anaer (bioMerieux® sa, Marcy I’Etoile, France) for 6, 12, 24, and 48 h. At these culture conditions, cardiac fibroblasts experienced varying levels of hypoxia in the presence or absence of methyl-β-cyclodextrin (MβCD, 2 mM) (Sigma, Inc., Germany), which binds cholesterol and disrupts caveolae.
Immunohistochemical staining
Immunohistochemistry was performed as described previously (35). The following primary antibodies were used for immunohistochemical staining: anti-caveolin-1-IgG (HuaAnBiotech, Inc., Hangzhou, China), anti-PTEN-IgG (Epitomics, Inc., California, USA), anti-α-SMA-Ig-G (Epitomics, Inc., California, USA).
The signal was enhanced by avidin-biotin-peroxidasecomplex (VectastainABCkit, Vector Laboratories, Burlingame, CA, USA), followed by visualization of the reaction with 3, 3’-diaminobenzidine tetrahydrochloride (DAB) solution (Peroxidase Sub-strateKit,Vector Laboratories, Burlingame, CA, USA). Controls were incubated with PBS in place of the primary antibody and no positive staining was observed (36).
Western blot analysis
Cells were collected in cold buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 g/mL leupeptin, 1 mM PMSF, and centrifuged at 11,000 rpm for 20 min. The protein extracts (10 g/lane) were electrophoresed on a 12% Tris HCl gel from Bio-Rad and transferred to a nitrocellulose membrane, which was stained by naphthol blue-black to confirm equal protein loading. The membranes were incubated in 5% skim milk for 2 h and then incubated with the following primary antibodies: caveolin-1 antibody (HuaAnBiotech, Inc., Hangzhou, China), PTEN antibody, phospho-Akt (pS129) antibody (Epitomics, Inc., California, USA), Akt (S473) antibody (Epitomics, Inc., California, USA), α-SMA antibody (Epitomics, Inc., California, USA), and Gapdh antibody (HuaAnBiotech, Inc., Hangzhou, China), and, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG and anti-mouse IgG secondary antibody and detection using Supersignal West picostable peroxide solution (Pierce, Rockford, IL) (37). Western blot was also performed as described above for examination of caveolin-1 expression from heart protein extracts.
Cell proliferation and viability assays
Cell proliferation was quantified using a colorimetric method based on the metabolic reduction of 3-(4, 5-dimethylthiozol-2-yl)-2, 5-diphenyltetrazo-lium bromide (MTT) dye to formazan, as described earlier (38). Briefly, cardiac fibroblasts were plated onto 96-multiwell plates at 8,000 cells/well. The next day, cells cultured under hypoxic conditions after 12, 24, or 48 hours were rinsed with PBS, and MTT was added. Four hours later, DMSO was added, and cells were incubated 15 minutes at 37 °C. Samples were measured at 570 nm.
Statistical analysis
Statistics were performed using GraphPad Prism software (GraphPad Software, Inc., CA, USA). All data was analyzed by SPSS 17.0 software (SPSS Inc, Chicago, Illinois, 2008). All experiments were repeated with at least three batches of cardiac fibroblasts and in all repetitions qualitatively similar data were obtained. Data were tested for significance using ANOVA or t-test, as appropriate. Results with P<0.05 were considered statistically significant.
Results
Collagen deposition increased and cardiac function attenuated following MI
To evaluate the process of cardiac fibrosis from ischemic injury, we induced myocardial infraction in a mouse model. Myocardial fibrosis was determined by qualitative evaluation of collagen deposition using Masson Trichrome Staining. Red staining indicated viable myocardium while blue staining indicated fibrosis due to infarction damage (Figure 1). We observed that the collagen fibers were disorganized in early MI, dominated by non-fibrillar collagen deposition, while in late MI, cross-linking matrix formed mature scar. Collagen deposition was found to be significantly increased at day 14 and 28 post-MI compared to day 7 (Figure 1). After MI, the mice also displayed cardiac dysfunction. The echocardiographic data from surviving mice after MI are shown in Table 1. LV contractile function (EF, FS) were significantly attenuated following MI. Interestingly, MI for 14, 28 days increased LV dimension and function as shown from LVIDd, LVISd, EF and FS compared with the day 7 group (all P<0.05). These results demonstrate that MI causes an increase in collagen deposition, as well as cardiac dysfunction. During the progression of cardiac fibrosis at day 14 and 28, increased collagen deposition and replacement fibrosis improved cardiac function.
Figure 1.

Myocardial fibrosis was determined by qualitative evaluation of collagen deposition using Masson Trichrome Staining in hearts from male C57BL/6 mice post-surgery groups and sham operated group (A). Red staining indicated viable myocardium, while blue staining indicated fibrosis. Original magnification: 400×. Percent of myocardial fibrosis in mice following myocardial infarction (B), *, P<0.05.
Table 1. Echocardiography in surviving male C57BL/6 mice after MI. LV contractile function (EF, FS) were significantly attenuated following MI. Interestingly, at 14 and 28 days after MI, there is increased LV dimension and function as shown from LVIDd, LVISd, EF and FS compared with day 7 group.
| Variable | Sham | Day 7 | Day 14 | Day 28 |
|---|---|---|---|---|
| IVSd (mm) | 0.82±0.08 (n=6) | 0.58±0.07 (n=10) | 0.62±0.16 (n=8) | 0.52±0.08 (n=6) |
| LVPWd (mm) | 0.90±0.10 (n=6) | 1.10±0.12 (n=10) | 1.00±0.12 (n=8) | 0.90±0.20 (n=6) |
| LVIDs (mm) | 1.90±0.22 (n=6) | 3.96±0.30 (n=10)* | 3.72±0.37 (n=8)# | 3.78±0.33 (n=6)# |
| LVIDd (mm) | 3.20±0.32 (n=6) | 4.88±0.38 (n=10)* | 4.68±0.30 (n=8)# | 4.68±0.32 (n=6)# |
| EF (%) | 77.98±1.40 (n=6) | 44.37±1.97 (n=10)* | 47.93±2.31 (n=8)# | 46.12±2.55 (n=6)# |
| FS (%) | 40.62±1.29 (n=6) | 18.63±0.92 (n=10)* | 20.46±1.17 (n=8)# | 19.53±1.83 (n=6)# |
LVIDd, Left Ventricular Internal Diastolic diameter; LVISd, Left Ventricular Internal Systolic diameter; EF, Ejection Fraction; FS, Fractional Shortening. (*: vs. Sham, P<0.05; #: vs. day 7, P<0.05).
Down regulation of caveolin-1 and α-SMA expression increased after MI
To evaluate the role of caveolin-1 in cardiac fibrosis, we measured caveolin-1 expression after MI in mouse models. We investigated the expression of caveolin-1 by Western blot in whole hearts. We have found that caveolin-1 levels in the infarct area were decreased post-MI. Compared to the 7 days post-MI, the protein expression of caveolin-1 at day 14 and 28 after MI were increased (Figure 2A). Western blotting assays confirmed a significant decrease in caveolin-1 protein at 7 days post MI (Figure 2B,C). And α-SMA was used as a marker of myofibroblasts, which indicated cardiac fibroblast phenotypic switch. The protein expression of α-SMA post-MI was increased compared to the sham operated control group. Furthermore, the expression of α-SMA protein at 7 days post-MI was greater than at 14 and 28 days post-MI (Figure 2D). Taken together, after myocardial infraction, both caveolin-1 and α-SMA protein expression peaks at 7 days post-MI and all these changes appeared in the infarct zone. This result indicates that caveolin-1 may inhibit cardiac fibroblasts from transforming to myofibroblasts.
Figure 2.

Immunohistochemical staining and western blotting of caveolin-1 (A-C) and α-SMA (D) expression in hearts from male C57BL/6 mice in the sham operated group and at 7, 14 and 28 days after MI. (A) Immunohistochemical staining of caveolin-1 expression; (B,C) western blotting of caveolin-1 expression, *, P<0.05; (D) immunohistochemical staining of α-SMA. Original magnification: 400×.
PTEN protein expression decreased in cardiac fibrosis following MI
We also demonstrated by immunohistochemical staining that membrane PTEN levels were decreased post-MI. Compared to 7 days post-MI, the protein expression of membrane PTEN at 14 days after MI was decreased (Figure 3). Previous studies have found that membrane PTEN activity inhibits the integrin/PI3K/Akt signal pathway. Therefore, reduced PTEN expression enhances the PI3K/Akt signal pathway to promote myocardial fibrosis.
Figure 3.

Immunohistochemical staining PTEN expression in hearts from male C57BL/6 mice at 7 and 14 days post-MI groups. PTEN protein expression was decreased at 14 days post-MI compared to 7 days post-MI. Original magnification: 400×. PTEN, phosphatase and tensin homolog.
Hypoxia promotes cardiac fibroblasts proliferation and increases α-SMA expression
To explore whether cardiac fibroblasts displayed proliferation under hypoxia, we used the MTT assay, a colorimetric determination of cell viability after hypoxia in vitro. Cell viability was increased at 12, 24, and 48 hours after hypoxia compared with the untreated group, and peaked at 24 hours after hypoxia (Figure 4). The data demonstrated that hypoxia could promote cardiac fibroblasts proliferation. We also observed that α-SMA, which is the marker of myofibroblasts, was gradually increased with increased hypoxia treatment time and was significantly increased at 24 and 48 hours P=0.0017 (Figure 5). Overall, these results indicate that under hypoxia stimulus, fibroblasts will proliferate and switch their phenotype to myofibroblasts (α-SMA positive cells).
Figure 4.

MTT cell proliferation assay of cardiac fibroblasts under hypoxia 12, 24 and 48 hours. *, P<0.05. Data are representative of three separate experiments.
Figure 5.
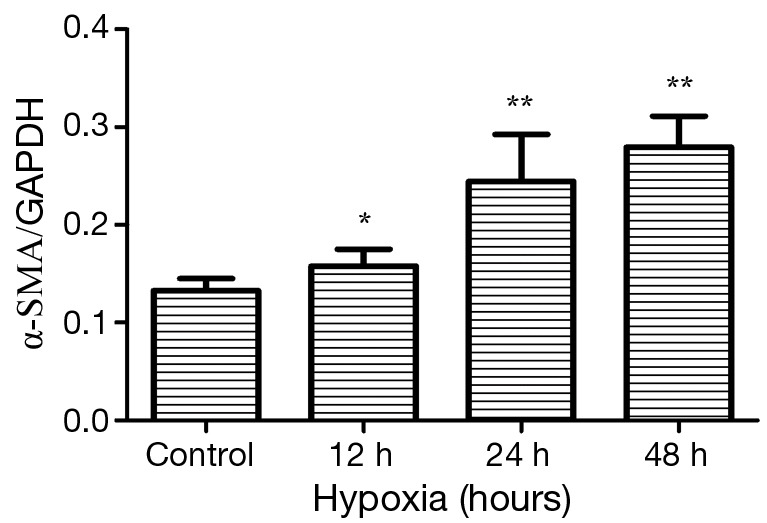
Expression of α-smooth muscle actin (α-SMA) in cardiac fibroblasts under normoxia and hypoxia conditions. Cardiac fibroblasts of hypoxia groups were exposed to GENbag anaer for 12, 24, and 48 hours. Hypoxia versus normoxia, *, P<0.05, **, P<0.005. Data are representative of three separate experiments.
Caveolin-1 and PTEN protein expression was reduced in fibroblasts under hypoxia in vitro
Pathological cardiac fibrosis can develop from a number of stimuli, including ischemia, inflammation, pressure overload and volume overload (39,40). A common feature of all these stimuli is tissue hypoxia, either directly or indirectly, because of increases in oxygen consumption by infiltrating inflammatory cells and activated resident cells. Prolonged local tissue hypoxia can lead to aberrant ventricular remodeling and cardiac fibrosis (40-43). In vivo experiments, we have verified that both caveolin-1 and PTEN protein expression are decreased in the infarct zone post-MI. To further investigate that result, we performed hypoxia experiments in vitro. We analyzed caveolin-1, PTEN, and p-AKT expression in myofibroblatsts derived from cardiac fibroblasts after culturing in hypoxia or normoxia conditions by western analysis. The protein expression of caveolin-1 and PTEN were gradually decreased and the expression of p-Akt was significantly increased after 12, 24 and 48 hours in hypoxia conditions (Figure 6).
Figure 6.
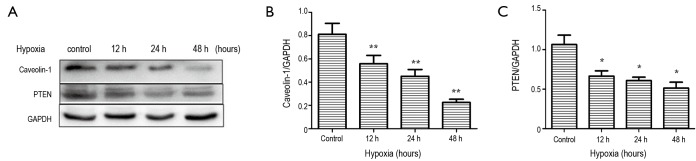
Expression of caveolin-1 and PTEN in cardiac fibroblasts of normoxia and hypoxia groups. Cardiac fibroblasts of hypoxia groups were exposed to GENbag anaer for 12, 24 and 48 hours. Hypoxia versus normoxia, *P<0.05, **P<0.005. (A) Western blotting; (B) expression of Caveolin-1, P=0.0019<0.005; (C) expression of PTEN, P=0.006<0.05. Data are representative of three separate experiments. PTEN, phosphatase and tensin homolog.
Disruption of caveolae with methyl-β-cyclodextrin (MβCD) enhanced p-Akt and α-SMA expression and fibroblast proliferation
MβCD depletes cholesterol and disrupts caveolae (44).
To further study that the mechanism of caveolae on caveolin-1/PTEN mediated signal pathway, we incubated cardiac fibroblasts with a 2 mM/L dose of MβCD for 2 hours before treatment with GENbag anaer. The p-Akt and α-SMA expression and cell viability increased after exposure to MβCD compared to cells not treated with MβCD after exposure to the same hypoxia conditions (Figure 7). We also showed that expression of caveolin-1 was higher in the MβCD treated groups. Therefore the caveolin-1/PTEN mediated signal pathway was dependent on caveolae formation. The MTT assay also demonstrated that cardiac fibroblasts displayed significant proliferation after exposure to hypoxia for 12, 24 and 48 hours in the presence of MβCD than in the untreated group (Figure 8). Over all, disruption of caveolae with MβCD leads to increased cardiac fibroblasts proliferation and conversion to myofibroblasts.
Figure 7.

Expression of Caveolin-1, PTEN, α-SMA and p-Akt in cardiac fibroblasts in the presence and absence of MβCD (2 Mm/L 2 hours) and under hypoxic conditions. Cardiac fibroblasts of hypoxia groups were incubated in 2 Mm/L doses of MβCD 2 hours before exposure to GENbag anaer for 12, 24 and 48 hours. *, P<0.05; **, P<0.005. (A) Western blotting; (B) expression of Caveolin-1; (C) expression of PTEN; (D) expression of α-SMA; (E) expression of p-Akt. Data are representative of three separate experiments. PTEN, phosphatase and tensin homolog; α-SMA, α-smooth muscle actin.
Figure 8.
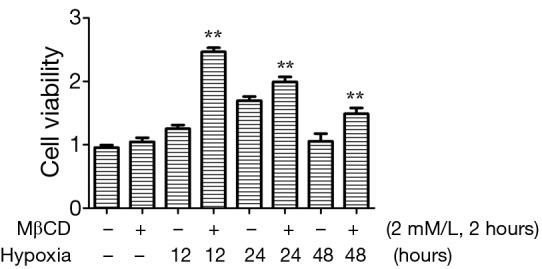
MTT cell proliferation assay of cardiac fibroblasts in absence or presence of MβCD after 12, 24 and 48 hours in hypoxic conditions. **, P<0.001. Data are representative of three separate experiments.
Discussion
Ischemia caused by coronary artery disease and MI leads to aberrant ventricular remodeling and cardiac fibrosis (40). Cardiac fibroblasts play a pivotal role in the development of cardiac fibrosis. We showed that ischemic injury induced phenotypic differentiation of cardiac fibroblasts into myofibroblasts which secreted more cardiac extracellular matrix (41,45-47). We also found that caveolin-1 protein expression was transiently down-regulated after MI, concomitant with inhibition of PTEN expression and therefore activation of the Integrin/PI3K/Akt signal pathway in cardiac fibroblasts. In vivo experiments found that hypoxia induced proliferation and phenotypic differentiation of cardiac fibroblasts. Furthermore, caveolin-1 and PTEN protein expression in cardiac fibroblast were gradually deceased under hypoxia conditions, accompanied by increased α-SMA expression. Interestingly, disruption of caveolae with MβCD also enhanced PI3K/Akt signal pathway and caused increased cardiac fibroblast proliferation and phenotypic differentiation. These results suggest that ischemic injury leads to enhanced fibrosis of the heart and caveolae play a key role in cardiac fibroblast proliferation and phenotypic switch under hypoxia, possibly mediated by the caveolin-1/PTEN signaling pathway to activate PI3K/Akt expression.
Following acute MI, cardiac fibroblasts in the heart become activated and rapidly proliferate (48). Our study showed that collagen deposition by Masson trichrome staining and myofibroblasts by immmuhistochemical staining gradually increased after permanent ligation of LAD at 7, 14 and 28 days after MI. MI injury induces cardiac fibroblasts to undergo a phenotypic switch to myofibroblasts which are central players in the profibrotic post-MI repair process (7,16,35). A trend toward increased α-SMA expression indicative of the transformation of fibroblasts to myofibroblasts was observed in the present study in infarcted regions, and is consistent with previous reports (9). We also found that systolic heart function at later stages after MI was improved than at earlier timepoints following MI. This result indicates that local collagen deposition may be beneficial for improvement of systolic cardiac function during heart remodeling.
Considerable data indicate that PTEN colocalizes with caveolin-1 at the plasma membrane as an Integrin/PTEN/caveolin-1 complex (15). We also found that caveolin-1 and PTEN expression in cardiac fibroblasts is significantly reduced during myofibroblast proliferation and collagen deposition in the formation of a mature scar after MI. Meanwhile in vitro, ischemic injury induced cardiac fibroblast differentiation and proliferation, and exhibited a similar decrease in caveolin-1 and PTEN protein levels. Here we provide several evidences that caveolae and the caveolin-1/PTEN signaling pathway are involved in the progression of cardiac fibrosis resulting from ischemic injury. First, low caveolin-1 expression in cardiac fibroblast peaked concurrently with augmented α-SMA levels in mouse models after MI. Immunohistochemical staining results indicates that myofibroblasts are a key component of the infarct zone and display low caveolin-1 expression at 7 days after MI, while caveolin-1 expression gradually returns to almost normal levels by 28 days post-MI, accompanied with lower α-SMA levels. Recent studies indicate that the extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium (49). Therefore, with the progress of collagen deposition, increased caveolin-1 expression results in inhibition of myofibroblast proliferation. Secondly, both in vivo and in vitro experiments revealed a decline in caveolin-1 and PTEN protein levels after hypoxia and leads to increases in cardiac fibroblast proliferation. Lastly, in vitro disruption of caveolae with MβCD enhanced cardiac fibroblast proliferation and phenotypic differentiation in hypoxia conditions. This result demonstrates that signal pathways such as PTEN/ PI3K/Akt and caveolin-1, which control cardiac fibroblast proliferation and phenotypic differentiation, are dependent on caveolae formation.
Numerous studies have revealed that caveolae, highly enriched in cholesterol and sphingolipids, play a pivotal role in regulating cell signaling (6,7). Membrane rafts and caveolae concentrate membrane proteins and other components involved in transport and signal transduction (10,11). Caveolins are the main structural and functional components of caveolae, cholesterol homeostasis, and cell signaling, and caveolin-1 is an essential constituent of adipocyte caveolae (50). Other studies have been confirmed that cholesterol can be depleted by MβCD treatment and therefore disrupt caveolae formation (51). Here we demonstrated that caveolin-1 and PTEN protein expression increased in cardiac fibroblasts groups exposed to MβCD compared to untreated controls. In addition, the protein levels of α-SMA and p-Akt both increased in cardiac fibroblasts treated with MβCD compared to the untreated groups under hypoxic conditions. Meanwhile, the MTT assay demonstrated that cardiac fibroblasts displayed greater proliferation after exposure to hypoxia for 12, 24 and 48 hours in the presence of MβCD (2 mM and 2 hours). Taken together, cholesterol depletion by MβCD induced disruption of caveolae up-regulated the activation of the PI3K/Akt pathway, thereby causing increased α-SMA and p-Akt protein expression and cell proliferation. We also have unexpectedly found that caveolin-1 and PTEN expression of fibroblasts in MβCD group was greater than in the untreated group. Recent studies suggests that down-regulation of caveolin-1 may enhance increased atrial fibrosis in atrial fibrillation patients (52). Therefore, cardiac fibroblasts proliferation and phenotypic differentiation may be resulting from disruption of the caveolin-1/PTEN complex, while increases in caveolin-1 and PTEN levels may be a feedback effect of enhanced fibrosis. Further studies will be needed to further explore how caveolin-1 and PTEN pathways modulate fibrosis from ischemic injury.
Acknowledgements
The present study was supported by Grants from Six Big Talent Peak of Jiangsu Province.
Disclosure: The authors declare no conflict of interest.
References
- 1.Lavine KJ, Kovacs A, Weinheimer C, et al. Repetitive myocardial ischemia promotes coronary growth in the adult mammalian heart. J Am Heart Assoc 2013;2:e000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayorga M, Bahi N, Ballester M, et al. Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J Biol Chem 2004;279:34882-9. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Yuan T, Du G, et al. The impact of 1,25-dihydroxyvitamin D3 on the expression of connective tissue growth factor and transforming growth factor-beta(1) in the myocardium of rats with diabetes. Diabetes Res Clin Pract 2014;104:226-33. [DOI] [PubMed] [Google Scholar]
- 4.Dean RG, Balding LC, Candido R, et al. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem 2005;53:1245-56. [DOI] [PubMed] [Google Scholar]
- 5.Gaspard GJ, MacLean J, Rioux D, et al. A novel beta-adrenergic response element regulates both basal and agonist-induced expression of cyclin-dependent kinase 1 gene in cardiac fibroblasts. Am J Physiol Cell Physiol 2014;306:C540-50. [DOI] [PubMed] [Google Scholar]
- 6.Ogata T, Naito D, Nakanishi N, et al. MURC/Cavin-4 facilitates recruitment of ERK to caveolae and concentric cardiac hypertrophy induced by alpha1-adrenergic receptors. Proc Natl Acad Sci U S A 2014;111:3811-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol 2007;8:185-94. [DOI] [PubMed] [Google Scholar]
- 8.Hansen CG, Nichols BJ. Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol 2010;20:177-86. [DOI] [PubMed] [Google Scholar]
- 9.Briand N, Dugail I, Le Lay S.Cavin proteins: New players in the caveolae field. Biochimie 2011;93:71-7. [DOI] [PubMed] [Google Scholar]
- 10.Glenney JR, Jr, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc Natl Acad Sci U S A 1992;89:10517-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothberg KG, Heuser JE, Donzell WC, et al. Caveolin, a protein component of caveolae membrane coats. Cell 1992;68:673-82. [DOI] [PubMed] [Google Scholar]
- 12.Scherer PE, Okamoto T, Chun M, et al. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U S A 1996;93:131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Z, Scherer PE, Okamoto T, et al. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem 1996;271:2255-61. [DOI] [PubMed] [Google Scholar]
- 14.Jasmin JF, Rengo G, Lymperopoulos A, et al. Caveolin-1 deficiency exacerbates cardiac dysfunction and reduces survival in mice with myocardial infarction. Am J Physiol Heart Circ Physiol 2011;300:H1274-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia H, Khalil W, Kahm J, et al. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol 2010;176:2626-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi F, Sottile J.Caveolin-1-dependent beta 1 integrin endocytosis is a critical regulator of fibronectin turnover. J Cell Sci 2008;121:2360-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Ren CH, Tahir SA, et al. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol 2003;23:9389-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherer PE, Lewis RY, Volonte D, et al. Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem 1997;272:29337-46. [DOI] [PubMed] [Google Scholar]
- 19.Song KS, Li Shengwen, Okamoto T, et al. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem 1996;271:9690-7. [DOI] [PubMed] [Google Scholar]
- 20.Jasmin JF, Malhotra S, Singh Dhallu M, et al. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res 2007;100:721-9. [DOI] [PubMed] [Google Scholar]
- 21.Mahmoudi M, Willgoss D, Cuttle L, et al. In vivo and in vitro models demonstrate a role for caveolin-1 in the pathogenesis of ischaemic acute renal failure. J Pathol 2003;200:396-405. [DOI] [PubMed] [Google Scholar]
- 22.Ratajczak P, Damy T, Heymes C, et al. Caveolin-1 and -3 dissociations from caveolae to cytosol in the heart during aging and after myocardial infarction in rat. Cardiovasc Res 2003;57:358-69. [DOI] [PubMed] [Google Scholar]
- 23.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci 2001;114:2375-82. [DOI] [PubMed] [Google Scholar]
- 24.Georgescu MM, Kirsch KH, Akagi T, et al. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A 1999;96:10182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stambolic V, Tsao MS, Macpherson D, et al. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res 2000;60:3605-11. [PubMed] [Google Scholar]
- 26.Lee JO, Yang H, Georgescu MM, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999;99:323-34. [DOI] [PubMed] [Google Scholar]
- 27.Tamura M, Gu J, Danen EH, et al. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 1999;274:20693-703. [DOI] [PubMed] [Google Scholar]
- 28.Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998;95:29-39. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y, Yu Q, Liu JH, et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem 2003;278:40057-66. [DOI] [PubMed] [Google Scholar]
- 30.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res 1997;57:2124-9. [PubMed] [Google Scholar]
- 31.Denley A, Gymnopoulos M, Kang S, et al. Requirement of phosphatidylinositol(3,4,5)trisphosphate in phosphatidylinositol 3-kinase-induced oncogenic transformation. Mol Cancer Res 2009;7:1132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degtyarev M, De Mazière A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 2008;183:101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Couet J, Li S, Okamoto T, et al. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 1997;272:6525-33. [DOI] [PubMed] [Google Scholar]
- 34.Wu W, Hu Y, Li J, et al. Silencing of Pellino1 improves postinfarct cardiac dysfunction and attenuates left ventricular remodeling in mice. Cardiovasc Res 2014;102:46-55. [DOI] [PubMed] [Google Scholar]
- 35.Savin S, Cvejic D, Isic T, et al. Thyroid peroxidase and galectin-3 immunostaining in differentiated thyroid carcinoma with clinicopathologic correlation. Hum Pathol 2008;39:1656-63. [DOI] [PubMed] [Google Scholar]
- 36.Janković J, Paskaš S, Marečko I, et al. Caveolin-1 expression in thyroid neoplasia spectrum: comparison of two commercial antibodies. Dis Markers 2012;33:321-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crisostomo PR, Wang Y, Markel TA, et al. Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B- but not JNK-dependent mechanism. Am J Physiol Cell Physiol 2008;294:C675-82. [DOI] [PubMed] [Google Scholar]
- 38.Ryhänen T, Mannermaa E, Oksala N, et al. Radicicol but not geldanamycin evokes oxidative stress response and efflux protein inhibition in ARPE-19 human retinal pigment epithelial cells. Eur J Pharmacol 2008;584:229-36. [DOI] [PubMed] [Google Scholar]
- 39.Swynghedauw B.Molecular mechanisms of myocardial remodeling. Physiol Rev 1999;79:215-62. [DOI] [PubMed] [Google Scholar]
- 40.Watson CJ, Collier P, Tea I, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet 2014;23:2176-88. [DOI] [PubMed] [Google Scholar]
- 41.van den Borne SW, Diez J, Blankesteijn WM, et al. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 2010;7:30-7. [DOI] [PubMed] [Google Scholar]
- 42.Barallobre-Barreiro J, Didangelos A, Schoendube FA, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 2012;125:789-802. [DOI] [PubMed] [Google Scholar]
- 43.Opie LH, Commerford PJ, Gersh BJ, et al. Controversies in ventricular remodelling. Lancet 2006;367:356-67. [DOI] [PubMed] [Google Scholar]
- 44.Kim S, Han J, Lee DH, et al. Cholesterol, a Major Component of Caveolae, Down-regulates Matrix Metalloproteinase-1 Expression through ERK/JNK Pathway in Cultured Human Dermal Fibroblasts. Ann Dermatol 2010;22:379-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porter KE, Turner NA. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacology & therapeutics 2009;123:255-78. [DOI] [PubMed] [Google Scholar]
- 46.Turner NA, Porter KE. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sedgwick B, Riches K, Bageghni SA, et al. Investigating inherent functional differences between human cardiac fibroblasts cultured from nondiabetic and Type 2 diabetic donors. Cardiovasc Pathol 2014;23:204-10. [DOI] [PubMed] [Google Scholar]
- 48.Deb A, Ubil E.Cardiac fibroblast in development and wound healing. J Mol Cell Cardiol 2014;70:47-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobaczewski M, de Haan JJ, Frangogiannis NG. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J Cardiovasc Transl Res 2012;5:837-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boscher C, Nabi IR. CAVEOLIN-1: Role in Cell Signaling. Adv Exp Med Biol 2012;729:29-50. [DOI] [PubMed] [Google Scholar]
- 51.Schönfelder U, Radestock A, Elsner P, et al. Cyclodextrin-induced apoptosis in human keratinocytes is caspase-8 dependent and accompanied by mitochondrial cytochrome c release. Exp Dermatol 2006;15:883-90. [DOI] [PubMed] [Google Scholar]
- 52.Yi SL, Liu XJ, Zhong JQ, et al. Role of caveolin-1 in atrial fibrillation as an anti-fibrotic signaling molecule in human atrial fibroblasts. PLoS One 2014;9:e85144. [DOI] [PMC free article] [PubMed] [Google Scholar]