

Alcid et al. developed a genetic screen using reconstituted RNAi in S. cerevisiae and systematically identified a large number of putative long noncoding RNA (lncRNA) repressors. They found that four highly conserved chromatin remodeling factors are global lncRNA repressors and identified antisense lncRNAs (CRRATs [chromatin remodeling-repressed antisense transcripts]) whose repression by these chromatin remodeling factors is required for the maintenance of normal levels of overlapping mRNA transcripts.
Keywords: chromatin, chromatin remodeling, lncRNAs, antisense RNAs
Abstract
Long noncoding RNAs (lncRNAs) are pervasively transcribed across eukaryotic genomes, but functions of only a very small subset of them have been demonstrated. This has led to active debate about whether many of them have any biological functions. In addition, very few regulators of lncRNAs have been identified. We developed a novel genetic screen using reconstituted RNAi in Saccharomyces cerevisiae and systematically identified a large number of putative lncRNA repressors. Among them, we found that four highly conserved chromatin remodeling factors are global lncRNA repressors that play major roles in shaping the eukaryotic lncRNA transcriptome. Importantly, we identified >250 antisense lncRNAs (CRRATs [chromatin remodeling-repressed antisense transcripts]) whose repression by these chromatin remodeling factors is required for the maintenance of normal levels of overlapping mRNA transcripts. Our results strongly suggest that regulation of mRNA through repression of antisense lncRNAs is far more broadly used than previously appreciated.
Recent advancement in genome-wide detection of RNA resulted in the unexpected finding that long noncoding RNAs (lncRNAs) are pervasively transcribed across the eukaryotic genomes (Djebali et al. 2012). It has been estimated that ∼75% of the human genome is transcribed (Djebali et al. 2012), despite the fact that ∼1.2% of the genome encodes protein-coding exons (Venter et al. 2001). Similarly, 75%–85% of the genome of the budding yeast Saccharomyces cerevisiae is estimated to be transcribed (Nagalakshmi et al. 2008). Although lncRNAs are generally transcribed at lower levels than mRNAs, these new findings showed that lncRNAs exceed mRNAs in terms of the number of transcription units on higher eukaryotic genomes. Some of these lncRNAs have critical regulatory roles. For example, Xist RNA is essential for mammalian X-chromosome inactivation, and its expression is precisely regulated by other lncRNAs transcribed from neighboring regions (Lee and Bartolomei 2013). HOTAIR is expressed from the human HOXC locus and regulates HOXD gene expression in trans by recruiting histone-modifying enzymes PRC2 and CoRest (Rinn et al. 2007; Gupta et al. 2010; Tsai et al. 2010). Indeed, an increasing number of lncRNAs have been shown to play critical roles in transcriptional regulation, cell type specification, and human diseases (Batista and Chang 2013; Flynn and Chang 2014). In budding yeast, transcription of several lncRNAs affects the abundance of the overlapping mRNA transcripts (Hongay et al. 2006; Camblong et al. 2007, 2009; Houseley et al. 2008; Castelnuovo et al. 2013). Therefore, lncRNA products or the act of lncRNA transcription itself can play regulatory roles, both of which are called “functional lncRNA transcription” here for simplicity. However, given the large number of lncRNAs transcribed from eukaryotic genomes, the functions of the vast majority of lncRNA transcription events remain unknown. This led to an active debate as to whether most instances of lncRNA transcription lack any functional roles (Louro et al. 2009; Doolittle 2013). However, it was recently shown in budding yeast (Rhee and Pugh 2012) and humans (Venters and Pugh 2013) that a large fraction of lncRNA transcription initiates from the sites where preinitiation complexes (PICs) are formed only for lncRNAs. This argues against the possibility that most lncRNAs are the unintended byproducts of stochastic initiation from mRNA start sites. Given that multiple basal transcription factors need to be targeted and assembled in an ordered fashion to form functional PICs, the fact that many PICs are dedicated for lncRNA transcripts is consistent with the possibility that eukaryotic cells intend to transcribe many of them. If this is the case, it is important to develop methods to systematically identify or enrich for lncRNAs or lncRNA transcription events that play important biological roles.
If functional lncRNA transcription is prevalent, another significant challenge is to elucidate regulatory mechanisms of lncRNA transcription. Although transcription factors and chromatin regulators that control mRNA transcription have been extensively studied, much less is known about regulators of lncRNA transcription. Our laboratory has previously found that a highly conserved ATP-dependent chromatin remodeling factor, Isw2, functions at the 3′ end of genes to repress antisense lncRNA (ASlncRNA) transcription in budding yeast (Whitehouse et al. 2007; Yadon et al. 2010). Recent genetic screens in yeast using reporter genes have identified histone deacetylases and histone chaperones as repressors of lncRNAs (Cheung et al. 2008; Marquardt et al. 2014). However, these factors affect only a small fraction of lncRNAs, and additional efforts are needed to identify regulators of lncRNA transcription.
In this study, we demonstrate that reconstitution of functional RNAi in S. cerevisiae alone does not cause strong growth defects but results in reduction of the abundance of mRNAs that have high levels of overlapping ASlncRNAs. We further found that global elevation of ASlncRNA levels in the presence of reconstituted RNAi in S. cerevisiae causes growth defects, likely due to destabilization of a large number of mRNAs and ASlncRNAs that overlap. Taking advantage of this finding, we systematically identified putative repressors of ASlncRNAs, including ATP-dependent chromatin remodeling factors, histone-modifying enzymes, the mediator complex subunits, and transcription factors. We demonstrate that the four ATP-dependent chromatin remodeling factors function as global ASlncRNA repressors and provide evidence that they play major roles in shaping the eukaryotic lncRNA transcriptome under a physiologically relevant condition (in the absence of RNAi). We further discovered that chromatin remodeling factors are targeted to initiation sites of a large fraction of these ASlncRNA and subsequently alter chromatin structure. Finally, we identified >250 AslncRNAs whose repression by chromatin remodeling factors is required for the maintenance of the normal level of overlapping mRNAs.
Results
Genetic interactions between RNAi and mutations that elevate lncRNA levels
It has been shown in both budding yeast (Neil et al. 2009; Xu et al. 2009) and mammalian cells (Core et al. 2008) that lncRNAs frequently initiate from bidirectional promoters, where mRNA and lncRNA initiate in opposite directions. Because lncRNAs are highly prevalent in both mammals (Djebali et al. 2012) and budding yeast (Xu et al. 2009), this means that, at gene-dense regions of mammalian genomes (Teif et al. 2012) and at many loci in yeast, lncRNAs often overlap with protein-coding genes in the antisense direction (Fig. 1A). This led us to hypothesize that global elevation of ASlncRNA levels in the presence of RNAi would cause significant growth defects due to destabilization of a large number of mRNAs and lncRNAs that are processed by RNAi (Fig. 1A).
Figure 1.

Genetic interactions between RNAi and mutations that elevate ASlncRNA levels. (A) The basis for the genetic interactions. If only the sense strand is transcribed (i) or even if both sense and antisense strands are transcribed in the absence of RNAi (ii), cells are fine, as the dsRNAs are not processed. In contrast, if the levels of ASlncRNAs are globally elevated in the presence of RNAi (iii), both mRNAs and ASlncRNAs are destabilized due to processing of dsRNAs by RNAi, causing growth defects. (B) Scatter plot of mRNA levels in wild-type and RNAi+ strains. mRNAs with high endogenous antisense levels (top 5%) are marked by violet dots. (C) An example of genetic interactions between RNAi and a mutation that stabilizes lncRNAs (rrp6). A fivefold dilution of saturated culture was spotted on YEPD and incubated at 37°C. The genotypes of the spotted strains are shown at the left. (D) The result of the proof of concept genetic screen.
To test our hypothesis, we reconstituted RNAi in S. cerevisiae by expressing Argonaute (AGO1) and Dicer (DCR1) genes of Saccharomyces castellii (Drinnenberg et al. 2009, 2011). This caused a dramatic decrease in the abundance of Ty1 retrotransposon mRNA, as reported (Supplemental Fig. S1A; Drinnenberg et al. 2009), showing that functional RNAi was reconstituted in our hands. Extensive phenotypic characterization by the Bartel laboratory (Drinnenberg et al. 2011) and our laboratory (data not shown) revealed that RNAi reconstitution in S. cerevisiae does not cause any detectable phenotypes other than a slight sensitivity to high temperature as well as lithium sensitivity, demonstrating that RNAi reconstitution alone does not significantly affect normal cell physiology. To test whether RNAi reconstitution leads to processing of endogenous dsRNA, we performed strand-specific RNA deep sequencing (ssRNA-seq) analysis of the S. cerevisiae strain with functional RNAi (hereafter RNAi+ strain) (Fig. 1B). Consistent with the modest phenotype, the RNA profiles of the RNAi+ and wild-type strains were, overall, quite similar (Spearman’s ρ = 0.986). Exceptions to this general trend are the genes with the highest levels of endogenous ASlncRNA (Fig. 1B, top fifth percentile shown, in violet), which exhibited a significant decrease in the mRNA abundance. This result demonstrates that the reconstituted RNAi can indeed process dsRNAs of the endogenous genes. We then crossed the RNAi+ strain with mutants that are known to globally stabilize lncRNAs—xrn1 (van Dijk et al. 2011), rrp6 (Davis and Ares 2006; Neil et al. 2009; Xu et al. 2009), and trf4 (Wyers et al. 2005)—as well as rpd3, which derepresses lncRNAs (Carrozza et al. 2005; Eden et al. 2009; Lickwar et al. 2009; Churchman and Weissman 2011). We also tested a histone variant mutant, htz1, which causes a global increase in lncRNAs due to failed transcriptional termination in Schizosaccharomyces pombe (Zofall et al. 2009). We observed strong synthetic growth defects between all of these mutations and the RNAi+ background (Fig. 1C,D). These results collectively show that global elevation of lncRNA levels can cause strong growth defects specifically in the presence of functional RNAi. Importantly, our results revealed that this synthetic RNAi system could be used as a tool to systematically identify genes that either repress or degrade ASlncRNAs.
RNAi as a tool for systematic identification of lncRNA repressors
To systematically identify repressors of ASlncRNAs, we conducted a genetic screen to identify genes whose mutations cause synthetic growth defects in the RNAi+ background. To this end, we performed synthetic genetic array (SGA) analysis (Tong et al. 2001) at three different conditions—at 25°C, at 37°C, and in the presence of 100 mM LiCl—because growth defects in the latter two conditions were common in the phenotype tests shown in Figure 1, C and D. Elevated lithium inhibits the activity of Xrn1 and stabilizes lncRNAs (van Dijk et al. 2011). Enriched (P < 0.001) gene ontology (GO) terms among the putative gene hits include “RNA processing and binding,” “ncRNA metabolic process,” and “ncRNA processing” (Fig. 2A; data not shown). These genes are likely involved in destabilization of ASlncRNAs. We also found a large number of genes involved in regulation of mRNA transcription, including many transcription repressors as well as subunits of the mediator complex. Several genes involved in histone modification were also among the hits, including SDS3, a subunit of the histone deacetylase complex Rpd3L, which has been shown to repress lncRNAs (Lickwar et al. 2009). Given the role of transcription termination factor Nrd1 complex in the attenuation of ASlncRNAs genome-wide (Schulz et al. 2013), it was initially surprising to not identify any genes involved in termination. However, all subunits of the Nrd1 complex are essential for cell viability and are not represented in the yeast deletion mutant library. Furthermore, we found eight gene-encoding subunits of four different ATP-dependent chromatin remodeling complexes—Isw2 (ITC1), Swr1 (SWR1, ARP6, and YAF9), Ino80 (IES2), and Rsc (RSC1, RSC2, and HTL1)—that are important for normal growth in the RNAi+ background (Fig. 2A). We confirmed genetic interactions between RNAi and mutants for these remodeling factors by creating mutations and performing genetic crosses in an independent genetic background (examples shown in Fig. 2B,C). Deletion of ITC1 and SWR1 genes is expected to completely abolish functions of the Isw2 and Swr1 complexes, respectively. The null mutations of the ATPase subunits for the Rsc and Ino80 complexes, sth1 and ino80, respectively, cause cell death and extreme growth defects and thus are not represented in the deletion mutant library. For the Rsc and Ino80 complexes, we created tetracycline-repressive alleles of the ATPase subunits STH1 and INO80 (tet-STH1 and tet-INO80), respectively, and confirmed strong genetic interactions of these alleles with RNAi (Fig. 2D,E; Supplemental Fig. S2). These alleles were used for the following analyses. It should be noted that, although RNAi is a highly useful tool to identify putative lncRNA repressors, it is not normally present in S. cerevisiae. Therefore, all subsequent analyses were done in the absence of RNAi.
Figure 2.

The results of the SGA screen using RNAi. (A) Cytoscape network plot of the genes that interact with RNAi. Nodes represent genes, and black edges represent a synthetic growth defect with RNAi. Nodes are grouped by statistically enriched GO terms. Gray dashed edges connecting genes to nodes at the right depict gene subunits of that chromatin remodeling complex. (B,C) Genetic interactions between RNAi and Δswr1-null (swr1 complex) (B) and Δitc1-null (Isw2 complex) (C) mutants. (D,E) Genetic interactions between RNAi and tet-STH1 (Rsc complex) (D) and tet-INO80 (Ino80 complex) (E).
Four highly conserved ATP-dependent chromatin remodeling complexes repress ASlncRNAs
Of the ATP-dependent chromatin remodeling complexes that we identified, only Isw2 had been shown to repress ASlncRNA transcript levels in S. cerevisiae at select loci (Whitehouse et al. 2007; Yadon et al. 2010). Despite this, genome-wide transcriptomic analysis of lncRNA has not been performed for an Isw2 complex mutant. We therefore investigated whether these four chromatin remodeling factors repress ASlncRNAs by ssRNA-seq (Parkhomchuk et al. 2009; Sultan et al. 2012). Because it is not well established how lncRNAs repressed by chromatin remodeling factors are processed, we did not perform any selection of RNA except for rRNA depletion (see the Materials and Methods). Our analyses of the lncRNA transcriptome in chromatin remodeling factor mutants identified a total of 1799 ASlncRNAs that are repressed, revealing that ATP-dependent chromatin remodeling factors play major roles in shaping the budding yeast ASlncRNA transcriptome (Fig. 3). ncRNAs longer than 200 base pairs (bp) are generally considered as lncRNAs (Rinn et al. 2007). However, we found that ASlncRNAs repressed by chromatin remodeling factors tend to be much longer than 200 bp (Fig. 3E–H; Supplemental Fig. S3). Among the four chromatin remodeling factors, Rsc (n = 545) (Fig. 3C,G) and Ino80 (n = 1155) (Fig. 3D,H) repress the largest number of ASlncRNAs, uncovering a novel function for these complexes. Swr1 has the most modest effects on the ASlncRNA transcriptome (n = 10) (Fig. 3A,E), but its mutation does derepress ASlncRNAs. Isw2 complex has intermediate effects (n = 89) (Fig. 3B,F). Importantly, 620 out of the 1799 ASlncRNAs repressed by chromatin remodeling factors have dedicated PICs around their transcription start sites (TSSs) (Rhee and Pugh 2012), suggesting that they are discrete transcription units rather than cryptic transcripts. Of the 620 PICs assigned to ASlncRNAs, 237 had been previously assigned to CUTs (cryptic unstable transcripts), and 168 had been assigned to SUTs (stable unannotated transcripts), while 152 were designated as “orphans,” and thus the lncRNAs controlled by these PICs have not been previously identified. Together, these results demonstrated that our genetic screen indeed identified ASlncRNS repressors as intended and that four highly conserved ATP-chromatin remodeling factors function as global repressors of ASlncRNAs under physiologically relevant conditions (in the absence of RNAi). Given that all four chromatin remodeling factors tested are ASlncRNA repressors, our results also suggest that many of the other genes that we identified in the screen likely function as ASlncRNAs repressors.
Figure 3.

Chromatin remodeling factors repress ASlncRNAs. (A–D) Representative strand-specific RNA-seq data in Δswr1 (A), Δitc1 (B), tet-STH1 (C), and tet-INO80 (D) mutants. Blue and purple signals denote Watson and Crick strand transcripts, respectively. The direction of the coding gene transcription is shown at the bottom of each panel. (E–H) ASlncRNA meta analyses. The ratio of ASlncRNA levels in in Δswr1 (E), Δitc1 (F), tet-STH1 (G), and tet-INO80 (H) mutants relative to wild-type cell levels is shown in log2 scale. The black lines represent the average signals, and the colored lines represent the RNA signals of the top 25% and bottom 25% changes in the mutants.
ATP-dependent chromatin remodeling factors repress unique sets of ASlncRNAs
The fact that Rsc, Isw2, Ino80, and Swr1 were identified as ASlncRNA repressors is intriguing, as they exhibit distinct biochemical activities. Rsc was previously reported to increase the size of NDRs (nucleosome-depleted regions) at gene promoters (Hartley and Madhani 2009), whereas Isw2 decreases NDR size (Whitehouse et al. 2007; Yadon et al. 2010). On the other hand, Swr1 is required for replacement of canonical histone H2A with the histone variant Htz1 (Kobor et al. 2004), whereas Ino80 is implicated in replacing Htz1 with H2A (Papamichos-Chronakis et al. 2011). We therefore investigated whether these remodeling factors have distinct or overlapping sets of ASlncRNA targets. Systematic comparison of ASlncRNAs repressed by these remodeling factors revealed that there is little overlap among the ASlncRNAs that are repressed (Fig. 4A; Supplemental Fig. S4). This result suggests that there are multiple distinct ways by which chromatin regulation can repress ASlncRNAs and that distinct sets of ASlncRNAs require different types of chromatin regulation for transcriptional repression.
Figure 4.

Chromatin remodeling factors repress unique sets of targets. (A) A heat map representation of the degree of overlap of ASlncRNAs repressed by four ATP-dependent chromatin remodeling factors by percentage. (B–D) Density ternary plots demonstrating a comparison of ASlncRNAs elevated in Isw2 (B), Rsc (C), and Ino80 (D) complexes versus in the exosome (rrp6) and xrn1 mutants. The dotted lines represent the threshold where the remodeling mutant is responsible for at least 50% of the total normalized fold change.
A significant fraction of ncRNAs is degraded by RNA surveillance mechanisms in vivo (Neil et al. 2009; Xu et al. 2009; van Dijk et al. 2011; Schulz et al. 2013), and previous studies identified ncRNAs based on their sensitivity (exosome-sensitive CUTs and Xrn1-sensitive unannotated transcripts [XUTs]) or insensitivity (SUTs) to RNA surveillance mutation. As a result, ncRNAs have been mainly characterized in RNA surveillance mutant backgrounds. To determine whether chromatin remodeling factor-repressed ASlncRNAs have been identified in these studies, we next tested how much overlap there is among ASlncRNAs repressed by chromatin remodeling factors and those that are degraded by the RNA surveillance mechanisms as well as SUTs. Because of the large number of ASlncRNAs derepressed in the mutants, we focused on Isw2, Rsc, and Ino80 complexes in the following studies. There are two predominant nucleases responsible for the degradation of lncRNAs, the exosome and Xrn1. The exosome is a nuclease with 3′–5′ exonuclease activity and is associated with the degradation of ∼1000 lncRNAs throughout the genome (Neil et al. 2009; Xu et al. 2009), and rrp6 mutation stabilizes these transcripts. Likewise, Xrn1 is a 5′–3′ cytoplasmic exonuclease associated with destabilizing a large number of lncRNAs (van Dijk et al. 2011). To visualize the extent to which the level of a particular lncRNA is dictated by chromatin remodeling, the exosome, and Xrn1, we constructed density ternary plots of lncRNAs regulated by each chromatin remodeler using the fold change of each ASlncRNA (see the Material and Methods). For Isw2-repressed lncRNAs, a vast majority of the transcripts is more strongly derepressed by mutation of Isw2 (itc1), suggesting that the levels of these lncRNAs are determined more by Isw2 than by the exosome (rrp6) and/or Xrn1 (xrn1). In contrast, for both Rsc- and Ino80-regulated lncRNAs, large fractions of each population of ASlncRNAs are strongly affected by the exosome (rrp6) and Xrn1 (xrn1), suggesting that these ASlncRNAs are also degraded by the exosome and/or Xrn1. Despite this finding, we also found a significant portion of ASlncRNAs regulated by both Rsc and Ino80, whose abundance is dictated more by chromatin remodeling factors than by the exosome and/or Xrn1 (Fig. 4B–D; Supplemental Fig. S4). In addition, a large fraction of ASlncRNAs that are derepressed in chromatin remodeling factor mutants exhibit a strong decrease in their abundance in rrp6 and/or xrn1 mutants (Fig. 4; Supplemental Fig. S4). We also found that, of the 1799 ASlncRNAs repressed by remodeling factors, 400 overlap with previously identified SUTs (Xu et al. 2009). Finally, we found that a large fraction of the 1799 ASlncRNAs repressed by chromatin remodeling factors are not significantly affected by Nrd1 (Supplemental Fig. S4). These results revealed that we identified a large number of previously unidentified lncRNAs.
Identification of ASlncRNAs directly repressed by chromatin remodeling factors
We next sought to identify ASlncRNAs that are likely directly repressed by chromatin remodeling factors. Our criteria for these RNAs are that the ASlncRNA level significantly increases in a chromatin remodeling factor mutant and that the chromatin remodeling factor is targeted to the TSS of the ASlncRNA. To this end, we analyzed genome-wide chromatin immunoprecipitation (ChIP) data of chromatin remodeling factors (Whitehouse et al. 2007; Yen et al. 2012) and identified ASlncRNAs repressed by remodeling factors whose TSSs are also proximally located to remodeling factor-bound sites (see the Materials and Methods). For Isw2, out of 89 repressed ASlncRNAs, 58 have Isw2 targeting around the ASlncRNS TSSs (Fig. 5A,D). For Rsc, we found that 216 ASlncRNAs out of 545 exhibit Rsc targeting around TSSs (Fig. 5B,E). Finally, for Ino80, we found 540 ASlncRNAs out of 1155 with proximal Ino80 targeting (Fig. 5C,F). Therefore, we found that ∼45% of ASlncRNAs repressed by chromatin remodeling factors have the corresponding remodeling factor specifically targeting TSSs. Given that these transcripts represent unique sets of lncRNAs, we named the ASlncRNAs directly repressed by chromatin remodeling factors transcripts CRRATs (chromatin remodeling-repressed antisense transcripts). Our cis-element search did not identify binding sites of any transcription factors whose binding sites are overrepresented around the TSSs of CRRATs (data not shown). Therefore, the mechanisms by which chromatin remodeling factors are targeted to these sites are currently unknown.
Figure 5.

Targeting and chromatin changes at ASlncRNAs regulated by chromatin remodeling factors. (A) Isw2 targeting and Isw2-dependent chromatin changes at an Isw2-repressed ASlncRNA. RNA-seq reads and nucleosome signals (MNase signals) (Yen et al. 2012) in wild-type and itc1 strains are shown. The orientation of sense and antisense transcripts are shown at the right. Isw2 ChIP signals (Whitehouse et al. 2007) are shown at the bottom. (B) Rsc targeting and Rsc-dependent chromatin changes at a Rsc-repressed ASlncRNA. RNA-seq reads and nucleosome signals (MNase signals) in wild-type and MET-STH1 strains (Van de Vosse et al. 2013) are shown. The orientation of sense and antisense transcripts are shown at the right. Rsc8 ChIP signals (Yen et al. 2012) are shown at the bottom. (C) Ino80 targeting and Ino80-dependent chromatin changes at an Ino80-repressed ASlncRNA. RNA-seq reads and Htz1 signals in wild-type and arp5 strains are shown (Yen et al. 2013). The orientation of sense and antisense transcripts are shown at the right. Ino80 ChIP signals (Yen et al. 2012) are shown at the bottom. (D) Meta analysis of Isw2 ChIP signals around the TSSs (position 0) of direct (n = 58; blue) and indirect (n = 31; orange) Isw2 targets. (E) Meta analysis of Rsc8 ChIP signals around the TSSs (position 0) of direct (n = 216; red) and indirect (n = 329; orange) Rsc targets. (F) Meta analysis of INO80 ChIP signals around the TSSs (position 0) of direct (n = 540; green) and indirect (n = 615; orange) Ino80 targets. (G, top) Ribbon plots of nucleosome signals around the TSSs (position 0) of direct Isw2 targets in the wild type (black) and isw2 mutant (blue). The lines represent the mean nucleosome signal, while the outer borders of the ribbon represent one standard error of the mean away from the mean. (Bottom) Ribbon plots (as above) of nucleosome signals around the TSSs (position 0) of indirect Isw2 targets in wild-type (black) and isw2 (orange) cells. (H, top) Ribbon plots (as in G) of nucleosome signals around the TSSs (position 0) of direct Rsc targets in wild-type (black) and MET-SHT1 (red) cells. (Bottom) Ribbon plots of nucleosome signals around the TSSs (position 0) of indirect Rsc targets in wild-type (black) and MET-SHT1 (orange) cells. (I, top) Htz1 signals around the TSSs (position 0) of direct (green) and indirect (orange) Ino80 targets in wild-type cells. (Bottom) Htz1 signals around the TSSs (position 0) of direct (green) and indirect (orange) Ino80 targets in arp5 cells.
We next examined how Isw2, Rsc, and Ino80 alter chromatin around TSSs of CRRATs. Isw2-dependent repression of mRNA transcription is generally associated with reduction in the size of the NDRs upstream of the mRNA initiation sites, which makes the TSSs covered by the upstream edges of the +1 nucleosomes (Whitehouse et al. 2007). Aligning all 58 Isw2-repressed lncRNAs at their putative TSSs in a wild-type background (Yen et al. 2012) revealed that, like mRNA targets, Isw2-repressed ASlncRNAs are associated with an upstream NDR (Fig. 5G). In contrast to mRNA targets, however, nucleosomes more deeply occlude the TSSs of Isw2-repressed ASlncRNAs in wild-type cells (Fig. 5G, top panel). In the absence of Isw2, nucleosomes shift away from NDRs, no longer occluding lncRNA TSSs. These results suggest that Isw2-repressed ASlncRNAs have a strong intrinsic tendency to exclude nucleosomes at their TSSs and that Isw2 represses them by sliding nucleosomes over their TSSs more deeply than at the mRNA TSSs. The lncRNAs that are derepressed in the itc1 mutant but do not have Isw2 targeting around their TSSs, likely indirect targets of Isw2, are generally associated with much smaller Isw2-dependent changes in nucleosome positioning (Fig. 5G, bottom panel), supporting our conclusion that they are indirectly affected by Isw2.
Rsc also functions around NDRs, but there have been reports on distinct chromatin regulation around NDRs for transcriptional regulation. Rsc was first reported to generally increase the size of NDRs to facilitate transcription (Hartley and Madhani 2009; Floer et al. 2010). However, it was recently reported that Rsc is also required for silencing of genes in subtelomeric regions and that its activity leads to higher signals of +1 and −1 nucleosomes (Van de Vosse et al. 2013). These reports suggest that Rsc may exhibit different chromatin remodeling activities depending on the context. Analysis of chromatin structure revealed that NDRs just upstream of ASlncRNA TSSs are wider and that the +1 nucleosome signals are much more discrete at direct Rsc targets as compared with indirect targets (Fig. 5H), suggesting that Rsc is targeted to the sites that have distinct chromatin structure. At direct targets, Rsc depletion generally causes the loss of the −1 nucleosome signals adjacent to lncRNA TSSs (Fig. 5H). This suggests that Rsc reduces the accessibility at the wide NDRs of its direct ASlncRNA targets by increasing the −1 nucleosome occupancy for lncRNA repression. In addition, Rsc depletion results in shifting of the +2 and +3 nucleosomes toward the TSSs. In contrast, the −1 nucleosome signals still remain, although at lower levels, upon Rsc depletion at indirect Rsc targets (Fig. 5H). In addition, at indirect targets, the +2 and +3 nucleosomes exhibit little shift in their positions, and instead the +1 nucleosome signals are reduced, and the +2 nucleosome signals are increased (Fig. 5H).
Ino80 acts at NDRs to remove the histone variant Htz1 at +1 nucleosomes of mRNA transcription units (Papamichos-Chronakis et al. 2011; Yen et al. 2013). To determine how Ino80 affects Htz1 levels for ASlncRNA repression, we examined the Htz1 level around the TSSs of 540 Ino80-repressed ASlncRNAs. In a wild-type background, levels of Htz1 around ASlncRNA TSSs, as determined by ChIP combined with deep sequencing (ChIP-seq), are similar at indirect and direct ASlncRNA targets of Ino80, although they are higher at the +1 and lower at −1 nucleosomes at direct targets (Fig. 5I; Albert et al. 2007). ARP5, encoding a subunit of the Ino80 complex, is required for Ino80 biochemical activity as well as DNA-binding activity of the complex. ChIP-exo analysis of Htz1 in an arp5 background demonstrated that there is a substantial accumulation of Htz1 at the 5′ ends of mRNA-coding genes (Yen et al. 2013). Analysis of Htz1 levels in an arp5 background at direct ASlncRNA targets of Ino80 revealed a substantial level of Htz1 enrichment at both −1 and +1 nucleosomes around TSSs that is much higher than neighboring regions (Fig. 5I). This result suggests that Ino80 represses ASlncRNA transcription by preventing the accumulation of abnormally high levels of Htz1 at the 3′ ends of a very large number of genes. Compared with direct targets, the level of Htz1 was lower at both −1 and +1 nucleosomes of indirect Ino80 target ASlncRNAs (Fig. 5I), again supporting the notion that their transcription is indirectly affected by Ino80.
These results collectively identified 814 CRRATs that are likely directly regulated by chromatin remodeling factors through chromatin regulation around their TSSs. The fact that much weaker or different chromatin changes were found at TSSs of the indirect targets of the remodeling factors despite the comparable level of ASlncRNA levels in the mutants (Supplemental Fig. 5) argues against the possibility that these chromatin changes are the results of elevated ASlncRNA transcription. Therefore, these results provide further support for the direct regulation of CRRATs by chromatin remodeling factors. Moreover, our results suggest that ASlncRNA can be repressed through diverse chromatin remodeling mechanisms.
Identification of ASlncRNAs with regulatory roles
Because lncRNAs are highly prevalent across eukaryotes, one important question is how many of them have functional roles in vivo (Louro et al. 2009). This is a highly significant issue, as only a handful of lncRNAs transcription units with regulatory roles have been identified in S. cerevisiae despite the fact that several thousand lncRNAs are transcribed across the genome (Hongay et al. 2006; Camblong et al. 2007, 2009; Houseley et al. 2008; Castelnuovo et al. 2013). Similarly, although ∼75% of the human genome is estimated to be transcribed by ncRNAs (Djebali et al. 2012), the functions of a very small fraction of them have been identified. Because known functional lncRNA transcription in S. cerevisiae is precisely regulated by environmental cues and cell type specificities (Hongay et al. 2006; Camblong et al. 2007, 2009; Houseley et al. 2008; Castelnuovo et al. 2013), we hypothesized that ASlncRNA transcription regulated by highly conserved chromatin remodeling factors (CRRATs) may be enriched for those with regulatory functions. Most known regulatory lncRNA transcription events in S. cerevisiae repress overlapping mRNAs. Consistent with this, visual inspection of individual CRRATs revealed many cases in which derepression of ASlncRNAs in chromatin remodeling factor mutants coincides with decreased levels of overlapping mRNAs (Fig. 5A–C). We therefore systematically identified CRRATs whose derepression in the mutant is associated with down-regulation of the overlapping mRNA levels (see the Materials and Methods for the details of RNA analyses). We identified a total of 259 such CRRATs (out of 814) that fit the criteria (Fig. 6A–C). Because these CRRATs are likely directly repressed by chromatin remodeling factors, our results suggest that repression of these ASlncRNAs by chromatin remodeling factors is required for the maintenance of the normal levels of overlapping mRNAs. In addition, mRNAs that do not overlap with CRRATs exhibit much smaller changes in their abundance in chromatin remodeling factor mutants, arguing against the possibility that the reduction in the mRNA levels of the 259 genes is due to random fluctuation (Supplemental Fig. 6). Together, these results support the possibility that chromatin remodeling factors regulate 259 genes through repression of ASlncRNA transcription. We found that most of the CRRATs in this class transcribe through the entire ORF of the overlapping genes and the promoter regions (Fig. 6A–C), which is unusually long for lncRNAs. This result is consistent with the earlier conclusion that CRRATs represent a unique set of lncRNA transcripts and suggests that production of long lncRNAs may be a common feature of lncRNA transcription events that play regulatory roles.
Figure 6.

Identification of ASlncRNAs whose repression by chromatin remodeling factors is required for the maintenance of the normal level of overlapping mRNAs. (A) Changes in the levels of Isw2-repressed CRRATs (dark blue) and overlapping mRNAs (light blue) in itc1 cells. The solid lines denote the mean, and the colored ribbon shows the RNA signals of the top 25% and bottom 25% values in the mutants. RNAs are aligned at the TSSs of mRNAs (left panel) and the TSSs of CRRATs (right panel). The Y-axis is in log2 scale, and a dotted line is at the value 0. (B) Changes in the levels of Rsc-repressed CRRATs (red) and overlapping mRNAs (light blue) in tet-STH1 cells. The solid lines denote the mean, and the colored ribbon shows the RNA signals of the top 25% and bottom 25% values in the mutants. RNAs are aligned at the TSSs of mRNAs (left panel) and the TSSs of CRRATs (right panel). The Y-axis is in log2 scale, and a dotted line is at the value 0. (C) Changes in the levels of Ino80-repressed CRRATs (green) and overlapping mRNAs (light blue) in tet-INO80 cells. The solid lines denote the mean, and the colored ribbon shows the RNA signals of the top 25% and bottom 25% values in the mutants. RNAs are aligned at the TSSs of mRNAs (left panel) and the TSSs of CRRATs (right panel). The Y-axis is in log2 scale, and a dotted line is at the value 0.
Discussion
Although lncRNAs are pervasively transcribed across eukaryotes, functions of the vast majority of them remain unknown. In addition, unlike mRNA transcription, very few regulators of lncRNAs have been identified. To address these crucial issues, we developed a novel genetic screen to systematically identify repressors of ASlncRNAs. This allowed us to identify a large number of ASlncRNA repressors as well as ASlncRNAs whose transcription likely have regulatory roles.
A novel genetic screen for ASlncRNA repressors
Previously, Cheung et al. (2008) identified genes that repress expression of cryptic transcripts from intragenic initiation sites using the HIS3 gene as a reporter. More recently, Marquardt et al. (2014) developed a genetic screen to isolate genes involved in repression of lncRNA transcription from divergent promoters using YFP and mCherry reporters. Curiously, both of these screens identified genes involved in chromatin assembly as well as histone genes among the strongest hits. These results revealed that chromatin assembly defects lead to elevated transcription of both intragenic cryptic RNAs and lncRNAs from some bidirectional promoters. Whether these transcripts have biological roles remains unknown. One interesting possibility is that these cryptic transcripts may signal to chromatin surveillance systems the location of chromatin defects, resulting in their timely repair. Interestingly, our screen identified a large number of genes known for regulation of mRNA transcription, which had only a few overlaps with those identified in these two previous screens. The difference in the results between our genetic screen and the other two is likely at least partly due to the fact that our screen did not use reporter genes and instead relied directly on genome-wide elevation of ASlncRNAs. What makes reporter assays particularly sensitive to chromatin assembly defects is an interesting question that needs to be addressed in the future. These results collectively demonstrate that different classes of lncRNAs regulated by diverse mechanisms exist and that they likely have distinct biological functions.
A large number of putative lncRNA repressors
So far, only a very small number of regulators for lncRNA transcription have been identified. However, the fact that all four chromatin remodeling factors identified in our screen are indeed ASlncRNA repressors suggest that many, if not all, other genes identified in our screen likely function as repressors of ASlncRNAs as well. This means that a large amount of resources is used to control ASlncRNA transcription. Among the identified putative repressors, the mediator, Paf1, and Rpd3 complex subunits were also identified by the genetic screen for suppressors of intragenic cryptic transcripts by Cheung et al. (2008) suggesting that they are likely involved in repression of multiple classes of lncRNAs. Most putative repressors identified in our screen also repress mRNA transcription, indicating that the dual roles of transcription repression on mRNA and lncRNAs are unexpectedly widespread. This means that the phenotypes of the mutants for these repressors need to be revisited, as they can be at least partly due to derepression of lncRNAs.
The current estimation for the number of lncRNAs transcribed in S. cerevisiae is based on the number of RNAs detected in wild-type cells as well as in mutants that stabilize or prevent premature termination of lncRNA transcripts, such as trf4 (Wyers et al. 2005) and rrp6 (Davis and Ares 2006) mutants. The estimated number of lncRNAs is already very large (∼900 CUTs [Xu et al. 2009], 800 SUTs [Xu et al. 2009], 1600 XUTs [van Dijk et al. 2011], and 1500 NUTs [Schulz et al. 2013]; note that there are overlaps between the classes), so it was unexpected that a significant fraction of ASlncRNAs repressed by chromatin remodeling factors is not strongly elevated in these mutants and thus is a novel class of lncRNAs. The fact that many uncharacterized putative ASlncRNA repressors exist suggests a possibility that the number of lncRNAs is currently underestimated, and a large number of currently unidentified lncRNAs will be found once the transcriptomes of the mutants of these repressors are analyzed.
Identification of lncRNAs with regulatory functions
Compared with the enormous number of lncRNAs transcribed in eukaryotic cells, the number of known functional lncRNA transcriptions is extremely small. This led to proposals that most lncRNAs transcribed in eukaryotes are nonfunctional (“noise” or “junk”) (Doolittle 2013). On the other hand, it has recently become clear that the small number of lncRNAs with regulatory functions play crucial roles in transcriptional control, cell type specification, and human diseases (Batista and Chang 2013; Lee and Bartolomei 2013; Flynn and Chang 2014), raising the possibility that many lncRNAs with important functional roles are yet to be identified. These opposing views have resulted in active discussions in the field as to how many lncRNAs have any biological functions (Kellis et al. 2014). One possible way to address this debate is to systematically identify lncRNAs with regulatory functions. However, a systematic screen for functional lncRNAs has been difficult, and discovery of functional lncRNAs has relied mostly on fortuitous events and locus-specific analyses. Based on the fact that transcription of lncRNAs with known regulatory functions is precisely regulated by environmental cues and/or cell type, we hypothesized that we would be able to enrich for ASlncRNAs with regulatory roles by focusing on those that are regulated by highly conserved ATP-dependent chromatin remodeling factors.
We indeed found that, in chromatin remodeling factor mutants, derepression of ∼32% (259 out of 814) of CRRATs is associated with a significant decrease in the level of mRNAs that they overlap. This strongly suggests that chromatin remodeling factors maintain the levels of these 259 mRNAs through repression of overlapping ASlncRNAs. This is most likely an underestimation of functional ASlncRNA transcription events, as only one growth condition (logarithmic growth in rich medium) was used for RNA analyses. Unexpectedly, ∼37% of mRNAs that overlap with ASlncRNAs that are likely indirect targets of chromatin remodeling factors (up-regulated in chromatin remodeling factor mutants but not targeted by them) are also associated with reduction of their mRNA levels. This result suggests that, irrespective of the underlying mechanisms, an increase in ASlncRNA levels can lead to a decrease in the level of overlapping mRNAs. Nonetheless, our results indicate that the mechanism to regulate mRNA levels through ASlncRNA control is far more widely used than currently appreciated. Moreover, given the many putative ASlncRNA repressors that we identified, it is highly likely that a very large number of cases in which ASlncRNA control is used to regulate mRNA levels in a similar fashion are yet to be discovered.
Materials and methods
Yeast strains
A list of all strains used in this study can be found in Supplemental Table S1. We carried out single-step gene deletions by standard lithium acetate transformation using KanMX, HygMX, and NatMX drug resistance markers as described for S. cerevisiae (Goldstein and McCusker 1999). Strains were also created using standard genetic crosses. For S. cerevisiae, genome sequences and annotations were downloaded from Ensembl or the Saccharomyces Genome Database.
Yeast growth conditions
Unless otherwise noted, strains were cultured at 30°C in either YPD or YC until OD600 = 0.4–0.7. For strains harboring Tet-repressible alleles, cells were cultured at 37°C until OD = 0.3, and then doxycycline was added to YC(-His) medium at a final concentration of 20 ng/mL. Cells were grown for 3 h before being harvested for RNA using standard hot acid phenol extraction.
Plasmid construction
pRS406-(AGO1-DCR1) was created by inserting restriction digest fragments containing the coding sequences and associated promoters of AGO1 and DCR1 from pRS404-PTEF-AGO1 and pRS405-PTEF-DCR1 (Drinnenberg et al. 2009) into pRS406. This plasmid was then used to integrate RNAi machinery into our laboratory strains.
Northern analysis
Ten micrograms to 20 µg of total RNA was added to 1.5× sample buffer (75% formamide, 13.4% formaline, 1× MOPS) and loaded onto a 1% agarose gel prepared with running buffer (1% agarose, 1× MOPS, 5% formalin). The RNA was transferred onto a GeneScreen membrane (Perkin Elmer) in 10× SSC overnight. The blot was then UV cross-linked and incubated with hybridization buffer (6× SSC, 0.1 mg/mL salmon sperm DNA, 0.5% SDS) for 1 h at 65°C. Radiolabeled probe to Ty1 or ACT1 was then added, and hybridization occurred overnight at 65°C. The membrane was then washed with 0.5× SSC for 30 min at 65°C. This wash was repeated once before exposure to film or phosphor screen.
Strand-specific library preparation and high-throughput RNA-seq
RNA-seq was done in the presence and absence of reconstituted RNAi in Figure 1. For transcriptome analyses of chromatin remodeling factor mutants, all experiments were done using mutants that do not have RNAi. Three micrograms of total RNA was depleted of ribosomal RNA species using Ribo-Zero magnetic rRNA removal kit (human/mouse/rat) (Epicentre). Strand-specific libraries were then prepared using the dUTP method combined with TruSeq (Illumina) as previously described (Parkhomchuk et al. 2009; Sultan et al. 2012). Fifty cycles of paired-end sequencing were performed on an Illumina HiSeq 2500 on either high-output mode or rapid run mode (Fred Hutchinson Cancer Research Center shared resources). All sequencing experiments were performed in biological duplicate.
RNA-seq analysis
Alignment
Reads were aligned to the S. cerevisiae genome (Saccharomyces_cerevisiae.EF4.69.dna.toplevel.fa) (Flicek et al. 2014) using TopHat2 (Kim et al. 2013) with the following settings: tophat2 -p 4 -G <gene_annotation_file> -I 2000–library-type=fr-firststrand -o <output_directory> <bowtie_index> <Read1.fastq> <Read2.fastq>. Reads were then trimmed of adapter sequences with a custom Python script using the Python module HTSeq (Anders et al. 2014).
Heuristic segmentation of RNA-seq data to identify putative transcript units
After reads were aligned, they were filtered such that only properly aligned, uniquely mapped reads were kept using a custom Python script and pysam (Li et al. 2009). Because replicates were highly reproducible (data not shown), reads for each replicate were combined to make per-base, strand-specific pileup files using pysam. Using this pileup file, putative transcript units were segmented by defining a minimum expression threshold, defined below. tRNAs, and rRNAs were excluded for every step in analysis.
Defining a threshold level using empirically determined tag density
For a known ORF, expression was calculated by the following equation:
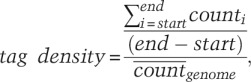 |
where i is the genomic position, count is the number of reads overlapping i, end is the last genomic position of the ORF, and start is the beginning position of the ORF. This was repeated for every ORF in the genome. The threshold was defined by the bottom fifth percentile expression value for transcripts longer than 250 bp (inclusive). For transcripts between 100 bp and 249 bp (inclusive), the threshold was the bottom 25th percentile expression value.
Heuristic segmentation of pileup files
Using the threshold defined above, putative transcripts were identified by computing the tag density within a 100-bp sliding window using a 1-bp step size. “Starts” and “ends” of transcript units were defined by whether the tag density exceeded the defined threshold and were at least 100 bp in length. Segments closer than 50 bp and less than twofold different in tag density were joined, which is commonly performed. See above for threshold differences based on length.
Identification of differentially expressed lncRNAs and mRNAs
Using the compiled putative transcript list, differentially expressed transcript units were defined by first enumerating the number of reads in each replicate that overlapped with each transcript and then using a negative binomial distribution (R-package DESeq) (Anders and Huber 2010) to determine differential expression. For remodeling mutants, putative transcripts that were up-regulated (≥1.25-fold) and had a P-value <0.05 were determined to be differentially expressed. Differentially expressed transcripts were then identified as ncRNAs by whether they overlapped with annotated features of the genome in a strand-specific manner. This was performed using custom scripts written in Python and BEDtools (Quinlan and Hall 2010). Fold change as well as absolute expression (in RPKM [reads per kilobase per million mapped reads] values) were determined using DESeq. Chromosome coordinates for all ASlncRNAs repressed by chromatin remodeling factors are listed in Supplemental Table S2.
Construction of heat maps and plots and statistical analysis
Heat maps, plots, and metagene plots were constructed in R using the packages “ggplot” or “ggtern.” To create ternary plots, for each lncRNA, the fold change was determined in the remodeling mutant, the exosome mutant, and the Xrn1 mutant. The fold changes were then normalized so that they summed to 1. These normalized fold changes were then determined for each lncRNA, and the resulting matrix was used in ggtern to make the plots.
Identification of CRRATs
All ASlncRNA TSSs were extended 100 bp in both the forward and reverse directions. The resulting 200-bp interval was then used to determine whether an ASlncRNA TSS was in proximity to a remodeler-bound nucleosome (Yen et al. 2012) using BEDtools (Quinlan and Hall 2010).
Differential expression analysis of mRNAs
mRNA annotations were downloaded from Ensembl (Saccharomyces_cerevisiae.EF4.65.gtf). After counting reads mapping to each mRNA using HTSeq, differential expression was determined using DESeq.
GO analysis
All GO analysis was performed using GOrilla (Eden et al. 2009).
Accession numbers
All RNA-seq data have been deposited at NCBI Sequence Read Archive under accession number SRP041297.
Acknowledgments
We thank H. Malik, I.A. Drinnenberg, B.F. Pugh, and the members of the Tsukiyama laboratory for helpful discussions; H. Malik and I.A. Drinnenberg for critical reading of the manuscript; F. Winston for yeast strains; and A. Marty and Fred Hutchinson Cancer Research Center shared resources for deep sequencing. This work was supported by grant RO1 GM058465 (to T.T.) and predoctoral fellowship F31 GM101944 (to E.A.A.). E.A.A. contributed in planning and performing experiments, analyzing and interpreting data, and writing the manuscript. T.T. contributed in planning experiments, interpreting data, and writing the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.250902.114.
References
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. 2007. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576 [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2014. HTSeq—A Python framework to work with high-throughput sequencing data. bioRxiv [DOI] [PMC free article] [PubMed]
- Batista PJ, Chang HY. 2013. Long noncoding RNAs: cellular address codes in development and disease. Cell 152: 1298–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. 2007. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell 131: 706–717 [DOI] [PubMed] [Google Scholar]
- Camblong J, Beyrouthy N, Guffanti E, Schlaepfer G, Steinmetz LM, Stutz F. 2009. Trans-acting antisense RNAs mediate transcriptional gene cosuppression in S. cerevisiae. Genes Dev 23: 1534–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592 [DOI] [PubMed] [Google Scholar]
- Castelnuovo M, Rahman S, Guffanti E, Infantino V, Stutz F, Zenklusen D. 2013. Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nat Struct Mol Biol 20: 851–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, Winston F. 2008. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol 6: e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall J, Lis J. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Ares M Jr. 2006. Accumulation of unstable promoter-associated transcripts upon loss of the nuclear exosome subunit Rrp6p in Saccharomyces cerevisiae. Proc Natl Acad Sci 103: 3262–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. 2013. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci 110: 5294–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, Bartel DP. 2009. RNAi in budding yeast. Science 326: 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Fink GR, Bartel DP. 2011. Compatibility with killer explains the rise of RNAi-deficient fungi. Science 333: 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2014. Ensembl 2014. Nucleic Acids Res 42: D749–D755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floer M, Wang X, Prabhu V, Berrozpe G, Narayan S, Spagna D, Alvarez D, Kendall J, Krasnitz A, Stepansky A, et al. 2010. A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 141: 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Chang HY. 2014. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 14: 752–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein G, McCusker J. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15: 1541–1553 [DOI] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. 2010. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464: 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137: 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongay CF, Grisafi PL, Galitski T, Fink GR. 2006. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell 127: 735–745 [DOI] [PubMed] [Google Scholar]
- Houseley J, Rubbi L, Grunstein M, Tollervey D, Vogelauer M. 2008. A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol Cell 32: 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. . 2014. Defining functional DNA elements in the human genome. Proc Natl Acad Sci 111: 6131–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobor MS, Venkatasubrahmanyam S, Meneghini MD, Gin JW, Jennings JL, Link AJ, Madhani HD, Rine J. 2004. A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol 2: E131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Bartolomei MS. 2013. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152: 1308–1323 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickwar CR, Rao B, Shabalin AA, Nobel AB, Strahl BD, Lieb JD. 2009. The Set2/Rpd3S pathway suppresses cryptic transcription without regard to gene length or transcription frequency. PLoS ONE 4: e4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louro R, Smirnova AS, Verjovski-Almeida S. 2009. Long intronic noncoding RNA transcription: expression noise or expression choice? Genomics 93: 291–298 [DOI] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S. 2014. A chromatin-based mechanism for limiting divergent noncoding transcription. Cell 157: 1712–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. 2009. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457: 1038–1042 [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL. 2011. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144: 200–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37: e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2012. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483: 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P. 2013. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155: 1075–1087 [DOI] [PubMed] [Google Scholar]
- Sultan M, Dokel S, Amstislavskiy V, Wuttig D, Sultmann H, Lehrach H, Yaspo ML. 2012. A simple strand-specific RNA-seq library preparation protocol combining the Illumina TruSeq RNA and the dUTP methods. Biochem Biophys Res Commun 422: 643–646 [DOI] [PubMed] [Google Scholar]
- Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Hofer T, Rippe K. 2012. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol 19: 1185–1192 [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368 [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Vosse DW, Wan Y, Lapetina DL, Chen WM, Chiang JH, Aitchison JD, Wozniak RW. 2013. A role for the nucleoporin Nup170p in chromatin structure and gene silencing. Cell 152: 969–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk EL, Chen CL, d’Aubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix-Ne P, Loeillet S, et al. 2011. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 475: 114–117 [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. 2001. The sequence of the human genome. Science 291: 1304–1351 [DOI] [PubMed] [Google Scholar]
- Venters BJ, Pugh BF. 2013. Genomic organization of human transcription initiation complexes. Nature 502: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. 2007. Chromatin remodelling at promoters suppresses antisense transcription. Nature 450: 1031–1035 [DOI] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle J-C, Dufour M-E, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B. 2005. Cryptic Pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737 [DOI] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457: 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadon AN, Van de Mark D, Basom R, Delrow J, Whitehouse I, Tsukiyama T. 2010. Chromatin remodeling around nucleosome-free regions leads to repression of noncoding RNA transcription. Mol Cell Biol 30: 5110–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149: 1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Pugh BF. 2013. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 154: 1246–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, Grewal SI. 2009. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461: 419–422 [DOI] [PMC free article] [PubMed] [Google Scholar]