

Abstract
Our group recently discovered that certain dietary nutrients possessing a trimethylamine (TMA) moiety, namely choline/phosphatidylcholine and L-carnitine, participate in the development of atherosclerotic heart disease. A meta-organismal pathway was elucidated involving gut microbiota–dependent formation of TMA and host hepatic flavin monooxygenase 3–dependent (FMO3-dependent) formation of TMA–N-oxide (TMAO), a metabolite shown to be both mechanistically linked to atherosclerosis and whose levels are strongly linked to cardiovascular disease (CVD) risks. Collectively, these studies reveal that nutrient precursors, gut microbiota, and host participants along the meta-organismal pathway elucidated may serve as new targets for the prevention and treatment of CVD.
Introduction
Cardiovascular disease (CVD) remains the leading cause of death in both the United States and industrialized societies, with growing incidence in developing countries (1). Factors contributing to CVD arise from genetic sources, environmental sources, or a combination of genetic and environmental sources (2). Despite extensive investigations in search of causal genetic variants, such as large-scale GWAS, less than one-fifth of attributable cardiovascular risk has been accounted for from genetic determinants (3, 4). At the same time, major advances in the treatment of atherosclerosis beyond high-potency lipid-lowering agents has not yet materialized (5). Indeed, even with high-potency statin therapy, at least a 50% residual risk remains (6), with the majority of events unchecked. Therefore, there is still ample room for improving our understanding of the processes contributing to CVD pathogenesis, and for improved prevention and treatment of CVD. While the recognizable contribution of genetics to CVD will no doubt significantly increase with time, a reassessment of environmental contributions to CVD pathogenesis and risks for adverse events is certainly worthwhile.
Our largest environmental exposure is what we eat. Technically speaking, food is a foreign object that we take into our bodies in kilogram quantities every day. From the latest National Health and Nutrition Examination Survey (NHANES, 2009–2010), the majority of individuals sampled achieved an intermediate or poor Healthy Eating Index (1). However, dietary composition is often difficult to assess, and even precise quantification of dietary intake may not necessarily reveal the many known and unknown factors that may influence the contributions of specific dietary nutrients to disease susceptibility (7). There has also been an overwhelming lack of appreciation at the bedside regarding the intricate and complicated processes that transform ingested food into the myriad metabolites that enter the circulation and fulfill or adversely affect various functional and metabolic processes in the body.
Over the past decade we have increasingly begun to appreciate the ecological diversity of microbes living symbiotically within us, a large proportion of which reside within our intestines. We now know that the human gut harbors more than 100 trillion microbial cells, far outnumbering the human host cells of the body (8). Indeed, a sobering fact is that Homo sapiens DNA is estimated to represent less than 10% of the total DNA within our bodies, due to the staggeringly large numbers of microbes that reside in and on us, primarily within our gut (9). Our microbial symbiont guests have coevolved with us and affect a wide range of physiologic and metabolic processes of the body. The major taxa present in gut microbiota consist primarily of 2 major bacterial phyla, Firmicutes and Bacteroidetes, whose proportions appear to remain remarkably stable over time within individuals and their family members (9, 10). However, the composition of the remaining gut microbiota is remarkably diverse and dynamic, with both acute and chronic dietary exposures significantly affecting the overall microbial community (8, 11). Hence, our metabolism and absorption of food occurs through the stable yet dynamic filter of trillions of bacteria that populate our digestive system, making the gut microbiota a filter of our largest environmental exposure. The composition of our gut microbial community is affected not only by dietary exposures, but also by genetic variants within the host — changes in the “terrarium” in which the gut microbes reside. Thus, there is a complex and somewhat dynamic interplay among acute and chronic dietary exposures, a fluid yet remarkably resilient gut microbial community, host genotype, and other external environmental exposures that collectively influence our meta-organismal metabolism, global physiology, and disease susceptibilities (12). The participation of gut microbiota in our health, immune function, and disease initiation and progression, remain largely unexplored areas.
There has been a long-standing understanding of the contribution of dysbiosis (abnormal changes in intestinal microbiota composition) to the pathogenesis of some diseases of altered intestinal health. Specifically, intestinal-derived endogenous endotoxins such as lipopolysaccharides (13), indoxyl sulfate (14), and para-cresyl sulfate (15) have been implicated to play important metabolic roles in conditions ranging from atherosclerosis to cardio-renal dysfunction (16–18). Yet recent advances have also revealed that in more subtle ways, differences in gut microbiota composition are associated with the development of complex metabolic disorders such as obesity and insulin resistance (19–24). It is now increasingly clear that gut microbiota play important metabolic roles in a far wider range of disease states, maintaining delicate balances and cross-talk within the human host and among themselves (12). The gut microbial ecosystem is arguably the largest endocrine organ in the body, capable of producing a wide range of biologically active compounds that, like hormones, may be carried in the circulation and distributed to distant sites within the host, thereby influencing different essential biological processes. This endocrine organ is plastic, generating distinct biologically active species depending upon both acute and chronic dietary exposures and a partially dynamic composition, but nonetheless fulfilling all of the requirements for definition as an endocrine organ.
This Review summarizes many recent developments in our understanding of the contributory role of gut microbiota as an active participant, through this endocrine function, in the development of atherosclerosis and its adverse complications of CVD. It also details strategies for targeting gut microbiota and the recently discovered meta-organismal pathway participants that contribute to trimethylamine–N-oxide (TMAO) formation for the potential prevention and treatment of CVD.
Discovery of the association between TMAO and CVD
Initial hypothesis-generating studies used untargeted metabolomic analyses of plasma samples to identify novel metabolites and their connecting pathways potentially associated with cardiovascular risk (25). An iterative process of case-control studies was performed using an initial learning cohort, a subsequent independent validation cohort, and then a third, larger independent validation cohort (n = 1,876 subjects), which identified a number of metabolites whose levels in plasma were reproducibly correlated with CVD risks (25). Structural validation revealed 3 of the metabolites were linked to phosphatidylcholine (PC; lecithin) metabolism — choline (m/z 104), betaine (m/z 116), and TMAO (m/z 76) (25). Betaine is a known direct oxidation product of choline (26, 27). TMAO had previously been suspected to arise from bacterial metabolism of choline via an intermediate, trimethylamine (TMA), and subsequent hepatic oxidation via flavin monooxygenase 3 (FMO3), thus forming TMAO (28–31). PC is the major dietary source of choline in omnivores (32–34), and direct ingestion of PC was shown to result in rises in choline, betaine, and TMAO levels (25). Further, our studies suggested that plasma levels of TMAO showed the strongest positive correlation with CVD risk (25). Thus, our initial metabolomics studies suggested that plasma levels of 3 metabolites of dietary PC and gut microbiota might be linked to CVD in humans (25).
Choline is an essential dietary nutrient. While we can synthesize much of our requirements, we still need to consume some choline in our diet or else develop a deficiency state, which is characterized by fatty liver, altered one-carbon methyl donor metabolic pathways, and neurologic disorder (19, 32, 35, 36). An obligatory role for gut microbiota in both TMA and TMAO formation from ingested PC was confirmed in animal model studies, which included germ-free mice (25), as well as human clinical investigations involving ingestion of egg yolk, isotope-labeled PC, and a cocktail of oral antibiotics (37). Recently, the association between acute egg yolk ingestion and increased plasma and urine TMAO concentrations was independently confirmed in humans (38). Additional studies have shown that L-carnitine, an alternative TMA-containing nutrient found almost exclusively in red meat, similarly serves as a dietary precursor to gut microbial production of TMA and TMAO in both mice and humans (ref. 39 and Figure 1). It is thus remarkable that the highest levels of choline and L-carnitine are often found in foods rich in cholesterol and fats, such as red meat, liver, and egg yolk. While numerous large-scale epidemiologic studies have linked red meat ingestion with heightened mortality and CVD risks, the relationship between egg ingestion and cardiovascular risks has shown conflicting results (40–49). Since other dietary nutrients possess a TMA moiety (e.g., glycerophosphocholine, betaine, various short- and long-chain acyl carnitines, and sphingomyelin), there are likely other gut microbe–dependent pathways that lead to the formation of TMA and TMAO, though these remain largely unexplored.
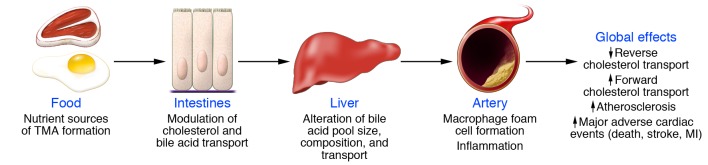
Figure 1. Nutrient/meta-organismal pathway associated with atherosclerosis and major adverse cardiovascular events.

Foods rich in cholesterol and fats are also often rich in the indicated dietary nutrients PC (lecithin), choline, and carnitine. Following ingestion, gut microbiota can use these nutrients as a carbon fuel source. While mammals do not have the enzyme, gut microbes have TMA lyases, which can cleave the C-N bond of these nutrients, releasing the TMA moiety as a waste product. Transport via the portal circulation brings the TMA to a cluster of hepatic enzymes, the FMOs (particularly FMO3), that efficiently oxidize TMA, thus forming TMAO. TMAO enters the circulation, where it is predominantly excreted by the kidneys. TMAO has been shown to affect cholesterol and sterol metabolism in animal models, enhancing macrophage cholesterol accumulation and atherosclerosis development. In multiple human studies, elevated TMAO has been independently associated with prevalent CVD and incident risks for MI, stroke, death, and revascularization. [O], oxidation.
TMAO is not a widely recognized metabolite in mammals, although its role as a marine bacterial nutrient is emerging (50). TMAO is abundant in some fish, in which it is used in the freeze avoidance response as a form of “antifreeze” (51). It is also known to possess small-molecule protein chaperone mimetic behavior (52), presumably owing to its small size and a combination of both hydrophobic and polar characteristics. Originally believed to be an inert nitrogenous waste product of protein degradation excreted in urine (53), more recent studies revealed that TMAO may have direct biological activity that facilitates the development or propagation of atherosclerosis and its adverse CVD events. TMAO supplementation of apolipoprotein E–null mice was observed to foster enhanced macrophage foam cell formation in both the artery wall and the peritoneal cavity, as well as to promote aortic root atherosclerotic plaque development (25). Moreover, enhanced protein surface levels were observed of previously implicated scavenger receptors (CD36 and SR-A1) on macrophages involved in cholesterol accumulation and foam cell formation (25). Direct dietary exposure to TMAO or its precursors, choline or L-carnitine (the latter 2 only in the presence of intact gut microbiota, which are required for TMAO formation), elicited significant reductions in reverse cholesterol transport in vivo in mouse models, as well as alterations in cholesterol and sterol metabolic pathways in multiple compartments including the artery wall, the liver, and the intestines (25, 39). Alterations were also observed in bile acid pool size and composition induced by TMAO (39). Further investigations revealed that TMAO levels help to explain a significant portion of atherosclerosis aortic root development across multiple different inbred strains of mice, and that TMAO levels are influenced by hepatic Fmo3, which is under complex control, including regulation by the farnesoid X receptor (FXR), a bile acid–activated nuclear receptor (54).
While plasma levels of all 3 metabolites (choline, betaine, and TMAO) identified in the original metabolomics study are associated with increased risk of CVD phenotypes in subjects presenting for cardiac risk evaluation (n = 1,876) (25), subsequent analyses in larger cohorts revealed that the prognostic value was largely confined to the formation of TMAO, especially from choline and L-carnitine (39, 55). In an alternative subsequent expansion study of over 4,000 subjects undergoing elective coronary angiography, elevated TMAO levels predicted major adverse cardiac events such as death, myocardial infarction (MI), and stroke over a 3-year period. Specifically, patients in the upper quartile for TMAO levels (compared with the lowest quartile) had a significant, 2.5-fold increased risk of experiencing a major adverse cardiac event (MI, stroke, or death), independent of traditional cardiovascular risk factors, renal function, and medication use, as well as overall poorer event-free survival (37).
In additional studies, dietary L-carnitine was similarly shown to foster accelerated atherosclerosis in mouse models, but only in the presence of intact gut microbiota and TMA/TMAO generation (39). Further, in a study examining plasma carnitine levels in sequential subjects undergoing elective diagnostic cardiac evaluation (n = 2,595), high carnitine levels were significantly associated with incident risks for MI, stroke, or death over a follow-up period of 3 years, but only in subjects with concurrently high TMAO levels (39). Thus, while elevated circulating carnitine, choline, or betaine levels (all substrates of gut microbiota and dietary precursors of TMA/TMAO production) were each associated with future risk of MI, stroke, or death independent of traditional risk factors, their prognostic values were primarily restricted to those with concomitantly elevated TMAO levels (39, 55). These observations highlight the obligatory role of intestinal microbiota in the generation of TMAO from multiple dietary nutrients, and TMAO as the proatherogenic species likely promoting the striking associations noted between plasma levels and both prevalent and incident CVD risks.
Mechanistic insights thus far observed (changes in bile acid and sterol metabolism in macrophage, hepatocyte and enterocyte compartments) demonstrate an interaction between the meta-organismal pathway that leads to TMAO generation and multiple well-established atherogenic processes (Figure 2). Nevertheless, direct molecular targets that mediate the many observed changes in expression levels of target genes and proatherosclerotic effect (i.e., what serve as “receptors” for TMAO) have not yet been identified and represent an important focus of future investigation for the field.
Figure 2. Effects of gut microbiota–dependent TMAO production on cholesterol and sterol metabolism and atherosclerosis.
Compartments and processes through which microbiota-generated TMAO affects cholesterol and sterol metabolism and atherosclerosis. The microbiota metabolizes dietary L-carnitine and choline to form TMA and TMAO, influencing cholesterol and sterol metabolism in macrophages, liver, and intestine (39).
Recent studies reveal that the potential pathogenic contribution of gut microbiota–dependent generation of TMAO may extend beyond the development and progression of atherosclerosis and its adverse complications (MI, stroke, or death). For example, we recently observed that circulating TMAO levels are higher in patients with heart failure compared with age- and gender-matched subjects without heart failure (56). Moreover, we observed a remarkably strong adverse prognostic value associated with elevated plasma TMAO levels among a cohort of stable patients with heart failure (n = 720) that was incremental to traditional risk factors, cardio-renal indices (B-type natriuretic peptide and glomerular filtration rate), and markers of systemic inflammation (C-reactive protein) (56). On the other hand, TMAO has been known to accumulate in the plasma of patients with impaired renal function, such as those with chronic kidney disease (CKD) and end-stage renal disease (57). Exciting new metabolomics data from the Framingham Heart Study indicated that TMAO was one of the few metabolites in the plasma of healthy subjects whose levels predicted incident development of CKD (58). Furthermore, results from recent studies in mice fed a high-fat diet also suggest that dietary TMAO may exacerbate impaired glucose tolerance, obstruct hepatic insulin signaling, and promote adipose tissue inflammation (59). The scope of biological processes affected by both TMAO and the TMAO production pathway is just starting to be appreciated, with multiple newly recognized links to chronic cardiometabolic disorders.
A diet/meta-organismal pathway is involved in TMAO formation
The precise mechanisms through which TMAO leads to CVD and adverse events are still under investigation. The striking associations between circulating TMAO levels and CVD risks observed in several large-scale clinical cohorts, coupled with the growing body of animal model data demonstrating direct mechanistic links with atherosclerosis and alterations in cholesterol/sterol metabolism (25, 37, 39, 54, 55, 60), collectively suggest that the meta-organismal pathway involving diet, gut microbes, TMA, liver FMO3, and TMAO may be an important new paradigm to consider for an improved understanding of atherosclerotic heart disease and perhaps other cardiometabolic disease processes. There may also exist other metabolites downstream of or in parallel with this pathway that might contribute to the observed findings with TMAO. More global approaches are warranted to study the “meta”-metabolome, and deciphering the functional participation, if any, of gut microbiota in diseases. Indeed, gut microbiota may influence disease susceptibility and responses to dietary exposures, as well as the magnitude of host genetic variant influences on disease phenotypes. The broader and more complex interrelationships between intestinal microbiota and their human host under differing environmental exposures needs to be considered as a potential contributor to the CVD process.
The studies described above thus reveal new potential dietary changes, gut microbial composition and enzyme pathways, and host enzymes (e.g., hepatic FMO3) as potential treatment targets to reduce cardiovascular risk (Figure 3). We will examine each step in this meta-organismal pathway and discuss potential interventions suggested for reducing TMAO generation.
Figure 3. Examples of microbial enzymes that generate TMA, using choline or carnitine as substrates.

Thus far, 2 distinct microbial enzyme systems have been identified in vitro that can produce TMA from either choline or carnitine as substrate (75, 77). The choline-utilizing enzyme cutC (catalytic polypeptide) and its partner cutD (regulatory polypeptide) make a complex that selectively uses choline as substrate and releases TMA (choline TMA lyase activity). Similarly, a microbial oxygenase (CntA) complexes with the microbial reductase (CntB) to form an active complex that selectively uses carnitine as substrate and forms TMA (carnitine TMA lyase activity). In theory, inhibition of microbial choline and/or carnitine TMA lyases may serve as a potential therapeutic target for CVD through the reduction of microbial TMA and subsequent host TMAO formation.
Modifying dietary substrate.
Modifying dietary nutrient intake is an obvious potential intervention that may influence the TMAO-producing meta-organismal pathway. Altered dietary exposure may potentially affect TMAO production by either reduction in the direct precursor substrate for TMA production or by altering gut microbial community composition, reducing the synthetic capacity to produce TMA from the different TMA-containing nutrients. TMA has long been recognized as an odorous byproduct of choline, PC, and L-carnitine during the decomposition of plants and animals, often by enterobacteria (31). Choline (free and esterified forms) and L-carnitine are the most common dietary nutrients ingested that produce TMA and TMAO (30, 37–39, 55).
Choline is present in most animal and some plant products, but is particularly abundant in egg yolk, meats, liver, fish, high-fat dairy products, and some nuts (34). The major dietary source of choline is PC (or lecithin), the primary phospholipid in membranes. Choline can be metabolized into betaine and function as a methyl donor that produces S-adenosylmethionine, influencing DNA and histone methylation (26, 32, 35). Severe choline deficiency causes neurologic impairment, and the targeting of choline metabolism enzymes is under investigation to promote tumor growth arrest (36, 61). In vivo studies in which trimethyl-d9–labeled betaine or PC were ingested showed a direct generation of d9-TMA and d9-TMAO within plasma and urine (37, 39, 55). Interestingly, only choline (and not betaine or lecithin) was found to produce a detectable rise in urine TMAO excretion in one small human study employing a variety of dietary exposures (30). More recent studies examining ingestion of either hard-boiled egg or egg yolk (37, 38, 62) or ingestion of trimethyl-d9-PC (37) have confirmed direct generation of TMAO from PC ingestion in humans. L-carnitine is a TMA nutrient that, in contrast to choline, is not required in our diets, since it is endogenously produced in mammals from dietary lysine, the single most abundant amino acid in plant and animal proteins (30, 63, 64). Omnivores obtain most L-carnitine from their diet. In cells, carnitine functions in the transport of fatty acids into mitochondria (65).
Epidemiologic studies have assessed the contributions of these dietary nutrients and CVD risks. High intake of choline and betaine was associated with diminished inflammation in one study (66, 67). However, an alternative report noted only a modest association with choline and betaine intake and CVD (68), which lost significance following adjustments for CVD risk markers (67, 69). Nevertheless, any nutritional study has a confounding factor of evaluating a broad mix of nutritional factors that may have opposing effects. Egg whites, for example, are large sources of immunmodulatory and antioxidant proteins (70), which are often ingested together with egg yolks if eggs are consumed.
As choline, PC, and carnitine are primary sources of gut microbiota–associated TMAO production, dietary modulation is a logical intervention strategy. We previously showed that vegetarians and vegans have markedly reduced synthetic capacity to make TMA and TMAO from dietary L-carnitine and have lower plasma TMAO levels than omnivores (39). Similarly, studies from our group (39) and others (71) show that gut microbial communities differ in vegetarians and vegans compared with omnivores. In animal model studies, chronic exposure to dietary L-carnitine increased TMA synthetic capacity by 10-fold (as monitored in a defined oral challenge of trimethyl-d9-carnitine), with a concurrent shift in gut microbial composition (39). Thus, chronic dietary exposure (e.g., omnivore vs. vegan/vegetarian among humans, or normal chow vs. chow plus L-carnitine in mouse studies) shifts gut microbial composition, with a selective advantage for bacterial species that prefer L-carnitine as a carbon fuel source to increase in proportion within the community and amplify the potential to produce TMA. Chronic dietary exposure can produce a profound effect on TMA synthetic capacity, whereas single (acute) dietary exposures affect TMA and TMAO production predominantly via modulation of precursor substrate availability.
While elimination of L-carnitine from the diet is a potentially attainable goal that may reduce some TMAO formation, because choline is an essential nutrient, its complete elimination from the diet is unwise. Further, bile has a very high total choline (PC) content, and the rapid turnover and sloughing of intestinal epithelial cells results in significant exposure of distal gut segments (and hence microbes) to choline, independent of dietary intake. Absorbent removal of TMA from the intestines by specific oral binders is a challenging but potentially feasible therapeutic approach for lowering TMA and TMAO levels. Such a strategy has been tested by the removal of gut microbiota–associated uremic toxins (72), and in animal models of renal dysfunction, the oral charcoal absorbent AST-120 significantly decreased necrotic areas and lessened aortic deposition of the uremic toxin indoxyl sulfate without affecting lesional macrophage or collagen content (73) while attenuating monocyte inflammation (74).
Regulating microbial metabolism: contributions of microbial enzymes.
Significant progress has recently been made in identifying some of the microbial enzymes that likely contribute to TMA formation from different dietary substrates. Thus far, 2 distinct classes of enzymes have been reported that differ in their overall catalytic strategy for cleaving the C-N bond in choline and carnitine (Figure 3 and refs. 75–77). TMA produced by these enzymes can actually be considered a bacterial waste product released by the microbial enzyme systems, with the remaining carbon fuel source liberated from the parent substrate (choline/PC or carnitine) being used. By identifying known microbial enzymes that can use the predicted cleavage product as substrate after TMA release, bioinformatics and genome mining approaches were used to identify a gene cluster responsible for anaerobic choline degradation within the genome of the choline-degrading bacterium Desulfovibrio desulfuricans (75, 76). Biochemical characterization of the microbial enzyme revealed a choline-specific TMA lyase that used an unusual glycyl radical enzyme intermediate. The microbial enzyme was found within a “choline utilization” (cut) gene cluster that contains several tightly clustered genes, including a catalytic unit (cutC) and a regulatory polypeptide (cutD), both of which were required for anaerobic TMA production from choline (75, 76). In other studies that used a similar bioinformatics approach with Acinetobacter baumannii as the model, an alternative microbial enzyme cluster (catalytic and regulatory protein CntA and CntB) specific for carnitine TMA lyase activity was also recently reported (77). Characterization of recombinant isolated CntA/B showed they formed a 2-component Rieske-type oxygenase/reductase complex (77).
The relative quantitative contributions of cutC/D- or CntA/B-type microbial enzyme clusters to TMA and TMAO production in humans is not yet known. Indeed, the existence of additional microbial enzyme strategies for cleaving and liberating TMA from dietary nutrients like choline, PC, and carnitine (as well as other TMA-containing species) remains to be established. Given the vast genetic diversity of gut microbes (10- to 100-fold greater than that found in Homo sapiens), it seems probable that additional microbial enzymes exist that can produce TMA from diverse nutrients. Targeting these enzymes as a potential therapeutic strategy to limit TMAO production represents an attractive though daunting task. Alternatively, modulating the microbial community through ingestion of a probiotic (a defined microbial composition) or prebiotic (a non-microbial ingested material that influences gut microbe community structure and composition) to elicit a reduction in TMA synthetic capacity and TMAO levels in the host is another therapeutic strategy.
Inhibiting host metabolism: contributions of hepatic FMOs.
The conversion from TMA to TMAO requires an oxidation step that is mediated by host enzyme machinery in the form of FMOs (54, 78–81). Gut microbe–produced TMA reaches the liver rapidly via the portal circulation, where a cluster of hepatic FMO enzymes efficiently oxidizes TMA into TMAO (54, 82). Previous studies have shown that subjects with a genetic defect in FMO3 can have markedly elevated TMA levels, leading to a noxious body odor that characterizes the condition (fish odor syndrome or trimethylaminuria [TMAU]) (83, 84). Given that TMAO, and not TMA, appears to be the culprit metabolite that fosters a proatherogenic effect, one might expect that subjects with this genetic disorder are protected from CVD. However, its rarity has thus far precluded report of any known cardiovascular phenotype. Interestingly, other than TMA, little is known about the endogenous substrates of the FMOs; rather more is known about the ability of FMOs to oxidize a wide range of xenobiotics and toxicants, including drugs such as barbiturates. Collectively, the FMOs are thought to oxidize a large number of endogenous amines (including TMA) as well as structurally diverse sulfur-, phosphorus-, and selenium-containing compounds. In recent studies each of the FMO family members were cloned and expressed, to determine which possessed synthetic capacity to use TMA as a substrate to generate TMAO. FMO1, FMO2, and FMO3 were all capable of forming TMAO, though the specific activity of FMO3 was at least 10-fold higher than that the other FMOs (54). Further, FMO3 overexpression in mice significantly increased plasma TMAO levels, while silencing FMO3 decreased TMAO levels (54). In both humans and mice, hepatic FMO3 expression was observed to be reduced in males compared with females (25, 54) and could be induced by dietary bile acids through a mechanism that involves FXR (54). Recent studies among multiple different inbred strains of mice in a “mouse diversity panel” linked variations in strain and sex to hepatic expression/activity of FMO3, TMAO levels, and atherosclerosis (54). Hence, further investigations using FMO3 loss- and gain-of-function experimental models to more directly probe this aspect of the TMAO pathway and cardiometabolic phenotypes are warranted.
In recent studies we examined natural genetic variants and hepatic expression levels of genes among multiple inbred strains of mice and humans to identify potential genetic determinants of TMAO production and their associations with CVD phenotypes (60). FMO3 and TMAO were significantly correlated, and TMAO levels accounted for 11% of the variation in atherosclerosis across the inbred strains of mice examined (60). We used a comparative GWAS approach to discover loci for plasma TMAO levels in mice and humans, and identified a locus for TMAO levels on chromosome 3 (P < 0.001) that co-localized with a highly significant (P < 0.001) cis-expression quantitative trait locus for solute carrier family 30 member 7 (Slc30a7) (60). Although this zinc transporter might represent a positional candidate gene responsible for the association signal at this locus in mice (60), these are weak associations in GWASs. Furthermore, GWASs for plasma TMAO levels in 1,973 humans identified 2 loci with suggestive evidence of association (P < 0.001) on chromosomes 1q23.3 and 2p12; however, genotyping of the lead variants at these loci in 1,892 additional subjects failed to replicate their association with plasma TMAO levels (60). The relatively limited genetic signals observed for TMAO levels in humans thus far is consistent with the concept that interpersonal differences in diet and the repertoire of gut microbial species, moreso than host genetic variants, likely serve as the primary determinants of plasma TMAO levels (60).
Pharmacologic inhibition of the FMOs in general and FMO3 specifically is expected to reduce TMAO production and potentially serve as a therapy for CVD risk reduction. However, given the known adverse effects of FMO3 inhibition from sufferers of fish odor syndrome, the untoward odorous side effects of inhibiting this enzyme make it a less attractive target.
Concluding remarks
Results of the studies described above suggest that a broader view of human metabolism, physiology, and disease must be considered. We are walking communities comprised not only of a Homo sapiens host, but also of trillions of symbiotic commensal microorganisms within the gut and on every other surface of our bodies. Gut microbiota serve as a filter for our largest environmental exposure — what we eat — and the microbial community within each of us significantly influences how we experience a meal. The global meta-metabolome within is remarkably complex and is influenced by dietary inputs, microbial composition, host genetic factors, and other external exposures. We need to appreciate that our gut microbial communities make up a large and plastic endocrine organ that may influence multiple metabolic and physiological processes. Simply cataloging the microbes within is not sufficient. Indeed, simply sequencing the microbial genes is insufficient because, as with eukaryotic cells, the presence of DNA alone does not necessarily translate into protein synthesis and function. Future studies need to focus on discovery and understanding at the functional level of specific microbial pathways and products that contribute to our physiology and that may contribute to disease processes. Gut microbiota represent a new target for therapeutic manipulation and targeting for the treatment and prevention of complex cardiometabolic diseases. The exploration of gut microbiota contributions to human host physiology and diseases represents an incredibly exciting and potentially fertile territory in biomedical research.
Acknowledgments
This research was supported by the NIH and the Office of Dietary Supplements (grants R01HL103866 and P20HL113452). The GeneBank study has been supported by NIH grants P01HL076491, P01HL098055, and R01HL103931 and by the Cleveland Clinic Clinical Research Unit of Case Western Reserve University Clinical Translational Science Award program (UL1TR 000439-06). S.L. Hazen is also partially supported by a gift from the Leonard Krieger endowment and by the Foundation LeDucq.
Footnotes
Conflict of interest: S.L. Hazen is named as co-inventor on pending patents held by the Cleveland Clinic relating to cardiovascular diagnostics. S.L. Hazen has been paid as a consultant for Abbott Laboratories, Cleveland HeartLab Inc., Esperion Therapeutics, Eli Lilly and Company, LipoScience Inc., Merck, Procter & Gamble, and Pfizer; has received research funds from Cleveland HeartLab Inc., LipoScience Inc., Procter & Gamble, Pfizer, and Takeda Pharmaceutical Company; and has the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Abbott Laboratories, Cleveland HeartLab Inc., Esperion Therapeutics, Frantz BioMarkers LLC, Siemens, and LipoScience Inc.
Reference information:J Clin Invest. 2014;124(10):4204–4211. doi:10.1172/JCI72331.
References
- 1.Go AS, et al. Heart disease and stroke statistics — 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nabel EG. Cardiovascular disease. N Engl J Med. 2003;349(1):60–72. doi: 10.1056/NEJMra035098. [DOI] [PubMed] [Google Scholar]
- 3.Ardissino D, et al. Influence of 9p21.3 genetic variants on clinical and angiographic outcomes in early-onset myocardial infarction. J Am Coll Cardiol. 2011;58(4):426–434. doi: 10.1016/j.jacc.2010.11.075. [DOI] [PubMed] [Google Scholar]
- 4.Ripatti S, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376(9750):1393–1400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 7.Freedman LS, Schatzkin A, Midthune D, Kipnis V. Dealing with dietary measurement error in nutritional cohort studies. J Natl Cancer Inst. 2011;103(14):1086–1092. doi: 10.1093/jnci/djr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Human Microbiome Project Consortium Structure, function diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faith JJ, et al. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;509(7500):357–360. doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 13.Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 14.Taki K, Tsuruta Y, Niwa T. Indoxyl sulfate and atherosclerotic risk factors in hemodialysis patients. Am J Nephrol. 2007;27(1):30–35. doi: 10.1159/000098542. [DOI] [PubMed] [Google Scholar]
- 15.Meijers BK, et al. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis. 2009;54(5):891–901. doi: 10.1053/j.ajkd.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 16.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13(11):790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 18.Collins SM. A role for the gut microbiota in IBS. Nat Rev Gastroenterol Hepatol. 2014;11(8):497–505. doi: 10.1038/nrgastro.2014.40. [DOI] [PubMed] [Google Scholar]
- 19.Dumas ME, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103(33):12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumas ME, Kinross J, Nicholson JK. Metabolic phenotyping and systems biology approaches to understanding metabolic syndrome and fatty liver disease. Gastroenterology. 2014;146(1):46–62. doi: 10.1053/j.gastro.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Ridaura VK, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587(pt 17):4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ueland PM, Holm PI, Hustad S. Betaine: a key modulator of one-carbon metabolism and homocysteine status. Clin Chem Lab Med. 2005;43(10):1069–1075. doi: 10.1515/CCLM.2005.187. [DOI] [PubMed] [Google Scholar]
- 27.Lever M, Slow S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin Biochem. 2010;43(9):732–744. doi: 10.1016/j.clinbiochem.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Barrett EL, Kwan HS. Bacterial reduction of trimethylamine oxide. Annu Rev Microbiol. 1985;39:131–149. doi: 10.1146/annurev.mi.39.100185.001023. [DOI] [PubMed] [Google Scholar]
- 29.Smith JL, Wishnok JS, Deen WM. Metabolism and excretion of methylamines in rats. Toxicol Appl Pharmacol. 1994;125(2):296–308. doi: 10.1006/taap.1994.1076. [DOI] [PubMed] [Google Scholar]
- 30.Zhang AQ, Mitchell SC, Smith RL. Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol. 1999;37(5):515–520. doi: 10.1016/S0278-6915(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 31.Bain MA, Fornasini G, Evans AM. Trimethylamine: metabolic, pharmacokinetic and safety aspects. Curr Drug Metab. 2005;6(3):227–240. doi: 10.2174/1389200054021807. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. J Lipid Res. 2008;49(6):1187–1194. doi: 10.1194/jlr.R700019-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Geiger O, Lopez-Lara IM, Sohlenkamp C. Phosphatidylcholine biosynthesis and function in bacteria. Biochim Biophys Acta. 2013;1831(3):503–513. doi: 10.1016/j.bbalip.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 34.USDA. USDA Database for the Choline Content of Common Foods: Release 2 (2008). USDA Web site.http://www.ars.usda.gov/Services/docs.htm?docid=6232. Updated May 17, 2013. Accessed on July 24, 2014
- 35.Ueland PM. Choline and betaine in health and disease. J Inherit Metab Dis. 2011;34(1):3–15. doi: 10.1007/s10545-010-9088-4. [DOI] [PubMed] [Google Scholar]
- 36.Zeisel SH. Nutritional importance of choline for brain development. J Am Coll Nutr. 2004;23(6):621S–626S. doi: 10.1080/07315724.2004.10719433. [DOI] [PubMed] [Google Scholar]
- 37.Tang WH, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller CA, et al. Effect of egg ingestion on trimethylamine-N-oxide production in humans: a randomized, controlled, dose-response study. Am J Clin Nutr. doi: 10.3945/ajcn.114.087692. [published online ahead of print June 18, 2014]. doi: 10.3945/ajcn.114.087692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koeth RA, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan A, et al. Red meat consumption and mortality: results from 2 prospective cohort studies. Arch Intern Med. 2012;172(7):555–563. doi: 10.1001/archinternmed.2011.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spence JD, Jenkins DJ, Davignon J. Egg yolk consumption and carotid plaque. Atherosclerosis. 2012;224(2):469–473. doi: 10.1016/j.atherosclerosis.2012.07.032. [DOI] [PubMed] [Google Scholar]
- 42.Kaluza J, Akesson A, Wolk A. Processed and unprocessed red meat consumption and risk of heart failure: prospective study of men. Circ Heart Fail. 2014;7(4):552–557. doi: 10.1161/CIRCHEARTFAILURE.113.000921. [DOI] [PubMed] [Google Scholar]
- 43.Kaluza J, Wolk A, Larsson SC. Red meat consumption and risk of stroke: a meta-analysis of prospective studies. Stroke. 2012;43(10):2556–2560. doi: 10.1161/STROKEAHA.112.663286. [DOI] [PubMed] [Google Scholar]
- 44.Larsson SC, Orsini N. Red meat and processed meat consumption and all-cause mortality: a meta-analysis. Am J Epidemiol. 2014;179(3):282–289. doi: 10.1093/aje/kwt261. [DOI] [PubMed] [Google Scholar]
- 45.Djousse L, Gaziano JM. Egg consumption in relation to cardiovascular disease and mortality: the Physicians’ Health Study. Am J Clin Nutr. 2008;87(4):964–969. doi: 10.1093/ajcn/87.4.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Djousse L, Gaziano JM. Egg consumption and risk of heart failure in the Physicians’ Health Study. Circulation. 2008;117(4):512–516. doi: 10.1161/CIRCULATIONAHA.107.734210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qureshi AI, Suri FK, Ahmed S, Nasar A, Divani AA, Kirmani JF. Regular egg consumption does not increase the risk of stroke and cardiovascular diseases. Med Sci Monit. 2007;13(1):CR1–CR8. [PubMed] [Google Scholar]
- 48.Scrafford CG, Tran NL, Barraj LM, Mink PJ. Egg consumption and CHD and stroke mortality: a prospective study of US adults. Public Health Nutr. 2011;14(2):261–270. doi: 10.1017/S1368980010001874. [DOI] [PubMed] [Google Scholar]
- 49.Tran NL, Barraj LM, Heilman JM, Scrafford CG. Egg consumption and cardiovascular disease among diabetic individuals: a systematic review of the literature. Diabetes Metab Syndr Obes. 2014;7:121–137. doi: 10.2147/DMSO.S58668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lidbury I, Murrell JC, Chen Y. Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc Natl Acad Sci U S A. 2014;111(7):2710–2715. doi: 10.1073/pnas.1317834111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raymond JA. Seasonal variations of trimethylamine oxide and urea in the blood of a cold-adapted marine teleost, the rainbow smelt. Fish Physiol Biochem. 1994;13(1):13–22. doi: 10.1007/BF00004115. [DOI] [PubMed] [Google Scholar]
- 52.Bennion BJ, Daggett V. Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: a chemical chaperone at atomic resolution. Proc Natl Acad Sci U S A. 2004;101(17):6433–6438. doi: 10.1073/pnas.0308633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Svensson BG, Akesson B, Nilsson A, Paulsson K. Urinary excretion of methylamines in men with varying intake of fish from the Baltic Sea. J Toxicol Environ Health. 1994;41(4):411–420. doi: 10.1080/15287399409531853. [DOI] [PubMed] [Google Scholar]
- 54.Bennett BJ, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(1):49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z, et al. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 2014;35(14):904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang WH, et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite, trimethylamine-N-oxide, in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 201361(10_S). doi:10.1016/S0735-1097(13)60750-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bell JD, Lee JA, Lee HA, Sadler PJ, Wilkie DR, Woodham RH. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: identification of trimethylamine-N-oxide. Biochim Biophys Acta. 1991;1096(2):101–107. doi: 10.1016/0925-4439(91)90046-c. [DOI] [PubMed] [Google Scholar]
- 58.Rhee EP, et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J Am Soc Nephrol. 2013;24(8):1330–1338. doi: 10.1681/ASN.2012101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao X, Liu X, Xu J, Xue C, Xue Y, Wang Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J Biosci Bioeng. 2014. p. S1389-1723(14)00081-4.. [DOI] [PubMed]
- 60.Hartiala J, et al. Comparative genome-wide association studies in mice and humans for trimethylamine N-oxide, a proatherogenic metabolite of choline and L-carnitine. Arterioscler Thromb Vasc Biol. 2014;34(6):1307–1313. doi: 10.1161/ATVBAHA.114.303252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeisel SH. Nutritional genomics: defining the dietary requirement and effects of choline. J Nutr. 2011;141(3):531–534. doi: 10.3945/jn.110.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.West AA, et al. Egg n-3 fatty acid composition modulates biomarkers of choline metabolism in free-living lacto-ovo-vegetarian women of reproductive age. J Acad Nutr Diet. 2014. p. S2212-2672(14)00190-7. [DOI] [PubMed]
- 63.Rebouche CJ, Seim H. Carnitine metabolism and its regulation in microorganisms and mammals. Annu Rev Nutr. 1998;18:39–61. doi: 10.1146/annurev.nutr.18.1.39. [DOI] [PubMed] [Google Scholar]
- 64.Bain MA, Faull R, Milne RW, Evans AM. Oral L-carnitine: metabolite formation and hemodialysis. Curr Drug Metab. 2006;7(7):811–816. doi: 10.2174/138920006778520561. [DOI] [PubMed] [Google Scholar]
- 65.Marcovina SM, et al. Translating the basic knowledge of mitochondrial functions to metabolic therapy: role of L-carnitine. Transl Res. 2013;161(2):73–84. doi: 10.1016/j.trsl.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Detopoulou P, Panagiotakos DB, Antonopoulou S, Pitsavos C, Stefanadis C. Dietary choline and betaine intakes in relation to concentrations of inflammatory markers in healthy adults: the ATTICA study. Am J Clin Nutr. 2008;87(2):424–430. doi: 10.1093/ajcn/87.2.424. [DOI] [PubMed] [Google Scholar]
- 67.Rajaie S, Esmaillzadeh A. Dietary choline and betaine intakes and risk of cardiovascular diseases: review of epidemiological evidence. ARYA Atheroscler. 2011;7(2):78–86. [PMC free article] [PubMed] [Google Scholar]
- 68.Bidulescu A, Chambless LE, Siega-Riz AM, Zeisel SH, Heiss G. Usual choline betaine dietary intake incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc Disord. 2007;7:20. doi: 10.1186/1471-2261-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dalmeijer GW, Olthof MR, Verhoef P, Bots ML, van der Schouw YT. Prospective study on dietary intakes of folate, betaine, and choline and cardiovascular disease risk in women. Eur J Clin Nutr. 2008;62(3):386–394. doi: 10.1038/sj.ejcn.1602725. [DOI] [PubMed] [Google Scholar]
- 70.Mine Y. Egg proteins and peptides in human health — chemistry, bioactivity and production. Curr Pharm Des. 2007;13(9):875–884. doi: 10.2174/138161207780414278. [DOI] [PubMed] [Google Scholar]
- 71.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lekawanvijit S, et al. The uremic toxin adsorbent AST-120 abrogates cardiorenal injury following myocardial infarction. PLoS One. 2013;8(12):e83687. doi: 10.1371/journal.pone.0083687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamamoto S, et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol Dial Transplant. 2011;26(8):2491–2497. doi: 10.1093/ndt/gfq759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ito S, et al. Reduction of indoxyl sulfate by AST-120 attenuates monocyte inflammation related to chronic kidney disease. J Leukoc Biol. 2013;93(6):837–845. doi: 10.1189/jlb.0112023. [DOI] [PubMed] [Google Scholar]
- 75.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A. 2012;109(52):21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9(7):1408–1413. doi: 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- 77.Zhu Y, et al. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc Natl Acad Sci U S A. 2014;111(11):4268–4273. doi: 10.1073/pnas.1316569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cashman JR, Zhang J. Human flavin-containing monooxygenases. Annu Rev Pharmacol Toxicol. 2006;46:65–100. doi: 10.1146/annurev.pharmtox.46.120604.141043. [DOI] [PubMed] [Google Scholar]
- 79.Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ. Flavin dependent monooxygenases. Arch Biochem Biophys. 2014;544:2–17. doi: 10.1016/j.abb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 80.Cashman JR. Role of flavin-containing monooxygenase in drug development. Expert Opin Drug Metab Toxicol. 2008;4(12):1507–1521. doi: 10.1517/17425250802522188. [DOI] [PubMed] [Google Scholar]
- 81.Zhang J, Cashman JR. Quantitative analysis of FMO gene mRNA levels in human tissues. Drug Metab Dispos. 2006;34(1):19–26. doi: 10.1124/dmd.105.006171. [DOI] [PubMed] [Google Scholar]
- 82.Cashman JR, et al. In vitro and in vivo inhibition of human flavin-containing monooxygenase form 3 (FMO3) in the presence of dietary indoles. Biochem Pharmacol. 1999;58(6):1047–1055. doi: 10.1016/S0006-2952(99)00166-5. [DOI] [PubMed] [Google Scholar]
- 83.Messenger J, Clark S, Massick S, Bechtel M. A review of trimethylaminuria: (fish odor syndrome). J Clin Aesthet Dermatol. 2013;6(11):45–48. [PMC free article] [PubMed] [Google Scholar]
- 84.Ayesh R, Mitchell SC, Zhang A, Smith RL. The fish odour syndrome: biochemical, familial, and clinical aspects. BMJ. 1993;307(6905):655–657. doi: 10.1136/bmj.307.6905.655. [DOI] [PMC free article] [PubMed] [Google Scholar]