

Abstract
Liraglutide is a glucagon-like peptide-1 (GLP-1) analog marketed for the treatment of type 2 diabetes. Besides lowering blood glucose, liraglutide also reduces body weight. It is not fully understood how liraglutide induces weight loss or to what degree liraglutide acts directly in the brain. Here, we determined that liraglutide does not activate GLP-1–producing neurons in the hindbrain, and liraglutide-dependent body weight reduction in rats was independent of GLP-1 receptors (GLP-1Rs) in the vagus nerve, area postrema, and paraventricular nucleus. Peripheral injection of fluorescently labeled liraglutide in mice revealed the presence of the drug in the circumventricular organs. Moreover, labeled liraglutide bound neurons within the arcuate nucleus (ARC) and other discrete sites in the hypothalamus. GLP-1R was necessary for liraglutide uptake in the brain, as liraglutide binding was not seen in Glp1r–/– mice. In the ARC, liraglutide was internalized in neurons expressing proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART). Electrophysiological measurements of murine brain slices revealed that GLP-1 directly stimulates POMC/CART neurons and indirectly inhibits neurotransmission in neurons expressing neuropeptide Y (NPY) and agouti-related peptide (AgRP) via GABA-dependent signaling. Collectively, our findings indicate that the GLP-1R on POMC/CART-expressing ARC neurons likely mediates liraglutide-induced weight loss.
Introduction
Most drugs that have been available to treat obesity are small molecules that cross the blood-brain barrier (BBB) and affect different neuronal networks. Several of those compounds have a rather broad spectrum of effects in the brain, sometimes leading to CNS side effects (1). New agents being considered for the treatment of obesity are analogs of the peripheral peptide hormones, like glucagon-like peptide-1 (GLP-1), peptide YY, and glucagon, and some are antagonists for receptors, like the ghrelin receptor (2, 3). These hormones are part of the gut-brain axis, and their respective receptors are often present in the periphery as well as in the brain (4–6). While many studies describe administration of hormones or analogs directly into the brain, surprisingly little is known about how and to what extent these physiologically secreted or peripherally administered peptide hormones gain access to the brain and how they may affect the key neuronal pathways that regulate energy balance, such as the neuropeptide Y/agouti-related peptide (NPY/AgRP) and proopiomelanocortin/cocaine- and amphetamine-regulated transcript (POMC/CART) neurons located in the arcuate nucleus (ARC) (7–11). In order to avoid on- or off-target CNS side effects, it would seem desirable that new drugs for the treatment of obesity specifically target those neurons.
During the past two decades the physiology and pharmacology of GLP-1 and GLP-1 analogs in glucose, food intake, and body weight control have been gradually dissected (12, 13). Both peripheral and brain GLP-1 receptors (GLP-1Rs) seem to be involved in mediating the specific effects (4). The physiology and pharmacology of GLP-1 are somewhat different. Physiologically, GLP-1 is a strong regulator of gastric emptying (GE), but this effect is subject to rapid tachyphylaxis upon continuous stimulation (14, 15). Pharmacologically, only short-acting GLP-1 analogs, like exenatide and lixisenatide, display a marked reduction of GE, which may contribute to short-term effects on food intake, while liraglutide and exenatide, formulated for slow release, have only a minor effect on GE, which then cannot be the mediator of the body weight effects (16, 17). The primary blood glucose-lowering effects of long-acting GLP-1 analogs are increases in glucose-dependent insulin secretion and lowering of glucagon secretion (18, 19). Apart from its effects to reduce blood glucose, peripherally circulating GLP-1 is believed to be a physiological satiety factor (20, 21). In the CNS, GLP-1 is a neurotransmitter in brain stem–hypothalamus pathways signaling satiety (4, 22, 23). The potential for peripherally administered GLP-1 as an antiobesity drug was first shown in humans in short-term studies with exogenous GLP-1, which showed reduced energy intake and effects on all components of appetite regulation: increased satiety and fullness and decreased hunger and prospective food consumption (24, 25).
As GLP-1 is a well-characterized neurotransmitter signaling satiety in the brain (22, 23), most studies aiming to elucidate the role of GLP-1 in appetite regulation have been based on administration of GLP-1 and analogs directly into the brain. Logically, peptides such as GLP-1 analogs would not be expected to readily cross the BBB and hence not readily be expected to be able to target GLP-1Rs in the brain. Nevertheless, some studies have shown that GLP-1 analogs seem to pass the BBB, although no clear details as to areas targeted or mechanisms have been reported (26, 27). GLP-1Rs are abundant in a number of circumventricular organs (CVOs), and it has been demonstrated that circulating GLP-1 can bind these receptors (28, 29). However, given the appetite- and weight-reducing effects of long-acting GLP-1 analogs, it is tempting to speculate that central GLP-1Rs behind the BBB can also be reached by peripherally circulating peptide-based GLP-1 analogs. Interestingly, ghrelin, which is another peripherally circulating peptide hormone known to activate receptors on NPY neurons behind the BBB, was recently proposed to gain direct access to the hypothalamus perhaps via fenestrated capillaries (30).
Liraglutide is the first GLP-1 analog that is under development for the obesity indication. Liraglutide dose dependently lowers body weight by reducing energy intake via an overall appetite reduction (31, 32). Phase 3 clinical trials have been completed, and applications to market liraglutide as a drug for treatment of obesity have been filed in the US and EU. Here, we show that liraglutide has important effects on CART as well as NPY/AgRP levels in the ARC. We used fluorescently labeled liraglutide injected peripherally to describe access to specific brain areas using a highly sensitive scanning technique. Different surgical lesioning techniques were used to assign the body weight–lowering effect to specific brain areas, and immunohistochemistry, in situ hybridization, and electrophysiology were used to suggest specific cellular and neuronal actions. Peripherally injected peptide-based analogs like liraglutide accessed not only CVOs but also neurons in the hypothalamus. Particularly, liraglutide directly influenced the balance in the orexigenic and anorexigenic pathways in the ARC. These data have important clinical implications, as they underline the ability of a pharmacological signal to act centrally to regulate energy balance.
Results
Liraglutide-induced lowering of appetite is independent of GLP-1R of the vagus nerve and the area postrema.
To address the role of the vagal nerve and area postrema (AP) GLP-1Rs in body weight regulation, liraglutide was dosed to rats (s.c., bidaily [BID], 200 μg/kg) subjected to subdiaphragmatic vagal afferent deafferentation (SDA) and to rats with surgically ablated AP (APx). 14-day liraglutide treatment reduced body weight gain by approximately 10% in both SDA and sham-operated animals (P < 0.001, sham vehicle vs. sham liraglutide, SDA vehicle vs. SDA liraglutide, Figure 1A). Food intake was also lowered by liraglutide (data not shown). There was no difference in the effect of liraglutide on food intake or body weight between sham and SDA animals, indicating that the vagal nerve does not mediate the effect of liraglutide on body weight lowering. Histological verification of the SDA lesion involved anterograde tracing using wheat germ agglutinin from the nodose ganglion on the lesioned side and i.p. fluorogold injection. Lesioned animals displayed a lack of anterogradely labeled fibers in the nucleus of the solitary tract (NTS) (Figure 1, C and D) and reduced retrograde fluorogold labeling in the right dorsal motor nucleus in truncally lesioned animals (Figure 1, G and H). Administration of cholecystokinin-8 was used as a positive control and was confirmed to reduce food intake in sham but not in SDA animals (Figure 1I). Ablation of AP in itself led to a reduction in body weight, as demonstrated in the AP-lesioned group dosed with vehicle (Figure 1B). However, 14-day liraglutide treatment reduced body weight gain to the same degree in both sham and AP-lesioned animals (P < 0.001, sham vehicle vs. sham liraglutide, APx vehicle vs. APx liraglutide). Ablation of AP was verified by histological staining of the brains sectioned around AP of both sham (Figure 1E) and APx-operated animals (Figure 1F). Thus, the vagal nerve and the AP seem not to be primary mediators for the liraglutide-induced weight loss.
Figure 1. Vagal and AP contributions to liraglutide body weight change.

(A) Liraglutide treatment reduced body weight gain significantly in both sham and SDA rats (*P < 0.001 sham vehicle vs. sham liraglutide; #P < 0.001 SDA vehicle vs. SDA liraglutide). (B) Whereas AP lesion changed the body weight set point, liraglutide treatment reduced body weight gain to the same degree in both sham and AP-ablated animals (*P < 0.001 sham vehicle vs. sham liraglutide; #P < 0.001 APx vehicle vs. APx liraglutide). (C and D) Wheat germ agglutinin (WGA) injected into the left nodose ganglion labeled afferent fibers in sham animals only (arrows). Note persistence of retrograde labeled dorsal motor nucleus neurons in both groups (double arrows). (E and F) Ablation of AP was verified histologically in (E) sham and (F) AP-ablated rats. Data are mean ± SEM, and statistical analyses are performed using 2-way repeated-measures ANOVA, with Bonferroni post-hoc analyses applied. (G and H) Efferent labeling of fluorogold injected i.p. was reduced in the gastrointestinal part of dorsal motor nucleus due to right truncal vagotomy (arrows). (I) CCK8 (8 μg/kg i.p.) reduced 30-minute food intake in sham rats but not in SDA-operated rats (*P < 0.01). Scale bars: 200 μm (C and D), 500 μm (E–H).
Liraglutide regulates neuronal expression of CART and NPY/AgRP.
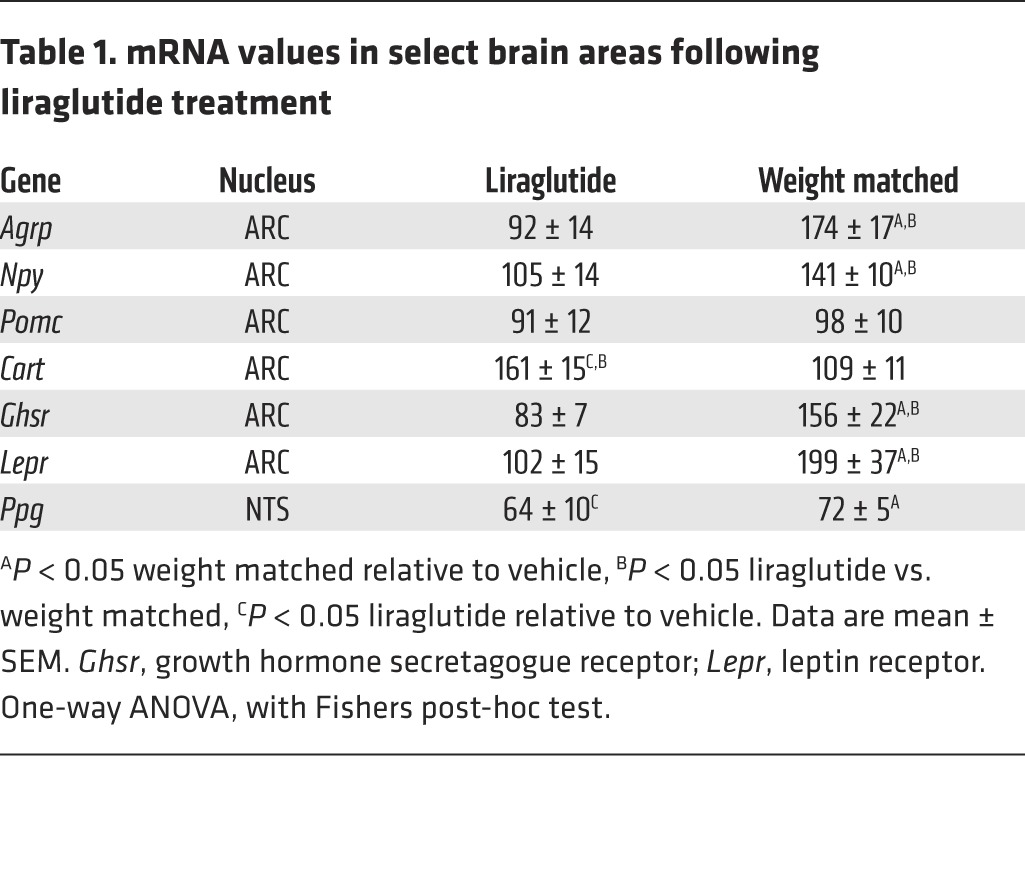
We next investigated the effects of long-term liraglutide treatment on ARC neuronal expression of POMC/CART and NPY/AgRP in male diet-induced obese (DIO) rats. Liraglutide treatment (s.c., BID, 200 μg/kg) for 28 days led to a significant reduction in food intake (P < 0.0001, vehicle vs. liraglutide, Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI75276DS1) and body weight (P < 0.001, vehicle vs. liraglutide, vehicle vs. weight matched, Supplemental Figure 1B), without significantly affecting energy expenditure (Supplemental Figure 1, C and D). Pair feeding was not sufficient to obtain the same body weight in vehicle-treated animals as in the liraglutide-treated animals; therefore, weight matching was used to obtain a group of vehicle-treated animals paired to have the same body weight as those on liraglutide. Subsequent evaluation of neuropeptide expression was performed by quantitative in situ hybridizations on hypothalamic brain sections (Figure 2, A and B). Liraglutide treatment significantly increased the mean Cart mRNA levels in the ARC 1.6-fold compared with both vehicle-treated, ad libitum–fed rats and weight-matched rats (P < 0.001, liraglutide vs. vehicle and vs. weight-matched), whereas Pomc expression was unaffected (Figure 2A). A 1.4-fold increase in Npy mRNA levels and a 1.7-fold increase in Agrp mRNA levels were observed in weight-matched rats, while levels were unaltered following treatment with liraglutide (P < 0.05, weight matched vs. vehicle and vs. liraglutide, Figure 2B), suggesting an ability of liraglutide to suppress the food-deprived hunger signals observed in the weight-matched control group. A follow-up study in male DIO rats fed a palatable chocolate-enriched diet and dosed for 28 days BID with liraglutide (200 μg/kg) showed the exact same pattern for these 4 ARC neuropeptides (data not shown). Although the ARC NPY/AgRP neurons did not express GLP-1R (Figure 2E), the marked suppression of NPY/AgRP expression that followed chronic liraglutide treatment prompted us to examine the effect of GLP-1R stimulation on NPY neurons. Brain slices from Npy-hrGFP mice were stimulated with GLP-1(7-36)amide, and NPY electrical responses were recorded. In 5 of 9 NPY cells (from 3 mice), GLP-1(7-36)amide (10 nM) caused a substantial outward current, with average current change of 20.8 ± 1.7 pA (Figure 2, C and F). As a positive control for cellular viability, only data from cells showing a positive inward current response from NMDA (100 μM) were used in this analysis. 12 cells from 8 animals were used in the presence of NMDA. Addition of the NMDA receptor agonist caused an inward current, with an average change of 41.1 ± 14.5 pA (Figure 2, C and F). To determine whether the inhibitory effect of GLP-1 on NPY neurons was presynaptic or postsynaptic, the effects of GLP-1(7-36)amide were determined in the presence of the GABA-A receptor antagonist bicuculline (20 μM). In 7 of 7 cells (from 5 mice), GLP-1(7-36)amide failed to change the current in the presence of bicuculline (Figure 2, D and F), demonstrating that GABA-A receptor activation was likely responsible for the inhibitory effects of GLP-1(7-36)amide on NPY neurons. Again, only data from cells responding to NMDA were used. In addition to the effects on the ARC neuropeptides, ghrelin and leptin receptor mRNA levels were significantly increased in weight-matched animals, and this was prevented by liraglutide treatment (P < 0.05, weight matched vs. vehicle and vs. liraglutide, Table 1). These changes in ghrelin and leptin receptor levels may also be involved in a circuit that overall leads to reduced appetite induced by GLP-1. Preproglucagon (Ppg) mRNA in the NTS was decreased in both weight-matched and liraglutide-treated rats compared with ad libitum–fed vehicle animals (P < 0.05, vehicle vs. weight matched and vs. liraglutide, Table 1). These data demonstrate that while NTS-produced GLP-1 is a physiological satiety factor; these neurons are not activated by liraglutide and thus possibly not involved in mediating the effects of long-acting GLP-1R agonists.
Figure 2. Liraglutide treatment regulates ARC gene expression and ARC neuronal activity.

(A) Liraglutide treatment for 28 days in DIO rats significantly increased mean Cart mRNA levels in the ARC (*P < 0.001 liraglutide vs. vehicle and vs. weight matched), whereas Pomc expression was unaffected. (B) Npy and Agrp mRNA levels were significantly increased in weight-matched rats — but not following treatment with liraglutide (#P < 0.05 weight matched vs. vehicle and vs. liraglutide). Data are mean ± SEM, and statistical analyses were performed using 1-way ANOVA, with Fishers post-hoc test. (C) Voltage-clamp recording of ARC-NPY neurons showed an increased outward current in the presence of GLP-1(7-36)amide (blue line) and an inward current with NMDA (red line). (D) Simultaneous GABA receptor inhibition by bicuculline (black line) showed a lack of change in the current with the addition of GLP-1(7-36)amide; however, NMDA retained the ability to cause an inward current. (E) The action of GLP-1(7-36) amide was not directly through GLP-1Rs on NPY/AgRP neurons, as no colocalization was observed between GLP-1R– (red, yellow arrows) and NPY/AgRP-positive (green, white arrows) neurons. Scale bars: 100 μm. (F) The effects of GLP-1(7-36)amide in the presence of bicuculline or NMDA on ARC-NPY neurons are summarized (mean ± SEM).
Table 1.
mRNA values in select brain areas following liraglutide treatment
Liraglutide gains access to hypothalamus following peripheral administration.
Due to the effects seen with liraglutide on CART and AgRP/NPY neuropeptides, we investigated whether peripherally administered liraglutide could access the brain directly. To test this, liraglutide was labeled with a fluorescent probe, VivoTag-S 750 (liraglutide750). The labeling of liraglutide retained the in vitro–binding abilities of the molecule; binding affinity (IC50) for the human GLP-1R was 0.5 and 0.7 nM for liraglutide and liraglutide750, respectively. Liraglutide750 lowered body weight in DIO mice by 20% following 4-day administration (s.c. BID, 400 μg/kg). For visualization of labeling in the brain, mice were dosed once s.c. with liraglutide750 (400 μg/kg) and killed after 6 hours. The brains were then processed, and the distribution of the fluorescent peptide in the brain was visualized using single plane illumination microscopy (SPIM). The sensitivity of this method is directly correlated to the number of GLP-1Rs on the cell membrane, since the fluorescent liraglutide will bind the receptor in a 1:1 ratio. In the brain, the signal intensity will therefore depend on the amount of GLP-1Rs on a given cell and the ability of the fluorescent ligand to reach this cell. Two criteria were used to assign a liraglutide signal: the 750-nm signal should differ significantly from the autofluorescence signal observed in the 680-nm channel and the 750-nm signal observed in wild-type mice should differ significantly from 750-nm signal observed in the Glp1r–/– mice. In the mouse brain, liraglutide750 was observed in all CVOs (Figure 3, A and B): in the zona interna of the median eminence (ME) (Figure 3, F and G), the AP (Figure 3, I and J), the subfornical organ, (Figure 4, D and E), the organum vasculosum of the lamina terminalis (Figure 4, A and B), and the choroid plexus (ChP) (Figure 4, J and K). Interestingly, liraglutide750 was also highly abundant within hypothalamic regions protected by the BBB, including the ARC (Figure 3, F and G), the paraventricular nucleus (PVN) (Figure 3, C and D), and the supraoptic nucleus and supraoptic decussation (Figure 4, G and H). Importantly, administration of liraglutide750 (s.c. 400 μg/kg) to Glp1r–/– mice revealed a loss of these nuclei-specific signals (Figure 3, E, H, and K, and Figure 4, C, F, I, and L), demonstrating that the accumulation of the liraglutide750 signal entirely depends on binding of liraglutide750 to the GLP-1R. Only the signal in ChP was also evident in mice lacking the GLP-1R (Figure 4L), possibly reflecting unspecific binding to this region. No liraglutide750 signal was observed in the NTS, which could indicate that peripheral liraglutide does not directly engage GLP-1R in this region (Figure 3J). A faint fluorescence signal was evident elsewhere in the brain stem, including NTS (Figure 3J); however, this signal had a complete overlap with an intense autofluorescent signal and could be observed in C57BL/6J and Glp1r–/– mice (Figure 3, J and K). Supplemental Video 1, reconstructed from individual scans, summarizes the access of liraglutide750 to the entire brain. The hypothalamic location of liraglutide750 was also assessed in rats, demonstrating the same distribution as in mice (Supplemental Figure 3, G–I). Furthermore, no fluorescent signal was observed in mice administered PBS (Supplemental Figure 4, B and E) or albagen conjugated to the fluorescent probe (Supplemental Figure 4, C and F), signifying that the fluorescent signal observed in the brain regions was not due to a property inherent to any fluorescently labeled peptide but rather a specific signal from the fluorescently labeled liraglutide. To further validate this, mice and rats were peripherally dosed with 3H-labeled liraglutide (400 μg/kg), and brain sections were generated to encompass some of the regions positive for liraglutide750. A radioactive signal was observed in both hypothalami and APs corresponding to the fluorescent signals observed in these regions (Supplemental Figure 3, D–F and J–L).
Figure 3. Distribution of fluorescently labeled liraglutide in the mouse brain.

Representative whole brain images viewed in the (A) dorsoventral or (B) sagittal plane from C57BL/6J mice administered with liraglutide750 (unspecific staining has been removed from the left side of the brain, as described in Supplemental Figure 2). The brain tissue was scanned at 620 nm and 710 nm, representing both autofluorescence from the tissue (gray) and specific signal (green). The red regions in C, F, and I are shown at higher magnification in D, G, and J, respectively. Images in D, E, G, H, J, and K show high-magnification views of a single section from (D, G, and J) C57BL/6J or (E, H, and K) Glp1r–/– mice administered liraglutide750. Liraglutide750 was detectable in (C and D) PVN, (F and G) ME and ARC, and (I and J) AP. (E, H, and K) In mice lacking a functional GLP-1R, no liraglutide750 signal could be detected in any of these regions. Scale bars: 200 μm (A, B, C, F, and I); 50 μm (D and E); 100 μm (G, H, J, and K).
Figure 4. Distribution of fluorescently labeled liraglutide in the mouse brain.
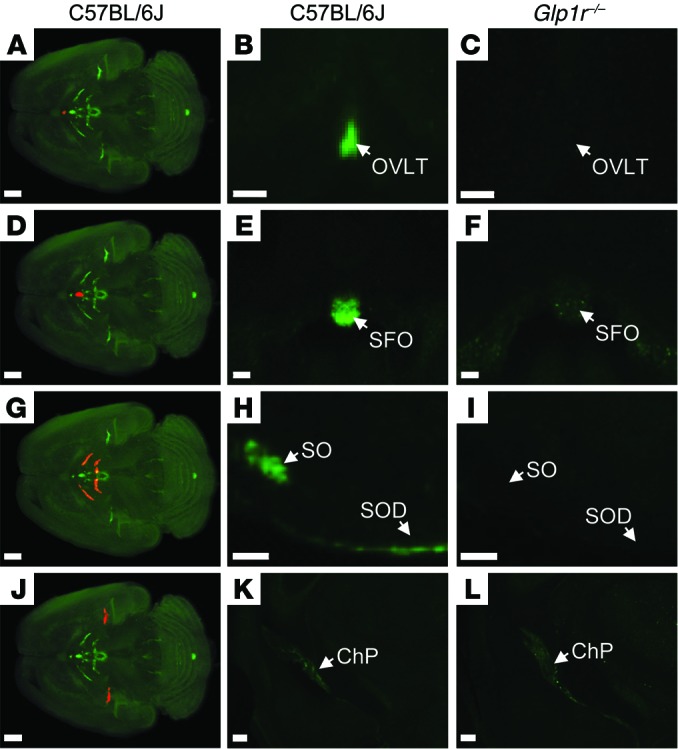
The brain tissue was scanned at 620 nm and 710 nm, representing both autofluorescence from the tissue (gray) and specific signal (green). The red regions in A, D, G, and J are shown at higher magnification in B, E, H and K, respectively (unspecific staining has been removed as described in Supplemental Figure 2). Images in the middle and right columns represent enlargements of a single section from (B, E, H, and K) C57BL/6J or (C, F, I, and L) Glp1r–/– mice administered with liraglutide750. Liraglutide750 was detectable in (A and B) organum vasculosum of the lamina terminalis, (D and E) subfornical organ, (G and H) supraoptic nucleus and supraoptic decussation, and (J and K) ChP. (C, F, I, and L) In mice lacking a functional GLP-1R, no liraglutide750 signal could be detected in any of these regions except from ChP. Scale bars: 200 μm (A, D, G, and J–L); 100 μm (B, C, E, F, H, and I). OVLT, organum vasculosum of lamina terminalis; SFO, subfornical organ; SO, supraoptic nucleus; SOD, supraoptic decussation.
The GLP-1R is internalized upon binding of liraglutide.
To test whether the fluorescently labeled liraglutide activates the GLP-1R, mice were dosed (s.c., 400 μg/kg) with either liraglutide594 or the GLP-1R antagonist exendin(9-39)Alexa Fluor 594 [exendin(9-39)594]. Pancreatic β cells were used as controls, as this is the most well-validated target for GLP-1R agonists and also because higher resolution allowed a clearer difference between internalized and membrane-retained fluorescence. In the pancreatic β cells, a fluorescent signal was observed in insulin-positive β cells in C57BL/6J mice following peripheral administration of liraglutide594 (Figure 5, A and B) and exendin(9-39)594 (Figure 5, C and D). These signals were absent in islets in Glp1r–/– mice (Figure 5, E–H), again demonstrating that the detection of the fluorescent signal in β cells depends on binding of the fluorescently labeled GLP-1 ligands to the GLP-1R. In C57BL/6J mice, liraglutide594 accumulated in the cytoplasm of the β cells (Figure 5, I and J). In contrast, exendin(9-39)594 was retained on the plasma membrane of the β cells (Figure 5, K and L). In hypothalamic ARC neurons, liraglutide appeared to be internalized, as in pancreatic β cells (Figure 5, N and J, ARC and pancreas, respectively), suggesting that GLP-1R also is internalized with an agonist ligand in neurons. Although more difficult to discriminate in neurons, exendin(9-39)594 appeared not to be internalized in neurons in the ARC either, as was observed with the antagonist in β cells (Figure 5, P and L, ARC and pancreas, respectively).
Figure 5. Distribution of liraglutide594 or exendin(9-39)594 in pancreas and brain.

(A–L) Representative images of mouse islets stained with Hoechst nuclear stain (blue), insulin (green), and liraglutide594/exendin(9-39)594 (red). (A–D) In C57BL/6J mice, both liraglutide594 and exendin(9-39)594 were detected in cells expressing insulin; (E–H) however, in mice lacking a functional GLP-1R, no liraglutide594 or exendin(9-39)594 signal could be detected in insulin expressing β cells. (I, J, and N) High-magnification images showed that liraglutide594 was internalized and the fluorescent signal was located in the cytoplasm, (K, L, and P) while exendin(9-39)594 remained at the plasma membrane. In the brain, (M and N) liraglutide594 had access to ARC, in which it bound the GLP-1R and internalized, (O and P) while exendin(9-39)594 labeled the same population of cells but without internalization. Scale bars: 100 μm (M and O), 50 μm (A–H), 10 μm (I–L, N, and P).
Liraglutide targets CART cells in the ARC.
To evaluate the distribution of GLP-1Rs on neurons in the ARC, double in situ hybridization/immunohistochemistry for GLP-1R and POMC was performed on sections from rat hypothalami. GLP-1R were expressed on virtually all POMC/CART neurons (Figure 6D). We then evaluated whether liraglutide594 targeted the GLP-1R–expressing POMC/CART cells directly by using double immunofluorescence confocal microscopy for CART and liraglutide594. Liraglutide594 was present specifically in the cytoplasm of cells positive for CART in the ARC (Figure 6, A–C). Whereas nearly all CART-positive cells were positive for liraglutide594, we also observed cells that were only positive for liraglutide594, indicating that one or more other cell types in the ARC are targeted by liraglutide. Thus, liraglutide594 targets the POMC/CART cells in the rodent ARC following peripheral injection and may adjust the neuronal activity following stimulation. To test this hypothesis, brain slices from Pomc-EGFP mice were used to examine the electrophysiological responses following direct application of GLP-1(7-36)amide to hypothalamic POMC neurons. Stimulation with GLP-1(7-36)amide dose dependently depolarized POMC neurons (5.3 ± 0.7 mV and 8.4 ± 1.4 mV for 10 and 100 nM, respectively) and increased the frequency of action potentials in 7 of 7 cells from 2 animals (Figure 6, E–G, P < 0.01 for RMP vs. GLP-1 10 nM and 100 nM, respectively). The same dose-dependent depolarization happened in the presence of presynaptic blockers, indicating that the effect is postsynaptic (4.1 ± 0.8 mV and 9.4 ± 1.4 mV for 10 and 100 nM, respectively, in 6 of 6 cells from 2 animals, Supplemental Figure 5). In voltage-clamp settings, GLP-1(7-36)amide caused an inward current, in contrast to the outward current caused by the GABA agonist baclofen (11.83 ± 7.2 pA and 22.67 ± 11.1 pA for 10 and 100 nM GLP-1 and 6.83 ± 1.1 pA for baclofen in 3 of 3 cells from 1 animal, Supplemental Figure 6). Interestingly, GLP-1(7-36)amide stimulation increased the frequency of GABAergic currents onto POMC neurons around 2-fold in 6 of 6 cells, which could oppose the direct depolarizing actions of GLP-1R stimulation (Supplemental Figure 7). Together with the data on the regulatory effects of GLP-1 on NPY-expressing cells (Figure 2), these data indicate that GLP-1 activates POMC/CART neurons directly at the level of the cell body and that the NPY/AgRP pathway is inhibited at the NPY/AgRP neurons via GABAergic interneurons, as illustrated in Figure 7.
Figure 6. Neuronal accumulation and activity following GLP-1R stimulation.

(A–C) Hypothalamic sections from rats injected with liraglutide594 (red) and stained with Hoechst nuclear stain (blue) and CART (green). (B and C) High-magnification confocal images revealed accumulation of fluoro liraglutide in the cytoplasm of CART-positive cells (arrows). (B) CART- and liraglutide594-positive cells. (C) The same image as in B with only liraglutide594 signal. (D) Double in situ hybridization/immunohistochemistry staining revealed that GLP-1R (red) colocalize (yellow arrows) with POMC/CART (green) in the ARC. (E) GLP-1 (10 nM and 100 nM) caused membrane depolarization and increased firing rate of spontaneous action potentials in POMC/CART cells. Dashed line indicates the resting membrane potential (RMP). The effects of increased concentrations of GLP-1(7-36)amide are summarized in F. (G) The effects of GLP-1(7-36)amide on firing rate of spontaneous action potentials in POMC/CART neurons. Results are shown as mean ± SEM. Scale bars: 25 μm (B and C); 100 μm (A and D). **P < 0.01 one-way ANOVA, post-hoc Bonferroni’s correction.
Figure 7. Proposed regulation of neuronal activation by liraglutide.

Summary diagram demonstrating the suggested regulatory pathway of GLP-1 on ARC NPY and POMC neurons. GLP-1 stimulates POMC neurons directly through the GLP-1R and is suggested to indirectly inhibit ARC-NPY neurons through an local inhibitory GABA neuron.
Liraglutide-induced body weight loss can be diminished by ARC but not PVN infusion of exendin(9-39) or PVN ablation.
Since liraglutide750 uptake was evident in both ARC and PVN, we assessed the relative importance of these nuclei in liraglutide-induced body weight loss. Exendin(9-39) (200 μg/d) was continuously infused bilaterally into the PVN (Figure 8A) and ARC (Figure 8B) of male SPD rats for 14 days in combination with BID peripheral injections of liraglutide (200 μg/kg). Whereas GLP-1R blockade in the PVN (Figure 8A) did not affect liraglutide-induced weight loss, the effect of antagonizing ARC (Figure 8B) seemed to attenuate liraglutide-induced weight loss, suggesting that ARC, more than PVN, is an important site for the long-term weight-reducing effects of liraglutide. To further assess the contribution of PVN to liraglutide-induced appetite regulation, PVN was electrolytically lesioned in rats (Figure 8C). The PVN lesion in itself led to a significant increase in body weight (P < 0.001, PVN lesion vehicle vs. sham vehicle), whereas 14 days of liraglutide treatment (200 μg/kg) reduced body weight gain significantly in both sham and PVN-lesioned animals (P < 0.01, sham vehicle vs. sham liraglutide; P < 0.001, PVN lesion vehicle vs. PVN lesion liraglutide), further indicating that the PVN is not the primary mediator of liraglutide-induced weight loss. If anything, the PVN lesion seemed to enhance the body weight–lowering effect of liraglutide. Correct lesion of the PVN was verified by histological staining of the brains of both sham (Figure 8D) and PVN-lesioned animals (Figure 8E).
Figure 8. PVN and ARC contributions to liraglutide-induced body weight change.

(A) Exendin(9-39) led to a significant increase in body weight when administered into the PVN, whereas liraglutide treatment reduced body weight gain significantly alone and in combination with exendin(9-39) (*P < 0.001, vehicle PVN + liraglutide s.c. vs. vehicle PVN + vehicle s.c.; †P < 0.001, vehicle PVN + vehicle s.c. vs. exendin(9-39) PVN + liraglutide s.c.; #P < 0.001, exendin(9-39) PVN + vehicle s.c. vs. vehicle PVN + vehicle s.c.). (B) Exendin(9-39) led to a slight but nonsignificant increase in body weight when administered into the ARC, whereas the effect of liraglutide treatment was attenuated when administered in combination with exendin(9-39) (*P < 0.001, vehicle ARC + liraglutide s.c. vs. vehicle ARC + vehicle s.c.; †P < 0.001, vehicle ARC + vehicle s.c. vs. exendin(9-39) ARC + liraglutide s.c). (C) Lesion of the PVN led to a significant increase in body weight (#P < 0.001, PVN lesion + vehicle vs. sham + vehicle), whereas animals with PVN lesions were fully responsive to the weight loss induced by liraglutide (*P < 0.01, sham vehicle vs. sham liraglutide; †P < 0.001, PVN lesion + vehicle vs. PVN lesion + liraglutide). The PVN lesion was histologically verified in (D) sham and (E) PVN-lesioned rats. Data are mean ± SEM, and statistical analyses are performed using 2-way repeated-measures ANOVA, with Bonferroni post-hoc analyses applied. Scale bars: 500 μm.
Discussion
Numerous comprehensive studies aim to describe the GLP-1R population responsible for GLP-1–induced energy intake reduction (20, 33–41). This task has been hampered by the widespread distribution of the GLP-1R in the brain and in the periphery on neurons that could rapidly signal to the brain. Physiologically, GLP-1 released from endocrine cells lining the gut acts peripherally (13, 42). In the brain, GLP-1 is a neurotransmitter in a brain stem–hypothalamic pathway involved in food intake and body weight regulation (4). While these two systems probably are separated in physiology, peripheral administration of pharmacological doses of GLP-1 analogs leads to persistent weight loss, indicating that under these circumstances brain GLP-1Rs may also be targeted. In the current study, we show that the long-acting GLP-1R agonist liraglutide, when injected peripherally, targets hypothalamic GLP-1Rs located on ARC neurons and that these neurons are likely mediators of liraglutide-induced weight loss as (a) they bind and internalize peripherally injected liraglutide, (b) they respond electrophysiologically to locally applied GLP-1 and their activity is regulated (measured as gene expression) by pharmacological dosing of liraglutide, and (c) local blockade of GLP-1Rs in the ARC attenuates liraglutide-induced weight loss.
Numerous studies using the canonical Glp1r–/– mice demonstrate that the single well-defined GLP-1R is the mediator of all of the classical actions of GLP-1 (43–45). The mice also have a complete loss of binding sites for GLP-1 in the brain, and no cFOS expression following dosing of structurally distinct long-acting GLP-1R agonists like exenatide and albiglutide (46, 47). Using a further refined model, it was recently shown that the brain GLP-1R was responsible for the weight-lowering effect of liraglutide, as liraglutide failed to cause weight loss in mice with nestin-Cre–mediated GLP-1R inactivation (48).
Most neuronal cell bodies are not in direct contact with the peripheral circulation, as they are protected by the BBB. However, several regions have fenestrated capillaries and allow for passage of certain compounds into the brain via CVOs. In the current study, we detected liraglutide in CVOs containing GLP-1Rs but also in the hypothalamic ARC and PVN. Both the ARC and PVN are localized behind the BBB, but they have been shown to have increased capillary density and possibly greater interaction with these capillaries (49). The PVN has the highest density of capillaries in the hypothalamus (50). While the ARC has a much lower concentration of capillaries, it does have specialized glial cells, tanycytes, which also may provide a unique interaction of neurons in the ARC with capillaries (51). The uptake of liraglutide was receptor dependent, as studies in Glp1r–/– mice showed no uptake. We furthermore found that liraglutide was internalized into CART neurons (expressing GLP-1Rs), as would be expected with a GPCR-mediated transport mechanism (52). Such a pattern could occur with passive diffusion across the BBB to those relevant neurons. However, although we cannot exclude passive diffusion as a mechanism, it seems an unlikely hypothesis, given the lack of signal in Glp1r–/– mice. Perhaps a more valid hypothesis is GLP-1R–mediated transport through CVOs, and perhaps other areas in the hypothalamus, via fenestrated capillaries and transport through specific localized compartments of the microenvironment of the cerebrospinal fluid (CSF), mediated by tanycytes (53). Such a mechanism was shown recently for leptin, gaining access to ARC via ME located tanycytes (54). We measured liraglutide in CSF and in brain parenchyma following capillary depletion (data not shown) and found concentrations above the detection limit but in the low-to-mid pM area. However, since liraglutide is highly protein bound in the circulation, and the total concentration is around 20 to 30 nM at relevant therapeutic doses, the relevance of such a low concentration is unknown (55, 56). Because of the high protein binding of liraglutide, and the technical difficulty in validating that isolated CSF and parenchyma are 100% free of contamination from blood during the procedure, it would be difficult to prove that any measured concentration in CSF and parenchyma could not be an artefact. An alternative but likely hypothesis is that passage into the ARC and the PVN happens via fenestrated capillaries directly from the ME, near the ME-ARC barrier (51). The ARC has been described to have a weak BBB and allow access to certain subpopulations of neurons (57). Yet another hypothesis is that neuronal terminals from ARC and PVN could reach as far as ME, and liraglutide could be transported in the soma of the neurons via axon terminals. ChP may be another way into the brain. Although the main function of ChP is considered to be CSF production (58), it has been described as a route into ARC for other hormones, including leptin (51). In ChP, the short form of the leptin receptor, has been described to function as a transport system providing access to CSF and specific neurons (59). Since the data presented here show the same access of liraglutide to ChP in normal and Glp1r–/– mice, ChP may not guide access to the brain for GLP-1R ligands but could be a result of the high protein binding of liraglutide.
Our data collectively indicate that GLP-1Rs in the ARC could play an important role as mediators of the long-term body weight–lowering effects of a peripherally administered long-acting GLP-1 analog — in this case liraglutide. However, this conclusion is somewhat contradictory to previous studies aiming to dissect the role of the central and peripheral GLP-1Rs in appetite and weight regulation. First, although our data show that the GLP-1Rs expressed on the vagal afferents are not necessary for liraglutide-induced anorexia or weight loss in rats, other studies have indicated that GLP-1Rs of vagal origin could play a role in food intake regulation. In a series of elegant experiments, Rüttiman et al. showed that, although peripheral administration of exenatide and liraglutide was still able to reduce food intake in vagally deafferentated rats, these animals showed a lower sensitivity to intraperitoneal administration of these compounds, indicating that vagal GLP-1Rs could play a role in the acute satiation process (34). Similarly, it was proposed recently by Plamboeck et al. that human vagotomized subjects showed a reduced sensitivity to the food intake inhibitory effects of peripherally injected GLP-1 (60). In our studies, we focused on the effect of long-term pharmacological dosing of liraglutide, and in this situation, neither vagal nor AP GLP-1Rs were necessary. This actually fits quite nicely with observations and conclusions from other studies that indicate a role for vagal GLP-1Rs in only short-term satiation processes (34, 61, 62) as well as with findings from a recent publication demonstrating that central GLP-1Rs, and not peripheral GLP-1Rs, including vagal GLP-1Rs, are necessary for liraglutide-induced weight loss (48). Our data from AP-lesioned animals are also in line with recent data from Baraboi et al. demonstrating that the anorectic response to a single injection of exendin-4 was intact in AP-lesioned rats (37). Although the most readily accessible GLP-1Rs (vagal and AP) do not appear to be responsible for the long-term weight-reducing effects of liraglutide, it should be noted that peripherally accessible GLP-1–binding sites have been detected in the NTS located just beneath the AP (28). We did not observe a clear entrance of fluorescently labeled liraglutide signal in the NTS, but we cannot, on the basis of this, exclude the possibility that GLP-1Rs in this nucleus also play a role in the appetite-reducing effects of peripherally circulating GLP-1. GLP-1Rs are expressed in the NTS, and it has been demonstrated that peripheral administration of exendin-4 activates neurons there (63, 64). Given the well-known role of NTS-produced GLP-1 in appetite regulation, it is tempting to speculate that peripheral administration of GLP-1 or analogs activates NTS GLP-1–positive neurons that in turn reduce appetite (22, 23). However, while peripheral GLP-1 activates neurons in the NTS, the PPG neurons here do not respond to GLP-1, and they do not express GLP-1Rs (65). PPG neurons in the NTS are important regulators of energy intake. They express leptin receptors and project to other brain areas that also express GLP-1R and have been shown to be important in food intake (40, 66). These neurons may be activated by peripheral signals, including GLP-1, via the vagal nerve but are unlikely to be directly activated by GLP-1, as they do not contain GLP-1Rs (65). Here, we show that PPG levels in the NTS are reduced both after weight reduction (weight matching) and liraglutide treatment, indicating that reduced weight and/or food intake lead to lower activity in the NTS PPG neurons. These data are in line with findings from other studies indicating that obesity can lead to increased PPG expression in the NTS (21, 67). Nevertheless, GLP-1R–expressing neurons in the NTS could still be involved in mediating the satiating and weight-reducing effects of circulating GLP-1 analogs. Hence, it has been shown that GLP-1Rs in the NTS are involved in mediating the acute anorectic effects of both directly applied GLP-1 (4th ventricular injection) analogs and peripherally administered GLP-1 analogs (68–70). These data indicate that brain stem GLP-1Rs can also be targeted by peripherally circulating GLP-1 in line with the aforementioned GLP-1R binding data (28), but whether these acute (0–24 hours) responses translate into lasting weight loss effects is less clear. Hence, although the AP is not necessary for liraglutide-induced weight loss, it cannot be excluded that GLP-1Rs in the underlying NTS could play a role.
In the hypothalamus, both the PVN and the ARC — also the two hypothalamic nuclei with the highest density of GLP-1Rs — were found to contain fluorescently labeled liraglutide following peripheral administration. Direct injection of GLP-1 into the PVN elicits a powerful anorectic response, and it is generally believed that the PVN is the primary site at which endogenous brain-derived GLP-1 exerts its anorectic/satiating effects (22, 33, 71–73). In line with this, we found that local blockade of GLP-1Rs directly in the PVN leads to hyperphagia and weight gain, but surprisingly, this blockade did not affect the feeding or weight loss response to liraglutide at all. On the contrary animals infused with exendin(9-39) in the PVN seemed to be more sensitive to the weight loss effects of peripheral liraglutide. The same pattern was seen when the PVN was lesioned, although lesioning is of course a much less sophisticated method of “blocking.” Although we could not block the reduction in food intake and body weight when we infused exendin(9-39) into the ARC, we nevertheless saw an attenuation of the effect of peripheral liraglutide, indicating that this nucleus could be involved in mediating the weight loss effects. It should be noted that the ARC is a very long (>5-mm) nucleus in the rat and that the blockade of GLP-1Rs in the ARC in the direct infusion experiment most likely was incomplete.
We have reported previously that rats with chemical lesions to the ARC induced by neonatal monosodium glutamate (MSG) injections respond to liraglutide with weight loss in much the same way as control animals (74). Although these data are somewhat contradictory to what we report in the current study, the MSG model is a rather complex lesioning model, as it affects a number of other brain areas that are accessible from the periphery during early brain development, including the CVOs (75). Importantly, it should also be noted that MSG treatment does not completely eradicate the arcuate POMC neurons, as a lateral population of POMC neurons has been shown to be present in adult MSG rats (76).
Overall, it seems entirely possible that the combined appetite-suppressing and weight-reducing effects of peripherally administered GLP-1 analogs are a mixture of acute anorectic effects elicited partly and initially by vagal GLP-1Rs and GLP-1Rs in the AP as well as in the underlying NTS. The long-term effects of liraglutide — the pharmacological effects — kick in later, and our data indicate that this involves GLP-1R located on key appetite- and glucose-regulating neurons in the ARC. It should be noted that a number of studies have assigned a role to ARC GLP-1Rs in glucose control; our data neither support nor reject this possibility — they merely indicate that these receptors are also involved in the long-term effects on body weight that is seen following pharmacological doses of this GLP-1 analog. It is intriguing to speculate that ARC GLP-1Rs are not only involved in controlling food intake and body weight (by increasing CART signaling and suppressing activity in the NPY/AgRP pathway) but also are involved in regulating glucose homeostasis, specifically by sensing enteric glucose, stimulating insulin secretion, and modulating hepatic and muscle glucose metabolism and possibly by redirecting peripheral blood flow, as has been suggested by several research groups (33, 77, 78). However, the pancreatic GLP-1R is mostly responsible for glucose lowering by peripherally injected GLP-1 analogs (19).
Activation of the central amygdala following peripheral injection of another long-acting GLP-1 analog, albiglutide, was shown already a decade ago (46), and we found the same pattern of neuronal activation measured by cFOS in AP, NTS, central amygdala, and the lateral parabrachial nucleus (data not shown). Liraglutide was not detected outside of CVOs and hypothalamus, so the activation in these areas except AP may be indirect. There likely are important effects of GLP-1R in areas of the brain associated with reward, as GLP-1R agonists have been show to lead to changed food preference away from typically rewarding foods (79), and exendin-4 has been shown to reduce basal as well as amphetamine-induced locomotor activity, and reduced alcohol intake has also been reported (80, 81).
A number of limitations in the data presented should be mentioned. We used only normal nonobese rodents. Recent studies document hypothalamic inflammation as an important pathophysiological component of obesity in both animals and humans, and such inflammation may lead to reduced sensitivity of ARC to respond to hormonal input (82, 83). Liraglutide effectively lowers body weight in nonobese and obese rats, obese pigs, and humans, and exendin-4 has been show to rapidly circumvent hypothalamic inflammation, all together indicating that nonobese rodents may be a suitable model to describe access to key appetite-regulating neurons (74, 79, 84–87). Also, a clinical study reported an increased hypothalamic connectivity using functional magnetic resonance imaging following a peripheral dose of exenatide (88). The use of neuronal lesion and ablation models has certain limitations, as they interrupt neuronal networks and can potentially induce compensatory responses or affect neuronal functions in distant connected areas. In the current study, we used the selective vagal deafferentation model that is the golden standard for selectively removing vagal afferent signaling. The SDA model is perhaps one of the most refined lesion methods, as it spares most of the vagal motor input to the gut and hence is believed to leave gut motility relatively undisturbed. However, it should be underscored that, in this model, half of the motor nerves are still lesioned, which may of course affect motility to a certain extent. While both the SDA and the AP and PVN lesional models indicate that these areas are not prime mediators for liraglutide-induced weight loss, they could still form part of the broad central network affected by GLP-1 analogs, as also indicated by the findings that liraglutide is binding to receptors in the AP and PVN. More refined models, like select neuronal adult-onset inducible knock out models, would give more precise information about the target neurons for the different central effects of GLP-1 analogs.
In conclusion, we have shown that GLP-1 analogs like liraglutide have access to specific brain areas relevant for appetite regulation. Liraglutide was measured in select CVOs and specific hypothalamic areas, and this signal was GLP-1R dependent. Furthermore, liraglutide was shown to have important effects on key primary hypothalamic neurons highly relevant in appetite regulation. The mode of action for peripherally administered GLP-1R agonists in body weight regulation seems consistent with a direct activation of CART/POMC neurons in the ARC, and an indirect GABA interneuron-dependent inhibition of NPY/AgRP neurons, consistent with regulation of these neuropeptides in the ARC. This is in line with the reported clinical effects of liraglutide to increase measures of satiety and decrease hunger.
Methods
Animals
All in vivo studies were conducted in accordance with approved national regulations in Denmark, which are fully compliant with internationally accepted principles for the care and use of laboratory animals, and with animal experimental licenses granted by the Danish Ministry of Justice. Animals were obtained from Taconic or The Jackson Laboratory and housed (rats, 2 per cage; mice, 5 per cage) in standard, temperature-controlled conditions with a 12-hour-light/dark cycle. The animals had ad libitum access to water and regular chow (no. 1324, Altromin, Brogaarden) unless otherwise stated.
Surgery models and compound administration
Subdiaphragmatic vagal deafferentation in male SPRD rats (SDA).
Sixteen rats underwent the SDA, and thirteen underwent a sham operation. The SDA in rats was performed as previously described (89), according to the original method (90). Further details are available in Supplemental Methods, along with functional verification of the SDA procedure by acute assessment of the effect of CKK on food intake. Animals were allowed to regain their presurgical body weight and were then randomized according to body weight into 4 experimental groups: group 1, vehicle sham, n = 6; group 2, vehicle SDA, n = 8; group 3, liraglutide sham, n = 7; group 4, liraglutide SDA, n = 8.
The experiment was started on day 0 with an acute GE test (data not shown). Animals were semifasted as they had access to only 50% of their previous 24-hour food intake and were administered with either liraglutide (100 μg/kg; 2 ml/kg) or vehicle by an i.v. tail injection. All animals were then dosed s.c. BID for 14 days with vehicle (Lonza DPBS buffer/Invitrogen Gibco PBS buffer) or liraglutide (200 μg/kg). Body weight and food and water intake were measured daily throughout the entire study period. At the end of the experiment, animals were euthanized in the morning and brains were excised and snap frozen in crushed dry ice for histological validations.
Specific ablation of the brain stem AP.
A total of 44 male SPRD rats (12–13 weeks of age) underwent APx or a sham surgery (sham). Rats were anesthetized with a mixture of ketamine (63 mg/kg) and xylazine (9.4 mg/kg) mixed 4:3 (0.1 ml/100 g body weight) and were maintained at 36°C to 37°C throughout surgery on a heating pad. The anesthetized rats were placed in a stereotaxic apparatus with the head ventroflexed. The skin and muscles were dissected along the midline, and the cisterna magna was opened to expose the dorsal surface of the medulla. The AP was visualized with an operating microscope and lesioned by aspiration using a blunt 23-gauge needle. Sham lesion surgeries only involved the exposure of the medulla. The muscles and skin were sutured, and rats were allowed to recover from anesthesia. Following the operational procedures, all animals were monitored closely and provided with analgesic (NSAID, buprenorphine/Rimadyl) for 4 days after operation. After surgery, the rats were allowed to fully recover their presurgical body weight and rate of body weight gain. Animals were then transferred into individual cages. A minimum of 5 days of habituation was allowed before first dose was administered. During these 5 days, animals were handled daily to accustom them to the experimental paradigm. Three animals died during surgery or early recovery. In addition, 4 animals were euthanized due to marked loss of body weight and low food intake or abnormal behavior in the preexperimental and early experimental period, leaving a total of 37 rats (24 APx, 13 sham) for final experiments. Following postsurgical recovery, the animals were randomized according to body weight into 4 experimental groups: group 1, vehicle sham, n = 6; group 2, vehicle APx, n = 10; group 3, liraglutide sham, n = 6; group 4, liraglutide APx, n = 10).
The experiment was started on day 0 with an acute GE test, as described above. All animals were then dosed s.c. BID for 21 days with vehicle (Lonza DPBS buffer/Invitrogen Gibco PBS buffer) or liraglutide (200 μg/kg). Body weight and food and water intake were measured daily throughout the entire study period. At the end of testing, all animals were euthanized and brains were removed and frozen on dry ice. The brain stems were cut into a series of 12-μm cryosections, stained in thionin, and histologically examined to verify the APx procedures. Histological verification of the APx procedure was performed for all animals, and only rats with a well-defined lesion were included in the final data.
PVN lesions.
A total of 48 male SPRD rats (~190 g) were anesthetized with a mixture of Hypnorm/Dormicum. An incision was made in the midline above the skull, and the skull was exposed. The rats were placed in the stereotaxic frame upon a heating pad (36°C–37°C). Using a drill, a small plate of the bone was removed and an electrode was inserted into the PVN (coordinates: –1.6; 0.5; –8.1). The electrode was attached to an electric current output (Digital Midgard Precision Current Source) and a 5-minute lesion at 10 μA was performed. This was done bilaterally. The bone plate was returned to the skull, and the surgical incision was closed with sutures. Sham surgery was performed with a method similar to the above description, but no current was applied. Following the operational procedures, all animals were monitored closely. Analgesic was provided by 1 daily s.c. dose of carprofen (0.1 ml/100 g body weight of Rimadyl at 50 mg/ml, diluted 1:9 in isotonic NaCl) for 3 days after operation. Rats were stratified according to body weight into 4 groups: group 1, vehicle sham, n = 7; group 2, liraglutide sham, n = 8; group 3, vehicle PVN lesion, n = 7; group 4, liraglutide PVN lesion, n = 6. The experiment was started on day 0 with an acute GE test as described above. All animals were then dosed s.c. BID for 14 days with vehicle (Lonza DPBS buffer/Invitrogen Gibco PBS buffer) or liraglutide (200 μg/kg). Body weight and food and water intake were measured daily throughout the entire study period. At the end of the experiment, animals were euthanized in the morning and brains were excised and snap frozen in crushed dry ice for histological verification of the lesion on brain sections counterstained with thionin. The PVN lesions were evaluated under microscope, and only rats with a well-defined lesion were included in the final data.
Other animal models and compound administration
Central and peripheral exendin(9-39) administration models.
A total of 2 cohorts of 48 male Sprague-Dawley rats (~190 g) were anesthetized with a mixture of Hypnorm/Dormicum combined with preoperative NSAID analgesia and were maintained at 36°C to 37°C throughout surgery on a heating pad. An incision was made in the midline above the skull. Three holes were drilled: 2 for anchoring s.c. screws and 1 for the cannula (coordinates: –1.0 caudal, 1.5 lateral to bregma). Acrylic cement was used to fix the cannula in place. The Alzet mini-pump (Alzet) was placed subcutaneously in the neck region of the animal. In addition, 1 Alzet mini-pump was placed in the subcutaneous space, inserted flow moderator first. The surgical incision was closed with sutures. Pumps were filled the day before the operation and “primed” (in 0.9% saline at 37°C) overnight according to the manufactures recommendation. All animals were provided with s.c. injections of carprofen (0.1 ml/100 g body weight of Rimadyl at 50 mg/ml, diluted 1:9 in isotonic NaCl) immediately prior to surgery. Following the operational procedures, all animals were provided with 1 daily s.c. dose of carprofen (0.1 ml/100 g body weight of Rimadyl at 50 mg/ml, diluted 1:9 in isotonic NaCl) for at least 3 days after operation. Animals were randomized into 8 different treatment groups (n = 6 per group) and provided with a cannula into the lateral ventricles: group 1, vehicle i.c.v. pump + vehicle s.c. pump + vehicle s.c.; group 2, vehicle i.c.v. pump + vehicle s.c. pump + liraglutide s.c.; group 3, vehicle i.c.v. pump + exendin(9-39) s.c. pump + vehicle s.c.; group 4, vehicle i.c.v. pump + exendin(9-39) s.c. pump + liraglutide s.c.; group 5, exendin(9-39) i.c.v. pump + vehicle s.c. pump + vehicle s.c. BID; group 6, exendin(9-39) i.c.v. pump + vehicle s.c. pump + liraglutide s.c. BID; group 7, exendin(9-39) i.c.v. pump + exendin(9-39) s.c. pump + vehicle s.c. BID; and group 8, exendin(9-39) i.c.v. pump + exendin(9-39) s.c. pump + liraglutide s.c. BID. Note that groups 7 and 8 are not included in Figure 8, A and B.
The experiments were started on day 0 with an acute GE test (data not shown). All animals were then dosed s.c. BID for 11 days with vehicle (Lonza DPBS buffer/Invitrogen Gibco PBS buffer) or liraglutide (200 μg/kg). Additionally, exendin(9-39) (200 μg/d) was administered throughout the experiment by s.c. or i.c.v. pump infusion. Body weight and food and water intake were measured daily throughout the entire study period. At the end of the experiment, animals were euthanized in the morning and brains were excised and snap frozen in crushed dry ice for histological validations.
mRNA analysis in the CNS following liraglutide administration.
A total of 30 selectively bred male DIO SPRD rats were included in the study (91). The animals were fed an energy-dense high-fat diet (12266B; Research Diets) and water ad libitum. At study start, animals were randomized according to body weight into 3 groups (n = 10 per group) and dosed s.c. once daily for 28 days with vehicle (PBS, BE 17-512F, Biowhitaker; 0.5 ml/kg) or liraglutide (100 μg/kg), which was gradually increased over the first 3 days from 50 to 100 μg/kg. A weight-matched group was offered a restricted amount of food (60%–90% of average food intake of the liraglutide group) to weight match these animals to the liraglutide-administered rats. Food intake and body weight were recorded daily from day –3. Water intake was recorded daily the first 2 weeks of the experimental period and then twice a week. Energy expenditure was analyzed on days 14 to 18 by indirect calorimetry performed at thermoneutrality (29°C in calorimetry cages; TSE system). Measurements were performed on 8 animals per group — the remaining 2 animals per group spent a similar amount of time in the calorimetry room as the remaining rats. The day before the test, 8 rats (randomly chosen across groups) and 2 of the remaining rats from each group were transferred to a temperature-controlled room (29°C). The morning of the next day, the rats were weighed and then transferred to air-tight plexiglas cages (food and water but no bedding). Airflow in and out was controlled by the calorimetry system. Oxygen consumption and CO2 production was measured every 20 minutes over the next 22 hours (data from the first 2-hour acclimatization period of the test was excluded). Respiratory exchange ratio (RQ; dCO2/dO2) and oxygen consumption (ml O2/h/kg lean body mass) were calculated for the light phase (6 hours) and the dark phase (12 hours). Following experimentation, rats were transferred to their home cages and given free access to food and water; however, the weight-matched animals received 90% of average food intake in the liraglutide group. On day 28 animals were euthanized by decapitation under CO2/O2 anesthesia. The brains were removed, frozen on dry ice, and kept at –80°C. For mRNA analysis, see In situ hybridization.
Distribution of fluorescently labeled GLP-1 analogs
Access of fluorescently labeled liraglutide to the brain.
To visualize the access and distribution of liraglutide in the brain and pancreas, fluorescently labeled liraglutide or exendin(9-39) was synthesized in-house by conjugating VivoTag-S 750 NIR FLUOROCHROME LABEL (Perkin Elmer) or Alexa Fluor 594 C5-maleimide (Molecular Probes, Life Technology) to the peptides. Receptor affinity and in vivo efficacy of liraglutide750 were performed as described in Supplemental Methods. For in vivo detection of the fluorescent peptides, mice (n = 5 per group, male, Glp1r–/–, C57BL/6J) or rats (n = 2 per group, male, SPRD) were single dosed s.c. (mice) or i.v. (rats) with 120 nmol/kg peptide750 dissolved in vehicle (PBS without calcium and magnesium, pH 7.5). The animals were anesthetized with isoflurane after 6 hours (or 4 hours in rats) following injection and transcardially perfused with heparinized (10 U/ml) saline (mice: 10 ml, rats: 90 ml) followed by 10% neutral buffered formalin (NBF) (mice: 10 ml, rats: 150 ml). Brains and pancreata were removed, immersed into 10% NBF, and stored at 4°C until further processed.
The brain tissue was dehydrated and cleared at room temperature, as described in Becker et al. (92). The tissue was dehydrated in increasing concentrations (50 vol%, 80 vol%, 96 vol%, 2 × 100 vol%) of tetrahydrofuran, at 3 to 12 hours per step. The dehydrated sections were then cleared until transparent by incubating with 3 × dibenzylether for 1 to 2 days in total. Visualization of fluorescence was performed with a light sheet ultramicroscope coupled to a SuperK EXTREME (EXR-15) laser system (LaVision). The samples were scanned in 5-μm steps at excitation/emission settings of 620/700 nm for autofluorescence signal and 710/775 nm for specific signal of liraglutide750. All samples were scanned with identical settings. Images were generated using Imaris Bitplane software (Imaris x64 7.5.1). For 2D imaging and immunohistochemistry, pancreas and brain tissue from mice dosed with either liraglutide594 or exendin(9-39)594 (as above) were saturated in sucrose (20%) and cryosectioned. Further methodology details on validation of liraglutide750, 3H-liraglutide, and albagen750 administration are given in the Supplemental Methods.
Immunohistochemistry.
Immunohistochemical staining for insulin in pancreas and CART in the ARC were performed on pancreas sections or brain sections. Brain tissue was saturated in sucrose 20% and frozen. Brains were sectioned into 30-μm cryosections covering the hypothalamus. All sections were collected and consecutively sampled into cryoprotectant. Pancreas sections were sectioned into 5-μm cryosections. Sections were blocked in TBS containing donkey serum or PBS/0.1% Triton X-100 and stained for insulin (1:500 guinea pig insulin antibody, Abcam ab7842), CART (rabbit anti-CART polyclonal antibody Ca7-OVA, described in Vrang et al., ref. 93), or IgG (Life Technology) diluted 1:500 in PBS/0.1% Triton X-100/0.2% BSA at 4°C overnight. Sections were rinsed in TBS (insulin) or PBS/0.1% Triton X-100 (CART) and incubated for 30 minutes with TNB buffer containing 1:500 donkey anti-guinea pig Cy2 (Jackson ImmunoResearch Laboratories) and 1:20,000 Hoechst nuclear stain (Invitrogen) for insulin staining or CSAII anti-rabbit linker HRP (Dako) for 15 minutes, followed by CSAII Amp. reagent diluted 1:1 in PBS (Dako) for CART staining. Brain sections were counterstained with Hoechst nuclear stain (1:10,000 μl in dH2O) and mounted onto slides. Images were obtained using a Zeiss AxioImager M2 microscope and a Fluoview FV10i confocal microscope (Olympus), and micrographs were digitally acquired from selected brain areas using ZEN 2011 software (Zeiss) for the AxioImager system and Fv10-ASW 2.1 (Olympus) for the Fluoview system.
Ex vivo and in vitro methods
In situ hybridization.
The brains were divided into 3 main regions by a ventrodorsal cut rostral to the optic chiasm and at level of the pons. The hypothalamus and hindbrain were mounted with Tissue-Tek in a cryostat, trimmed, and eventually cut into at least 12 systematic uniform random series of 12-μm coronal sections. Sections from the hypothalamus were sampled from the rostral part of the hypothalamic PVN to the caudal extension of the ARC. Sections from the dorsal vagal complex covered the full rostrocaudal extension of the NTS. Sections were allowed to dry at room temperature and kept at –80°C until hybridizations were performed. In situ hybridizations were performed using 33P-labeled RNA riboprobes or digoxigenin-labeled RNA probes (CART and NPY for dual labeling in situ hybridization histochemistry) directed against specific cDNAs. In situ hybridization was performed on hypothalamic or brain stem sections. Antisense and sense probes were generated by in vitro transcription from linearized plasmid DNA (provided by Novo Nordisk) containing specific cDNA clones. The transcription mixture contained (per 24 μl) 5x transcription buffer (Promega; 5 μl), RNAse inhibitor (1 μl), CAG stock (10 mM CTP, ATP, and GTP; 4 μl), 33P-UTP (1 mCi/ml; 10 μl), linear DNA template (1 μg; approximately 2 μl), and RNA polymerase (20 U/μl; 2 μl). Transcription was carried out at 37°C for 2 hours followed by phenol/chloroform extraction and ethanol/ammonium acetate precipitation to isolate the RNA probe. To reduce average probe length to approximately 100 bp all probes were subjected to a limited alkali hydrolysis. The digoxigenin-labeled RNA probes were synthesized using T3 and T7 polymerases (Promega). Sections were fixed in 4% paraformaldehyde in PBS, acetylated in triethanolamine (0.1 M), and dehydrated through an ethanol gradient (from water to absolute ethanol). Hybridization mixture containing the RNA probe (or a mixture of the 33P-labeled GLP-1R antisense [RNA] probe and a digoxigenin-labeled RNA probe against CART and NPY) was added to the dry sections (36 μl per slide per 18 μl per section), after which the sections were coverslipped and incubated at 47°C overnight. Washes after hybridization were performed twice for 60 minutes at 62°C and 67°C in 50% formamide. Following hybridization, sections for quantitative mRNA analyses were exposed to autoradiographic films for 1 to 4 days and developed in Kodak D19 developer. The hybridization signals were evaluated using NIH image software. The hybridization signals were estimated quantitatively using an oval frame covering the nuclei under examination. The signals were quantified as the product of frame area (in square millimeters) and mean pixel intensity within the actual frame. Data were normalized according to vehicle expression levels and presented as relative values. Local background subtraction method was applied. Sections for dual labeling in situ hybridization histochemistry were washed 5 times following hybridization in PBS (0.05 M)/0.1% Triton X-100 (PBS-X), incubated in a blocking buffer consisting of 5% BSA in PBS-X, and subsequently incubated overnight in sheep anti-digoxigenin antibody (1:1,000, Roche). The next day hypothalamic sections were incubated with a donkey anti-sheep antibody (Fab2 fragment, Jackson ImmunoResearch Laboratories) diluted 1:1,000 in blocking buffer. Sections were rinsed, incubated in avidin-biotin-peroxidase complex (Vector elite kit), rinsed again, and incubated with biotinylated tyramine. Finally, hypothalamic sections were reacted with streptavidin–Alexa Fluor 488 (Molecular Probes), dehydrated, and dipped in K5 emulsion (AgFa). Sections were exposed for 1 week, developed in D19 developer (Kodak), mounted in Pertex, and then examined using a Nikon E1000 microscope.
Electrophysiology.
Whole-cell patch-clamp recordings were made from the soma of POMC or NPY neurons. Tissue originated from homozygous Npy-hrGFP male mice on a C57BL/6J background was purchased from The Jackson Laboratory (stock 006417) for breeding (94). Pomc-EGFP mice were generated as previously described (95).
Brain slices (250 μm) containing ARC were continuously perfused with 95% O2, 5% CO2 aCSF (124 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2.6 mM NaH2PO4, 26 mM NaHCO3, 2 mM MgSO4, 2 mM CaCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4) using a gravity-fed perfusion system with a flow rate of 1 to 3 ml per minute. The effects of adding GLP-1(7-36)amide (10 or 100 nM) were investigated by either whole-cell voltage clamp in NPY and POMC neurons or whole-cell current clamp in POMC neurons. Furthermore, inhibitory postsynaptic currents in POMC neurons were recorded in voltage-clamp mode using a chloride-based internal solution (140 mM CsCl, 10 mM HEPES, 5 mM MgCl2, 1 mM BAPTA, 5 mM MgATP, and 0.3 mM NaGTP). NMDA (100 μM) or bicuculline (20 μM) was made from a 10- or 100-mM stock, respectively. Tetrodotoxin (1 μM) was prepared from a stock of 2 mM (Alomone Labs). 6-cyano-7-nitroquinoxaline-2,3-dione (10 μM) and DL-2-amino-5-phosphonovaleric acid (50 μM) were prepared from a 10-mM and 50-mM stock, respectively. Microelectrodes had resistances of 2 to 5 ⁄M when filled with an internal solution: 125 mM Kgluconate, 2 mM KCl, 5 mM MgATP, 0.3 mM NaGTP, 10 mM EGTA, 5 mM HEPES, and 0.05% neurobiotin, adjusted to pH 7.4 with NaOH or KOH. Neurons were recorded at a holding potential of –65 mV. Whole-cell capacitance and resistance were electronically compensated. Adequate whole-cell access (Ra <20 M⁄) and membrane resistance (>500 M⁄) were verified at the beginning and the end of recording. Data acquisition was performed using either the Axopatch 200B or MultiClamp 700B Amplifier (Molecular Devices). Data were collected using the computer interface Digidata 1322 and pCLAMP software (9.2 and 10; Molecular Devices) at a sample frequency of 20 kHz, with low-pass filtering at 2 kHz. Electrophysiological recordings were analyzed with Clampfit-10 software (Molecular Devices). For current clamp experiments, the liquid junction potential of –5 mV was corrected in the analysis.
Statistics
All data were entered into Excel 5.0 or 2003 spread sheets and subsequently subjected to statistical analyses using GraphPad Prism or Statview Software. Statistical significance was set to P < 0.05. Statistical evaluation of the data was carried out using 1- or 2-way repeated-measures ANOVA, with Fishers post-hoc analysis (1-way) or Bonferroni post-hoc analysis (2-way) between control and treatment groups in cases in which statistical significance was established. Specifically for electrophysiological recordings, a paired t test was used to compare the action potential firing percentage of NPY neurons between control and treatment duration. For current clamp recordings in POMC neurons, 1-way ANOVA was used to analyze the change in membrane potential before and after treatment (GraphPad Software). All data are expressed as mean ± SEM. For event analysis of spontaneous action potential firing under current clamp conditions, Clampfit 10 software (Molecular Devices) was used. Recordings were analyzed for action potential firing during treatment with GLP-1. To quantify the frequency of the number of events before and after the application of GLP-1, the calculated events were divided by bin size (time).
Supplementary Material
Acknowledgments
Arian F. Baquero is funded by an ADA minority fellowship. All other work was fully funded by Novo Nordisk.
Footnotes
Conflict of interest: Novo Nordisk markets liraglutide for the treatment of diabetes. Anna Secher, Jacob Hecksher-Sørensen, Charles Pyke, Kirsten Raun, Lauge Schäffer, Mads Tang-Christensen, Brent M. Witgen, and Lotte Bjerre Knudsen are all full-time employees of Novo Nordisk and hold minor share portions as part of their employment. Kevin L. Grove and Michael A. Cowley consult for and have research funding from Novo Nordisk. Niels Vrang consults for Novo Nordisk.
Reference information:J Clin Invest. 2014;124(10):4473–4488. doi:10.1172/JCI75276.
See the related Commentary beginning on page 4223.
References
- 1.Nathan PJ, O’Neill BV, Napolitano A, Bullmore ET. Neuropsychiatric adverse effects of centrally acting antiobesity drugs. CNS Neurosci Ther. 2011;17(5):490–505. doi: 10.1111/j.1755-5949.2010.00172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki K, Jayasena CN, Bloom SR. Obesity and appetite control. Exp Diabetes Res. 2012;2012:824305. doi: 10.1155/2012/824305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong CX, Brubaker PL. Ghrelin, the proglucagon-derived peptides and peptide YY in nutrient homeostasis. Nat Rev Gastroenterol Hepatol. 2012;9(12):705–715. doi: 10.1038/nrgastro.2012.185. [DOI] [PubMed] [Google Scholar]
- 4.Vrang N, Larsen PJ. Preproglucagon derived peptides GLP-1, GLP-2 and oxyntomodulin in the CNS: role of peripherally secreted and centrally produced peptides. Prog Neurobiol. 2010;92(3):442–462. doi: 10.1016/j.pneurobio.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Babilon S, Mörl K, Beck-Sickinger AG. Towards improved receptor targeting: anterograde transport, internalization and postendocytic trafficking of neuropeptide Y receptors. Biol Chem. 2013;394(8):921–936. doi: 10.1515/hsz-2013-0123. [DOI] [PubMed] [Google Scholar]
- 6.Sivertsen B, Holliday N, Madsen AN, Holst B. Functionally biased signalling properties of 7TM receptors — opportunities for drug development for the ghrelin receptor. Br J Pharmacol. 2013;170(7):1349–1362. doi: 10.1111/bph.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kristensen P, et al. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature. 1998;393(6680):72–76. doi: 10.1038/29993. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 9.Woods SC, Schwartz MW, Baskin DG, Seeley RJ. Food intake and the regulation of body weight. Annu Rev Psychol. 2000;51:255–277. doi: 10.1146/annurev.psych.51.1.255. [DOI] [PubMed] [Google Scholar]
- 10.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essentials for feeding in adult mice but can be ablated in neonates. Science. 2005;310(5748):683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 11.Cansell C, Denis RG, Joly-Amado A, Castel J, Luquet S. Arcuate AgRP neurons and the regulation of energy balance. Front Endocrinol (Lausanne). 2012;3:169. doi: 10.3389/fendo.2012.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kieffer TJ, Habener JF. The glucagon-like peptides. Endocr Rev. 1999;20(6):876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- 13.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 14.Nauck MA, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273(5 pt 1):E981–E988. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- 15.Nauck MA, Kemmeries G, Holst JJ, Meier JJ. Rapid tachyphylaxis of the glucagon-like peptide 1-induced deceleration of gastric emptying in humans. Diabetes. 2011;60(5):1561–1565. doi: 10.2337/db10-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- 17.Jelsing J, Vrang N, Hansen G, Raun K, Tang-Christensen M, Bjerre Knudsen L. Liraglutide: short-lived effect on gastric emptying -- long lasting effects on body weight. Diabetes Obes Metab. 2012;14(6):531–538. doi: 10.1111/j.1463-1326.2012.01557.x. [DOI] [PubMed] [Google Scholar]
- 18.Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ.The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes. 201059:176571765–1770. 10.2337/db09-1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamont BJ, Li Y, Kwan E, Brown TJ, Gaisano H, Drucker DJ. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J Clin Invest. 2012;122(1):388–402. doi: 10.1172/JCI42497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology. 2009;150(4):1680–1687. doi: 10.1210/en.2008-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrera JG, Jones KR, Herman JP, D’Alessio DA, Woods SC, Seeley RJ. Hyperphagia and increased fat accumulation in two models of chronic CNS glucagon-like peptide-1 loss of function. J Neurosci. 2011;31(10):3904–3913. doi: 10.1523/JNEUROSCI.2212-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turton MD, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379(6560):69–72. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]
- 23.Tang-Christensen M, et al. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol. 1996;271(4 pt 2):R848–R856. doi: 10.1152/ajpregu.1996.271.4.R848. [DOI] [PubMed] [Google Scholar]
- 24.Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101(3):515–520. doi: 10.1172/JCI990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdich C, et al. A meta-analysis of the effect of glucagon-like peptide-1 (7-36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab. 2001;86(9):4382–4389. doi: 10.1210/jcem.86.9.7877. [DOI] [PubMed] [Google Scholar]
- 26.Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 2002;18(1–2):7–14. doi: 10.1385/JMN:18:1-2:07. [DOI] [PubMed] [Google Scholar]
- 27.McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31(17):6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orskov C, Poulsen SS, Moller M, Holst JJ. Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes. 1996;45(6):832–835. doi: 10.2337/diab.45.6.832. [DOI] [PubMed] [Google Scholar]
- 29.Morita S, Miyata S. Different vascular permeability between the sensory and secretory circumventricular organs of adult mouse brain. Cell Tissue Res. 2012;349(2):589–603. doi: 10.1007/s00441-012-1421-9. [DOI] [PubMed] [Google Scholar]
- 30.Schaeffer M, et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci U S A. 2013;110(4):1512–1517. doi: 10.1073/pnas.1212137110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Astrup A, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374(9701):1606–1616. doi: 10.1016/S0140-6736(09)61375-1. [DOI] [PubMed] [Google Scholar]
- 32.van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WH. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite, energy metabolism in obese, non-diabetic adults. Int J Obes (Lond). 2014;38(6):784–793. doi: 10.1038/ijo.2013.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes. 2008;57(8):2046–2057. doi: 10.2337/db07-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rüttiman EB, Arnold M, Hillebrand JJ, Geary N, Langhans W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology. 2009;150(3):1174–1181. doi: 10.1210/en.2008-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayes MR, Bradley L, Grill HJ. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology. 2009;150(6):2654–2659. doi: 10.1210/en.2008-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barrera JG, D’Alessio DA, Drucker DJ, Woods SC, Seeley RJ. Differences in the central anorectic effects of glucagon-like peptide-1 and exendin-4 in rats. Diabetes. 2009;58(12):2820–2827. doi: 10.2337/db09-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baraboi ED, Smith P, Ferguson AV, Richard D. Lesions of area postrema and subfornical organ alter exendin-4-induced brain activation without preventing the hypophagic effect of the GLP-1 receptor agonist. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1098–R1110. doi: 10.1152/ajpregu.00326.2009. [DOI] [PubMed] [Google Scholar]
- 38.Baraboi ED, St-Pierre DH, Shooner J, Timofeeva E, Richard D. Brain activation following peripheral administration of the GLP-1 receptor agonist exendin-4. Am J Physiol Regul Integr Comp Physiol. 2011;301(4):R1011–R1024. doi: 10.1152/ajpregu.00424.2010. [DOI] [PubMed] [Google Scholar]
- 39.Dossat AM, Lilly N, Kay K, Williams DL. Glucagon-like peptide 1 receptors in nucleus accumbens affect food intake. J Neurosci. 2011;31(41):14453–14457. doi: 10.1523/JNEUROSCI.3262-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alhadeff AL, Rupprecht LE, Hayes MR. GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. Endocrinology. 2012;153(2):647–658. doi: 10.1210/en.2011-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalvi PS, Nazarians-Armavil A, Purser MJ, Belsham DD. Glucagon-like peptide-1 receptor agonist, exendin-4, regulates feeding-associated neuropeptides in hypothalamic neurons in vivo and in vitro. Endocrinology. 2012;153(5):2208–2222. doi: 10.1210/en.2011-1795. [DOI] [PubMed] [Google Scholar]
- 42.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 43.Scrocchi LA, et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med. 1996;2(11):1254–1258. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- 44.During MJ, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9(9):1173–1179. doi: 10.1038/nm919. [DOI] [PubMed] [Google Scholar]
- 45.Kim M, et al. GLP-1 receptor activation Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. 2013;19(5):567–575. doi: 10.1038/nm.3128. [DOI] [PubMed] [Google Scholar]
- 46.Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes. 2004;53(9):2492–2500. doi: 10.2337/diabetes.53.9.2492. [DOI] [PubMed] [Google Scholar]
- 47.Tatarkiewicz K, Sablan EJ, Polizzi CJ, Villescaz C, Parkes DG. Long-term metabolic benefits of exenatide in mice are mediated solely via the known glucagon-like peptide 1 receptor. Am J Physiol Regul Integr Comp Physiol. 2014;306(7):R490–R498. doi: 10.1152/ajpregu.00495.2013. [DOI] [PubMed] [Google Scholar]
- 48.Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J Clin Invest. 2014;124(6):2456–2463. doi: 10.1172/JCI72434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ciofi P. The arcuate nucleus as a circumventricular organ in the mouse. Neurosci Lett. 2011;487(2):187–190. doi: 10.1016/j.neulet.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 50.Abernethy WB, Bell MA, Morris M, Moody DM. Microvascular density of the human paraventricular nucleus decreases with aging but not hypertension. Exp Neurol. 1993;121(2):270–274. doi: 10.1006/exnr.1993.1095. [DOI] [PubMed] [Google Scholar]
- 51.Rodríguez EM, Blázquez JL, Guerra M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: the former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides. 2010;31(4):757–776. doi: 10.1016/j.peptides.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Roed SN, et al. Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol Cell Endocrinol. 2014;382(2):938–949. doi: 10.1016/j.mce.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 53.Langlet F, Mullier A, Bouret SG, Prevot V, Dehouck B. Tanycyte-like cells form a blood-cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. J Comp Neurol. 2013;521(15):3389–3405. doi: 10.1002/cne.23355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balland E, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014;19(2):293–301. doi: 10.1016/j.cmet.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacobsen LV, Hindsberger C, Robson R, Zdravkovic M. Effect of renal impairment on the pharmacokinetics of the GLP-1 analogue liraglutide. Br J Clin Pharmacol. 2009;68(6):898–905. doi: 10.1111/j.1365-2125.2009.03536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plum A, Jensen LB, Kristensen JB. In vitro protein binding of liraglutide in human plasma determined by reiterated stepwise equilibrium dialysis. J Pharm Sci. 2013;102(8):2882–2888. doi: 10.1002/jps.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Norsted E, Gömüç B, Meister B. Protein components of the blood–brain barrier (BBB) in the mediobasal hypothalamus. J Chem Neuroanat. 2008;36(2):107–121. doi: 10.1016/j.jchemneu.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 58.Mortazavi MM, et al. The choroid plexus: a comprehensive review of its history, anatomy, function, histology, embryology, and surgical considerations. Childs Nerv Syst. 2014;30(2):205–214. doi: 10.1007/s00381-013-2326-y. [DOI] [PubMed] [Google Scholar]
- 59.Devos R, et al. et al. OB protein binds specifically to the choroid plexus of mice and rats. Proc Natl Acad Sci U S A. 1996;93(11):5668–5673. doi: 10.1073/pnas.93.11.5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plamboeck A, et al. The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastroint Liver Physiol. 2013;304(12):G1117–G1127. doi: 10.1152/ajpgi.00035.2013. [DOI] [PubMed] [Google Scholar]
- 61.Hayes MR, et al. The common hepatic branch of the vagus is not required to mediate the glycemic and food intake suppressive effects of glucagon-like-peptide-1. 2011;301(5):R1479. doi: 10.1152/ajpregu.00356.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Labouesse MA, Stadlbauer U, Weber E, Arnold M, Langhans W, Pacheco-López G. Vagal afferents mediate early satiation and prevent flavour avoidance learning in response to intraperitoneally infused exendin-4. J Neuroendocrinol. 2012;24(12):1505–1516. doi: 10.1111/j.1365-2826.2012.02364.x. [DOI] [PubMed] [Google Scholar]
- 63.Merchenthaler I, Lane M, Shughrue P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol. 1999;403(2):261–280. doi: 10.1002/(SICI)1096-9861(19990111)403:2<261::AID-CNE8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto H, et al. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. J Clin Invest. 2002;110(1):43–52. doi: 10.1172/JCI15595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hisadome K, Reimann F, Gribble FM, Trapp S. Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like Peptide 1 neurons. Diabetes. 2010;59(8):1890–1898. doi: 10.2337/db10-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scott MM, Williams KW, Rossi J, Lee CE, Elmquist JK. Leptin receptor expression in hindbrain Glp-1 neurons regulates food intake and energy balance in mice. J Clin Invest. 2011;121(6):2413–2421. doi: 10.1172/JCI43703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vrang N, et al. Upregulation of the brainstem preproglucagon system in the obese Zucker rat. Brain Res. 2008;1187:116–124. doi: 10.1016/j.brainres.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 68.Hayes MR, Skibicka KP, Grill HJ. Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology. 2008;149(8):4059–4068. doi: 10.1210/en.2007-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayes MR, et al. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab. 2011;13(3):320–330. doi: 10.1016/j.cmet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao S, Kanoski SE, Yan J, Grill HJ, Hayes MR. Hindbrain leptin and glucagon-like-peptide-1 receptor signaling interact to suppress food intake in an additive manner. Int J Obes (Lond). 2012;36(12):1522–1528. doi: 10.1038/ijo.2011.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMahon LR, Wellman PJ. PVN infusion of GLP-1-(7-36) amide suppresses feeding but does not induce aversion or alter locomotion in rats. Am J Physiol. 1998;274(1 pt 2):R23–R29. doi: 10.1152/ajpregu.1998.274.1.R23. [DOI] [PubMed] [Google Scholar]
- 72.Tang-Christensen M, Larsen PJ, Thulesen J, Rømer J, Vrang N. The proglucagon-derived peptide, glucagon-like peptide-2, is a neurotransmitter involved in the regulation of food intake. Nat Med. 2000;6(7):802–807. doi: 10.1038/77535. [DOI] [PubMed] [Google Scholar]
- 73.Barrera JG, Sandoval DA, D’Alessio DA, Seeley RJ. GLP-1 and energy balance: an integrated model of short-term and long-term control. Nat Rev Endocrinol. 2011;7(9):507–516. doi: 10.1038/nrendo.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Larsen PJ, Fledelius C, Knudsen LB, Tang-Christensen M. Systemic administration of the long-acting GLP-1 derivative NN2211 induces lasting and reversible weight loss in both normal and obese rats. Diabetes. 2001;50(11):2530–2539. doi: 10.2337/diabetes.50.11.2530. [DOI] [PubMed] [Google Scholar]
- 75.Bunyan J, Murrell EA, Shah PP. The induction of obesity in rodents by means of monosodium glutamate. Br J Nutr. 1976;35(1):25–39. doi: 10.1079/BJN19760005. [DOI] [PubMed] [Google Scholar]
- 76.Reyes TM, Sawchenko PE. Involvement of the arcuate nucleus of the hypothalamus in interleukin-1-induced anorexia. J Neurosci. 2002;22(12):5091–5099. doi: 10.1523/JNEUROSCI.22-12-05091.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knauf C, et al. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest. 2005;115(12):3554–3563. doi: 10.1172/JCI25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knauf C, et al. Role of central nervous system glucagon-like Peptide-1 receptors in enteric glucose sensing. Diabetes. 2008;57(10):2603–2612. doi: 10.2337/db07-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raun K, von-Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes. 2007;56(1):8–15. doi: 10.2337/db06-0565. [DOI] [PubMed] [Google Scholar]
- 80.Erreger K, Davis AR, Poe AM, Greig NH, Stanwood GD, Galli A. Exendin-4 decreases amphetamine-induced locomotor activity. Physiol Behav. 2012;106(4):574–578. doi: 10.1016/j.physbeh.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davis JF, et al. Gastric bypass surgery attenuates ethanol consumption in ethanol-preferring rats. Biol Psychiatry. 2012;72(5):354–360. doi: 10.1016/j.biopsych.2012.01.035. [DOI] [PubMed] [Google Scholar]
- 82.Thaler JP, Guyenet SJ, Dorfman MD, Wisse BE, Schwartz MW. Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 2013;62(8):2629–2634. doi: 10.2337/db12-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thaler JP, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raun K, von VP, Knudsen LB. Liraglutide, a once-daily human glucagon-like peptide-1 analog, minimizes food intake in severely obese minipigs. Obesity (Silver Spring). 2007;15(7):1710–1716. doi: 10.1038/oby.2007.204. [DOI] [PubMed] [Google Scholar]
- 85.Astrup A, Rossner S, Van Gaal L. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374(9701):1606–1616. doi: 10.1016/S0140-6736(09)61375-1. [DOI] [PubMed] [Google Scholar]
- 86.Wadden TA, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: The SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443–1451. doi: 10.1038/ijo.2013.120. [DOI] [PubMed] [Google Scholar]
- 87.Gao Y, et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia. 2014;62(1):17–25. doi: 10.1002/glia.22580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schlögl H, et al. Exenatide-induced reduction in energy intake is associated with increase in hypothalamic connectivity. Diabetes Care. 2013;36(7):1933–1940. doi: 10.2337/dc12-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Madsen AN, Jelsing J, van de Wall EH, Vrang N, Larsen PJ, Schwartz GJ. Rimonabant induced anorexia in rodents is not mediated by vagal or sympathetic gut afferents. Neurosci Lett. 2009;449(1):20–23. doi: 10.1016/j.neulet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 90.Norgren R, Smith GP. A method for selective section of vagal afferent or efferent axons in the rat. Am J Physiol. 1994;267(4 pt 2):R1136–R1141. doi: 10.1152/ajpregu.1994.267.4.R1136. [DOI] [PubMed] [Google Scholar]
- 91.Levin BE, Dunn-Meynell AA, McMinn JE, Alperovich M, Cunningham-Bussel A, Chua SC. A new obesity-prone, glucose-intolerant rat strain (F.DIO). Am J Physiol Regul Integr Comp Physiol. 2003;285(5):R1184–R1191. doi: 10.1152/ajpregu.00267.2003. [DOI] [PubMed] [Google Scholar]
- 92.Becker K, Jährling N, Saghafi S, Weiler R, Dodt HU. Chemical clearing and dehydration of GFP expressing mouse brains. PLoS One. 2012;7(3):e33916. doi: 10.1371/journal.pone.0033916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vrang N, Tang-Christensen M, Larsen PJ, Kristensen P. Recombinant CART peptide induces c-Fos expression in central areas involved in control of feeding behaviour. Brain Res. 1999;818(2):499–509. doi: 10.1016/s0006-8993(98)01349-3. [DOI] [PubMed] [Google Scholar]
- 94.Draper S, et al. Differential gene expression between neuropeptide Y expressing neurons of the dorsomedial nucleus of the hypothalamus and the arcuate nucleus: microarray analysis study. Brain Res. 2010;1350:139–150. doi: 10.1016/j.brainres.2010.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heisler LK, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51(2):239–249. doi: 10.1016/j.neuron.2006.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.