

Abstract
Parkinson’s disease (PD) and multiple system atrophy (MSA) are progressive neurodegenerative disorders classified as synucleinopathies, which are defined by the presence of α-synuclein protein pathology. Genetic studies have identified a total of 18 PARK loci that are associated with PD. The SNCA gene encodes the α-synuclein protein. The first pathogenic α-synuclein p.A53T substitution was discovered in 1997; this was followed by the identification of p.A30P and p.E46K pathogenic substitutions in 1998 and 2004, respectively. In the last year, two possible α-synuclein pathogenic substitutions, p.A18T and p.A29S, and two probable pathogenic substitutions, p.H50Q and p.G51D have been nominated. Next-generation sequencing approaches in familial PD have identified mutations in the VPS35 gene. A VPS35 p.D620N substitution remains the only confirmed pathogenic substitution. A second synucleinopathy, MSA, originally was considered a sporadic condition with little or no familial aggregation. However, recessive COQ2 mutations recently were nominated to be the genetic cause in a subset of familial and sporadic MSA cases. Further studies on the clinicogenetics and pathology of parkinsonian disorders will facilitate clarification of the molecular characteristics and pathomechanisms underlying these disorders.
Keywords: SNCA, VPS35, PD, MSA, Genetics, Familial
Introduction
The synucleinopathies are group of disorders characterized by the presence of α-synuclein protein aggregation at autopsy [1]. Parkinson’s disease (PD) is the major synucleinopathy and the most common neurodegenerative movement disorder. Patients with PD typically present with bradykinesia, muscle rigidity, resting tremor, and postural instability; they respond well to L-dopa therapy. PD is characterized pathologically by Lewy bodies and Lewy neurites in which the α-synuclein protein accumulates. Although approximately 15% of PD patients report a family history of PD [2], large families with Mendelian inheritance are rare. To date, various genetic methods have been applied to multiplex familial forms of PD revealing a total of 18 genomic loci, which now includes seven known genes. Among these seven genes, four (SNCA, LRRK2, VPS35, and EIF4G1) are associated with autosomal dominant PD [3]. Multiple system atrophy (MSA) is a rare neurodegenerative movement disorder. Patients with MSA typically present with dysautonomia accompanied by a combination of parkinsonism, cerebellar ataxia, and pyramidal signs. MSA is characterized pathologically by glial cytoplasmic inclusions composed of α-synuclein protein [4]. MSA originally was considered a sporadic condition; however, an international collaborative group recently nominated a potential gene for this condition [5].
This paper focuses on the recent genetic research on PD with regards to novel SNCA and VPS35 mutations; in addition, we also introduce our series of familial forms of clinical MSA.
Familial Parkinsonism related to α-synuclein pathology
PD and MSA are major neurodegenerative parkinsonian disorders related to α-synuclein pathology [4]. In this section, we discuss the current status of clinical and genetic research on PD with SNCA gene mutations and on MSA.
Parkinson’s disease with SNCA mutations
The first point mutation in the SNCA gene, resulting in α-synuclein p.A53T substitution, was identified in a Greek-Italian family in 1997 [6]; thereafter, two additional mutations in the SNCA gene, resulting in α-synuclein p.A30P and p.E46K substitutions, were reported in German and Spanish families, respectively [7, 8]. In addition to these point mutations, genomic multiplications of the entire SNCA gene also cause PD [9, 10]. Although α-synuclein protein appears to play a central role in the pathogenesis of PD, mutations in the SNCA gene have been identified very rarely. However, recently four additional α-synuclein substitutions (p.A18T, p.A29S, p.H50Q, and p.G51D) have been identified. In this section, we review these newly discovered mutations.
α-Synuclein p.A18T and p.A29S
Hoffmann et al. screened the SNCA gene for 629 Polish PD patients, including 169 with early-onset PD and 460 with late-onset PD [11]. Thirteen percent of the patients had a familial form of PD. They identified p.A18T and p.A29S substitutions in the α-synuclein protein. These two substitutions were absent in 630 healthy controls and in the Exome Variant Database (http://evs.gs.washingont,edu/EVS/). One patient carrying the α-synuclein p.A18T substitution presented with a typical PD phenotype at age 60, later developing mild dysautonomia and cognitive impairment. The patient did not have a family history of neurodegenerative disorders. His parents were not available for genetic sequencing; however, one of the unaffected siblings did not carry the substitution. The other patient carrying an α-synuclein p.A29S substitution experienced rapidly progressive PD beginning at age 60 years, which was followed by the appearance of restless legs syndrome and psychiatric symptoms, including anxiety and depression. The patient did not have family history of neurodegenerative disorders. The brain of the patient with the α-synuclein p.A29S substitution showed characteristic pathological findings of PD. The parents of the patient were not available for genetic sequencing.
Overall, the clinical phenotype of patients with α-synuclein p.A18T and p.A29S substitutions is characterized by a relatively typical PD phenotype. The pathogenicity of these two substitutions still remains unclear; therefore, further genetic and functional analyses on other patients with the mutation are warranted.
α-Synuclein p.H50Q
Apple-Cresswell et al. performed Sanger sequencing on the coding region of the SNCA gene for 110 PD patients [12]. Sixty-six percent of patients had a familial form of PD. They identified the α-synuclein p.H50Q substitution in a single PD patient. The mutation was absent in an additional 1,105 PD patients, 875 healthy controls, and a publicly available next-generation sequencing database (http://main.genome-browser.bx.psu.edu/). The patient developed L-dopa responsive familial PD at age 60 years, which was followed by cognitive impairment, including apathy and dementia. His mother had parkinsonism, and his aunt, as well as a sibling, had dementia. Proukakis et al. [13] performed genetic sequencing on the coding region of the SNCA gene for five cases from the Queen Square PD Brain Bank. They identified the α-synuclein p.H50Q substitution in a single sporadic PD patient. The mutation was absent in the database of single nucleotide polymorphisms and in 450 healthy controls. The patient developed L-dopa responsive PD at age 71, which was followed by forgetfulness. His brain showed characteristic pathological findings of PD, which was accompanied by mild Alzheimer’s pathology. Overall, the clinical phenotype of patients with the α-synuclein p.H50Q substitution is characterized by a PD phenotype accompanied by cognitive impairment. Of note, the patients described by the two groups shared a common ancestral founder [12].
α-Synuclein p.G51D
Lesage et al. [14] performed whole exome sequencing on three PD patients from a three generation French family. They identified the α-synuclein p.G51D substitution in these four patients. The mutation was absent in 236 neurologically normal controls and 200 additional index patients with autosomal dominant PD. The clinical features of these four patients are characterized by early-onset and rapidly progressive PD with mild-to-moderate responsiveness to L-dopa therapy. Three of the patients had pyramidal signs, and two had severe psychiatric symptoms, such as anxiety and depression. The brain of one of the patients showed a wide distribution of α-synuclein Lewy body pathology, similar to that of patients with SNCA multiplications. Cytoplasmic inclusions in the cortex were p62 and ubiquitin positive but Aβ and TDP-43 negative. Kiely et al. [15] performed Sanger sequencing on three family members from a two generation British family. They identified the α-synuclein p.G51D heterozygous substitution in all of the affected patients. The mutation was absent in 4,500 normal controls. The proband developed PD at age 19; progressive cognitive impairment, visual hallucinations, dysautonomia, limb myoclonus, and pyramidal signs subsequently appeared. He eventually had seizures and died following a disease duration of 29 years. His father had a similar illness and his sibling developed L-dopa responsive PD with occasional visual hallucinations. At autopsy, the patient’s brain showed extensive α-synuclein pathology, accompanied by mild Alzheimer’s pathology. The Lewy body pathology included neuronal α-synuclein positive inclusions as well as glial cytoplasmic inclusions, which more typically are seen in cases of MSA. Cytoplasmic inclusions were p62 positive. TDP-43 positive neurocytoplasmic inclusions were present in moderate intensity in the basal ganglia. Overall, the clinical phenotype of patients with the α-synuclein p.G51D substitution is characterized by early-onset, rapidly progressive PD accompanied by pyramidal signs and psychiatric symptoms with the absence of cognitive impairment in the early stage of illness. These features are distinguishable from those of patients with the other SNCA mutations.
Characteristic clinical and pathological features of the SNCA mutations are summarized in Table 1.
Table 1.
Demographic, clinical, and pathological characteristics of SNCA mutations
| Mutation | Country | AAO/MDD (years) | Clinical phenotype | Pathology |
|---|---|---|---|---|
| A18T | Poland | 50/NA | PD, dysautonomia, dementia | NA |
| A29S | Poland | 60/17 | PD, restless legs syndrome, psychiatric symptoms | LB pathology |
| A30P | Germany | 54–76 | PD (sustained response to L-dopa therapy), dementia (mild)*, hallucination* | LB pathology, tau pathology |
| E46K | Spain (Basque) | 50–69 | PD, cognitive impairment, visual hallucination, REM behavior disorder | LB pathology |
| H50Q | England | 60–71/12 | PD, cognitive impairment, psychiatric symptoms | LB pathology, tau pathology |
| G51D | France | 19–60/6 | PD (early-onset, rapidly progressive), pyramidal signs, psychiatric symptoms, | LB pathology, tau pathology, TDP-43 pathology |
| A53T | Greece, Italia, Sweden, Australia, Korea | 20–85/12 | PD (early-onset, rapidly progressive), dementia, dysautonomia*, myoclonus*, psychiatric symptoms* | LB pathology, tau pathology, TDP-43 pathology |
| Duplication | France, Italy, Japan, Sweden, Korea | 31–77/15 | PD, hallucination, cognitive impairment, dysautonomia*, myoclonus* | LB pathology, tau pathology |
| Triplication | Sweden, Japan | 20–50/7 | PD, cognitive impairment, dysautonomia, psychiatric symptoms, hallucination, myoclonus*, postural tremor* | LB pathology |
AAO=age at onset; MAA=mean age at onset; MDD=mean disease duration; LB=Lewy body; NA=not available; PD=Parkinson’s disease;
uncommon
Structures of α-synuclein and substitutions
The sequence of the α-synuclein protein can be divided into three major domains: the N-terminal amphipathic region, the central NAC domain (non-amyloid b-component) and the C-terminal, highly acidic region containing several phosphorylation sites [16]. All six SNCA point mutations have been identified in the N-terminal amphipathic region of α-synuclein protein. The structure of N-terminal amphipathic region can be subdivided into two α-helix stretches (positions 3–30 and 45–92), which are divided by Helix-Turn-Helix in the secondary structure (Figure 1A) [12, 13, 16]. α-Synuclein p.A18T, p.A29S and p.A30P substitutions are in the first helical fragment; p.E46K, p.G50Q, p.G51D and p.A53T substitutions are in the second helical fragment. The clinical phenotype of newly identified p.A18T and p.A29S carriers resembles the phenotype observed in patients with the previously reported pathogenic p.A30P substitution [8]. These three mutations are associated with a typical late-onset form of PD [11]. However, the phenotypes exhibited by α-synuclein p.E46K, p.G50Q,p.G51D and A53T substitutions show severe and rather rapidly progressive parkinsonism, cognitive decline and sometimes visual hallucinations, suggesting a clinical presentation for these substitutions more reminiscent of dementia with Lewy bodies [7, 12–15]. Patients with α-synuclein p.G51D and p.A53T substitutions have earlier disease onset than patients with the other five substitutions [12–15].
Figure 1.
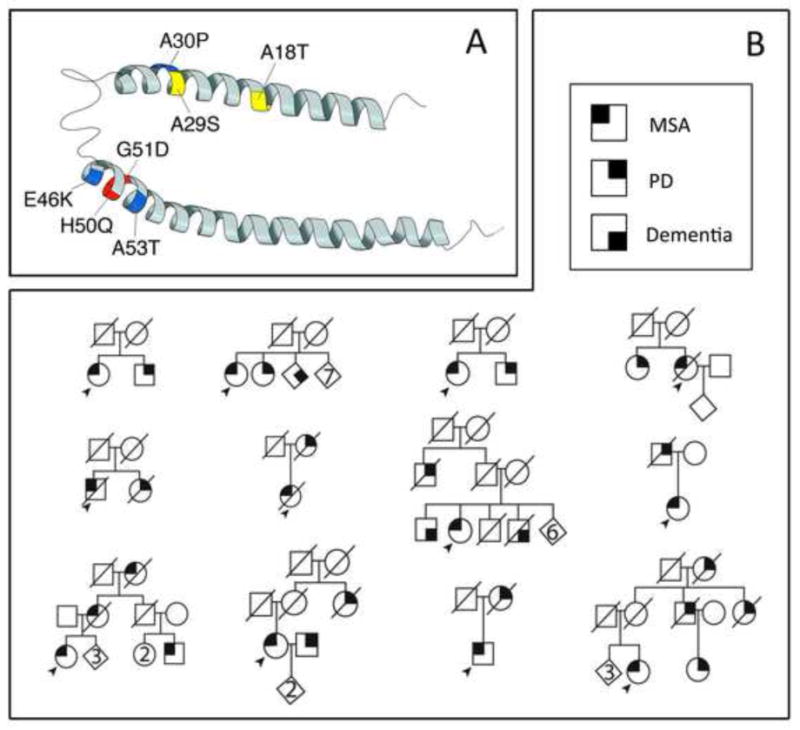
Secondary structure of α-synuclein protein (A). Positions for previously known pathogenic substitutions, p.A30P, p.E46K, and p.A53T (in blue); positions for newly discovered probable pathogenic substitutions, p.H50Q and p. G51D (in red), and positions for newly discovered possible pathogenic substitutions, p.A18T and p.A29S, (in yellow). Pedigrees of the multiple system atrophy family from the Mayo Clinic Florida in Jacksonville, Florida (B). Standard pedigree symbols were used. Round symbols indicate females; squares indicate males; and diagonal lines indicate that the individual is deceased. Diamonds were used to disguise gender. Numbers in diamonds indicate the number of siblings. The solid arrowhead indicates the proband. * indicates autopsy patients.
Multiple System Atrophy
MSA is a rare neurodegenerative disorder characterized clinically by dysautonomia with various combinations of poorly L-dopa responsive parkinsonism, cerebellar ataxia, and pyramidal signs [17]. MSA originally was considered a sporadic condition. However, several small familial aggregations of MSA have been reported and, more commonly, familial aggregates of α-synucleinopathy, (i.e. PD and MSA) in the same kindreds. Müllner et al. [18, 19] first reported a German family in which two members were diagnosed with MSA. Subsequently, seven families with MSA with detailed clinical information have been reported from two independent Japanese groups [20, 21]. Within these eight families, probands presented with definite or probable MSA, and the other family members manifested with MSA or PD. We have collected 61 MSA cases that were diagnosed based on the current diagnostic criteria for MSA [17]. Among these cases, 12 index cases have family members who were affected by MSA or PD. Pedigrees of these families are shown in Figure 1B. Most of pedigrees showed possible autosomal dominant with reduced penetrance or recessive patterns of inheritance. The clinical diagnosis in seven probands was probable MSA-P (MSA with predominant parkinsonism), four were possible MSA-P, and one was probable MSA-P+C (MSA of mixed type parkinsonism-cerebellar ataxia). The mean age at symptomatic onset was 65 years old with poor L-dopa responsiveness in most of the probands. The clinical features of previously reported MSA families and of our familial cases are summarized in Table 2. Of note, variants of the SNCA gene [22] and gene polymorphisms in the IL-1A [23], IL-8 [24], and ACT [25] genes, and copy number loss of SHC2 [26] genes have been reported to be associated with increased risk of MSA.
Table 2.
Summary of clinical features of familial multiple system atrophy
| Family | Ethnicity | Diagnosis (age at onset (years)) | Suggested inheritance | L-dopa responsiveness in MSA cases | References |
|---|---|---|---|---|---|
| Previous cases | |||||
| 1 | Germany | Definite MSA-P (68)*, probable MSA-P (46) | Autosomal dominant | Good†/NA‡ | Wüllner, et al. [19, 20] |
| 2 | Japan | Probable MSA-P (57)*, probable MSA-C (65) | Autosomal dominant | Not treated†/NA‡ | Soma, et al. [21] |
| 3 | Probable MSA-C (46)*, PD (74) | Autosomal dominant | NA | ||
| 4 | Probable MSA-P (65)*, PD (67) | Autosomal dominant | Poor | ||
| 5 | Japan | Definite MSA-P (68), Definite MSA-P (62) | Autosomal recessive | Poor†/poor‡ | Hara, et al. [22] The MSA Research Collaboration [5] |
| 6 | Probable MSA-P (72), probable MSA-P (63) | Autosomal recessive | Poor†/poor‡ | ||
| 7 | Probable MSA-P+C (68), probable MSA-P+C (67) | Autosomal recessive | Poor†/poor‡ | ||
| 8 | Probable MSA-P (69), probable MSA-C (58) | Autosomal recessive | Poor†/poor‡ | ||
| 9 | Japan | Probable MSA-P (53), Definite MSA-P (52), PD, PD | Autosomal recessive | Poor†/poor‡ | The MSA Research Collaboration [5] |
| 10 | Probable MSA-C (50), probable MSA-C (44) | Autosomal recessive | NA†/NA‡ | ||
| Our cases | |||||
| 1 | United States | Possible MSA-P (66)*, MSA© | Autosomal dominant | Poor†/NA‡ | This study |
| 2 | Probable MSA-P (64)*, MSA (Shy-Drager syndrome) | Autosomal dominant | NA†/NA‡ | ||
| 3 | Probable MSA-P (58)*, PD | Autosomal dominant | Moderate | ||
| 4 | Probable MSA-P (64)*, PD (79) | Autosomal dominant | Poor | ||
| 5 | Probable MSA-P (59)*, PD | Unclear | NA | ||
| 6 | Probable MSA-P (83)*, PD | Unclear | NA | ||
| 7 | Probable MSA-P+C (58)*, PD | Autosomal dominant | NA | ||
| 8 | Possible MSA-P (61)*, PD, AD | Autosomal dominant | NA | ||
| 9 | Possible MSA-P (49)*, PD | Autosomal dominant | NA | ||
| 10 | Probable MSA-P (70)*, PD | Unclear | NA | ||
| 11 | Possible MSA-P*, PD | Autosomal dominant | NA | ||
| 12 | Probable MSA-P (72)*, PD | Autosomal dominant | Poor | ||
MSA= multiple system atrophy; MSA-C=multiple system atrophy with predominant cerebellar ataxia; MSA-C+P=multiple system atrophy of mixed type; MSA-P=multiple system atrophy with predominant Parkinsonism; NA=not available; PD=Parkinson’s disease;
proband;
L-dopa responsiveness for proband;
L-dopa responsiveness for the other family members with MSA;
type of MSA was unknown due to lack of clinical information
Mutations in the COQ2 gene in MSA
The MSA Research Collaboration group [5] recently has nominated the COQ2 gene as a causative gene for a small subset of Japanese MSA cases. They performed a combination of linkage analysis and whole-genome sequencing on a multiplex family and discovered a homozygous COQ2 p.M78V-M343A substitution. Mutational analysis of the COQ2 gene on another MSA family revealed compound heterozygous substitutions, p.R337X and p.V343A, in two affected patients. The mutations also were found in two unaffected siblings; however, neither variant was observed in 180 Japanese controls. Subsequent mutational analysis of the COQ2 gene and functional analyses of COQ2 protein identified eight heterozygous or compound heterozygous variants, which are mildly or severely deleterious, in 758 sporadic MSA patients from Japanese, European, and North American series. Those variants were not found in 1129 controls. The p.V343A variant is relatively common in Japanese populations, but not in European or North American populations. The investigators concluded that the p.V343A variant and other heterozygous variants in the COQ2 genes found in this study could be susceptibility factors for MSA rather than causal factors. Two patients of Alzheimer’s disease without any characteristic features of MSA also were found to carry the homozygous COQ2 p.V343A substitution. The Mayo Clinic Florida MSA (α-synucleinopathy) pedigrees presented here (Figure 1B) all were screened for the entire coding region of the COQ2 gene but no mutations were observed (unpublished data).
Familial parkinsonism with unknown pathology
In several familial parkinsonian disorders with known pathogenic gene mutations, including PD with mutations in the DJ-1 and VPS35 genes, the pathological characteristics are still uncertain. In this section, we discuss the current status of clinical and genetic research on PD with VPS35 mutations.
Parkinson’s disease with VPS35 mutations
Mutations in the VPS35 gene are known to cause PD. The pathological characteristics of such mutations are unknown, although given the clinical presentation, an α-synucleinopathy is probable. VPS35 mutations were first reported in two large families with autosomal dominant PD by independent groups in 2011 [27, 28]. So far, fifteen reports assessing the frequency of VPS35 mutations in familial and sporadic PD have been published (Table 2). In this section, we summarize the previous reports related to VPS35 mutations. Vilariño-Güell et al. [27] applied next-generation sequencing technologies to a previously reported Swiss family with late-onset PD [29]. They identified the VPS35 p.D620N substitution in all six affected family members who were available for genetic testing. They also identified the mutation in three families from the USA, Tunisia, and Israel, as well as in one sporadic patient from Israel. The mutation was absent in 4,326 multi-ethnic subjects in a case-control PD series and in 3,309 neurologically normal controls. All affected patients presented clinically with tremor predominant, L-dopa responsive PD. At the same time, Zimprich et al. [28] performed exome sequencing on a large, Austrian family with late-onset PD. They identified the VPS35 p.D620N substitution in all seven affected family members who were available for genetic testing. They also identified the mutation in two additional Austrian families. The mutation was absent in 1,014 controls. The affected patients presented clinically with L-dopa responsive PD that occasionally was accompanied by action tremor.
So far, 13 other studies have been performed on the VPS35 gene for PD and other Lewy body diseases [Supplemental references 1–13]. Affected patients carrying VPS35 mutations present clinically with at least 3 of 4 cardinal symptoms of PD, including bradykinesia, rigidity, resting tremor, and postural instability. Patients occasionally have action tremor and mild cognitive impairment. So far, all of the reported patients have responded to L-dopa therapy. At this time, only the VPS35 p.D620N substitution has been confirmed as pathogenic. The VPS35 p.D620N substitution has been observed predominantly in families of Caucasian descent with autosomal dominant PD [27, 28] [Supplemental references 1–5]. In contrast, the mutation has been rare in Asian populations, with the exception of Japanese populations [Supplemental references 6–11]. The frequency of the VPS35 p.D620N substitution in patients with familial PD and sporadic PD are estimated to be 0–1.3% and 0–0.3%, respectively. Further genetic and functional studies are warranted for several other VPS35 substitutions, such as p.P316S, p.Y507F, p.R524W, p.I560T, p.M571I, p.H599R, p.M607V, and p.E787K that have not been identified in controls [27, 28] [Supplemental references 12, 13].
The results of previous analyses for the VPS35 p.D620N substitution are summarized in Table 2.
Conclusion
Various genetic analytical methods, such as linkage analysis and exome sequencing, have uncovered causative genetic factors in a number of multiplex monogenic forms of PD and parkinsonian disorders [1]. Genome-wide association studies have identified a substantial number of susceptibility genes and genetic risk factors for these disorders. Henceforth, these techniques are being gradually replaced by next-generation DNA sequencing techniques [30]. These techniques allow researchers to sequence the entire human genome with the ability to detect single nucleotide variants and small insertions and deletions, as well as large scale genomic deletions, multiplications and rearrangements. These techniques are more cost-effective and can be performed faster than conventional genetic screening techniques. Given that most neurodegenerative disorders have a genetic basis, there are still many other familial forms of Parkinsonism for which genetic causes or genetic risk factors remain to be discovered. Identification of causative genetic factors and genetic risk factors will contribute to the clarification of the underlying mechanisms of disorders, which may in turn lead to the development of useful biomarkers, curative therapies, and preventive measures, and also to the establishment of systems for appropriate genetic testing and genetic counseling. Looking ahead, there is no doubt that the new-generation DNA sequencing techniques will be applied to clinical practice. These techniques certainly will aid physicians in making clinical diagnoses. However, before these techniques can be applied to general clinical practice, there are various issues to be resolved: improvement of sensitivity and specificity of the genetic sequencing, certain ethical matters, training of specialists on genetic counseling, reinforcement on management of personal genetic information, and other issues. Additionally, physicians need to constantly update their knowledge of clinico-genetic information of various disorders in order to appropriately select genetic testing for each patient.
Supplementary Material
Table 3.
Summary of previous studies on VPS D620N mutations
| Country/group | Total number of subjects | Number of mutation carriers | References |
|---|---|---|---|
| Switzerland | 2 fPD† | 2 | [28] |
| United States | 1600 PD | 1 | |
| Norway | 688 PD | 0 | |
| Canada | 574 PD | 0 | |
| Taiwan | 406 PD | 0 | |
| Ireland | 377 PD | 0 | |
| Poland | 362 PD | 0 | |
| Tunisia | 199 PD | 1 | |
| Israel | 120 PD | 2 | |
| Australia | 2 fPD† | 2 fPD | [29] |
| Australia | 486PD | 2 fPD | |
| Australia and Germany | 860 PD | 2 | |
| United Kingdom | 160 fPD, 175 EOPD, 262 sPD | 1 fPD | [1]® |
| Italy | 475 fPD | 0 | [2]® |
| Belgium | 520 PD, 85 LBD, 75 PDD | 0 | [3]® |
| China | 32 fPD, 512PD | 0 | [4]® |
| European origin | 246 fPD | 3 fPD | [5]® |
| China | 27 fPD, 1011 sPD | 0 | [6]® |
| China | 72 fPD, 130 sPD | 0 | [7]® |
| Germany | 276 fPD, 416 sPD | 1 fPD | [8]® |
| Serbia | 71 fPD, 347 sPD | 0 | |
| United States | 441 sPD | 0 | |
| Chile | 192 fPD, 31 sPD | 0 | |
| Japan | 300 fPD, 433 sPD | 4 (3 fPD, 1sPD) | [9]® |
| GEOPD consortium | 8870 PD | 7 (5 fPD, 2 sPD) | [10]® |
| China | 609 PD | 0 | [11]® |
| India | 69 fPD, 251 sPD | 0 | [12]® |
| United States | 213 PD | 0 | [13]® |
EOPD=early-onset PD; fPD=familial Parkinson’s disease; GEO-PD=Genetic Epidemiology of Parkinson’s Disease; PD=Parkinson’s disease; sPD=sporadic Parkinson’s disease;
each case is from a single multiplex family;
references are available as supplemental material.
Acknowledgments
This manuscript is based on the invited lecture provided by ZKW during the 20th World Congress on Parkinson’s Disease and Related Disorders, Geneva, Switzerland on December 9th, 2013. PT is supported by the Max Kade Foundation. ZKW is partially supported by the NIH/NINDS P50 NS072187, Mayo Clinic Center for Regenerative Medicine, and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch. ZKW serves as editor-in-chief of Parkinsonism and Related Disorders and was therefore recused from the editorial processes involving this article.
We would like to thank Kelly E. Viola and Victoria L. Jackson for their editorial assistance, and we would also like to thank Margaret A. McKinney for her illustrative support.
Footnotes
Conflicts of Interest
All authors have no conflict of interest in relation to this manuscript.
References
- 1.Puschmann A, Bhidayasiri R, Weiner WJ. Synucleinopathies from bench to bedside. Parkinsonism & related disorders. 2012;18 (Suppl 1):S24–7. doi: 10.1016/S1353-8020(11)70010-4. [DOI] [PubMed] [Google Scholar]
- 2.McDonnell SK, Schaid DJ, Elbaz A, Strain KJ, Bower JH, Ahlskog JE, et al. Complex segregation analysis of Parkinson’s disease: The Mayo Clinic Family Study. Ann Neurol. 2006;59:788–95. doi: 10.1002/ana.20844. [DOI] [PubMed] [Google Scholar]
- 3.Sundal C, Fujioka S, Uitti RJ, Wszolek ZK. Autosomal dominant Parkinson’s disease. Parkinsonism & related disorders. 2012;18 (Suppl 1):S7–10. doi: 10.1016/S1353-8020(11)70005-0. [DOI] [PubMed] [Google Scholar]
- 4.Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. 2012:2. doi: 10.1101/cshperspect.a009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med. 2013;369:233–44. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- 6.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 7.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of neurology. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 8.Kruger R, Kuhn W, Leenders KL, Sprengelmeyer R, Muller T, Woitalla D, et al. Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology. 2001;56:1355–62. doi: 10.1212/wnl.56.10.1355. [DOI] [PubMed] [Google Scholar]
- 9.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 10.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman-Zacharska D, Koziorowski D, Ross OA, MM, Poznanski J, Jurek M, et al. Novel A18T and pA29S substitutions in α-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat Disord. 2013 doi: 10.1016/j.parkreldis.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28:811–3. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 13.Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–4. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013 doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- 15.Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–69. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 17.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wullner U, Abele M, Schmitz-Huebsch T, Wilhelm K, Benecke R, Deuschl G, et al. Probable multiple system atrophy in a German family. J Neurol Neurosurg Psychiatry. 2004;75:924–5. doi: 10.1136/jnnp.2003.025155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wullner U, Schmitt I, Kammal M, Kretzschmar HA, Neumann M. Definite multiple system atrophy in a German family. J Neurol Neurosurg Psychiatry. 2009;80:449–50. doi: 10.1136/jnnp.2008.158949. [DOI] [PubMed] [Google Scholar]
- 20.Soma H, Yabe I, Takei A, Fujiki N, Yanagihara T, Sasaki H. Heredity in multiple system atrophy. J Neurol Sci. 2006;240:107–10. doi: 10.1016/j.jns.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, et al. Multiplex families with multiple system atrophy. Archives of neurology. 2007;64:545–51. doi: 10.1001/archneur.64.4.545. [DOI] [PubMed] [Google Scholar]
- 22.Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, et al. SNCA variants are associated with increased risk for multiple system atrophy. Annals of neurology. 2009;65:610–4. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Combarros O, Infante J, Llorca J, Berciano J. Interleukin-1A (-889) genetic polymorphism increases the risk of multiple system atrophy. Movement disorders: official journal of the Movement Disorder Society. 2003;18:1385–6. doi: 10.1002/mds.10540. [DOI] [PubMed] [Google Scholar]
- 24.Infante J, Llorca J, Berciano J, Combarros O. Interleukin-8, intercellular adhesion molecule-1 and tumour necrosis factor-alpha gene polymorphisms and the risk for multiple system atrophy. J Neurol Sci. 2005;228:11–3. doi: 10.1016/j.jns.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 25.Furiya Y, Hirano M, Kurumatani N, Nakamuro T, Matsumura R, Futamura N, et al. Alpha-1-antichymotrypsin gene polymorphism and susceptibility to multiple system atrophy (MSA) Brain Res Mol Brain Res. 2005;138:178–81. doi: 10.1016/j.molbrainres.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki H, Emi M, Iijima H, Ito N, Sato H, Yabe I, et al. Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Mol Brain. 2011;4:24. doi: 10.1186/1756-6606-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–7. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wider C, Skipper L, Solida A, Brown L, Farrer M, Dickson D, et al. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord. 2008;14:465–70. doi: 10.1016/j.parkreldis.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 30.Foo JN, Liu J, Tan EK. Next-generation sequencing diagnostics for neurological diseases/disorders: from a clinical perspective. Hum Genet. 2013;132:721–34. doi: 10.1007/s00439-013-1287-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.