

Background: Recent focus has been given to anti-amyloidogenic approaches for Alzheimer disease therapy.
Results: Methylene blue prevented behavioral impairment and mitigated Alzheimer disease-like pathology.
Conclusion: Methylene blue may be prophylactic for Alzheimer disease by inhibiting β-secretase activity and mitigating brain pathology.
Significance: Oral methylene blue treatment modulates β-secretase.
Keywords: Alzheimer Disease, Amyloid, Amyloid Precursor Protein (APP), Animal Model, Secretase, β-Secretase, Anti-amyloidogenic, Methylene Blue, Transgenic Mouse
Abstract
Amyloid precursor protein (APP) proteolysis is required for production of amyloid-β (Aβ) peptides that comprise β-amyloid plaques in the brains of patients with Alzheimer disease (AD). Here, we tested whether the experimental agent methylene blue (MB), used for treatment of methemoglobinemia, might improve AD-like pathology and behavioral deficits. We orally administered MB to the aged transgenic PSAPP mouse model of cerebral amyloidosis and evaluated cognitive function and cerebral amyloid pathology. Beginning at 15 months of age, animals were gavaged with MB (3 mg/kg) or vehicle once daily for 3 months. MB treatment significantly prevented transgene-associated behavioral impairment, including hyperactivity, decreased object recognition, and defective spatial working and reference memory, but it did not alter nontransgenic mouse behavior. Moreover, brain parenchymal and cerebral vascular β-amyloid deposits as well as levels of various Aβ species, including oligomers, were mitigated in MB-treated PSAPP mice. These effects occurred with inhibition of amyloidogenic APP proteolysis. Specifically, β-carboxyl-terminal APP fragment and β-site APP cleaving enzyme 1 protein expression and activity were attenuated. Additionally, treatment of Chinese hamster ovary cells overexpressing human wild-type APP with MB significantly decreased Aβ production and amyloidogenic APP proteolysis. These results underscore the potential for oral MB treatment against AD-related cerebral amyloidosis by modulating the amyloidogenic pathway.
Introduction
Alzheimer disease (AD)3 is a progressive neurodegenerative disorder that is characterized by cognitive and psychiatric disturbances, eventually leading to an inability to perform activities of daily living independently and safely (1). Histopathological hallmarks include extracellular deposits of amyloid-β (Aβ) peptides as amyloid plaques in the brain parenchyma, intracellular neurofibrillary tangles formed by cytoplasmic aggregates of hyperphosphorylated microtubule-associated Tau protein, neuronal cell death, synaptic degeneration and loss, and gliosis (2). According to the World Alzheimer Report, there were 35.6 million people living with dementia in 2010, and this is expected to increase to 65.7 million in 2030 and to as many as 115.4 million by 2050. The cumulative incidence of dementia is predicted to reach 682 million people in the next 40 years (3), illustrating that AD has become a worldwide public health crisis. Shockingly, the global care cost for dementia patients is currently 1% of the global gross domestic product (3). Therefore, it is becoming increasingly evident that effective disease treatment or prevention is needed. There is still uncertainty surrounding whether mis-metabolism of amyloid precursor protein (APP) leading to cerebral Aβ accumulation (amyloid cascade hypothesis), abnormal Tau phosphorylation (Tau hypothesis), aberrant glial cytokine cycle (neuroinflammation hypothesis), increased reactive oxygen species (free radical hypothesis), or reduced acetylcholine synthesis (cholinergic hypothesis) is the root cause of AD. Based on the “amyloid cascade hypothesis” of AD, which theorizes that cerebral Aβ accumulation sets a toxic downstream cascade into motion (2, 4, 5), a surge of research activity has been directed toward anti-amyloid therapeutics. Specific approaches have included the following: preventing the self-assembly of Aβ into soluble oligomers and insoluble deposits, enhancing Aβ degradation, directly or indirectly targeting Aβ neurotoxicity, preventing or reducing Aβ production, and improving Aβ efflux from the brain into the peripheral blood (6).
Despite continued efforts from academia and the pharmaceutical industry, no new drugs for AD have made it to the clinic since 2003 (7). These failures have largely been due to toxicity and efficacy issues in preclinical rodent models and in clinical trials. Moreover, currently available pharmacotherapeutics (i.e. acetylcholinesterase inhibitors or N-methyl d-aspartate antagonists) show precious little symptomatic benefit, and none have been successful at preventing, treating, or curing the disease. Additional clinical trials are currently underway based on the amyloid cascade hypothesis, and we await results from those studies. Nonetheless, this inconvenient truth has led the field to explore alternative approaches to designer drugs for AD prevention and treatment.
Methylene blue (MB; 3,7-bis(dimethylamino)-phenothiazin-5-ium chloride) is a heterocyclic aromatic chemical compound that belongs to the phenothiazine class of compounds. Current indications for MB include treatment for enzymopenic hereditary methemoglobinemia and acute acquired methemoglobinemia, ifosfamide-induced encephalopathy, urinary tract infections in elderly patients, vasoplegic adrenaline-resistant shock, and pediatric malaria. Additionally, MB is routinely employed as a diagnostic tracer dye in surgical procedures (8, 9). In the context of neurological diseases, the beneficial effects of MB have been demonstrated for treatment of psychosis in both preclinical (10, 11) and clinical studies (12–14). Moreover, MB may also have beneficial effects on cognitive performance in AD patients (8). In this regard, promising results from a phase II clinical trial of MB (methylthioninium chloride, RemberTM) for mild-to-moderate AD patients showed significantly improved cognitive function after 6 months of treatment compared with placebo control, and this led to stabilized AD progression over the course of a year (15, 16). This finding triggered a wave of comments, indicating that MB may be useful as an AD therapeutic agent. Despite these reports, its target(s) and mechanism(s) of action in the context of AD remain to be elucidated. This knowledge gap prompted us to evaluate whether MB might improve AD-like pathology in a transgenic mouse model of cerebral amyloidosis.
To test this, we orally administered the compound to an accelerated mouse model of cerebral amyloidosis (bearing APP “Swedish” APPK595N/M596L (APPswe) and presenilin 1 (PS1) exon 9-deleted mutant human transgenes; designated PSAPP mice) (17, 18) for 3 months, commencing at 15 months of age, and examined behavioral impairment, AD-like pathology, and APP processing at 18 months of age. Parallel analyses were performed in vitro utilizing Chinese hamster ovary cells stably expressing human wild-type APP (CHO/APPWT cells) (19).
EXPERIMENTAL PROCEDURES
Ethics Statement
All experiments were performed in accordance with the guidelines of the National Institutes of Health, and all animal studies were approved by the Saitama Medical University Institutional Animal Care and Use Committee. Animals were humanely cared for during all experiments, and all efforts were made to minimize animal suffering. Animals were anesthetized with sodium pentobarbital (50 mg/kg) and euthanized by transcardial perfusion with ice-cold physiological saline containing heparin (10 units/ml).
Mice
Male B6.Cg-Tg(APPswe, PSEN1dE9)85Dbo/Mmjax mice (bearing APPswe and PS1 exon 9-deleted mutant human transgenes) on the congenic C57BL/6J background (designated PSAPP mice) were obtained from the Jackson Laboratory (Bar Harbor, ME) and were bred with female C57BL/6J mice to yield PSAPP and WT offspring. PSAPP mice overproduce human Aβ1–40 and Aβ1–42 peptides and develop progressive cerebral β-amyloid deposits as well as learning and memory impairment (17, 18, 20–23). All mice were characterized by PCR genotyping for mutant human APP and PS1 transgenes as described elsewhere (21). We strictly used PSAPP and WT littermates obtained from this breeding strategy for all analyses. Thus, all mice used in this study are genetically comparable.
MB was obtained from Merck Millipore, resuspended in distilled water, and orally administered to 12 PSAPP mice (PSAPP-MB mice; six males and six females). As a vehicle control, 12 additional PSAPP mice received distilled water vehicle (PSAPP-V mice; six males and six females). Moreover, 24 WT littermates received MB (WT-MB mice; six males and six females) or distilled water (WT-V mice, six males and six females). In parallel, to examine whether MB treatment prevented versus reversed kinetics of cerebral amyloid accumulation, eight untreated PSAPP mice at 15 months of age (PSAPP-15M, four males and four females) were included for analysis of β-amyloid pathology. Baseline cognitive status was determined in untreated PSAPP versus WT mice at 15 months of age just prior to dosing. Subsequently, beginning at 15 months of age, animals were gavaged with MB (3 mg/kg) or vehicle once daily for 3 months. Mice were housed in a specific pathogen-free barrier facility under a 12:12-h light/dark cycle with ad libitum access to food and water.
Behavioral Analyses
Exploratory activity was evaluated by individually placing mice into a novel environment (the left corner of a white polyethylene chamber: 54 × 39 × 20 cm) (24–26). Their activity was recorded for 20 min by an overhead video camera (BL-C131, Panasonic, Fukuoka, Japan) connected to a Windows PC, and horizontal locomotion and rearing scores were counted for each 2-min time bin.
Novel object recognition and memory retention were also assessed (27). Briefly, each mouse was habituated in a cage for 4 h, and then two objects of different shapes were concurrently provided to the mouse for 10 min. The number of times that the mouse explored the familiar object (defined as number of instances where a mouse directed its nose 2 cm or less distance from the object) that was later replaced by a novel object was counted for the initial 5 min of exposure (training phase). To test memory retention on the following day, one of the familiar objects was replaced with a different shaped novel object and then explorations of the novel object were counted for 5 min (retention test). The recognition index, taken as an index of episodic memory, is reported as frequency (%) of explorations of the novel versus familiar objects.
Subsequently, Y-maze total arm entry and spontaneous alternation were assessed to measure exploratory activity and spatial working memory (20, 26). Briefly, mice were individually placed in one arm of a radially symmetric Y-maze made of opaque gray acrylic (arms: 40 cm long and 4 cm wide; walls, 30 cm tall), and the sequence of arm entries and total number of entries were recorded over a period of 8 min, beginning when the animal first entered the central area. Percent alternation was defined as entries into sequentially different arms on consecutive occasions using the following formula: % alternation = number of alternations/(number of total arm entries − 2) × 100%.
Finally, the radial arm water maze (RAWM) test was performed to assess spatial reference learning and memory (28). This test optimizes sample size requirements when compared with other typical rodent memory tasks (20). Briefly, the RAWM test was conducted over 2 days and consisted of triangular wedges in a circular pool (80 cm diameter) configured to form swim lanes that enclose a central open space. A circular pool was filled with water maintained at 23–26 °C. According to this design, the maze can be organized to independently detect errors in working and reference memory. After a minimum of 20 min of habituation to the room, mice naïve to the test were placed in the pool and allowed to search for the platform for 60 s. Subsequently, the mouse was dropped into a random start arm (predetermined on a score sheet) and allowed to swim until it located and climbed onto the platform (goal) over a period of 60 s. Errors accrued when the mouse failed to enter an arm for 15 s. If the mouse was unable to find the platform after 60 s, it was guided to the platform. Times (latency) to locate the platform and error numbers were recorded. Once the platform was found, either by self-discovery or by guidance, the mouse was allowed to rest there for 15 s. Subsequently, the mouse was then removed, towel-dried, and replaced in a cage with a heat lamp slightly overhead for a 30-min rest period. Another mouse was selected, and the process was repeated until all mice were tested. Each mouse was given a total of 15 trials. On day 1, the goal alternated between being visible and hidden as the trials proceeded for each mouse; although on day 2, the goal was always hidden. All data were organized as individual blocks of 3 trials each for further analyses. The goal arms remained in the same location for both days, whereas the start arm was randomly altered, with the position recorded on score sheets. All behavioral tests were performed in a room (6 m × 4.5 m) with indirect lighting and multiple visible cues on the walls. The examiner determined the time of swimming until the mouse reached the platform (latency) using a stopwatch. In addition, trials were recorded using an overhead video camera and were analyzed using customized macro software in Microsoft Excel. All trials were performed at the same time of day (±1 h), during the animals' light phase. To avoid interference with behavioral testing, MB or vehicle treatment was carried out 1 h after concluding behavioral testing.
Tissue Preparation
At 18 months of age, animals were anesthetized with sodium pentobarbital (50 mg/kg) and euthanized by transcardial perfusion with ice-cold physiological saline containing heparin (10 units/ml). Brains were isolated and quartered (sagitally at the level of the longitudinal fissure of the cerebrum and then coronally at the level of the anterior commissure) using a mouse brain slicer (Muromachi Kikai, Tokyo, Japan). Right anterior cerebral quarters were weighed and snap-frozen at −80 °C for Western blot analyses. Right posterior cerebral quarters were weighed and snap-frozen at −80 °C. These cerebral quarters were sequentially extracted in the following: 1) tris-buffered saline (TBS) (25 mm Tris-HCl, pH 7.4, 150 mm NaCl); 2) 2% SDS; and 3) 5 m guanidine-HCl for sandwich enzyme-linked immunosorbent assays (ELISAs). Left anterior cerebral quarters were weighed and immersed in RNA stabilization solution (RNAlater®, Applied Biosystems, Foster City, CA) and then snap-frozen at −80 °C for quantitative real time-PCR (QRT-PCR) analyses. Left posterior cerebral quarters were immersion-fixed in 4% paraformaldehyde in 0.1 m phosphate buffer at 4 °C overnight and routinely processed in paraffin for immunohistochemical analyses.
Immunohistochemistry
For paraffin blocks, we sectioned five coronal sections (per set) with a 100-μm interval and a thickness of 5-μm for each brain region (for retrosplenial cortex (RSC), entorhinal cortex (EC), and hippocampus (H), bregma −2.92 to −3.64 mm) (29). One set of five sections was prepared for each brain region for analyses of Aβ deposits/β-amyloid plaques (for burden and plaque morphometry analyses). Immunohistochemical staining was conducted according to the manufacturer's protocol using a Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA) coupled with the diaminobenzidine reaction, except that the biotinylated secondary antibody step was omitted for Aβ immunohistochemical staining. A biotinylated human Aβ17–24 monoclonal antibody (4G8; 1:200, Covance Research Products, Emeryville, CA) was used as a primary antibody. Using additional sets of five sections, normal mouse serum (isotype control) or 0.1 m phosphate-buffered saline (PBS, pH 7.4) was used instead of primary or ABC reagent as a negative control.
Image Analysis
Images were acquired as digitized tagged-image format files (to retain maximum resolution) using a BX60 microscope with an attached CCD camera system (DP-70, Olympus, Tokyo, Japan), and digital images were routed into a Windows PC for quantitative analyses using SimplePCI software (Hamamatsu Photonics, Hamamatsu, Shizuoka, Japan). We captured images of five 5-μm sections through each anatomic region of interest (RSC, EC, and H) based on anatomic criteria defined by Franklin and Paxinos (29), and we obtained a threshold optical density that discriminated staining from background. Each anatomic region of interest was manually edited to eliminate artifacts. Selection bias was controlled for by analyzing each region of interest in its entirety. For Aβ burden analysis, data are reported as the percentage of labeled area captured (positive pixels) divided by the full area captured (total pixels).
For β-amyloid plaque morphometric analyses, diameters (based on maximum length) of β-amyloid plaques were measured, and numbers of β-amyloid plaques falling into three mutually exclusive categories (<25, 25–50, or >50 μm) were blindly tabulated. Results are presented as mean plaque number per mouse for each brain region surveyed. For cerebral amyloid angiopathy (CAA) morphometric analysis, we blindly counted numbers of Aβ antibody-positive cerebral vessels in each anatomic region of interest. Those data are represented as mean CAA deposit numbers per mouse.
Cell Culture
CHO/APPWT cells were kindly provided by Drs. Stefanie Hahn and Sascha Weggen (University of Heinrich Heine, Düsseldorf, Germany), and cell culture was performed in accordance with previously described methods (19). Briefly, CHO/APPWT cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1 mm sodium pyruvate, and 100 units/ml penicillin and streptomycin. CHO/APPWT cells were seeded in 24-well tissue culture plates at 2 × 105 cells per well for 24 h before treatment. Cultured cells were treated with MB (1.56, 3.13, 6.25, 12.5, or 25 μm) or 0.1 m PBS (pH 7.4; control) for 6 h in the same media prior to analysis.
Cell-free β-Site APP-cleaving Enzyme 1 (BACE1) Activity Assay
To directly test the effect of MB on BACE1 activity, we used available kits based on secretase-specific peptides conjugated to DABCYL/EDANS fluorogenic reporter molecules (Cayman Chemical, Ann Arbor, MI) in accordance with the manufacturer's instructions. Briefly, appropriate amounts of 1× assay buffer, BACE1 enzyme, and various concentrations of MB (1.56, 3.13, 6.25, 12.5, or 25 μm) were added to a 96-well plate. Subsequently, the reactions were initiated by adding 2.5 μl of fluorogenic substrate to all wells being used. The plate was carefully shaken for 10 s to mix, covered with a plate cover, and incubated for 40 min at room temperature prior to reading fluorescence values. Following incubation, fluorescence (335–345 nm excitation and 485–510 nm emission) was read at 25 °C using an SH-9000 microplate fluorimeter with SF6 software (CORONA ELECTRIC, Hitachinaka, Ibaraki, Japan). Background was determined from negative controls (omission of BACE1 enzyme and fluorogenic substrate). All data are shown as % of BACE1 activity with all relative fluorescence units divided by 100% BACE1 activity data (fluorescent emission of the substrate incubated with BACE1 enzyme alone).
ELISA
We separately quantified Aβ1–40 and Aβ1–42 in brain homogenates and cultured CHO/APPWT cell supernatants by sandwich ELISAs. Brain Aβ1–40 and Aβ1–42 species were detected by a three-step extraction protocol (30, 31). Briefly, brains were homogenized using TissueLyser LT (Qiagen, Valencia, CA; two times for 1 min at 50 Hz) in TBS solution containing protease inhibitor mixture (Sigma) and centrifuged at 18,800 × g for 60 min at 4 °C, and supernatants were removed (representing the TBS-soluble fraction). Resulting pellets were treated with 2% SDS in H2O with the same protease inhibitors and homogenized using TissueLyser LT (once for 1 min at 50 Hz). We then centrifuged the homogenates at 18,800 × g for 60 min at 4 °C and collected supernatants (comprising the 2% SDS-soluble fraction). Finally, the remaining pellets were treated with 5 m guanidine HCl and dissolved by occasional mixing on ice for 30 min and centrifuged at 18,800 × g for 60 min at 4 °C, and supernatants were then collected (representing the guanidine HCl-soluble fraction).
Aβ1–40 and Aβ1–42 species were separately quantified in individual samples in duplicate using ELISA kits (catalog number 27718 for Aβ1–40 and number 27712 for Aβ1–42; IBL, Fujioka, Gunma, Japan) in accordance with the manufacturer's instructions (32). We also quantified Aβ oligomers in the 2% SDS-soluble fraction in duplicate individual samples by Aβ oligomer ELISA (catalog number 27725; IBL) according to the manufacturer's instructions (33). All samples fell within the linear range of the standard curve. For brain homogenates, Aβ1–40 and Aβ1–42 ELISA values were reported as picomolar or nanomolar, and Aβ oligomer concentration was reported as picomolar. For cultured CHO/APPWT cell supernatants, Aβ1–40 and Aβ1–42 ELISA values were reported as picograms/ml.
Western Blot
Cultured CHO/APPWT cells were treated with various doses of MB (1.56, 3.13, 6.25, 12.5, or 25 μm) or 0.1 m PBS (pH 7.4; control) for 12 h, and lysed in ice-cold lysis buffer (containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and 1 mm PMSF). Mouse brain homogenates were lysed in TBS solution containing protease inhibitor mixture (Sigma) followed by TNE buffer (10 mm Tris-HCl, 1% Nonidet P-40, 1 mm EDTA, and 150 mm NaCl), and aliquots corresponding to 10 μg of total protein were electrophoretically separated using 10 or 15% Tris glycine gels based on target protein molecular weights. Electrophoresed proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad) that were blocked in blocking buffer (1% (w/v) nonfat dry milk in TBS containing 0.1% (v/v) Tween 20) for 1 h at ambient temperature. Membranes were then hybridized for 1 h at ambient temperature with primary antibodies as follows: a carboxyl-terminal APP polyclonal antibody (1:400, IBL); a carboxyl-terminal PS1 monoclonal antibody (PS1-loop; 1:500, Merck Millipore); a carboxyl-terminal BACE1 polyclonal antibody (1:400, IBL); a carboxyl-terminal BACE monoclonal antibody (61-3E7, 1:1,000, Merck Millipore); an amino-terminal Aβ1–16 monoclonal antibody (82E1; 1:150, IBL), an amino-terminal Aβ1–17 monoclonal antibody (6E10; 1:1,000, Merck Millipore); a nicastrin polyclonal antibody (corresponds to amino acid residues 688–708 from human nicastrin) (1:1000, Thermo Fisher Scientific, Waltham, MA); an amino-terminal presenilin enhancer 2 (PEN-2) polyclonal antibody (1:250, Thermo Fisher Scientific); an actin polyclonal antibody (1:500, Santa Cruz Biotechnology, Santa Cruz, CA) as a loading control, or a β-actin monoclonal antibody (AC-74, 1:4,000, Sigma) as a loading control. Membranes were then rinsed three times for 30 min each in TBS containing 0.1% (v/v) Tween 20 and incubated for 1 h at ambient temperature with appropriate horseradish peroxidase-conjugated secondary antibodies. After additional rinsing as above, membranes were incubated for 5 min at ambient temperature with enhanced chemiluminescence substrate (SuperSignal West Femto Extended Duration Substrate, Thermo Fisher Scientific), exposed to film, and developed.
QRT-PCR
We quantified BACE1 and β-actin mRNA levels by QRT-PCR analysis. Total RNA was extracted using an RNeasy mini kit (Qiagen), and first strand cDNA synthesis was carried out using the QuantiTect reverse transcription kit (Qiagen) in accordance with the manufacturer's instructions. We diluted cDNA 1:1 in H2O and carried out QRT-PCR for all genes of interest using cDNA-specific TaqMan primer/probe sets (TaqMan Gene Expression Assays, Applied Biosystems) on an ABI 7500 fast real time PCR instrument (Applied Biosystems). Each 20-μl reaction mixture contained 2 μl of cDNA with 1 μl of TaqMan gene expression assay reagent, 10 μl of TaqMan Fast Universal PCR master mix (Applied Biosystems), and 7 μl of H2O. Thermocycler conditions consisted of the following: 95 °C for 15 s, followed by 40 cycles of 95 °C for 1 s and 60 °C for 20 s. TaqMan probe/primer sets were as follows: mouse BACE1 (number Mm00478664_m1) and mouse β-actin (number Mm00607939_s1; used as an internal reference control) (Applied Biosystems). Samples that were not subjected to reverse transcription were run in parallel as negative controls to rule out genomic DNA contamination, and a no template control was also included for each primer set (data not shown). The cycle threshold number (CT) method (34) was used to determine relative amounts of initial target cDNA in each sample. Results are expressed relative to vehicle-treated WT mice.
Statistical Analysis
All experiments were performed by an examiner blinded to sample or subject identities, and the code was not broken until the analyses were completed. Data are presented as the mean ± 1 S.E. A hierarchical analysis strategy was used for time-dependent behavioral data in which the first step was a repeated-measures analysis of variance (ANOVA) to assess the significance of the main effects and interactive terms. If significant, post hoc testing was done with Tukey's HSD or Dunnett's T3 methods, and appropriate p values were reported based on adjustment according to Levene's test for equality of the variance. For all other data, in instances of single mean comparisons, Levene's test followed by t test for independent samples was performed. In instances of multiple mean comparisons, one-way ANOVA was used, followed by post hoc comparison of the means using Bonferroni's or Dunnett's T3 methods (where appropriateness was determined by Levene's test for equality of the variance). A p value of less than 0.05 was considered to be significant. All analyses were performed using the Statistical Package for the Social Sciences (SPSS), release 19.0 (IBM, Armonk, NY).
RESULTS
Methylene Blue Treatment Reverses Behavioral Benefits in Aged PSAPP Mice
We started by orally administering MB or vehicle to PSAPP or WT mice at 15 months of age, for a period of 3 months. Baseline cognitive status was determined in untreated PSAPP versus WT mice at 15 months of age just prior to dosing, and PSAPP mice disclosed cognitive impairment in a comprehensive testing battery. Behavioral evaluation was also made at the conclusion of treatment (18 months of age).
When placed into a novel environment, PSAPP-V mice showed hyperactivity as measured by higher locomotion and rearing scores compared with the other three groups of animals (Fig. 1A). This behavioral phenotype has been reported in cerebral amyloidosis mouse models (e.g. Tg2576 or PSAPP mice) (20, 22, 23, 26, 35), and it may reflect disinhibition because of cortical and/or hippocampal damage (25). Overall ANOVA disclosed the main effects of genotype (p < 0.001 for both locomotion and rearing scores) and treatment (p < 0.001 for both locomotion and rearing scores), and repeated-measures ANOVA followed by post hoc comparison revealed statistically significant differences between PSAPP-V mice and the other three groups of mice for both locomotion and rearing scores (Fig. 1A, **, p < 0.01 for PSAPP-V versus PSAPP-MB, WT-V, or WT-MB mice). The hyperactivity phenotype was completely reversed in PSAPP-MB mice, as they did not statistically differ from WT-V or WT-MB animals (p > 0.05).
FIGURE 1.

Oral methylene blue treatment completely reverses behavioral impairment in PSAPP mice. Data were obtained from PSAPP mice treated with vehicle (PSAPP-V, n = 12) or with MB (PSAPP-MB, n = 12) and also wild-type mice treated with vehicle (WT-V, n = 12) or with MB (WT-MB, n = 12) for 3 months beginning at 15 months of age and subjected to behavioral testing at 18 months of age. A, locomotion (upper panel) and rearing (lower panel) scores obtained from open field activity testing are shown. B, recognition index (%) in the object recognition test is shown from training (upper panel) and retention test phases (lower panel). C, Y-maze test data are represented as total arm entries (upper panel) and spontaneous alternation (lower panel). D, 2-day radial arm water maze test data are shown with five blocks per day for errors (upper panel) and escape latency (lower panel). All statistical comparisons were performed among PSAPP-V, PSAPP-MB, WT-V, and WT-MB mice (*, p < 0.05; **, p < 0.01 for PSAPP-V versus PSAPP-MB, WT-V, and WT-MB mice).
We then tested learning and memory by novel object recognition test in the same cohort of mice. If mice remember an initial encounter with a novel object, they tend to preferentially explore the new versus familiar object, typically operationalized as “recognition index” (27). All four groups showed similar recognition index (51.0–52.4%) during the training phase of the test, whereas in the retention phase, one-way ANOVA followed by post hoc comparison revealed statistically significant differences on recognition index between PSAPP-V mice and the other three mouse groups (Fig. 1B, *, p < 0.05 for PSAPP-V versus PSAPP-MB, WT-V, or WT-MB mice). Of note, PSAPP-MB mice had significantly increased novel object exploration frequency by 74.3% versus PSAPP-V animals (54.1%) (Fig. 1B), but did not significantly differ from WT-V (65.6%) or WT-MB (66.0%) animals (p > 0.05), demonstrating complete improvement of novel object recognition impairment in PSAPP mice related with APP mutant transgene expression by MB treatment.
Exploratory activity and spatial working memory were further examined by Y-maze total arm entry and spontaneous alternation tests. One-way ANOVA followed by post hoc comparison disclosed statistically significant differences on Y-maze total arm entries between PSAPP-V mice and the other three mouse groups (Fig. 1C, PSAPP-V (27.6), PSAPP-MB (21.5), WT-V (19.4), or WT-MB (19.7) mice, as shown in parentheses by the number of arms entered; *, p < 0.05 for PSAPP-V versus PSAPP-MB, WT-V, or WT-MB mice). Increased exploratory activity phenotype was totally alleviated in PSAPP-MB mice, as they did not statistically differ from WT-V or WT-MB animals (p > 0.05).
Mice tend to spontaneously alternate arm entries in the Y-maze, such that they will visit the three arms in sequence more frequently than would occur by chance (50%, see dashed line in Fig. 1C); this is generally interpreted as a measure of spatial working memory. One-way ANOVA followed by post hoc testing disclosed statistically significant differences on Y-maze spontaneous alternation between PSAPP-V mice and the other three mouse groups (Fig. 1C, PSAPP-V (46.5%), PSAPP-MB (58.9%), WT-V (63.3%), or WT-MB (61.1%) mice, as shown in parentheses by alternation (%); *, p < 0.05 for PSAPP-V versus PSAPP-MB, WT-V, or WT-MB mice). Importantly, PSAPP-V mice showed less tendency to alternate, whereas PSAPP-MB animals had significantly increased alternation behavior (Fig. 1C) but did not significantly differ from WT-V or WT-MB groups (p > 0.05), revealing that 3-month oral MB treatment completely improved defective spatial working memory in PSAPP mice.
The same cohort of animals was further evaluated in the RAWM test, an assay of hippocampus-dependent spatial reference learning and memory in rodents (20, 28). On day 1, overall ANOVA disclosed the main effects of block (p < 0.001 for both errors and escape latency) and genotype (p < 0.05 for both errors and escape latency). Repeated-measures ANOVA followed by post hoc comparison did not reveal statistically significant differences between PSAPP-V mice and the other three mouse groups, although PSAPP-V mice showed a tendency to increase errors and a longer escape latency to reach the visible or hidden platform location than the other three groups of animals. By contrast, for day 2, overall ANOVA disclosed the main effects of block (p < 0.01 for errors and p < 0.001 for escape latency), treatment (p < 0.001 for errors and p < 0.05 for escape latency), and genotype (p < 0.001 for errors and p = 0.001 for escape latency), and repeated-measures ANOVA followed by post hoc comparison showed statistically significant differences between PSAPP-V mice and the other three mouse groups (Fig. 1D, **, p = 0.01 for errors, *, p < 0.05 for escape latency; as compared PSAPP-V with PSAPP-MB, WT-V, or WT-MB mice). PSAPP-V mice had increased errors and longer latencies to reach the hidden platform location than the other three mouse groups, whereas PSAPP-MB mice showed significantly less errors and shorter latencies, and their behavior did not significantly differ from WT-V or WT-MB mice, demonstrating that 3-month MB treatment completely remediated mutant APP transgene-associated spatial reference learning and memory impairment.
We ruled out the possibility that behavioral differences in the RAWM test were due to motivational issues or to locomotor impairment, as there were no significant between-group differences (p > 0.05) on swim speed during 2 days of the test. Moreover, the degree of thigmotaxis could indicate anxiety and impact interpretation of results in the RAWM test. In this regard, however, we did not observe thigmotaxis, operationalized as prolonged movement of the mice along the pool circumference, in any animals examined during 2 days of the test. Finally, for all of the behavioral tests conducted, we used multiple ANOVA models with gender as a categorical covariate, but we did not detect significant gender main effects or interactive terms (p > 0.05). We also stratified by gender and found a similar pattern of results as above in both males and females (data not shown).
Methylene Blue Treatment Attenuates Alzheimer-like Pathology in Aged PSAPP Mice
To determine whether oral MB treatment altered Aβ/β-amyloid pathology in aged PSAPP mice, we performed the following: 1) conventional β-amyloid “burden” analysis using a monoclonal antibody against Aβ17–24 (4G8); 2) β-amyloid plaque morphometric analysis; and 3) separate Aβ1–40 and Aβ1–42 ELISAs. Aged PSAPP mice had typical cerebral β-amyloid deposits (17, 18, 22, 23). In fact, PSAPP-V mouse brains had 7.1–8.6% of cerebral β-amyloid burden at 18 months of age that distributed diffusely throughout RSC, EC, and H regions and was markedly and significantly reduced by 3.1–5.8% in all three PSAPP-MB brain regions, with greatest reduction in the EC (Figs. 2 and 3A, ***, p < 0.001, % reduction for RSC, 30.1%; EC, 56.3%; H, 36.1%). It is noteworthy that PSAPP mice at 15 months of age (when dosing started) had 6.2–7.5% cerebral β-amyloid burden (Figs. 2 and 3A). Thus, a 3-month treatment with MB actually reversed cerebral amyloid deposition when compared with untreated baseline (15 months of age) (Figs. 2 and 3A), an effect that was independent of gender (data not shown).
FIGURE 2.
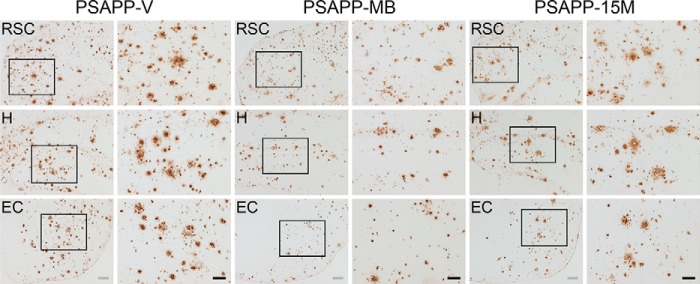
Amelioration of cerebral parenchymal β-amyloid deposits in PSAPP mice treated with methylene blue. Representative photomicrographs were obtained from PSAPP mice treated with vehicle (PSAPP-V) or with MB (PSAPP-MB) for 3 months starting at 15 months of age (mouse age at sacrifice = 18 months) as well as 15-month-old PSAPP mice (PSAPP-15M). Immunohistochemistry using an anti-Aβ17–24 monoclonal antibody (4G8) is depicted, demonstrating cerebral β-amyloid deposits in PSAPP-V, PSAPP-MB, and PSAPP-15M mice. Brain regions shown include the following: RSC (top), H (middle), and EC (bottom). In images from PSAPP-V and PSAPP-MB mice as well as PSAPP-15M mice, each right-hand panel is a higher magnification image from left panel insets. Scale bars, 200 μm (gray) and 100 μm (black).
FIGURE 3.

Cerebral parenchymal and vascular β-amyloid deposits and Aβ levels are reduced in methylene blue-treated PSAPP mice. Data were obtained from PSAPP mice treated with vehicle (PSAPP-V, n = 12) or with MB (PSAPP-MB, n = 12) for 3 months commencing at 15 months of age (mouse age at sacrifice = 18 months) for A–D as well as 15-month-old PSAPP mice (PSAPP-15M), n = 8 for A. A, quantitative image analysis for Aβ burden using an anti-Aβ17–24 monoclonal antibody (4G8) is shown, and each brain region is indicated on the x axis (RSC, H, and EC). B, morphometric analysis of cerebral parenchymal β-amyloid deposits is shown in PSAPP-V and PSAPP-MB mice. Coronal brain sections were stained with 4G8 antibody, and deposits were blindly counted based on maximum diameter and assigned to one of three mutually exclusive categories as follows: small (<25 μm; top), medium (between 25 and 50 μm; middle), or large (>50 μm; bottom). Mean plaque subset number per mouse is shown on the y axis, and each brain region is represented on the x axis. C, severity of cerebral amyloid angiopathy (mean CAA deposit number per mouse) is shown on the y axis with brain region indicated on the x axis. D, TBS-soluble, 2% SDS-soluble, and 5 m guanidine HCl-extractable fractions from three-step extracted brain homogenates were separately measured by sandwich ELISA for human Aβ1–40 and Aβ1–42. Statistical comparisons for A–D are within each brain region and/or Aβ species, and between PSAPP-V and PSAPP-MB mice (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
To examine whether reduced Aβ burden was specific to a particular plaque size subset or occurred more generally, morphometric analysis of β-amyloid deposits was done in both MB- and vehicle-treated PSAPP mice. According to previously described methods (22, 23, 26, 36, 37), β-amyloid deposits were assigned to one of three mutually exclusive categories according to maximum diameter as follows: small (< 25 μm), medium (between 25 and 50 μm), or large (> 50 μm). Mean numbers of deposits in all three subsets showed statistically significant decreases in PSAPP-MB versus PSAPP-V mice across all three brain regions examined, with the greatest reduction in the large sized subset, especially in the EC (Figs. 2 and 3B, **, p < 0.01; ***, p < 0.001, % reduction for EC, 41.5–64.6% (small, 17.7–45.8%; medium, 28.1–41.5%; and large, 46.0–64.6%); RSC, 17.7–46.0%; H, 25.5–47.5%). Stratification by gender revealed the same pattern of results in both males and females (data not shown).
It is now established that 83% of AD brains suffer from cerebral vascular β-amyloid deposits (CAA) (38), and PSAPP mice also develop vascular β-amyloid deposits with age (17, 18, 22, 23). In PSAPP-V mice, cerebral vascular β-amyloid deposits were frequently observed in walls of penetrating arteries at the pial surface in the RSC and EC regions and in walls of small arteries at the hippocampal fissure and brachium of the superior colliculus in the H region. We blindly scored Aβ17–24 monoclonal antibody (4G8)-stained cerebral vascular deposits in RSC, EC, and H regions of PSAPP-V and PSAPP-MB mice and found significant reductions in PSAPP-MB mouse brains in all three brain regions examined (Fig. 3C, **, p < 0.01; ***, p < 0.001).
Biochemical analysis of Aβ species in brain homogenates corroborated that PSAPP-MB mice had statistically significant reductions in both Aβ1–40 and Aβ1–42 levels in the detergent-soluble fraction (Fig. 3D, 26.1 and 22.2%, respectively; *, p < 0.05), whereas only Aβ1–42 abundance was statistically significantly reduced in the TBS-soluble fraction (Fig. 3D, 49.8%; **, p < 0.01). Moreover, the guanidine HCl-soluble fraction, which most closely reflects Aβ deposits detected by immunohistochemistry, revealed statistically significant reductions in PSAPP-MB mice for both Aβ1–40 and Aβ1–42 (Fig. 3D, 23.0 and 58.1%, respectively; *, p < 0.05; ***, p < 0.001).
Modulation of β-Secretase in PSAPP Mouse Brains after Methylene Blue Treatment
We reasoned that reversal of cerebral amyloidosis in PSAPP-MB mice could be due to the following: 1) increased brain-to-blood Aβ efflux (39); 2) dampened APP or PS1 transgene expression; or 3) inhibition of amyloidogenic APP processing. We started by analyzing peripheral blood samples from PSAPP-V and PSAPP-MB mice at 18 months of age for plasma Aβ1–40 and Aβ1–42 species, but we did not detect between-group differences (data not shown). To clarify the possibility that delayed progression of cerebral amyloidosis in MB-treated PSAPP mice was caused by decreased expression of transgene-derived APP or PS1, brain homogenates from PSAPP-V and PSAPP-MB mice were probed with an amino-terminal APP polyclonal or a carboxyl-terminal PS1 monoclonal antibody. Densitometry showed comparable band intensities for APP and PS1 holoproteins, showing that MB treatment did not affect APP or PS1 expression (Fig. 4A).
FIGURE 4.

Methylene blue modulates amyloidogenic amyloid precursor protein processing via inhibition of β-site APP-cleaving enzyme 1 in PSAPP mice. A, Western blots are shown using an amino-terminal APP polyclonal antibody (pAb N-APP; holo-APP is shown) or a carboxyl-terminal BACE1 polyclonal antibody (pAb BACE1). An amino-terminal amyloid-β1–17 (Aβ) monoclonal antibody (mAb 82E1), which detects various amyloidogenic APP cleavage fragments, including Aβ monomer and oligomers as well as phospho-C99 (P-C99) and nonphospho-C99 (C99), is also shown. Actin is included as a loading control for each blot, and densitometry data are represented as ratios of actin below each lane. B, densitometry analyses are shown for ratios of C-99 or P-C99 to actin. C, in the 2% SDS-soluble brain homogenate fraction, Aβ oligomers were measured by sandwich ELISA. D, densitometry analysis is shown for the ratio of BACE1 to actin. Representative Western blots for A were obtained from PSAPP mice treated with vehicle (PSAPP-V, n = 3) or with MB (PSAPP-MB, n = 3). Data for B–D were obtained from PSAPP mice treated with vehicle (PSAPP-V, n = 12) or with MB (PSAPP-MB, n = 12) for 3 months beginning at 15 months of age. All statistical comparisons are between PSAPP-V and PSAPP-MB mice (*, p < 0.05).
We moved on to test the possibility that MB modulated amyloidogenic APP metabolism. Thus, we probed brain homogenates from PSAPP-V and PSAPP-MB mice with an amino-terminal Aβ1–16 monoclonal antibody (82E1) that reacts with amyloidogenic β-carboxyl-terminal fragment (β-CTF, C99) and phospho-β-CTF (P-C99) as well as monomeric and oligomeric Aβ species. Densitometry confirmed that APP metabolism to C99 and P-C99 was significantly deceased in PSAPP-MB mice (Fig. 4, A and B, *, p < 0.05). In parallel, we noted decreased abundance of Aβ species between 25 and 75 kDa (presumed Aβ oligomers), and the 4-kDa monomeric Aβ band was also attenuated in PSAPP-MB brains (Fig. 4A). Reduced expression of Aβ oligomers was also observed by sandwich ELISA in PSAPP-MB versus PSAPP-V brains (Fig. 4C, *, p < 0.05).
Given the above evidence, we hypothesized that MB could directly or indirectly target β-secretase cleavage of APP. BACE1 (β-secretase) is a type I transmembrane aspartyl protease primarily responsible for proteolysis of APP to produce soluble APP-β (sAPP-β) and amyloidogenic β-CTF (C99). C99 is then cleaved by the γ-secretase complex, releasing Aβ species of various lengths (40–44). To examine whether 3-month MB treatment could modulate β-secretase, we probed brain homogenates from PSAPP-V and PSAPP-MB mice with a carboxyl-terminal BACE1 polyclonal antibody and found reduced BACE1 expression in PSAPP-MB mouse brain homogenates (Fig. 4A). Moreover, densitometry confirmed significantly decreased BACE1 protein expression (Fig. 4D, *, p < 0.05). To further verify whether MB altered BACE1 expression at the transcriptional level, relative expression levels of BACE1 mRNA were assayed in mouse brain homogenates by QRT-PCR, but no significant between-group differences were found (expressed as mean relative fold over WT-V mice ± 1 S.E.; PSAPP-V mice, 0.90 ± 0.05; PSAPP-MB mice, 0.92 ± 0.06; WT-MB mice, 0.99 ± 0.06). When taken together, these data suggest that MB operates at the protein level to destabilize BACE1.
Amyloidogenic APP Metabolism Is Attenuated in CHO/APPWT Cells after Methylene Blue Treatment
Mitigated cerebral amyloidosis and reduced amyloidogenic APP metabolism in PSAPP-MB mice could be due to direct or indirect modes of MB action. To determine whether MB could directly modulate APP metabolism, CHO/APPWT cells were treated with a dose range of MB. CHO/APPWT cells stably express human wild-type APP (19). A key advantage of using an APPWT cell line is that the vast majority of AD patients do not harbor APP/PS1/PS2 mutations. Strikingly, separate sandwich ELISAs showed that MB inhibited both Aβ1–40 and Aβ1–42 release into the CHO/APPWT cell culture media (Fig. 5A). Significant reduction for both Aβ species was evident even at the lowest dose (1.56 μm) of MB (*, p < 0.05; **, p < 0.01; ***, p < 0.001 for each dose versus PBS control) (Fig. 5A).
FIGURE 5.

Inhibition of amyloidogenic precursor protein metabolism in CHO/APPWT cells by methylene blue. A, Aβ1–40 and Aβ1–42 species were separately measured in cell supernatants from CHO/APPWT cells by sandwich ELISAs. B, inhibition of amyloidogenic APP processing in CHO/APPWT cells treated with various doses of MB. Western blots using an amino-terminal Aβ1–17 monoclonal antibody (mAb 6E10) or a carboxyl-terminal BACE monoclonal antibody (mAb BACE) show holo-APP, carboxyl-terminal fragment generated by amyloidogenic APP cleavage (C99, β-CTF), and BACE, respectively. β-actin is included as an internal reference control, and densitometry data are shown as ratios of β-actin below each lane. C, densitometry results are shown as ratios of C99 to β-actin at various MB doses. All statistical comparisons are between 0 μm and various doses of MB (*, p < 0.05; **, p < 0.01; ***, p < 0.001), and similar results were observed in 3–4 independent experiments.
To determine whether these effects were due to reduced amyloidogenic APP metabolism, Western blots were performed with an amino-terminal Aβ1–17 monoclonal antibody (6E10) that detects amyloidogenic C99 as well as Aβ oligomers (25–75 kDa) (Fig. 5B). Qualitatively, results showed less abundance of C99 with increasing doses of MB, whereas holo-APP expression remained unaltered, even at the highest dose (25 μm) of MB. Significant differences were found when comparing PBS-treated CHO/APPWT cells (control) to cells that were challenged with 3.13–25 μm of MB (Fig. 5C, **, p < 0.01; ***, p < 0.001). To determine whether BACE1 protein levels were reduced by MB treatment, MB-treated CHO/APPWT cell lysates were probed with a carboxyl-terminal BACE1 monoclonal antibody (Fig. 6A). Significant differences were quantitatively found when comparing PBS-treated (control) CHO/APPWT cells to cells that were treated with 1.56–25 μm MB (Fig. 6B, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
FIGURE 6.

Methylene blue modulates BACE1 expression and activity in CHO/APPWT cells. A, modulation of BACE1 expression in CHO/APPWT cells treated with various doses of MB. Western blots are shown using a carboxyl-terminal BACE monoclonal antibody (mAb BACE), a nicastrin polyclonal antibody (pAb nicastrin), a carboxyl-terminal PS1 monoclonal antibody (pAb PS1), and an amino-terminal presenilin enhancer 2 (PEN-2) polyclonal antibody (pAb PEN-2). β-actin is included as an internal reference control, and densitometry data are shown as ratios of β-actin below each lane. B, densitometry results are shown as ratios of BACE to β-actin at various MB doses. C, cell-free BACE1 activity assay results are displayed, and % of BACE1 activity is shown on the y axis. All statistical comparisons are between 0 μm and various doses of MB (*, p < 0.05; **, p < 0.01; ***, p < 0.001), and similar results were observed in 3–4 independent experiments.
To clarify whether MB directly or indirectly inhibited BACE1 activity, we employed a BACE1 activity assay consisting of combining a dose range of MB in a cell-free system with BACE1 enzyme and fluorogenic reporters. The result was positive in that significant differences were detected with 1.56–25 μm MB (Fig. 6C, 65.1 to 74.5% of 100% BACE1 activity, *, p < 0.05). Of note, significant reduction in BACE1 activity was evident even at the lowest dose (1.56 μm) of MB used (Fig. 6C, *, p < 0.05). Moreover, to determine whether the effects of MB were confined to β-secretase, MB-treated CHO/APPWT cell lysates were probed with antibodies against three major γ-secretase components as follows: nicastrin, PS1, and PEN-2. Densitometry disclosed comparable band intensities for nicastrin, PS1, and PEN-2 proteins at any MB dose examined, supporting selective β-secretase modulation by MB treatment (Fig. 6A). Importantly, CHO/APPWT cells treated with escalating doses of MB did not evidence cellular toxicity, as examined by lactate dehydrogenase release cytotoxicity assay (data not shown). Together, these results show that MB reduces β-secretase cleavage and consequent amyloidogenic metabolism of APPWT protein.
DISCUSSION
We report that oral MB treatment of aged PSAPP mice for 3 months improves behavioral impairment, attenuates cerebral amyloidosis, and dampens amyloidogenic APP metabolism by inhibiting BACE1 expression and activity. Parallel results from cultured CHO/APPWT cells supported MB attenuation of Aβ generation and inhibition of β-secretase cleavage. Furthermore, cell-free BACE1 activity assay confirmed a direct mode of MB action on inhibiting BACE1 activity. Collectively, the present results provide preclinical evidence that long term MB treatment is a promising AD-modifying therapy.
MB, a cationic dye with the molecular formula C16H18N3SCl, belongs to a class of compounds known as phenothiazines. MB is water-soluble, and its color is deep blue in the oxidized state (maximum absorption at wavelengths of 609 and 668 nm), and colorless when in the reduced state (leuco-MB). Both MB and leuco-MB exist as a redox couple in equilibrium, forming a reversible oxidation-reduction system or electron donor-acceptor couple (8, 9). MB is currently approved for a number of therapies, including treatment of inborn methemoglobinemia and acquired methemoglobinemia induced by overexposure to drugs (i.e. nitrophenol, nitrate, cyanide compound, etc.), ifosfamide-induced encephalopathy, urinary tract infections in elderly patients, vasoplegic adrenaline-resistant shock, and pediatric malaria. Moreover, the drug has been employed as a dye in neuroanatomy, neuropathology, and bacteriology. MB has also been used as a tracer dye for intraoperative visualization of nerve tissues and endocrine glands and for the detection of fluid leaks in gastrointestinal and urinary systems as well as pathological fistulae (8, 9).
MB has pleiotropic biological activities and targets a number of molecules (i.e. monoamine oxidase A, nitric-oxide synthase (NOS), guanylate cyclase, methemoglobin (metHb), acetylcholinesterase (AChE), and disulfide reductases such as glutathione reductase or dihydrolipoamide dehydrogenase (45, 46)). Given this bioactivity profile, we hypothesized that long term oral MB treatment to aged cerebral amyloidosis model mice may show disease-modifying effects and also delay the progression of AD-like pathology, even if administered late in the course of disease. To test this, MB was orally given to 15-month-old PSAPP mice at 3 mg/kg/day via gavage, as this treatment strategy more precisely delivers agents compared with ad libitum access to drinking water or chow.
In the clinic, the recommended safe dose of MB is between 1 and 4 mg/kg (47). The LD50 of orally administered MB is as high as 1,180 mg/kg in rats and 3,500 mg/kg in mice (8), and the dose that we administered to aged mice (3 mg/kg/day) is orders of magnitude lower. The tolerable daily intake in humans can be extrapolated from rodent LD50 threshold data (48). Assuming that the default uncertainty factor accounting for interspecies variation is 10 (49), the tolerable daily intake calculated from mouse LD50 is 21.0 g of MB for a 60-kg human, which is considerably above the dose administered in this study.
However, adverse effects must be taken into consideration for any therapeutic agent, as such effects have the potential to arrest clinical trials, regardless of efficacy. Thus, it is worth mentioning that we did not detect adverse events, including occurrence of atypical behavior, altered food/water intake, or mortality associated with long term MB treatment, except for pale blue coloration of urine and feces as well as the mucosal surface of the urinary bladder, stomach, and duodenum. These findings reinforce the notion that the dose of MB used in aged mice is safe, although toxicology analyses for long term MB treatment would be necessary to conduct in humans.
As to how MB exerts its biological effects, the compound has a low molar mass (319.86 g/mol) in anhydrous form. MB passes the blood-brain barrier irrespective of the administration route and has been shown to be highly bioavailable in the brain (50, 51). The oral absorption of MB is reported to be 53–97% with plasma peak concentrations after 30–60 min (52). About 65–85% of MB is reduced in erythrocytes and peripheral tissues to the leuco-MB form (52). However, given its hydrophilicity and its positive charge, it is not likely that MB passes lipid bilayer membranes. In this regard, the presence of a sulfur atom has been suggested as the reason for its high affinity for neural tissues (53). Moreover, cationic MB is reduced to its uncharged leuco-MB form by a plasma membrane reductase immediately before crossing the blood-brain barrier, followed by intracellular redoxidation to the oxidized form. Both MB and leuco-MB exist as a redox couple in equilibrium, forming a reversible oxidation-reduction system or electron donor-acceptor couple. Importantly, leuco-MB is uncharged, lipophilic, and enters cells by diffusion across the plasma membrane where it can be re-oxidized and thus sequestered within neuronal cells (8, 9). Future study is warranted to examine which form of MB mediates its biological actions on mitigating Aβ pathology.
Our results show that long term MB treatment significantly improves transgene-associated behavioral impairment, including hyperactivity, decreased object recognition, and defective spatial working and reference memory, but does not alter nontransgenic mouse behavior. Our data are consistent with promising results from a phase II clinical trial of MB (methylthioninium chloride, RemberTM) for mild to moderate AD patients, showing significant improvement of cognitive decline after a 6-month treatment as compared with placebo controls, and stabilized AD progression over the course of a year (15, 16). Moreover, it has been reported that MB treatment in 3× Tg-AD model mice rescues learning and memory deficits and reduces Aβ levels by increasing chymotrypsin- and trypsin-like activities of the proteasome; but has no effect on Tau accumulation, phosphorylation, and mislocalization in the brain (54). Furthermore, it has been reported that PSAPP mice treated with MB have reduced cognitive impairment, cerebral amyloid load, and mitochondrial dysfunction (55). As to how MB exerts its beneficial effects on cognitive impairment, there is evidence that the cholinergic system plays an important role in the regulation of learning and memory, and it is well-known that MB can inhibit AChE activity. Failure of the cholinergic system leads to a shift toward the amyloidogenic pathway, and the cholinergic hypothesis holds that loss of acetylcholine contributes to cognitive decline in AD (56). Based on this hypothesis, AChE inhibitors are currently prescribed as pharmacotherapeutics in parallel with N-methyl d-aspartate antagonists.
Our results further show that brain parenchymal and cerebral vascular β-amyloid deposits as well as levels of various Aβ species, including oligomers, are mitigated in MB-treated PSAPP mice. Thus, the relationship between behavioral benefit and ameliorated cerebral amyloidosis after MB treatment deserves consideration. There are extensive reports that selective removal of Aβ species by immunotherapeutic approaches remediates behavioral impairment in AD mouse models (57–59); yet, it has been shown that cerebral β-amyloid deposits do not correlate well with behavioral impairment (60, 61). This conflicting evidence has led to the speculation that Aβ oligomers are the critical neurotoxic species (62–64). In support, there is a strong correlation between abundance of soluble Aβ oligomers and cognitive disturbance in mouse models of cerebral amyloidosis (58, 61). In this regard, we found decreased oligomeric Aβ by Western blot and by sandwich ELISA in brain homogenates from MB-treated PSAPP mice. These observations may be owed to less amyloidogenic β-secretase APP proteolysis. Consistent with our results, MB has been reported to inhibit the formation of Aβ oligomers (65), and extracellular accumulation of a 56-kDa soluble Aβ assembly (Aβ*56) perpetrates memory deficits in Tg2576 mice (66). Collectively, although Aβ is generally considered to be responsible for behavioral deficits in transgenic mouse models of cerebral amyloidosis, it should be noted that the primary result of reduced β-cleavage of APP proteolysis by MB treatment is reduced C99 and sAPP-β. Thus, it remains possible that MB reduction of any combination of amyloidogenic APP metabolites (including C99 and sAPP-β) could at least partially contribute to cognitive benefit in PSAPP mice.
MB was originally reported to be a potential inhibitor of Tau aggregation in vitro (67, 68). However, this earlier result was not confirmed in a zebrafish model system (69). It was later reported that MB treatment of 3× Tg-AD mice rescued defective learning and memory and reduced Aβ abundance by increasing chymotrypsin- and trypsin-like activities of the proteasome, but those authors did not find any effect on Tau accumulation, phosphorylation, or mislocalization (54). Although these reports apparently contradict earlier in vitro studies showing that MB can inhibit Tau aggregation (67, 68), there have been accumulating positive reports using Tau transgenic mice and other systems (e.g. primary neuronal or organotypic slice cultures) that confirm the original observations (70, 71). Given this apparent effect of MB on Tau, the compound's use as an AD therapeutic has been evaluated. Results from a phase II clinical trial of MB (methylthioninium chloride, RemberTM) for mild to moderate AD patients showed significant improvement of cognitive decline compared with placebo. Additionally, MB slowed AD progression over the course of a year (15, 16).
Our results also demonstrate that MB treatment attenuates amyloidogenic β-secretase APP cleavage, both in vivo and in vitro. Of interest, we found that MB targets BACE1 protein stability (without affecting BACE1 mRNA expression) and β-secretase activity, resulting in decreased abundance of β-secretase cleavage metabolites (C99/P-C99 CTFs) and subsequent Aβ species, including oligomeric forms. Thus, MB exerts its effects on BACE1 both by limiting protein abundance and also by directly reducing enzymatic activity, as shown in a cell-free BACE1 activity assay. Furthermore, data from this cell-free BACE1 activity assay clearly indicate that MB competitively inhibits the binding of the BACE1 substrate (H-RE(EDANS)EVNLDAEFK(Dabcyl)R-OH in DMSO) to BACE1. Regarding the selectivity of MB as a β-secretase modulator, MB treatment of PSAPP mice did not alter PS1 expression in brain homogenates (Fig. 4A). Moreover, expression of three key γ-secretase components: nicastrin, PS1, and PEN-2 proteins, remained unaffected in CHO/APPWT cell lysates treated with escalating doses of MB, supporting selective β-secretase modulation by MB treatment (Fig. 6A). Amyloidogenic β-secretase cleavage of APP molecules is thought to be a rate-limiting step for Aβ generation (40–44), and both α- and β-secretases compete for APP proteolysis (72). Thus, reducing β-secretase activity and limiting its protein abundance could theoretically polarize toward nonamyloidogenic α-secretase cleavage, and this has generally been the therapeutic rationale for β-secretase modulators.
Collectively, our data indicate that mitigated cerebral amyloidosis in PSAPP mice and decreased Aβ secretion from CHO/APPWT cells following MB treatment are, at least partially, because of reduced amyloidogenic β-secretase cleavage of APP molecules. Nonetheless, it remains possible that reduced BACE1 levels/activity would also impact processing of other BACE1 substrates. In this regard, long term modulation of BACE1 activity has been indirectly shown to affect alternative BACE1 substrates (i.e. the cell adhesion protein P-selectin glycoprotein ligand-1 (73), the APP homolog proteins APP-like protein 1/2 (74, 75), the low density lipoprotein receptor-related protein (76), the β-subunit of voltage-gated sodium channels (77), and the control protein of myelination (78, 79). Relevant to this issue, BACE1-null mice are reported to be viable and to have a normal phenotype, suggesting that inhibition of this enzyme could be clinically feasible with few side effects (80–82). By contrast, it should be noted that recent reports demonstrated that BACE1-null mice have several abnormalities (83–86), raising a concern that complete inhibition or entire absence of BACE1 function may not be free of unwanted side effects. More to this point, it is known that MB is pleiotropic and targets other substrates, including monoamine oxidase A, NOS, guanylate cyclase, metHb, AChE, and disulfide reductases (45, 46). Therefore, long term administration of MB for AD treatment could significantly impact normal brain function. Thus, further translational study is warranted to examine whether long term MB therapy has effects on proteins other than BACE1 and is safe for use in the clinic.
In conclusion, our data demonstrate that long term oral MB treatment in aged Alzheimer PSAPP mice for 3 months remediates behavioral impairment while concomitantly ameliorating cerebral amyloidosis by modulating β-secretase. MB has low toxicity, low molecular mass, high water solubility, high bioavailability, ability to cross the blood-brain barrier, and is already approved for use in the clinic. If cerebral amyloid pathology in this transgenic mouse model mirrors the clinical syndrome, then long term MB therapy could be a safe and promising disease-modifying therapy to advanced AD with no discernible side effects.
Acknowledgments
We thank Drs. Stefanie Hahn and Sascha Weggen (University of Heinrich Heine, Düsseldorf, Germany) for generously gifting the CHO/APPWT cells.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01NS076794-01 from NINDS (to T. T.). This work was also supported by Grant-in-aid for Scientific Research (C) from the Japan Society for the Promotion of Science 22500320 (to T. M.), Alzheimer's Association Zenith Fellows Award ZEN-10-174633 (to T. T.), American Federation of Aging Research/Ellison Medical Foundation Julie Martin Mid-Career Award in Aging Research Grant M11472 (to T. T.), and generous start-up funds from the Zilkha Neurogenetic Institute of the Keck School of Medicine at the University of Southern California (to T. T.).
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- Aβ
- amyloid-β
- MB
- methylene blue
- APPswe
- APP “Swedish” APPK595N/M596L
- PS1
- presenilin 1
- CHO/APPWT cells
- Chinese hamster ovary cells stably expressing human wild-type APP
- RAWM
- radial arm water maze
- RSC
- retrosplenial cortex
- EC
- entorhinal cortex
- H
- hippocampus
- CAA
- cerebral amyloid angiopathy
- BACE1
- β-site APP-cleaving enzyme 1
- PEN-2
- presenilin enhancer 2
- QRT-PCR
- quantitative real time-PCR
- ANOVA
- analysis of variance
- β-CTF
- β-carboxyl-terminal fragment
- sAPP-β
- soluble APP-β
- NOS
- nitric-oxide synthase
- metHb
- methemoglobin
- AChE
- acetylcholinesterase.
REFERENCES
- 1. Brookmeyer R., Gray S. (2000) Methods for projecting the incidence and prevalence of chronic diseases in aging populations: application to Alzheimer's disease. Stat. Med. 19, 1481–1493 [DOI] [PubMed] [Google Scholar]
- 2. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 3. Alzheimer's Disease International (2012) Overcoming the Stigma of Dementia, World Alzheimer Report 2012. Alzheimer's Disease International, London [Google Scholar]
- 4. Rozemuller J. M., Eikelenboom P., Stam F. C., Beyreuther K., Masters C. L. (1989) A4 protein in Alzheimer's disease: primary and secondary cellular events in extracellular amyloid deposition. J. Neuropathol. Exp. Neurol. 48, 674–691 [DOI] [PubMed] [Google Scholar]
- 5. Hardy J., Allsop D. (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388 [DOI] [PubMed] [Google Scholar]
- 6. Bates K. A., Verdile G., Li Q. X., Ames D., Hudson P., Masters C. L., Martins R. N. (2009) Clearance mechanisms of Alzheimer's amyloid-β peptide: implications for therapeutic design and diagnostic tests. Mol. Psychiatry 14, 469–486 [DOI] [PubMed] [Google Scholar]
- 7. Gravitz L. (2011) Drugs: a tangled web of targets. Nature 475, S9–S11 [DOI] [PubMed] [Google Scholar]
- 8. Oz M., Lorke D. E., Hasan M., Petroianu G. A. (2011) Cellular and molecular actions of methylene blue in the nervous system. Med. Res. Rev. 31, 93–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schirmer R. H., Adler H., Pickhardt M., Mandelkow E. (2011) “Lest we forget you-methylene blue …” Neurobiol. Aging 32, 2325.e7–16 [DOI] [PubMed] [Google Scholar]
- 10. Klamer D., Engel J. A., Svensson L. (2004) Phencyclidine-induced behaviour in mice prevented by methylene blue. Basic Clin. Pharmacol. Toxicol. 94, 65–72 [DOI] [PubMed] [Google Scholar]
- 11. Riha P. D., Bruchey A. K., Echevarria D. J., Gonzalez-Lima F. (2005) Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur. J. Pharmacol. 511, 151–158 [DOI] [PubMed] [Google Scholar]
- 12. Narsapur S. L., Naylor G. J. (1983) Methylene blue. A possible treatment for manic depressive psychosis. J. Affect. Disord. 5, 155–161 [DOI] [PubMed] [Google Scholar]
- 13. Naylor G. J., Martin B., Hopwood S. E., Watson Y. (1986) A two-year double-blind crossover trial of the prophylactic effect of methylene blue in manic-depressive psychosis. Biol. Psychiatry 21, 915–920 [DOI] [PubMed] [Google Scholar]
- 14. Deiana S., Harrington C. R., Wischik C. M., Riedel G. (2009) Methylthioninium chloride reverses cognitive deficits induced by scopolamine: comparison with rivastigmine. Psychopharmacology (Berl) 202, 53–65 [DOI] [PubMed] [Google Scholar]
- 15. Gura T. (2008) Hope in Alzheimer's fight emerges from unexpected places. Nat. Med. 14, 894. [DOI] [PubMed] [Google Scholar]
- 16. Wischik C., Staff R. (2009) Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of Tau-aggregation inhibitor therapy. J. Nutr. Health Aging 13, 367–369 [DOI] [PubMed] [Google Scholar]
- 17. Borchelt D. R., Ratovitski T., van Lare J., Lee M. K., Gonzales V., Jenkins N. A., Copeland N. G., Price D. L., Sisodia S. S. (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19, 939–945 [DOI] [PubMed] [Google Scholar]
- 18. Garcia-Alloza M., Robbins E. M., Zhang-Nunes S. X., Purcell S. M., Betensky R. A., Raju S., Prada C., Greenberg S. M., Bacskai B. J., Frosch M. P. (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524 [DOI] [PubMed] [Google Scholar]
- 19. Hahn S., Brüning T., Ness J., Czirr E., Baches S., Gijsen H., Korth C., Pietrzik C. U., Bulic B., Weggen S. (2011) Presenilin-1 but not amyloid precursor protein mutations present in mouse models of Alzheimer's disease attenuate the response of cultured cells to γ-secretase modulators regardless of their potency and structure. J. Neurochem. 116, 385–395 [DOI] [PubMed] [Google Scholar]
- 20. Arendash G. W., King D. L., Gordon M. N., Morgan D., Hatcher J. M., Hope C. E., Diamond D. M. (2001) Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 891, 42–53 [DOI] [PubMed] [Google Scholar]
- 21. Jankowsky J. L., Slunt H. H., Gonzales V., Jenkins N. A., Copeland N. G., Borchelt D. R. (2004) APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol. Aging 25, 885–892 [DOI] [PubMed] [Google Scholar]
- 22. Mori T., Rezai-Zadeh K., Koyama N., Arendash G. W., Yamaguchi H., Kakuda N., Horikoshi-Sakuraba Y., Tan J., Town T. (2012) Tannic acid is a natural β-secretase inhibitor that prevents cognitive impairment and mitigates Alzheimer-like pathology in transgenic mice. J. Biol. Chem. 287, 6912–6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mori T., Koyama N., Guillot-Sestier M. V., Tan J., Town T. (2013) Ferulic acid is a nutraceutical β-secretase modulator that improves behavioral impairment and Alzheimer-like pathology in transgenic mice. PLoS ONE 8, e55774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laghmouch A., Bertholet J. Y., Crusio W. E. (1997) Hippocampal morphology and open-field behavior in Mus musculus domesticus and Mus spretus inbred mice. Behav. Genet. 27, 67–73 [DOI] [PubMed] [Google Scholar]
- 25. Kim K. S., Han P. L. (2006) Optimization of chronic stress paradigms using anxiety- and depression-like behavioral parameters. J. Neurosci. Res. 83, 497–507 [DOI] [PubMed] [Google Scholar]
- 26. Town T., Laouar Y., Pittenger C., Mori T., Szekely C. A., Tan J., Duman R. S., Flavell R. A. (2008) Blocking TGF-β-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 14, 681–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Rosa R., Garcia A. A., Braschi C., Capsoni S., Maffei L., Berardi N., Cattaneo A. (2005) Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 102, 3811–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alamed J., Wilcock D. M., Diamond D. M., Gordon M. N., Morgan D. (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc. 1, 1671–1679 [DOI] [PubMed] [Google Scholar]
- 29. Franklin K. B. J., Paxinos G. (2001) The Mouse Brain in Stereotaxic Coordinates, Academic Press, San Diego [Google Scholar]
- 30. Kawarabayashi T., Younkin L. H., Saido T. C., Shoji M., Ashe K. H., Younkin S. G. (2001) Age-dependent changes in brain, CSF, and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J. Neurosci. 21, 372–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jankowsky J. L., Slunt H. H., Gonzales V., Savonenko A. V., Wen J. C., Jenkins N. A., Copeland N. G., Younkin L. H., Lester H. A., Younkin S. G., Borchelt D. R. (2005) Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med. 2, e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Horikoshi Y., Sakaguchi G., Becker A. G., Gray A. J., Duff K., Aisen P. S., Yamaguchi H., Maeda M., Kinoshita N., Matsuoka Y. (2004) Development of Aβ terminal end-specific antibodies and sensitive ELISA for Aβ variant. Biochem. Biophys. Res. Commun. 319, 733–737 [DOI] [PubMed] [Google Scholar]
- 33. Xia W., Yang T., Shankar G., Smith I. M., Shen Y., Walsh D. M., Selkoe D. J. (2009) A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 66, 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monney L., Sabatos C. A., Gaglia J. L., Ryu A., Waldner H., Chernova T., Manning S., Greenfield E. A., Coyle A. J., Sobel R. A., Freeman G. J., Kuchroo V. K. (2002) Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 [DOI] [PubMed] [Google Scholar]
- 35. King D. L., Arendash G. W., Crawford F., Sterk T., Menendez J., Mullan M. J. (1999) Progressive and gender-dependent cognitive impairment in the APPSW transgenic mouse model for Alzheimer's disease. Behav. Brain Res. 103, 145–162 [DOI] [PubMed] [Google Scholar]
- 36. Mori T., Town T., Tan J., Yada N., Horikoshi Y., Yamamoto J., Shimoda T., Kamanaka Y., Tateishi N., Asano T. (2006) Arundic acid ameliorates cerebral amyloidosis and gliosis in Alzheimer transgenic mice. J. Pharmacol. Exp. Ther. 318, 571–578 [DOI] [PubMed] [Google Scholar]
- 37. Mori T., Koyama N., Arendash G. W., Horikoshi-Sakuraba Y., Tan J., Town T. (2010) Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer's disease. Glia 58, 300–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ellis R. J., Olichney J. M., Thal L. J., Mirra S. S., Morris J. C., Beekly D., Heyman A. (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology 46, 1592–1596 [DOI] [PubMed] [Google Scholar]
- 39. DeMattos R. B., Bales K. R., Cummins D. J., Paul S. M., Holtzman D. M. (2002) Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 295, 2264–2267 [DOI] [PubMed] [Google Scholar]
- 40. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 41. Sinha S., Lieberburg I. (1999) Cellular mechanisms of β-amyloid production and secretion. Proc. Natl. Acad. Sci. U.S.A. 96, 11049–11053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 43. Vassar R., Kovacs D. M., Yan R., Wong P. C. (2009) The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 29, 12787–12794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan R., Bienkowski M. J., Shuck M. E., Miao H., Tory M. C., Pauley A. M., Brashier J. R., Stratman N. C., Mathews W. R., Buhl A. E., Carter D. B., Tomasselli A. G., Parodi L. A., Heinrikson R. L., Gurney M. E. (1999) Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537 [DOI] [PubMed] [Google Scholar]
- 45. Buchholz K., Schirmer R. H., Eubel J. K., Akoachere M. B., Dandekar T., Becker K., Gromer S. (2008) Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob. Agents Chemother. 52, 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harvey B. H., Duvenhage I., Viljoen F., Scheepers N., Malan S. F., Wegener G., Brink C. B., Petzer J. P. (2010) Role of monoamine oxidase, nitric oxide synthase and regional brain monoamines in the antidepressant-like effects of methylene blue and selected structural analogues. Biochem. Pharmacol. 80, 1580–1591 [DOI] [PubMed] [Google Scholar]
- 47. Clifton J., 2nd, Leikin J. B. (2003) Methylene blue. Am. J. Ther. 10, 289–291 [DOI] [PubMed] [Google Scholar]
- 48. Barnes D. G., Dourson M. (1988) Reference dose (RfD): description and use in health risk assessments. Regul. Toxicol. Pharmacol. 8, 471–486 [DOI] [PubMed] [Google Scholar]
- 49. Dourson M. L., Felter S. P., Robinson D. (1996) Evolution of science-based uncertainty factors in noncancer risk assessment. Regul. Toxicol. Pharmacol. 24, 108–120 [DOI] [PubMed] [Google Scholar]
- 50. Peter C., Hongwan D., Küpfer A., Lauterburg B. H. (2000) Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharmacol. 56, 247–250 [DOI] [PubMed] [Google Scholar]
- 51. Walter-Sack I., Rengelshausen J., Oberwittler H., Burhenne J., Mueller O., Meissner P., Mikus G. (2009) High absolute bioavailability of methylene blue given as an aqueous oral formulation. Eur. J. Clin. Pharmacol. 65, 179–189 [DOI] [PubMed] [Google Scholar]
- 52. DiSanto A. R., Wagner J. G. (1972) Pharmacokinetics of highly ionized drugs. II. Methylene blue-absorption, metabolism, and excretion in man and dog after oral administration. J. Pharm. Sci. 61, 1086–1090 [DOI] [PubMed] [Google Scholar]
- 53. Müller T. (1998) Methylene blue supravital staining: an evaluation of its applicability to the mammalian brain and pineal gland. Histol. Histopathol. 13, 1019–1026 [DOI] [PubMed] [Google Scholar]
- 54. Medina D. X., Caccamo A., Oddo S. (2011) Methylene blue reduces Aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 21, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Paban V., Manrique C., Filali M., Maunoir-Regimbal S., Fauvelle F., Alescio-Lautier B. (2014) Therapeutic and preventive effects of methylene blue on Alzheimer's disease pathology in a transgenic mouse model. Neuropharmacology 76, 68–79 [DOI] [PubMed] [Google Scholar]
- 56. Nordberg A. (2006) Mechanisms behind the neuroprotective actions of cholinesterase inhibitors in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 20, S12–S18 [DOI] [PubMed] [Google Scholar]
- 57. Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 [DOI] [PubMed] [Google Scholar]
- 58. Kotilinek L. A., Bacskai B., Westerman M., Kawarabayashi T., Younkin L., Hyman B. T., Younkin S., Ashe K. H. (2002) Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J. Neurosci. 22, 6331–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mouri A., Noda Y., Hara H., Mizoguchi H., Tabira T., Nabeshima T. (2007) Oral vaccination with a viral vector containing Aβ cDNA attenuates age-related Aβ accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J. 21, 2135–2148 [DOI] [PubMed] [Google Scholar]
- 60. Holcomb L. A., Gordon M. N., Jantzen P., Hsiao K., Duff K., Morgan D. (1999) Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav. Genet. 29, 177–185 [DOI] [PubMed] [Google Scholar]
- 61. Westerman M. A., Cooper-Blacketer D., Mariash A., Kotilinek L., Kawarabayashi T., Younkin L. H., Carlson G. A., Younkin S. G., Ashe K. H. (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 22, 1858–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 [DOI] [PubMed] [Google Scholar]
- 63. Cleary J. P., Walsh D. M., Hofmeister J. J., Shankar G. M., Kuskowski M. A., Selkoe D. J., Ashe K. H. (2005) Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84 [DOI] [PubMed] [Google Scholar]
- 64. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Necula M., Breydo L., Milton S., Kayed R., van der Veer W. E., Tone P., Glabe C. G. (2007) Methylene blue inhibits amyloid Aβ oligomerization by promoting fibrillization. Biochemistry 46, 8850–8860 [DOI] [PubMed] [Google Scholar]
- 66. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 67. Wischik C. M., Edwards P. C., Lai R. Y., Roth M., Harrington C. R. (1996) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. U.S.A. 93, 11213–11218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Taniguchi S., Suzuki N., Masuda M., Hisanaga S., Iwatsubo T., Goedert M., Hasegawa M. (2005) Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 280, 7614–7623 [DOI] [PubMed] [Google Scholar]
- 69. van Bebber F., Paquet D., Hruscha A., Schmid B., Haass C. (2010) Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol. Dis. 39, 265–271 [DOI] [PubMed] [Google Scholar]
- 70. Congdon E. E., Wu J. W., Myeku N., Figueroa Y. H., Herman M., Marinec P. S., Gestwicki J. E., Dickey C. A., Yu W. H., Duff K. E. (2012) Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hosokawa M., Arai T., Masuda-Suzukake M., Nonaka T., Yamashita M., Akiyama H., Hasegawa M. (2012) Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS ONE 7, e52389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gandhi S., Refolo L. M., Sambamurti K. (2004) Amyloid precursor protein compartmentalization restricts β-amyloid production: therapeutic targets based on BACE compartmentalization. J. Mol. Neurosci. 24, 137–143 [DOI] [PubMed] [Google Scholar]
- 73. Lichtenthaler S. F., Dominguez D. I., Westmeyer G. G., Reiss K., Haass C., Saftig P., De Strooper B., Seed B. (2003) The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J. Biol. Chem. 278, 48713–48719 [DOI] [PubMed] [Google Scholar]
- 74. Li Q., Südhof T. C. (2004) Cleavage of amyloid-β precursor protein and amyloid-β precursor-like protein by BACE 1. J. Biol. Chem. 279, 10542–10550 [DOI] [PubMed] [Google Scholar]
- 75. Pastorino L., Ikin A. F., Lamprianou S., Vacaresse N., Revelli J. P., Platt K., Paganetti P., Mathews P. M., Harroch S., Buxbaum J. D. (2004) BACE (β-secretase) modulates the processing of APLP2 in vivo. Mol. Cell Neurosci. 25, 642–649 [DOI] [PubMed] [Google Scholar]
- 76. von Arnim C. A., Kinoshita A., Peltan I. D., Tangredi M. M., Herl L., Lee B. M., Spoelgen R., Hshieh T. T., Ranganathan S., Battey F. D., Liu C. X., Bacskai B. J., Sever S., Irizarry M. C., Strickland D. K., Hyman B. T. (2005) The low density lipoprotein receptor-related protein (LRP) is a novel β-secretase (BACE1) substrate. J. Biol. Chem. 280, 17777–17785 [DOI] [PubMed] [Google Scholar]
- 77. Wong H. K., Sakurai T., Oyama F., Kaneko K., Wada K., Miyazaki H., Kurosawa M., De Strooper B., Saftig P., Nukina N. (2005) β subunits of voltage-gated sodium channels are novel substrates of β-site amyloid precursor protein-cleaving enzyme (BACE1) and γ-secretase. J. Biol. Chem. 280, 23009–23017 [DOI] [PubMed] [Google Scholar]
- 78. Willem M., Garratt A. N., Novak B., Citron M., Kaufmann S., Rittger A., DeStrooper B., Saftig P., Birchmeier C., Haass C. (2006) Control of peripheral nerve myelination by the β-secretase BACE1. Science 314, 664–666 [DOI] [PubMed] [Google Scholar]
- 79. Hu X., He W., Diaconu C., Tang X., Kidd G. J., Macklin W. B., Trapp B. D., Yan R. (2008) Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 22, 2970–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Luo Y., Bolon B., Kahn S., Bennett B. D., Babu-Khan S., Denis P., Fan W., Kha H., Zhang J., Gong Y., Martin L., Louis J. C., Yan Q., Richards W. G., Citron M., Vassar R. (2001) Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 4, 231–232 [DOI] [PubMed] [Google Scholar]
- 81. Ohno M., Sametsky E. A., Younkin L. H., Oakley H., Younkin S. G., Citron M., Vassar R., Disterhoft J. F. (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41, 27–33 [DOI] [PubMed] [Google Scholar]
- 82. Nishitomi K., Sakaguchi G., Horikoshi Y., Gray A. J., Maeda M., Hirata-Fukae C., Becker A. G., Hosono M., Sakaguchi I., Minami S. S., Nakajima Y., Li H. F., Takeyama C., Kihara T., Ota A., Wong P. C., Aisen P. S., Kato A., Kinoshita N., Matsuoka Y. (2006) BACE1 inhibition reduces endogenous Aβ and alters APP processing in wild-type mice. J. Neurochem. 99, 1555–1563 [DOI] [PubMed] [Google Scholar]
- 83. Wang H., Song L., Lee A., Laird F., Wong P. C., Lee H. K. (2010) Mossy fiber long-term potentiation deficits in BACE1 knock-outs can be rescued by activation of α7 nicotinic acetylcholine receptors. J. Neurosci. 30, 13808–13813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cai J., Qi X., Kociok N., Skosyrski S., Emilio A., Ruan Q., Han S., Liu L., Chen Z., Bowes Rickman C., Golde T., Grant M. B., Saftig P., Serneels L., de Strooper B., Joussen A. M., Boulton M. E. (2012) β-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol. Med. 4, 980–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hitt B., Riordan S. M., Kukreja L., Eimer W. A., Rajapaksha T. W., Vassar R. (2012) β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J. Biol. Chem. 287, 38408–38425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yan R., Vassar R. (2014) Targeting the β secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 13, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]