

Background: The α6β4 integrin assembles via an unknown mechanism with receptor tyrosine kinases.
Results: HER2-dependent activation of α6β4 depends on capture of the β4 cytoplasmic domain by syndecan-1, whereas HER1 (EGFR) relies on syndecan-4.
Conclusion: Cell invasion and survival mediated by the α6β4 integrin depend on its assembly with kinase-specific syndecans.
Significance: These novel interactions may provide targets for new therapeutics to combat carcinogenesis.
Keywords: Carcinogenesis, Cell Invasion, Epidermal Growth Factor Receptor (EGFR), Integrin, Laminin, Syndecan, Wound Healing
Abstract
Epithelial cells are highly dependent during wound healing and tumorigenesis on the α6β4 integrin and its association with receptor tyrosine kinases. Previous work showed that phosphorylation of the β4 subunit upon matrix engagement depends on the matrix receptor syndecan (Sdc)-1 engaging the cytoplasmic domain of the β4 integrin and coupling of the integrin to human epidermal growth factor receptor-2 (HER2). In this study, HER2-dependent migration activated by matrix engagement is compared with migration stimulated by EGF. We find that whereas HER2-dependent migration depends on Sdc1, EGF-dependent migration depends on a complex consisting of human epidermal growth factor receptor-1 (HER1, commonly known as EGFR), α6β4, and Sdc4. The two syndecans recognize distinct sites at the extreme C terminus of the β4 integrin cytoplasmic domain. The binding motif in Sdc1 is QEEXYX, composed in part by its syndecan-specific variable (V) region and in part by the second conserved (C2) region that it shares with other syndecans. A cell-penetrating peptide containing this sequence competes for HER2-dependent epithelial migration and carcinoma survival, although it is without effect on the EGFR-stimulated mechanism. β4 mutants bearing mutations specific for Sdc1 and Sdc4 recognition act as dominant negative mutants to block cell spreading or cell migration that depends on HER2 or EGFR, respectively. The interaction of the α6β4 integrin with the syndecans appears critical for it to be utilized as a signaling platform; migration depends on α3β1 integrin binding to laminin 332 (LN332; also known as laminin 5), whereas antibodies that block α6β4 binding are without effect. These findings indicate that specific syndecan family members are likely to have key roles in α6β4 integrin activation by receptor tyrosine kinases.
Introduction
The α6β4 integrin is localized to hemidesmosomes at the basal surface of epithelial cells where it mediates the anchorage to laminin 332 (LN332, also known as laminin-5 (1)) in the underlying basal lamina. This aids the cell in resisting frictional forces, especially in stratified epidermis where LN332 associates with collagen VII anchoring fibrils that extend into the underlying dermis (2). The unusually long cytoplasmic domain of the β4 integrin subunit is key to this anchorage role; consisting of over 1,000 amino acids, it links the integrin to the intermediate filament cytoskeleton within cells (2–4). However, when receptor tyrosine kinases are activated during epithelial wound healing, or in tumorigenesis, this stable anchorage is disrupted, leading to hemidesmosome breakdown (5, 6). EGFR,2 HER2, c-Met, and Ron kinase are known to associate with the integrin when it is released from hemidesmosomes (4, 7–18). Assembly of these kinases with the integrin initiates tyrosine phosphorylation of the β4 cytoplasmic domain, most notably in tumors, providing docking sites for signaling effectors that drive cell proliferation, invasion, and survival (4, 6, 9, 11, 19–27).
Work utilizing a number of mammary carcinoma cell lines, focusing mostly on HER2+ cells, shows that HER2/α6β4 signaling is critical for invasion and survival of these tumors (13, 14, 21). Other work has shown a strong link between α6β4 expression and “basal-like” or triple negative breast cancer that is characterized by high EGFR expression (28). Complementing their expression in the tumors, HER2 and EGFR are also expressed in endothelial cells, especially those induced by tumors (29–31), and couple with the α6β4 integrin during tumor-induced angiogenesis (32).
The distal third of the β4 tail containing the tyrosine phosphorylation sites necessary for this signaling has been termed the β4 “signaling domain” (21). In studies using the murine mammary tumor virus-Neu mouse model of HER2+ breast cancer, replacement of native β4 with a β4 mutant (β41355T) lacking this signaling domain acts as a suppressor of breast cancer (21), suggesting that the wild-type β4 receptor couples with HER2 in vivo to drive tumorigenesis. This mutant also reduces tumor-induced angiogenesis in several tumor models and reduces tumor progenitor cell formation in prostate cancer (32, 33).
The means by which receptor tyrosine kinases physically associate with and activate the α6β4 integrin are not well understood. However, our recent work suggests a possible role for syndecans, a four-member family of heparan sulfate proteoglycans that acts as matrix receptors. A number of studies have suggested a link between α6β4 integrin and syndecans in cell migration and tumorigenesis. The phosphorylated and “activated” α6β4 integrin redistributes to the leading edges of invading keratinocytes or tumors; these leading edges overexpress the unprocessed form of LN332 that retains the LG4,5 heparin-binding region that engages syndecans (34–37). Syndecan (Sdc)-1 expression is highly up-regulated in keratinocytes at the margins of wounds (38), and unprocessed LN332 retaining the LG4,5 syndecan-binding domain causes Sdc1-dependent keratinocyte attachment, spreading, and migration (35, 39). Intriguingly, Rouselle and co-workers (40) recently demonstrated that Sdc1 and Sdc4 bind differently to the LG4,5 domain, suggesting that engagement of these two syndecans with LN332 leads to different cell behaviors. Sdc1 has also been shown to bind the γ2 chain on LN332, with this interaction suppressing phosphorylation of the integrin β4 subunit (41). It is likely that processing of LN332, which removes these domains (42), influences these syndecan-regulated activities.
Our recent work shows that all four syndecans engage the cytoplasmic domain of the β4 integrin (43). In HaCat keratinocytes and A431 cervical carcinoma cells, Sdc1 is found in a complex with α6β4 integrin, HER2, and the Src family kinase Fyn (43). Clustering of this receptor complex upon matrix engagement, which can be mimicked by clustering antibodies, causes autophosphorylation of HER2, activation of Fyn, and Fyn-mediated phosphorylation of the β4 cytoplasmic domain leading to cell spreading on LN332 and cell survival. Capture of the β4 integrin cytoplasmic domain by Sdc1 is essential for phosphorylation of the integrin; because the syndecan cytoplasmic domain is very short, it appears likely that this coupling positions the integrin cytoplasmic signaling domain near the membrane where it is phosphorylated by Fyn.
Although our finding that Sdc1 forms a complex with HER2 and α6β4 integrin provided new insight into the integrin activation mechanism by HER2, it also presented additional puzzles. First, phosphorylation of the integrin in response to matrix engagement or antibodies in HaCat keratinocytes or A431 carcinoma cells depends strictly on HER2, despite the fact that the cells also express c-Met and EGFR that are also known to assemble with and activate the α6β4 integrin. Second, the HER2-specific activation of the integrin is abolished by silencing Sdc1 expression, indicating a high degree of reliance on this single syndecan; other syndecans expressed by the cells, namely Sdc2 and Sdc4, do not appear to participate despite the fact that they also bind the integrin cytoplasmic domain. This suggests that the activation of α6β4 integrin by HER2 is specific for Sdc1 and that other syndecan family members may regulate activation by the other kinases.
To address this hypothesis, we now extend our analysis to specifically question the role of different syndecans in α6β4-dependent migration of keratinocytes and mammary epithelial cells induced by HER2 or EGFR. We find that HER2-coupled migration depends on engagement of the β4 integrin cytoplasmic domain by Sdc1, whereas EGFR-stimulated migration depends on Sdc4.
EXPERIMENTAL PROCEDURES
Antibodies
Anti-integrin antibodies used were as follows: mouse mAb 3E1 (hybridoma facility, Memorial Sloan Kettering, New York); ASC-3 and ASC-8 against the β4 integrin extracellular domain; rabbit polyclonal antibody AB1922 to the β4 cytoplasmic domain; P1B5 to the α3 and β1 integrin extracellular domains (Millipore, Billerica, MA); and rat mAb GoH3 to the α6 integrin extracellular domain (BD Biosciences). Rabbit antibody BM165 to the integrin-binding site in the laminin α3 LG1–3 domain was kindly provided by Dr. Peter Marinkovich, Stanford University. Protein tag antibodies were as follows: anti-pentahistidine antibody (Qiagen,Valencia, CA); goat anti-biotin antibody (Vector Laboratories, Burlingame, CA); mouse anti-biotin mAb212.26.A2 (Jackson ImmunoResearch, West Grove, PA); rabbit anti-hemagglutinin (HA) mAb C29F4 (Cell Signaling, Danvers, MA); and mouse anti-HA mAb 12CAS (Roche Applied Science). Human Sdc4 and Sdc1 were detected by mouse mAb F94-8G3 (kindly provided by Dr. Guido David, University of Leuven, Belgium) and B-A38 (Accurate Chemical & Scientific, Westbury, NY), respectively. Mouse Sdc4 was detected by mAb KY8.2 (generously provided by Dr. Paul W. Kincade, Oklahoma Medical Research Foundation). Goat polyclonal anti-EGFR (1005), rabbit polyclonal anti-HER2/Neu (c-18) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and mouse anti-EGFR mAb EGFR.1 and anti-HER2 mAb 9C6 (BD Biosciences) were used against human EGFR and HER2.
Reagents
Dulbecco's modified Eagle's medium (DMEM) and rhodamine-conjugated phalloidin were from Invitrogen; glutathione-conjugated Sepharose beads were from GE Healthcare; human recombinant epidermal growth factor was from Sigma; HER2 inhibitor (AG825) was from Chemicon; and EGFR inhibitor (Iressa) was kindly provided by Dr. Deric Wheeler (University of Wisconsin). Two siRNAs (target sequence, 1213AGGAGGAATTCTATGCCTGA1232 and 197CAGGAATCTGATGACTTTGAG217) specific for human Sdc1 and Sdc4 were designed by Ambion (Invitrogen). Oleoyl-l-α-lysophosphatidic acid sodium salt (LPA) and mouse anti-biotin-Sepharose were from Sigma. Sdc1 C-terminal peptides were synthesized by Genscript (Piscataway, NJ). LN332 was from KeraFast (Boston, MA).
Constructs
The expression of Sdc1 and Sdc4 in pcDNA3 vector, with or without the C2 domain, was described previously (43). The cDNA for human integrin β4 was also expressed in the pcDNA3 vector. Mutations were introduced using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Cells were transfected as described previously (43).
Fusion Protein Expression and Purification
The cDNA for integrin β4 encoding amino acids 1677–1752 was inserted into pTRC-His A vector; site mutations and deletions were generated by using the QuikChange site-directed mutagenesis kit. His6-tagged β4 cytoplasmic domain fusion protein was expressed in BL21 (DE3) pLysE Escherichia coli (Invitrogen) by isopropyl 1-thio-β-d-galactopyranoside induction and purified on nickel-nitrilotriacetic acid beads following cell lysis in 100 mm NaH2PO4, 10 mm Tris base, and 8 m urea. Native or mutagenized GST-syndecan-1 cytoplasmic domain (S1CD) and GST-S4CD in pGEX vector were expressed in E. coli and purified on glutathione-Sepharose beads following cell lysis in 150 mm NaCl, 20 mm sodium phosphate (pH 7.4), and 1% Triton X-100.
Cell Culture
Human HaCat keratinocytes and A431 carcinoma cells were cultured as described previously (43). SKBr3 mammary carcinoma cells were grown in DMEM, supplemented with 10% calf serum or 10% fetal bovine serum (Hyclone Laboratories, Logan, UT), 4 mm l-glutamine (Sigma), and 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen) at 37 °C and 92.5% air, 7.5% CO2. MCF10A mammary epithelial cells were grown in DMEM/F-12 50:50 plus 15 mm HEPES, l-glutamine, 5% horse serum, 10 μg/ml insulin, 0.5 μg/ml hydrocortisone, and 0.02 μg/ml EGF.
Cell Spreading and Motility Assays
LN332 diluted to 0.1 or 1 μg/ml in calcium/magnesium-free phosphate-buffered saline (CMF-PBS: 135 mm NaCl, 2.7 mm KCl, 10.2 mm Na2HPO4·7H2O, and 1.75 mm KH2PO4 (pH 7.4)) was pre-coated on 96-well plates (Fisher) at 37 °C for 2 h and blocked with serum-free 50 mm HEPES-buffered DMEM (pH 7.4) containing 0.1% heat-denatured bovine serum albumin (plating medium) at 37 °C for 30 min. Cells were lifted with trypsin (0.25% w/v), washed with DMEM, and regenerated in suspension for 1 h at 37 °C in DMEM containing 10% calf serum. Cells were then plated in plating medium at 5 × 103 cells/ml and allowed to adhere and spread for 2 h at 37 °C. Unfixed cells were photographed using a PlanApo 20 (0.75 numerical aperture) objective and a Photometrics CoolSnap ES camera on a Nikon Eclipse TE2000U microscopy system.
For motility assays, MCF10A cells (5 × 103/well) were plated in 96-well plates pre-coated with 1 μg/ml LN332 for 2 h at 37 °C. Cells were cultured for 10 h on the stage of a Nikon Eclipse TE2000U microscope in a temperature- and humidity-controlled environmental chamber. Images were acquired at 5-min intervals using a PlanApo 20 (0.75 numerical aperture) objective and a Photometrics CoolSnap ES camera. Cell migration data were analyzed by using track object in MetaMorph® Imaging Software (Molecular Devices, Inc.).
Immunostaining
Cells were washed twice in CMF-PBS and fixed for 12 h in CMF-PBS + 2% paraformaldehyde at 4 °C, followed by three washes in PBS and blocking with PBS containing 10% goat serum for 2 h at room temperature. For permeabilization, the cells were treated with PBS containing 0.5% Triton X-100 for 3 min at room temperature. Cells were incubated with 5 μg/ml mAb 3E1 diluted in blocking buffer for 2 h at room temperature, washed four times with PBS, and incubated for 1 h with anti-mouse IgG (2 μg/ml) conjugated with Alexa-488 in blocking buffer, followed by five washes with PBS before mounting in Immu-mount (Shandon). Images were acquired by using a PlanApo 20 (0.75 numerical aperture) objective.
siRNA Treatment and Flow Cytometry
siRNA oligonucleotides specific for human Sdc1 or Sdc4 were used as described previously (43). To measure cell surface syndecan expression, suspended cells were incubated for 1 h on ice with 1 μg of primary antibody per 3 × 105 cells, then washed and counterstained by Alexa-488-conjugated secondary antibodies, and scanned on a FACSCalibur bench top cytometer. Cell scatter and propidium iodide staining profiles were used to gate live single-cell events and data analysis.
Wound Healing Assay
HaCat or MCF10A cells or cells transfected with mutant β4 were grown to confluence on 48-well plates and then were starved for 8 h by serum deprivation followed by introduction of a scratch wound in the monolayer using a 200-μl pipette tip. Cells were cultured up to an additional 15–18 h in serum-free DMEM containing LPA (3 μm), mAb 3E1 (10 μg/ml), and goat anti-mouse IgG (30 μg/ml) or EGF (10 ng/ml) in the presence or absence of inhibitors. Images were acquired using a PlanApo 20 (0.75 numerical aperture) objective and a Photometrics CoolSnap ES camera on a Nikon Eclipse TE2000U microscopy system, and wound closure was quantified using Metamorph®.
Immunoprecipitation and Bead Capture Assays
Immunoprecipitations were carried out as described previously (43). To assess cell surface protein, intact cells were labeled with sulfo-NHS-LC biotin (Pierce/Thermo Scientific, Rockford, IL) in accordance with the manufacturer's instructions. Biotinylated protein was isolated using anti-biotin-agarose and eluted in 0.1 m glycine-HCl (pH 2.5). The eluate was neutralized with 1.0 m Tris-HCl (pH 8.5) prior to immunoprecipitation of β4 integrin. For bead capture assays, lysates of cells or His6-tagged β4 cytoplasmic domain were incubated at 4 °C overnight with 100 μl of glutathione-Sepharose 4B beads, GST-S4CD, or GST-S1CD in the presence or absence of competing peptides. Samples were resolved by electrophoresis under reduced conditions on a 15% Laemmli gel, transferred to Immobilon P, and probed with primary antibody followed by an alkaline phosphatase-conjugated secondary antibody. Visualization of immunoreactive bands was performed by using ECF reagent (GE Healthcare) and scanned on a Typhoon Trio Variable Mode Imager (GE Healthcare).
RESULTS
Migration Depends on LN332 and α6β4 Integrin Activation
Regardless of the substratum on which they are plated, the migration of keratinocytes to close a wound relies on a collaboration between the α6β4 and α3β1 integrins, which deposit an oriented matrix of LN332 and engage this substratum to provide signaling necessary for the directional migration of the cells (44, 45). The participation of the α6β4 integrin in this process relies on its physical coupling to receptor tyrosine kinases, among which are HER2 and EGFR, that cause tyrosine phosphorylation of the β4 integrin cytoplasmic domain. We have shown previously that tyrosine phosphorylation of the β4 integrin when clustered either by plating cells on LN332 or by directly clustering the integrin with an antibody sandwich depends (i) specifically on HER2, and (ii) capture of the β4 integrin cytoplasmic domain via the cytoplasmic domain of Sdc1, allowing phosphorylation of the integrin by Fyn kinase downstream of activated HER2 (43).
To test whether HER2-mediated activation of the integrin causes Sdc1-dependent cell migration, we relied upon our antibody clustering technique to specifically cluster and activate α6β4 and analyzed the ability of serum-starved HaCat keratinocytes to close a scratch wound. Somewhat surprisingly, we found that clustering the integrin is not sufficient to cause cell migration (Fig. 1A). Because LPA has been shown to stimulate α6β4-dependent migration (46–48), ostensibly by promoting increased deposition of LN332 that the cells engage to migrate (49), we tested whether LPA would stimulate our mechanism. Although we found that LPA alone was not sufficient to cause migration, it did so when combined with integrin clustering, with maximal migration observed in the presence of 3 μm LPA (Fig. 1A). Migration is disrupted by mAb BM165 that targets the integrin-binding site in the LG1–3 domains of the α3 chain of LN332 (50) confirming that migration requires a LN332 substratum deposited by the cells (Fig. 1B). Nonetheless, because clustering is mediated by mAb 3E1, a β4 integrin blocking antibody (51), it is unlikely that α6β4 participates in this adhesion. This is confirmed by the failure of either GoH3 or ASC-8, two additional blocking antibodies that target α6 or β4 integrin, respectively, to block migration when added along with the clustering antibodies (51–53). However, migration is blocked by mAb P1B5, a blocking antibody specific for α3β1 integrin (53), suggesting that it alone is responsible for adhesion to the LN332. The LPA-dependent migration also relies on the activation of HER2, as it is blocked by 10 μm tyrphostin AG825 specific for HER2 and is not blocked by 1 μm Iressa (gefitinib) specific for EGFR (Fig. 1B). It is also blocked by 0.1 μm PP2, a general Src kinase inhibitor shown previously to block phosphorylation of β4 integrin by Fyn downstream of HER2 (43).
FIGURE 1.

HER2 and EGFR are involved in distinct α6β4-dependent cell motility mechanisms. A, confluent HaCat keratinocytes are starved and then stimulated to migrate for 15–18 h in a scratch wound assay by the addition of 10 ng/ml EGF (EGF chemokinesis) or by clustering of the integrin using mAb3E1 plus a secondary anti-mouse IgG in the presence of 1–30 μm LPA to mimic matrix chemokinesis. Maximal haptotactic migration is observed in the presence of 3 μm LPA, which is used for all remaining experiments. B, HaCat keratinocytes are stimulated to undergo matrix chemokinesis (clustered α6β4) or EGF-stimulated chemokinesis in the presence or absence of mAb P1B5 to block binding by α3β1 integrin, mAb GoH3, or ASC-8 to block α6β4 integrin binding, BM165 to block the integrin-binding sites in LN332, 1 μm Iressa to block EGFR, and 10 μm tyrphostin AG825 to block HER2 or the general Src kinase inhibitor PP2. C, HaCat human keratinocytes or MCF10A mammary epithelial cells are plated on 0.1 or 1 μg/ml LN332. D, HaCat or MCF10A cells are treated with EGF, or α6β4 integrin is clustered to activate the EGFR- or HER2-dependent integrin activation mechanisms. GoH3 or P1B5 is used to inhibit α6β4 or α3β1 binding to the laminin. E, MCF-10A cells are plated on 1 μg/ml LN332 under serum-free conditions, and the motility of individual cells is tracked over 10 h in the presence or absence of EGF. Relative migration distances are shown on spatial plot maps. Cells are treated with 10 μm AG825 to block HER2, 1 μm Iressa to block EGFR, or a combination of both inhibitors.
To compare the HER2-dependent mechanism with integrin activation by EGFR, we analyzed the closure of scratch wounds by keratinocytes stimulated by EGF. Serum-starved HaCat cells stimulated with 10 ng/ml EGF migrate to completely close a scratch wound in 15–18 h (Fig. 1, A and B). Not surprisingly, EGF-induced migration is disrupted by 1 μm Iressa (Fig. 1B). Correspondingly, it is not affected by inhibition of HER2 with 10 μm tyrphostin AG825 (Fig. 1B). Migration is disrupted by mAb BM165, confirming that EGF-stimulated migration also requires a LN332 substratum (Fig. 1B). Also, similar to HER2-stimulated migration, adhesion appears mediated solely by α3β1 integrin, as migration is blocked by P1B5 specific for α3β1 and not by ASC-8 or GoH3 (Fig. 1B). ASC-3, another β4-specific antibody, also fails to block migration (data not shown). Unlike HER2-dependent migration, 0.1 μm PP2 is without effect, suggesting that the mechanism of integrin activation downstream of EGFR is distinct from HER2.
Motility Is Dependent on the Amount of Deposited LN332
To further validate this assay, we tested the response of the cells to different concentrations of exogenous LN332. HaCat keratinocytes or MCF10A mammary epithelial cells plated on substrata coated with 0.1 μg/ml LN332 adhere but fail to spread (Fig. 1C). However, spreading does take place on 0.1 μg/ml LN332 if the cells are stimulated either with EGF or α6β4-clustering antibodies (Fig. 1D), and this spreading is dependent on the α3β1 integrin, but not the α6β4 integrin, engaging the laminin. When higher amounts of exogenous LN332 are provided (1 μg/ml), the cells spread (Fig. 1C) and migrate (Fig. 1E) without additional stimulation. This migration is blocked by AG825 but not by Iressa (Fig. 1E), indicating that HER2 is the central receptor tyrosine kinase that is activated by laminin-driven chemokinesis (matrix chemokinesis). In the presence of EGF (EGF-driven chemokinesis), the migration of the cells on 1 μg/ml LN332 is not blocked by AG825 alone, rather, blockade requires a combination of AG825 and Iressa (Fig. 1E), presumably to inhibit the matrix- and EGF-chemokinetic mechanisms.
Involvement of Sdc1 and Sdc4 in α6β4-dependent Migration
To further analyze the HER2- and EGFR-dependent mechanisms, we elected to use the scratch wound assay employing cultured cells, as this gave us the opportunity to activate one or the other mechanism using exogenous stimuli. Because we have shown previously that HER2-dependent activation of the α6β4 integrin depends on engagement of the β4 integrin cytoplasmic domain by Sdc1 (43), we tested whether this syndecan is essential for HER2-dependent and/or EGFR-dependent cell migration. HaCat cells treated with human Sdc1-specific siRNA show a reduction in expression of over 90% (data not shown). Silencing Sdc1 expression leads to nearly 90% inhibition of HER2-dependent migration caused by clustering the α6β4 integrin (Fig. 2A); this is rescued by expression of mouse Sdc1 (mSdc1) that evades the human-specific siRNA, but it cannot be rescued by mSdc1ΔC2 (Fig. 2A), a mutant that lacks the C-terminal C2 region (EFYA sequence) in its cytoplasmic domain necessary for engaging the β4 integrin cytoplasmic domain (Fig. 3A) (43).
FIGURE 2.

Sdc1 is required for HER2-dependent haptotaxis, whereas Sdc4 is required for EGFR-dependent chemotaxis. A, potential roles for Sdc1 or Sdc4 in HaCat wound closure are tested by silencing expression of the endogenous human syndecans with siRNA, together with rescue from either wild-type mouse Sdc1 or Sdc4, or mouse mutants unable to engage the β4 cytoplasmic domain (mSdc1ΔC2 or mSdc4ΔC2). Migration data represent the mean of six wells ± S.D. B, lysates of HaCat cells expressing either mSdc4 or the mSdc4ΔC2 mutant were subjected to immunoprecipitation (IP) using mAb KY8.2 specific for mouse Sdc4, P1B5 specific for α3β1 integrin, or 3E1 specific for α6β4 integrin. Immunoprecipitates were analyzed on Western blots probing for wild-type Sdc4 or mutant mSdc4ΔC2 with KY8.2. C, HaCat cell lysates were subjected to immunoprecipitation using mAb B-A38 specific for human Sdc1 (hSdc1), F94-8G3 specific for human Sdc4, 3E1 specific for β4 integrin, and anti-neu (c-18) for HER2 or anti-EGFR (1005). Blots were probed for HER2 using anti-neu (c-18) or anti-EGFR (1005).
FIGURE 3.

Sdc1 and Sdc4 engage distinct sites at the C terminus of the β4 integrin cytoplasmic domain. A, complete cytoplasmic domains of human Sdc1 (S1CD) and Sdc4 (S4CD) are shown, denoting the juxtamembrane region conserved across the syndecan family (C1), the distal conserved region (C2) composed of the amino acids EFYA, and the V region unique to each syndecan family member. Also shown are the C-terminal 32 amino acids of the β4 integrin subunit contained in a His6-tagged β4 integrin construct (β41677–1752) used in binding studies. Double-headed arrows denote Glu-1729 and Arg-1733 (in bold), which were mutated to alanine to disrupt syndecan-specific binding to this construct. An arrow also denotes the site at which the construct is truncated to remove the last 24 amino acids (His6-β4(1677–1728)) necessary for syndecan binding. B, conservation of amino acids 1727–1736 in the β4 integrin cytoplasmic domain across species is shown, with conserved Glu-1729 and Arg-1733 in bold. Asterisks denote threonines that disrupt binding of the β4 cytoplasmic domain to plectin when phosphorylated (66). C, glutathione beads preincubated with 2 μm GST-S1CD, GST-S4CD, GST-S1CDΔC2, or GST-S4CDΔC2 were tested for their ability to capture 2 μm His6-β4(1677–1752), detected on Western blots with anti-penta-His. D–F, glutathione beads preloaded with GST-S1CD or S4CD are tested for the capture of either 2 or 4 μm His6-β4(1677–1752) or His6-β4(1677–1728) (D), or constructs in which point mutations have been introduced at Arg-1733 (His6-β4R1733A) (E) or Glu-1729 (His6-β4E1729A) (F).
In contrast to the HER2 mechanism, silencing Sdc1 leads to only a modest decrease in EGF-induced chemokinesis (Fig. 2A). However, because all four syndecans contain the C-terminal EFYA sequence known to bind β4 integrin (43), we tested the possibility that a different syndecan participates in the EGF-stimulated mechanism. Among the remaining syndecans expressed by these cells, Sdc2 and Sdc4, silencing Sdc2 expression has no effect on EGF-stimulated migration (data not shown). However, silencing Sdc4 leads to a significant blockade of EGF-induced chemokinesis, which can be rescued by mSdc4 but not by mSdc4ΔC2 (Fig. 2A). Note that silencing Sdc4 expression also disrupts HER2-dependent migration. But this is rescued by mSdc4ΔC2 as well as by mSdc4, suggesting that its role in HER2-dependent migration does not trace to its engagement of β4 integrin. Although we have not investigated this additional role of Sdc4 further as part of this study, this may be due to an association with the α3β1 integrin, which actively participates in both matrix chemokinesis and EGF chemokinesis (cf. Fig. 1B). Mouse Sdc4 co-immunoprecipitates with both α6β4 integrin and α3β1 integrins, and although α6β4 binding is lost when the C2 domain of mSdc4 is removed, mSdc4ΔC2 is still able to co-immunoprecipitate with α3β1 (Fig. 2B).
Taken together, these findings suggest that HER2 and EGFR form two independent complexes with the α6β4 integrin, one dependent on Sdc1 and the other on Sdc4. To test this possibility, we immunoprecipitated the two syndecans and probed their potential association with HER2 or EGFR (Fig. 2C). HER2 co-immunoprecipitates with Sdc1 and the α6β4 integrin. EGFR, in contrast, co-immunoprecipitates with Sdc4 and the α6β4 integrin. We also detect heterodimers of HER2 or EGFR when either is immunoprecipitated. This is not unexpected, as EGFR and HER2 are well known to form heterodimers in the presence of EGF. This conclusion is supported by the finding that starved cells fail to exhibit these heterodimers, which re-appear after 30 min of stimulation with EGF (data not shown). However, heterodimers are not readily detected in the syndecan immunoprecipitates.
Yeast two-hybrid analysis previously localized the syndecan-binding site to the C-terminal third (amino acids 1473–1752) of the β4 cytoplasmic domain (43). To identify the binding site(s) more definitively, overlapping fragments representing the N-terminal (amino acids 1452–1572), middle (amino acids 1552–1693), or C-terminal (amino acids 1677–1752) thirds of this region were expressed as recombinant His6-tagged proteins in bacteria and used for binding studies. No capture of the N-terminal or middle third was observed when using recombinant Sdc1 or Sdc4 cytoplasmic domains expressed as GST fusion proteins (GST-S1CD or GST-S4CD) (data not shown). However, the 76-amino acid fragment consisting of the C terminus of the integrin (His6-β4(1677–1752)) is captured by either GST-S1CD or GST-S4CD, and capture is significantly reduced when the C2 region (see Fig. 3A) is removed from either syndecan construct (Fig. 3C), although capture is not completely abolished.
Sdc1 and Sdc4 Recognize Different Motifs in the β4 Integrin Cytoplasmic Domain
To more narrowly define the potential syndecan-binding site, we analyzed the β4 sequence for amino acid conservation across species and found regions of high conservation within the last 24 amino acids of the C terminus (Fig. 3A). A β4 C-terminal fragment in which these 24 amino acids have been removed (His6-β4(1677–1728)) is no longer captured by either GST-S1CD or GST-S4CD (Fig. 3D), indicating that their interaction site is likely within this region. Next, we set out to mutate individual amino acids to identify the specific site(s). Mutation of Arg-1733 in the C-terminal fragment abolishes its capture by S1CD (Fig. 3E). However, this does not disrupt capture by S4CD. Instead, mutation of Glu-1729 abolishes capture by GST-S4ED and has no effect on capture by S1CD (Fig. 3F). A scan of this binding sequence across vertebrate species shows exact conservation of Glu-1729 and only occasional substitution of Arg-1733 with another basic amino acid (His) or with Gln, an amino acid with a similar sized side group also capable of forming hydrogen bonds (Fig. 3B).
These findings suggest that although the syndecan C2 region is required for β4 integrin capture, Sdc1 and Sdc4 are likely to rely on adjoining amino acids in their V regions (Fig. 3A), which are unique to each syndecan, to provide additional specificity for the binding interaction. To test this hypothesis, we used a peptide competition assay to identify the binding sequence in the cytoplasmic domain of Sdc1. We see effective competition in our in vitro pulldown assay using the peptide KQEEFYA (Fig. 4A), which is composed of three amino acids from the V region of Sdc1 together with its C2 region (cf. Fig. 3A). Next, the amino acids within the Sdc1 peptide necessary to bind the integrin fragment were assessed. Note that the EFYA sequence that comprises the C2 region has been shown to be a PDZ domain-binding motif, depending on Phe (P−2) and Ala (P0) (54). Tyr at P−1 can be mutated to Phe or Ala, but it cannot be phosphorylated without disrupting PDZ domain binding (54, 55). To test whether β4 integrin binding obeys these same rules, we tested peptides in which the Phe or Tyr is mutated, or Tyr is phosphorylated. Unlike PDZ binding, neither Phe (P−2) nor Ala (P0) is required, and phosphorylation of Tyr (P−1) is tolerated (Fig. 4B). However, mutation of Tyr (P−1) to Ala, or even its conservative mutation to Phe, blocks the binding activity. A further alanine screen across all seven amino acids in the KQEEFYA sequence shows the binding motif necessary for competing S1CD binding to β4 cytoplasmic domain to be -QEE-Y- (Fig. 4C), which consists of two amino acids from the V region and two from the C2 region of Sdc1.
FIGURE 4.
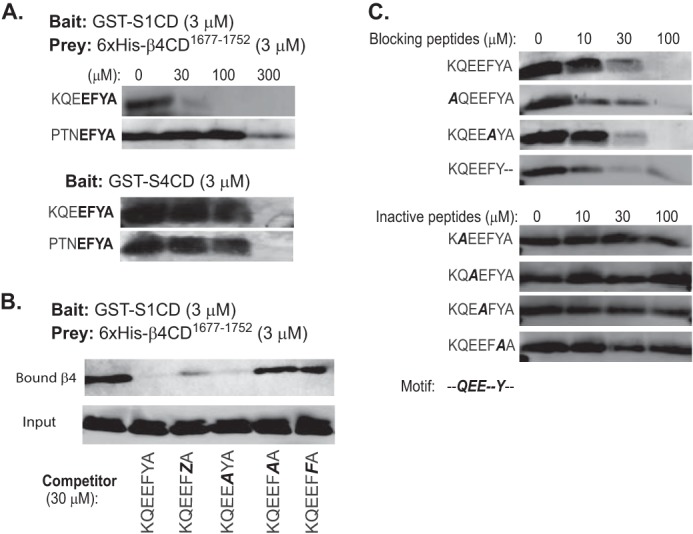
Distinct cytoplasmic motifs in Sdc1 and Sdc4 engage the β4 cytoplasmic domain. The glutathione bead pulldown assay is used to assess the capture of His6-β4(1677–1752) (prey) either by S1CD or S4CD (bait) in the presence of competing peptides. Captured β4 construct is visualized on Western blots by staining with anti-penta-His. A, capture by GST-S1CD or GST-S4CD in the presence of 0–300 μm KQEEFYA peptide sequence derived from the Sdc1 or PTNEFYA derived from Sdc4. The conserved C2 region (EFYA) is shown in bold. B, capture by GST-S1CD in the presence of Sdc1-specific peptide in which Phe-308 is mutated to Ala (A), or Tyr-309 is phosphorylated (Z), or is mutated to Ala or Phe (F). C, capture by GST-S1CD in the presence of Sdc1-specific peptide in which individual amino acids are mutated to Ala or Ala-310 is deleted.
The -QEE-Y- motif is wholly conserved in the C terminus of Sdc1 or Sdc3 but is not conserved apart from the two amino acids in the C2 region in Sdc2 (TKEEFYA) or Sdc4 (PTNEFYA), the other two syndecans expressed along with Sdc1 in HaCat cells. To test whether or not the KQEEFYA peptide competes for Sdc4 binding, we tested the capture of β4(1677–1752) by GST-S4CD over a range of peptide concentrations (Fig. 4A). Although competition is observed, it requires 300 μm concentrations, 10-fold higher than similar inhibition of Sdc1. This competition only at higher peptide concentrations is mimicked by a PTNEFYA representing the last seven amino acids of Sdc4. It competes for Sdc1 capture of His6-β4(1677–1752) only at 300 μm (Fig. 3A), likely reflecting the EFYA sequence that it shares with KQEEFYA. But its competition for capture by Sdc4 also requires 300 μm, suggesting that competition is again due only to the shared EFYA sequence and that higher affinity Sdc4 binding is likely to require interaction(s) elsewhere in its V region. Although this binding site has not been analyzed as part of this work, the finding confirms that Sdc1 and Sdc4 rely on distinct interaction motifs.
Cell Penetrating TAT Peptide Containing the Sdc1-binding Motif Disrupts HER2-dependent Integrin Signaling
Having confirmed the syndecan-specific capture of the β4 cytoplasmic domain in vitro, we returned to our bioassays to assess their importance in HER2- or EGFR-stimulated migration. First, we used a cell-penetrating TAT-KQEEAYA peptide, based on the cell-penetrating sequence of the HIV transactivator of transcription (TAT) protein (56, 57) coupled to the Sdc1-binding motif to target β4 capture by Sdc1. Phe (P−2) was mutated to Ala to minimize potential disruption of PDZ domain interaction by the peptide. This peptide competes effectively for His6-β4(1677–1752) capture by GST-S1CD in pulldown assays, as expected, and fails to compete for capture by GST-S4CD (Fig. 5A). A control peptide (TAT-KQEAAA), in which the essential Tyr (P−1) is mutated, fails to compete for capture by either syndecan (Fig. 5A).
FIGURE 5.

Cell-penetrating peptide containing the Sdc1-specific β4-binding motif disrupts HER2-dependent haptotaxis and cell survival. TAT peptide is constructed containing the Sdc1-specific binding motif in which Phe-308 necessary for PDZ binding but not for β4 binding is mutated (KQEEAYA) (active peptide) or Phe-308 is mutated along with Tyr-309 necessary for β4 binding (KQEEAAA) (inactive control peptide). A, active or control TAT peptides are tested as competitors of His6-β4(1677–1752) capture by GST-S1CD or GST-S4CD. B, HaCat keratinocytes are stimulated with LPA and α6β4 integrin clustering antibodies to mimic HER2-dependent matrix chemokinesis or stimulated with EGF to undergo EGFR-dependent chemokinesis in the presence of 0–10 μm TAT peptides. C, MCF10A human breast epithelial cells are stimulated to undergo matrix chemokinesis in the presence of TAT peptides ranging from 0 to 10 μm. D, HaCat keratinocytes, MCF10A mammary epithelium, A431 cervical carcinoma, or HER2-positive SKBr3 breast carcinoma cells were grown for 1 week in the presence of TAT peptide ranging from 0 to 30 μm. Cell death was quantified by trypan blue exclusion. Data represent the mean of six wells ± S.D. (* indicates difference between control and active peptide >0.05.)
The active and control TAT peptides were next tested in the HaCat scratch wound assay. The active TAT-KQEEAYA peptide displayed a concentration-dependent inhibition of matrix chemokinesis mimicked by clustered α6β4 integrin (Fig. 5B). In contrast, the peptide had no effect on stimulation by EGF. The active peptide also blocked the α6β4-dependent migration of MCF10A mammary epithelial cells (Fig. 5C). The peptide also caused cell death in SKBr3 cells, a HER2+ breast carcinoma cell line in which α6β4 activation by overexpressed HER2 has been linked to cell survival (Fig. 5D) (58, 59), suggesting that the linkage between α6β4 integrin activation and HER2 in this apoptotic mechanism is also mediated by the cytoplasmic domain of Sdc1. The peptide had no effect on nontransformed HaCat and MCF10A cells and had minimal effect on the A431 carcinoma cells.
β4 Integrin Constructs Bearing Sdc1- or Sdc4-specific Mutations Act as Dominant Negative Mutants
In a second approach to demonstrate the importance of the syndecan-specific regulation, we introduced the wild-type β4 integrin construct, or integrin constructs bearing the R1733A or E1729A mutations predicted to disrupt specific syndecan binding, into A431 cervical carcinoma cells, which display a β4-dependent spreading phenotype when plated on LN332 (43). The cell surface expression of Sdc1, Sdc4, HER2, EGFR, α3β1 integrin, or α6β4 integrin was evaluated in these cells and found to be equivalent in each of the three cell strains (Fig. 6A).
FIGURE 6.

α6β4R1733A and α6β4E1729A integrin constructs act as dominant negative mutants to block the HER2- and EGFR-dependent mechanisms, respectively, during A431 cell spreading. A, A431 cells expressing HA-tagged β4WT, β4R1733A, or β4E1729A constructs were subjected to flow cytometry to quantify relative expression of cell surface α6β4 integrin (3E1), Sdc1 (B-A38), and Sdc4 (F4–8G3), α3β1 integrin (ASC-1), and HER2 (9C6) or EGFR (EGFR.1). B, A431 cells expressing the β4WT, β4R1733A, or β4E1729A constructs are plated on 1 μg/ml LN332 for 2 h to activate the HER2-dependent signaling mechanism (spread cells denoted by arrowheads) with or without addition of 10 μm AG825 or 1 μm Iressa to inhibit HER2 or EGFR, respectively. The percentage of spread cells in triplicate wells from two experiments is quantified. C, A431 cells expressing β4 receptor constructs are plated on 1 μg/ml LN332 for 2 h in the presence of 10 ng/ml EGF with or without AG825 or Iressa and quantified as in B. D, A431 parental cells, β4WT-expressing cells, or β4R1733A-expressing cells are fixed, permeabilized, and stained for β4 integrin with mAb 3E1. E, relative amounts of endogenous α6β4 and HA-tagged α6β4R1733A (mutant) integrin at the cell surface is determined by biotinylation of parental or α6β4R1733A-expressing (R1733A) cells, followed by immunoprecipitation with mAb 3E1 to obtain “total β4,” with anti-HA (mAb 12CAS) to obtain “mutant β4,” or 3E1 precipitation of integrin remaining in the supernatant after anti-HA precipitation to quantify native endogenous β4. The relative amounts of total α6β4, HA-tagged α6β4R1733A, and biotinylated (cell surface) α6β4 integrin in each pool are assessed by staining a single blot with anti-β4 (AB1922), anti-HA (C29F4), and anti-biotin (212.26.A2) by stripping and reprobing.
Cells expressing wild-type integrin display a highly spread phenotype when plated on LN332, reflecting constitutive activation of the HER2-dependent mechanism identified in a previous report (43). This is confirmed by inhibition of the spreading by 10 μm tyrphostin AG825 (Fig. 6B). Surprisingly, we also found that spreading is greatly reduced by expression of β4R1733A, suggesting that the β4R1733A integrin acts as a dominant negative mutant to disrupt the Sdc1-dependent mechanism (Fig. 6B). Spreading on LN332 is not affected by the β4E1729A mutant even in the presence of EGFR inhibitor (Fig. 6B). In contrast, stimulation of the cells with EGF overcomes inhibition by the β4R1733A mutant, even when supplemented by the HER2 inhibitor AG825 (Fig. 6C). However, supplementation of AG825 with Iressa, such that both the HER2 and EGFR mechanisms are inhibited, affects the cell's ability to spread on the laminin. Similarly, β4E1729A blocks spreading induced by EGF if supplemented with AG825 to block the constitutively active HER2 mechanism. Thus, the HER2- and EGFR-dependent mechanisms are inhibited by expression of the β4R1733A and β4E1729A mutants, respectively.
The fact that overexpression of the β4 mutants does not increase total α6β4 integrin at the cell surface (cf. Fig. 6A) is likely explained by competition of the β4 subunits for a limiting pool of α6 integrin subunit, leading to the dominant negative phenotype that we observed. Testing this hypothesis, we found that staining permeabilized cells for β4 integrin expression shows high levels of overexpressed protein in perinuclear structures, likely the endoplasmic reticulum/Golgi, in A431 cells expressing β4WT or β4R1733A constructs compared with parental cells (Fig. 6D). Biotin labeling of cell surface integrin in β4R1733A-expressing cells confirms that the majority of the cell surface integrin contains the mutant construct (Fig. 6E). Cell surface proteins were isolated using anti-biotin beads following biotinylation of intact cells, and either total β4 integrin was precipitated from this pool using mAb 3E1 or the mutant β4 alone was precipitated using its HA tag. Finally, endogenous or “native” integrin was captured from the anti-HA supernatant. Note that the total amount of biotinylated cell surface α6β4 integrin detected on Western blots is the same when comparing parental A431 and R1733A mutant cells, confirming the flow cytometry results (cf. Fig. 6A). Additionally, the HA-tagged (mutant) integrin includes the majority of biotinylated (cell surface) integrin in the mutant cells (Fig. 6E), compared with the amount of biotinylated native integrin.
β4 Dominant Negative Mutants Specific for Sdc1 or Sdc4 Binding Blocks Matrix or EGF Chemokinesis, Respectively
The dominant negative role of the β4R1733A and β4E1729A mutants provides a tool to further evaluate the role of Sdc1 and Sdc4 in HER2- and EGFR-dependent migration. This assay used HaCat keratinocyte and MCF10A mammary epithelial cell lines transfected with cDNA for β4WT, β4R1733A, or β4E1729A integrins. Analysis of receptor expression in the HaCat cells again demonstrates that overexpression of the β4 constructs does not change the relative cell surface expression levels of α6β4 integrin as expected, nor does it change the cell surface expression of Sdc1 or Sdc4, HER2 or EGFR, or the α3β1 integrin (Fig. 7A).
FIGURE 7.

Dominant negative α6β4R1733A integrin disrupts HER2-dependent haptotaxis and α6β4E1729A disrupts EGFR-dependent chemotaxis of HaCat and MCF10A cells. A, HaCat cells expressing β4WT, β4R1733A, or β4E1729A mutants are subjected to flow cytometry to determine relative cell surface expression of α6β4 integrin, Sdc1 and Sdc4, α3β1 integrin, and HER2 or EGFR. B, either total or HA-tagged β4 integrin was immunoprecipitated (IP) with mAb 3E1 or anti-HA from lysates of HaCat cells expressing HA-β4WT, HA-β4R1733A, or HA-β4E1729A mutants. Immunoprecipitates divided into duplicate samples were probed for co-immunoprecipitation of integrin and either HER2 or EGFR. C, confluent HaCat cells expressing either β4WT, β4R1733A, or β4E1729A integrin subunits are stimulated to close a scratch wound during a 15–18-h treatment with 3 μm LPA, 10 μg/ml 3E1, and 30 μg/ml anti-mouse secondary antibody to stimulate HER2-dependent haptotaxis. Bar, 40 μm. D, quantification of scratch wound closure by either parental HaCat cells or cells transfected with β4 integrin constructs and stimulated to undergo haptotaxis or chemotaxis in response to 10 ng/ml EGF. The migration of parental cells in the absence of stimulation or stimulated with LPA alone is shown as a control. E, quantification of scratch wound closure by either parental MCF10A cells or cells transfected with β4 integrin constructs and stimulated as described in C. Data represent the mean of six wells ± S.D.
We next examined the effect of the β4 mutations on assembly of the syndecan-coupled receptor complexes (Fig. 7B). HER2 and Sdc1 were observed to co-immunoprecipitate with β4 from HaCat cells overexpressing HA-tagged wild-type β4 (HA-β4WT). However, little or no HER2 or Sdc1 was observed to co-immunoprecipitate with β4 if the cells are overexpressing the β4 mutant deficient in Sdc1 binding (HA-β4R1733A). A similar finding was observed for cells overexpressing HA-β4E1729A, namely capture of EGFR and Sdc4 with the integrin is dramatically reduced (Fig. 7B). This confirms that Sdc1 and Sdc4 engagement with the β4 cytoplasmic domain was necessary for association of HER2 and EGFR with α6β4 integrin, respectively.
These findings provide a mechanistic explanation for the roles of Sdc1 and Sdc4 in epithelial cell migration. In scratch wound assays, the HaCat cells expressing β4R1733A display significant inhibition of matrix chemokinesis stimulated by clustering the β4 integrin in the presence of LPA, whereas the β4E1729A mutant is without effect (Fig. 7, C and D). The same behavior is observed with the MCF10A cells (Fig. 7E). In contrast, EGF-stimulated chemokinesis is inhibited by the β4E1729A dominant negative mutant and not by β4R1733A (Fig. 7, D and E), demonstrating the clear dependence on Sdc1 and Sdc4 in HER2- and EGFR-mediated migration, respectively.
DISCUSSION
Our findings demonstrate a fundamental role for syndecans in linking receptor tyrosine kinases to the signaling activity of α6β4 integrin during matrix- and EGF-stimulated chemokinesis of epithelial cells. These findings extend our previous work, which identified a role for Sdc1 in HER2-mediated phosphorylation of the β4 integrin cytoplasmic domain during cell attachment and spreading. In this study, we again find that Sdc1 is required for HER2-mediated activation of the α6β4 integrin in wound healing assays or tumor cell survival, but we find that Sdc4 is necessary for activation of the integrin during chemokinesis induced by EGF. Our studies reveal a direct interaction between the syndecans and the β4 integrin cytoplasmic domain that is syndecan type-specific.
The β4 cytoplasmic domain is over 1,000 amino acids long, consisting of a juxtamembrane Na+-Ca+ exchanger motif, two pairs of FNIII repeats separated by a connecting segment, and a C-terminal tail. In type I hemidesmosomes found in stratified epithelia, the β4 integrin cytoplasmic domain binds to plectin, the cytoplasmic domain of bullous pemphigoid antigen 180 (BP180 or type XVII collagen) and BP230 (60). In type II hemidesmosomes found in simple epithelia, the β4 integrin subunit interaction is limited to plectin (61). The plectin interaction involves the first set of FNIII repeats and a portion of the connecting segment (49, 62, 63). This is supplemented by an interaction of the β4 C terminus, which folds up to interact with the connecting segment and the plako domain of plectin (64, 65). Signaling from activated receptor tyrosine kinases, such as HER2 or EGFR, causes disruption of plectin binding both by phosphorylation of serines 1356, 1360, 1364, and 1424 in the connecting segment of the β4 cytoplasmic domain by ERK1/2, protein kinase C, protein kinase A, and potentially other kinases and by phosphorylation of threonine 1736 at the β4 C terminus (5, 6, 66–68). This threonine, as well as the adjacent threonine 1727, is within a PKD1 phosphorylation consensus site, and mutation of either threonine to a phosphomimetic aspartate residue disrupts plectin binding (66). However, the failure of PKD1 inhibitor to block this suggests that other Ca2+/calmodulin-dependent protein kinase-related kinases are likely to carry out this phosphorylation as well (66). Our data now suggest that this same region of the integrin C terminus associates with syndecans, presumably when dissociated from plectin. Direct binding was initially identified by performing pulldown studies using recombinant syndecan cytoplasmic domains to capture either native or truncation mutants of recombinant β4 integrin cytoplasmic domain. This identified critical amino acids Arg-1733 and Glu-1729, located within the distal 24 amino acids of the integrin tail, that are specific for the binding of Sdc1 or Sdc4, respectively. The importance of these amino acids is demonstrated by β4 constructs in which one or the other of these amino acids is mutated to alanine; these constructs act as dominant negative mutants to disrupt HER2- or EGFR-dependent cell migration that depends on Sdc1 and Sdc4, respectively. As these amino acids are flanked by amino acids (cf. Fig. 3A) that are targeted for phosphorylation to disrupt plectin binding (66), it raises the question for future study whether or not threonine phosphorylation affects syndecan binding, potentially enhancing the interaction while disrupting plectin interactions.
It is interesting to note that although the syndecans have the exactly conserved C2 (EFYA) motif at their C terminus, and utilize parts of this motif for binding, their interaction with the integrin is syndecan type-specific, potentially providing a mechanism for syndecan-specific regulation. The C2 region of the syndecans also binds to type II PDZ domains, studied most extensively in syntenin and Tiam-1 (54, 57, 69, 70). Where information is available, key differences are seen between Sdc1 engaging the β4 integrin, syntenin, and Tiam-1. Binding to syntenin relies on an XFXA motif and is disrupted if Tyr (P−1) is phosphorylated (54, 55, 70). Binding to the Tiam-1 PDZ domain appears to rely on KXEXFXX composed partly of the C2 region (EFYA) and KXE from the syndecan V region. This motif is shared by Sdc1 and Sdc3, which show strongest binding to Tiam1 of the four syndecans. Unlike syntenin, phosphorylation of Tyr (P−2) in the syndecan is tolerated by this PDZ domain (69). In contrast, our current work shows that β4 binding relies on QEEXYX; the Tyr can be phosphorylated but cannot be mutated to Phe, identifying an important role for the hydroxyl group on the tyrosine, likely for hydrogen bonding. This binding motif is also composed of two amino acids (XQE) from its V region, a region that is exactly conserved across species but is distinct for each syndecan family member. The failure of the analogous PTNEFYA peptide derived from Sdc4 to compete specifically for Sdc4 binding to β4 suggests that the capture site in Sdc4 extends further into the V region and may be more complex. The Sdc4 V region has been implicated in other activities, notably binding of PIP2 and capture of active PKCα to enhance integrin signaling when the syndecan is localized to focal adhesions (71–73). Whether these interactions compete for or enhance Sdc4-specific capture of the β4 integrin is not currently known.
We find that the α3β1 integrin co-immunoprecipitates with Sdc4 from HaCat cell lysates, suggesting that its activity may depend on Sdc4 as well. As such, Sdc4 and determinants in its V region may have dual functions in epithelial cell migration, participating in signaling mediated by α3β1 integrin, as well as in the activation of α6β4 integrin if the migration depends on EGFR (cf. Fig. 2). Araki et al. (74) have shown that PE75, a peptide derived from LG4 domain in the LN α3 chain, promotes clustering of Sdc4 with β1 integrins on HaCat cells and β1 integrin activation. In addition, the Sdc4 transmembrane domain is known to cause its homodimerization (33, 75), which may enhance its clustering and signaling activity. Whether or not α3β1 and α6β4 integrin are part of a single receptor complex that relies on Sdc4 remains to be examined.
The presence of the syndecans as obligate partners in the α6β4 integrin activation mechanism provides additional insight into previous studies in which a β4 truncation mutant (β41355T) has been used in models of tumorigenesis and tumor-induced angiogenesis. This truncation removes the second pair of FNIII repeats and C-terminal tail of the integrin cytoplasmic domain containing tyrosines critical for integrin signaling. It is found to disrupt Neu-induced breast tumorigenesis, tumor-induced angiogenesis in a number of cancer types, and the HER2- and c-Met-mediated persistence of tumor progenitor cells in a prostate cancer model (21, 32, 76). Although our findings do not reduce the impact and overall conclusions of these important studies, they do offer a modified view of how this mutant may act, as this truncation deprives the integrin of the Sdc1 and Sdc4 engagement sites normally present at its C terminus. Thus, not only are the tyrosines that are targets of HER2- and EGFR-mediated integrin activation removed, but the mechanism by which HER2 or EGFR cause phosphorylation of those tyrosines, together with other signals that may depend on Sdc1 and Sdc4 coupling with the integrin, is disrupted as well.
Whereas the work from Guo et al. (21) suggests that the HER2-dependent mechanism is active in vivo when HER2 is expressed as the Neu oncogene, and we find that the mechanism appears constitutively necessary for the survival of HER2-overexpressing SKBr3 mammary carcinoma cells, it does not appear to be constitutively active on nontransformed HaCat or MCF10A epithelia grown in culture. However, plating the cells on increased LN332 concentrations does activate the mechanism, as does clustering the α6β4 integrin with antibodies when on lower LN332 concentrations. Our previous work demonstrates that this engagement causes autophosphorylation of HER2, downstream activation of Fyn, phosphorylation of β4, and activation of PI3K (43). These findings suggest that this mechanism is activated in vivo either by overexpression of HER2 or kinase-activating mutations of the receptor, or by enhanced deposition of LN332. Modulation of LN332 expression by LPA may partly explain the tumorigenic activity of this lipid (46, 77). Our data also suggest, however, that α6β4 integrin binding to the LN332 is not required; thus, its primary role may be to act as a signaling scaffold when bound by the syndecans, rather than having a role in clustering the receptor complex by engaging the matrix. Activation of the complex does depend on α3β1 integrin, however, suggesting the possibility that α3β1 integrin is present in the same receptor complex or, at a minimum, in a common membrane signaling domain that causes clustering and activation of HER2 when engaged with LN332. The syndecan is also a candidate for engaging the LN332. In preliminary studies, we find that denuding the syndecans of their heparan sulfate chains has no effect on cell attachment and spreading on LN332 (data not shown). However, Rouselle and co-workers (40) have published intriguing findings that Sdc4 engages LN332 directly via protein determinants in its extracellular domain, potentially revealing another means by which these receptor complexes may be activated.
The migration of epithelial cells has been shown to depend on their deposition of an LN332 substratum (44). It is widely accepted that α3β1 integrin plays an important role in the motility on this substratum, but the exact role of α6β4 is subject to uncertainty (44, 45). However, the motility defect observed with β4-deficient PA-JEB cells reveals an essential role for this integrin (44, 45). One intriguing study suggests that α6β4 is not required for motility per se, rather, it is necessary to orient newly deposited LN332 into linear arrays that enhance directional migration, together with α6β4-dependent activation of Rac1 and remodeling of the actin cytoskeleton (44). Another study analyzing the EGF-stimulated migration of keratinocytes plated on a variety of matrices (collagen I or IV, FN, or LN111) found that migration on newly deposited LN332 can occur as long as either the α6β4 or the α3β1 integrin is ligated (45). As in our studies, blocking antibodies to the α6β4 integrin did not disrupt migration. However, α6β4 expression was nonetheless required. Our studies support and extend these previous findings, suggesting that although direct engagement of the α6β4 integrin with the matrix is not required, engagement of the cytoplasmic domain of the β4 subunit with Sdc1 or Sdc4 is necessary for it to participate as a signaling scaffold during cell migration on LN332 promoted by HER2 or EGFR, respectively.
Acknowledgments
We thank the University of Wisconsin Carbone Cancer Center for the use of its shared services, supported by National Institutes of Health Grant P30 CA014520 from NCI.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-CA109010 and R01-CA163662 (to A. C. R.).
This article was selected as a Paper of the Week.
- EGFR
- epidermal growth factor receptor
- HER2
- human epidermal growth factor receptor-2
- LN332
- laminin 332
- Sdc
- syndecan
- LPA
- oleoyl-l-α-lysophosphatidic acid sodium salt
- FN
- fibronectin
- mSdc
- mouse syndecan
- TAT
- transactivator of transcription
- V
- variable.
REFERENCES
- 1. Aumailley M., Bruckner-Tuderman L., Carter W. G., Deutzmann R., Edgar D., Ekblom P., Engel J., Engvall E., Hohenester E., Jones J. C., Kleinman H. K., Marinkovich M. P., Martin G. R., Mayer U., Meneguzzi G., Miner J. H., Miyazaki K., Patarroyo M., Paulsson M., Quaranta V., Sanes J. R., Sasaki T., Sekiguchi K., Sorokin L. M., Talts J. F., Tryggvason K., Uitto J., Virtanen I., von der Mark K., Wewer U. M., Yamada Y., Yurchenco P. D. (2005) A simplified laminin nomenclature. Matrix Biol. 24, 326–332 [DOI] [PubMed] [Google Scholar]
- 2. Nievers M. G., Schaapveld R. Q., Sonnenberg A. (1999) Biology and function of hemidesmosomes. Matrix Biol. 18, 5–17 [DOI] [PubMed] [Google Scholar]
- 3. Hopkinson S. B., Jones J. C. (2000) The N terminus of the transmembrane protein BP180 interacts with the N-terminal domain of BP230, thereby mediating keratin cytoskeleton anchorage to the cell surface at the site of the hemidesmosome. Mol. Biol. Cell 11, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilhelmsen K., Litjens S. H., Sonnenberg A. (2006) Multiple functions of the integrin α6β4 in epidermal homeostasis and tumorigenesis. Mol. Cell. Biol. 26, 2877–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rabinovitz I., Tsomo L., Mercurio A. M. (2004) Protein kinase C-α phosphorylation of specific serines in the connecting segment of the β4 integrin regulates the dynamics of type II hemidesmosomes. Mol. Cell. Biol. 24, 4351–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilhelmsen K., Litjens S. H., Kuikman I., Margadant C., van Rheenen J., Sonnenberg A. (2007) Serine phosphorylation of the integrin β4 subunit is necessary for epidermal growth factor receptor-induced hemidesmosome disruption. Mol. Biol. Cell 18, 3512–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agazie Y. M., Hayman M. J. (2003) Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 23, 7875–7886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mainiero F., Pepe A., Yeon M., Ren Y., Giancotti F. G. (1996) The intracellular functions of α6β4 integrin are regulated by EGF. J. Cell Biol. 134, 241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mariotti A., Kedeshian P. A., Dans M., Curatola A. M., Gagnoux-Palacios L., Giancotti F. G. (2001) EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 155, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertotti A., Comoglio P. M., Trusolino L. (2005) β4 integrin is a transforming molecule that unleashes Met tyrosine kinase tumorigenesis. Cancer Res. 65, 10674–10679 [DOI] [PubMed] [Google Scholar]
- 11. Bertotti A., Comoglio P. M., Trusolino L. (2006) β4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J. Cell Biol. 175, 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bon G., Folgiero V., Di Carlo S., Sacchi A., Falcioni R. (2007) Involvement of α6β4 integrin in the mechanisms that regulate breast cancer progression. Breast Cancer Res. 9, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falcioni R., Antonini A., Nisticò P., Di Stefano S., Crescenzi M., Natali P. G., Sacchi A. (1997) α6β4 and α6β1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp. Cell Res. 236, 76–85 [DOI] [PubMed] [Google Scholar]
- 14. Gambaletta D., Marchetti A., Benedetti L., Mercurio A. M., Sacchi A., Falcioni R. (2000) Cooperative signaling between α6β4 integrin and ErbB-2 receptor is required to promote phosphatidylinositol 3-kinase-dependent invasion. J. Biol. Chem. 275, 10604–10610 [DOI] [PubMed] [Google Scholar]
- 15. Santoro M. M., Gaudino G., Marchisio P. C. (2003) The MSP receptor regulates α6β4 and α3β1 integrins via 14-3-3 proteins in keratinocyte migration. Dev. Cell 5, 257–271 [DOI] [PubMed] [Google Scholar]
- 16. Trusolino L., Bertotti A., Comoglio P. M. (2001) A signaling adapter function for α6β4 integrin in the control of HGF-dependent invasive growth. Cell 107, 643–654 [DOI] [PubMed] [Google Scholar]
- 17. Tsuruta D., Kobayashi H., Imanishi H., Sugawara K., Ishii M., Jones J. C. (2008) Laminin-332-integrin interaction: a target for cancer therapy? Curr. Med. Chem. 15, 1968–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giancotti F. G. (2007) Targeting integrin β4 for cancer and anti-angiogenic therapy. Trends Pharmacol. Sci. 28, 506–511 [DOI] [PubMed] [Google Scholar]
- 19. Mainiero F., Murgia C., Wary K. K., Curatola A. M., Pepe A., Blumemberg M., Westwick J. K., Der C. J., Giancotti F. G. (1997) The coupling of α6β4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 16, 2365–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaw L. M., Rabinovitz I., Wang H. H., Toker A., Mercurio A. M. (1997) Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell 91, 949–960 [DOI] [PubMed] [Google Scholar]
- 21. Guo W., Pylayeva Y., Pepe A., Yoshioka T., Muller W. J., Inghirami G., Giancotti F. G. (2006) β4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126, 489–502 [DOI] [PubMed] [Google Scholar]
- 22. Merdek K. D., Yang X., Taglienti C. A., Shaw L. M., Mercurio A. M. (2007) Intrinsic signaling functions of the β4 integrin intracellular domain. J. Biol. Chem. 282, 30322–30330 [DOI] [PubMed] [Google Scholar]
- 23. Dutta U., Shaw L. M. (2008) A key tyrosine (Y1494) in the β4 integrin regulates multiple signaling pathways important for tumor development and progression. Cancer Res. 68, 8779–8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Datta S. R., Brunet A., Greenberg M. E. (1999) Cellular survival: a play in three Akts. Genes Dev. 13, 2905–2927 [DOI] [PubMed] [Google Scholar]
- 25. Dans M., Gagnoux-Palacios L., Blaikie P., Klein S., Mariotti A., Giancotti F. G. (2001) Tyrosine phosphorylation of the β4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J. Biol. Chem. 276, 1494–1502 [DOI] [PubMed] [Google Scholar]
- 26. Shaw L. M. (2001) Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the α6β4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell. Biol. 21, 5082–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang X., Dutta U., Shaw L. M. (2010) SHP2 mediates the localized activation of Fyn downstream of the α6β4 integrin to promote carcinoma invasion. Mol. Cell. Biol. 30, 5306–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu S., Simin K., Khan A., Mercurio A. M. (2008) Analysis of integrin β4 expression in human breast cancer: association with basal-like tumors and prognostic significance. Clin. Cancer Res. 14, 1050–1058 [DOI] [PubMed] [Google Scholar]
- 29. Amin D. N., Hida K., Bielenberg D. R., Klagsbrun M. (2006) Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 66, 2173–2180 [DOI] [PubMed] [Google Scholar]
- 30. Bruns C. J., Solorzano C. C., Harbison M. T., Ozawa S., Tsan R., Fan D., Abbruzzese J., Traxler P., Buchdunger E., Radinsky R., Fidler I. J. (2000) Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 60, 2926–2935 [PubMed] [Google Scholar]
- 31. Kedar D., Baker C. H., Killion J. J., Dinney C. P., Fidler I. J. (2002) Blockade of the epidermal growth factor receptor signaling inhibits angiogenesis leading to regression of human renal cell carcinoma growing orthotopically in nude mice. Clin. Cancer Res. 8, 3592–3600 [PubMed] [Google Scholar]
- 32. Nikolopoulos S. N., Blaikie P., Yoshioka T., Guo W., Giancotti F. G. (2004) Integrin β4 signaling promotes tumor angiogenesis. Cancer Cell 6, 471–483 [DOI] [PubMed] [Google Scholar]
- 33. Choi S., Lee E., Kwon S., Park H., Yi J. Y., Kim S., Han I. O., Yun Y., Oh E. S. (2005) Transmembrane domain-induced oligomerization is crucial for the functions of syndecan-2 and syndecan-4. J. Biol. Chem. 280, 42573–42579 [DOI] [PubMed] [Google Scholar]
- 34. Utani A., Nomizu M., Matsuura H., Kato K., Kobayashi T., Takeda U., Aota S., Nielsen P. K., Shinkai H. (2001) A unique sequence of the laminin α3 G domain binds to heparin and promotes cell adhesion through syndecan-2 and -4. J. Biol. Chem. 276, 28779–28788 [DOI] [PubMed] [Google Scholar]
- 35. Okamoto O., Bachy S., Odenthal U., Bernaud J., Rigal D., Lortat-Jacob H., Smyth N., Rousselle P. (2003) Normal human keratinocytes bind to the α3LG4/5 domain of unprocessed laminin-5 through the receptor syndecan-1. J. Biol. Chem. 278, 44168–44177 [DOI] [PubMed] [Google Scholar]
- 36. Goldfinger L. E., Hopkinson S. B., deHart G. W., Collawn S., Couchman J. R., Jones J. C. (1999) The α3 laminin subunit, α6β4 and α3β1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J. Cell Sci. 112, 2615–2629 [DOI] [PubMed] [Google Scholar]
- 37. Goldfinger L. E., Stack M. S., Jones J. C. (1998) Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J. Cell Biol. 141, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elenius K., Vainio S., Laato M., Salmivirta M., Thesleff I., Jalkanen M. (1991) Induced expression of syndecan in healing wounds. J. Cell Biol. 114, 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bachy S., Letourneur F., Rousselle P. (2008) Syndecan-1 interaction with the LG4/5 domain in laminin-332 is essential for keratinocyte migration. J. Cell. Physiol. 214, 238–249 [DOI] [PubMed] [Google Scholar]
- 40. Carulli S., Beck K., Dayan G., Boulesteix S., Lortat-Jacob H., Rousselle P. (2012) Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin α3 LG45 protein domain. J. Biol. Chem. 287, 12204–12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ogawa T., Tsubota Y., Hashimoto J., Kariya Y., Miyazaki K. (2007) The short arm of laminin γ2 chain of laminin-5 (laminin-332) binds syndecan-1 and regulates cellular adhesion and migration by suppressing phosphorylation of integrin β4 chain. Mol. Biol. Cell 18, 1621–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marinkovich M. P., Lunstrum G. P., Burgeson R. E. (1992) The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J. Biol. Chem. 267, 17900–17906 [PubMed] [Google Scholar]
- 43. Wang H., Leavitt L., Ramaswamy R., Rapraeger A. C. (2010) Interaction of syndecan and α6β4 integrin cytoplasmic domains: Regulation of ErbB2-mediated integrin activation. J. Biol. Chem. 285, 13569–13579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sehgal B. U., DeBiase P. J., Matzno S., Chew T. L., Claiborne J. N., Hopkinson S. B., Russell A., Marinkovich M. P., Jones J. C. (2006) Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J. Biol. Chem. 281, 35487–35498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Russell A. J., Fincher E. F., Millman L., Smith R., Vela V., Waterman E. A., Dey C. N., Guide S., Weaver V. M., Marinkovich M. P. (2003) α6 β 4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of α3β1 integrin. J. Cell Sci. 116, 3543–3556 [DOI] [PubMed] [Google Scholar]
- 46. Jourquin J., Yang N., Kam Y., Guess C., Quaranta V. (2006) Dispersal of epithelial cancer cell colonies by lysophosphatidic acid (LPA). J. Cell. Physiol. 206, 337–346 [DOI] [PubMed] [Google Scholar]
- 47. O'Connor K. L., Chen M., Towers L. N. (2012) Integrin α6β4 cooperates with LPA signaling to stimulate Rac through AKAP-Lbc-mediated RhoA activation. Am. J. Physiol. Cell Physiol. 302, C605–C614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Connor K. L., Shaw L. M., Mercurio A. M. (1998) Release of cAMP gating by the α6β4 integrin stimulates lamellae formation and the chemotactic migration of invasive carcinoma cells. J. Cell Biol. 143, 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geerts D., Fontao L., Nievers M. G., Schaapveld R. Q., Purkis P. E., Wheeler G. N., Lane E. B., Leigh I. M., Sonnenberg A. (1999) Binding of integrin α6β4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 147, 417–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rousselle P., Aumailley M. (1994) Kalinin is more efficient than laminin in promoting adhesion of primary keratinocytes and some other epithelial cells and has a different requirement for integrin receptors. J. Cell Biol. 125, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weaver V. M., Petersen O. W., Wang F., Larabell C. A., Briand P., Damsky C., Bissell M. J. (1997) Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 137, 231–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Egles C., Huet H. A., Dogan F., Cho S., Dong S., Smith A., Knight E. B., McLachlan K. R., Garlick J. A. (2010) Integrin-blocking antibodies delay keratinocyte re-epithelialization in a human three-dimensional wound healing model. PLoS One 5, e10528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Byron A., Humphries J. D., Askari J. A., Craig S. E., Mould A. P., Humphries M. J. (2009) Anti-integrin monoclonal antibodies. J. Cell Sci. 122, 4009–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grootjans J. J., Zimmermann P., Reekmans G., Smets A., Degeest G., Dürr J., David G. (1997) Syntenin, a PDZ protein that binds syndecan cytoplasmic domains. Proc. Natl. Acad. Sci. U.S.A. 94, 13683–13688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sulka B., Lortat-Jacob H., Terreux R., Letourneur F., Rousselle P. (2009) Tyrosine dephosphorylation of the syndecan-1 PDZ binding domain regulates syntenin-1 recruitment. J. Biol. Chem. 284, 10659–10671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saito Y., Imazeki H., Miura S., Yoshimura T., Okutsu H., Harada Y., Ohwaki T., Nagao O., Kamiya S., Hayashi R., Kodama H., Handa H., Yoshida T., Fukai F. (2007) A peptide derived from tenascin-C induces β1 integrin activation through syndecan-4. J. Biol. Chem. 282, 34929–34937 [DOI] [PubMed] [Google Scholar]
- 57. Shepherd T. R., Klaus S. M., Liu X., Ramaswamy S., DeMali K. A., Fuentes E. J. (2010) The Tiam1 PDZ domain couples to syndecan1 and promotes cell-matrix adhesion. J. Mol. Biol. 398, 730–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Folgiero V., Avetrani P., Bon G., Di Carlo S. E., Fabi A., Nisticò C., Vici P., Melucci E., Buglioni S., Perracchio L., Sperduti I., Rosanò L., Sacchi A., Mottolese M., Falcioni R. (2008) Induction of ErbB-3 expression by α6β4 integrin contributes to tamoxifen resistance in ERβ1-negative breast carcinomas. PLoS One 3, e1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Folgiero V., Bachelder R. E., Bon G., Sacchi A., Falcioni R., Mercurio A. M. (2007) The α6β4 integrin can regulate ErbB-3 expression: implications for α6β4 signaling and function. Cancer Res. 67, 1645–1652 [DOI] [PubMed] [Google Scholar]
- 60. Borradori L., Sonnenberg A. (1999) Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol. 112, 411–418 [DOI] [PubMed] [Google Scholar]
- 61. Orian-Rousseau V., Aberdam D., Fontao L., Chevalier L., Meneguzzi G., Kedinger M., Simon-Assmann P. (1996) Developmental expression of laminin-5 and HD1 in the intestine: epithelial to mesenchymal shift for the laminin γ-2 chain subunit deposition. Dev. Dyn. 206, 12–23 [DOI] [PubMed] [Google Scholar]
- 62. Niessen C. M., Hulsman E. H., Rots E. S., Sánchez-Aparicio P., Sonnenberg A. (1997) Integrin α6β4 forms a complex with the cytoskeletal protein HD1 and induces its redistribution in transfected COS-7 cells. Mol. Biol. Cell 8, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. de Pereda J. M., Lillo M. P., Sonnenberg A. (2009) Structural basis of the interaction between integrin α6β4 and plectin at the hemidesmosomes. EMBO J. 28, 1180–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Koster J., van Wilpe S., Kuikman I., Litjens S. H., Sonnenberg A. (2004) Role of binding of plectin to the integrin β4 subunit in the assembly of hemidesmosomes. Mol. Biol. Cell 15, 1211–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rezniczek G. A., de Pereda J. M., Reipert S., Wiche G. (1998) Linking integrin α6β4-based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the β4 subunit and plectin at multiple molecular sites. J. Cell Biol. 141, 209–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Frijns E., Kuikman I., Litjens S., Raspe M., Jalink K., Ports M., Wilhelmsen K., Sonnenberg A. (2012) Phosphorylation of threonine 1736 in the C-terminal tail of integrin β4 contributes to hemidesmosome disassembly. Mol. Biol. Cell 23, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Frijns E., Sachs N., Kreft M., Wilhelmsen K., Sonnenberg A. (2010) EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin β4. J. Biol. Chem. 285, 37650–37662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Germain E. C., Santos T. M., Rabinovitz I. (2009) Phosphorylation of a novel site on the β4 integrin at the trailing edge of migrating cells promotes hemidesmosome disassembly. Mol. Biol. Cell 20, 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu X., Shepherd T. R., Murray A. M., Xu Z., Fuentes E. J. (2013) The structure of the Tiam1 PDZ domain/phospho-syndecan1 complex reveals a ligand conformation that modulates protein dynamics. Structure 21, 342–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zimmermann P., Tomatis D., Rosas M., Grootjans J., Leenaerts I., Degeest G., Reekmans G., Coomans C., David G. (2001) Characterization of syntenin, a syndecan-binding PDZ protein, as a component of cell adhesion sites and microfilaments. Mol. Biol. Cell 12, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee D., Oh E. S., Woods A., Couchman J. R., Lee W. (1998) Solution structure of a syndecan-4 cytoplasmic domain and its interaction with phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 273, 13022–13029 [DOI] [PubMed] [Google Scholar]
- 72. Woods A., Couchman J. R. (1998) Syndecans: synergistic activators of cell adhesion. Trends Cell Biol. 8, 189–192 [DOI] [PubMed] [Google Scholar]
- 73. Woods A., Longley R. L., Tumova S., Couchman J. R. (2000) Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch. Biochem. Biophys. 374, 66–72 [DOI] [PubMed] [Google Scholar]
- 74. Araki E., Momota Y., Togo T., Tanioka M., Hozumi K., Nomizu M., Miyachi Y., Utani A. (2009) Clustering of syndecan-4 and integrin β1 by laminin α3 chain-derived peptide promotes keratinocyte migration. Mol. Biol. Cell 20, 3012–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Oh E. S., Woods A., Couchman J. R. (1997) Multimerization of the cytoplasmic domain of syndecan-4 is required for its ability to activate protein kinase C. J. Biol. Chem. 272, 11805–11811 [DOI] [PubMed] [Google Scholar]
- 76. Yoshioka T., Otero J., Chen Y., Kim Y. M., Koutcher J. A., Satagopan J., Reuter V., Carver B., de Stanchina E., Enomoto K., Greenberg N. M., Scardino P. T., Scher H. I., Sawyers C. L., Giancotti F. G. (2013) β4 Integrin signaling induces expansion of prostate tumor progenitors. J. Clin. Invest. 123, 682–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yamashita H., Tripathi M., Jourquin J., Kam Y., Liu S., Weidow B., Quaranta V. (2010) Lysophosphatidic acid upregulates laminin-332 expression during A431 cell colony dispersal. J. Oncol. 2010, 10.1155/2010/107075 [DOI] [PMC free article] [PubMed] [Google Scholar]