

Background: Stop codon frequencies in 4684 bacterial genomes are analyzed.
Results: With increasing genomic GC content, TAA% decreases, and TGA% increases reciprocally, but TAG% remains almost unchanged (∼20%). The TAG:TGA ratio matches well with the RF1:RF2 ratio.
Conclusion: TAG is the minor stop codon, and expression of genes with TAG is correlated with the RF1 level.
Significance: Our work establishes the correlation between stop codon frequency and RF1/RF2 abundance.
Keywords: Bacteria, Protein Synthesis, Ribosome, Translation, Translation Release Factor, Codon Usage, Stop Codon
Abstract
We present a comprehensive analysis of stop codon usage in bacteria by analyzing over eight million coding sequences of 4684 bacterial sequences. Using a newly developed program called “stop codon counter,” the frequencies of the three classical stop codons TAA, TAG, and TGA were analyzed, and a publicly available stop codon database was built. Our analysis shows that with increasing genomic GC content the frequency of the TAA codon decreases and that of the TGA codon increases in a reciprocal manner. Interestingly, the release factor 1-specific codon TAG maintains a more or less uniform frequency (∼20%) irrespective of the GC content. The low abundance of TAG is also valid with respect to expression level of the genes ending with different stop codons. In contrast, the highly expressed genes predominantly end with TAA, ensuring termination with either of the two release factors. Using three model bacteria with different stop codon usage (Escherichia coli, Mycobacterium smegmatis, and Bacillus subtilis), we show that the frequency of TAG and TGA codons correlates well with the relative steady state amount of mRNA and protein for release factors RF1 and RF2 during exponential growth. Furthermore, using available microarray data for gene expression, we show that in both fast growing and contrasting biofilm formation conditions, the relative level of RF1 is nicely correlated with the expression level of the genes ending with TAG.
Introduction
Many bacteria and eukaryotic organisms show bias in their usage of synonymous codons. For the 61 sense codons, this phenomenon has been studied in depth (1, 2). Sense codon frequency is believed to correlate well with tRNA abundance (2, 3) and thus might influence the rate and accuracy of protein synthesis as well as folding of the individual domains (4–6). In the standard bacterial codon table, there are three stop codons, TAG, TGA, and TAA (UAG, UGA, and UAA on mRNA), which are recognized by two class I release factors, RF13 and RF2. The release factors initiate termination of translation by binding to the stop codons at the decoding center and releasing the nascent peptide from the peptidyl-tRNA at the peptidyltransferase center. The stop codons are not perfectly synonymous as UAG is recognized solely by RF1, UGA is recognized solely by RF2, and UAA is recognized by both factors (7). However, the existence of three stop codons raises the question of whether or not there is bias in their usage.
There are not many studies on stop codon usage in bacteria to date. In an earlier study, Sharp and Bulmer (8) analyzed the stop codon bias in Escherichia coli, Bacillus subtilis, and Saccharomyces cerevisiae. In the few coding sequences available at that time, TAA was observed to be the most abundant stop codon. The authors concluded that this strong bias toward TAA might be due to the fact that it can be recognized by both release factors, which was suggested in other related studies as well (9, 10). In a later work, Sun et al. (11) analyzed more than 70,000 genes from eukaryotes including fungi, plant, and human. They found TAA to be the most abundant stop codon in lower eukaryotes, whereas in higher eukaryotes, TGA was the most abundant. In a more detailed investigation, stop codon determinants in six prokaryotic and five eukaryotic genomes were studied with the same conclusions (12). In other earlier works, the context of surrounding nucleotides, especially those immediately after the stop codon, in translation termination has been discussed (10, 13, 14). Also, tools such as TRANSTERM were prepared to investigate the up- and downstream regions of stop codons in a given species (9). This tool also allowed clarification of the relevance of tandem stop codons in E. coli (15). There is only one example to date where a large scale analysis of stop codon usage in bacteria (736 species) has been performed (16). This work clearly demonstrated the pattern of variation of the stop codons with genomic GC content and reached the conclusion that the bacterial stop codons are not selectively equivalent. The authors, however, did not make the detailed data for stop codon distribution in a particular species available to the general readers. Also, although they correctly identified TAG as the least frequent stop codon, no biochemical experiments were performed to elucidate the molecular mechanisms behind negative selection on TAG stop codon.
Here we present a complementary large scale analysis of stop codon usage in bacteria involving over 8.5 million coding sequences from 4684 bacterial genome sequences. We intentionally limited our analysis to bacteria and did not explore the eukaryotic cellular organelles of bacterial origin because the assigned stop codons for the organelles (e.g. mitochondria) are not fully established (17). We constructed a publicly available stop codon usage database for common bacterial genomic sequences where information regarding a specific bacterium can be easily extracted from an alphabetically arranged list. Our results, reconfirming earlier reports (16, 18), demonstrate strong bias in stop codon usage in different bacteria and show that the distribution of TGA and TAA, but not of TAG, is likely driven by genomic GC content. Next, we analyzed the region immediately downstream of the stop codon for all genes in E. coli, B. subtilis, and Mycobacterium smegmatis for additional stop codons.
Because in bacteria three stop codons are read by two release factors in a semispecific manner, we asked the additional question whether there exists a correlation between the usage of the stop codons and the abundance of RF1 and RF2. Previous results showed that in exponentially growing E. coli RF2 is about 5 times more abundant than RF1 (19, 20). In the present study, we analyzed three model bacteria, E. coli, M. smegmatis, and B. subtilis, for RF1 and RF2 abundance using qPCR and Western blotting. Furthermore, we analyzed the gene types associated with the three stop codons and investigated stop codon usage in E. coli based on available gene expression microarray data. We observed that the level of expression of the genes ending with TAG correlates very well with the level of RF1 in different physiological conditions.
EXPERIMENTAL PROCEDURES
Sequences
The gene sequences used in this work were obtained from the National Center for Biotechnology Information (NCBI) database. In total, 4684 genomes (including bacterial plasmids) were analyzed and gathered in a database (which can be downloaded upon request). Throughout the text, TAA, TAG, and TGA are used as stop codons irrespective of the DNA or mRNA context to simplify the discussion.
The “Stop Codon Counter”
The stop codon counter is a custom program written in the Java programming language. Given a file with coding sequences in the FASTA format, the program determines how often a set of specified codons (called “criteria”) appear as the last codon in a gene. The three canonical stop codon sequences (TAA, TAG, and TGA) are set as the default to be counted. The set of counted codons can be extended by using the “add criteria” function in the program. Once the files containing the sequences are loaded into the program and analyzed, the stop codon counter generates a separate output file for each input file where the genes are categorized by their stop codons. Additionally, the frequency and absolute count of each codon are recorded. The program and the database are freely available upon request.
Determination of Additional Stop Codons
To identify additional stop codons after a primary stop codon, we scanned all 3′-untranslated regions (UTRs) in the chromosome of E. coli K12, B. subtilis (substrain 168), and M. smegmatis (MC2 155). The UTRs were extracted using the powerful web-based tool TRANSTERM (9). A shell script was applied to search for the stop codons in the 3′-UTRs. The frequency of occurrence was estimated by dividing the number of genes with additional stop codons by the total number of genes.
Gene Expression Analysis
The expression levels from aerobically growing (21) or under biofilm-forming (22) E. coli cells were obtained from microarray data and analyzed with GENEVESTIGATOR (23). The genes were categorized according to their affiliation of biological processes using the PANTHER database (24).
Absolute Quantitative Real Time PCR
Log phase cultures of E. coli BL21(DE3), M. smegmatis MC2 155, and B. subtilis 168 were harvested, and growth was stopped by adding of the starting volume of stop solution (5% water saturated phenol and 95% ethanol). The cells were pelleted by centrifugation, suspended in lysis buffer (100 mm Tris-HCl, 40 mm EDTA, 200 mm NaCl, and 0.5% sodium dodecyl sulfate), and incubated for 5 min at 65 °C. The cell walls of M. smegmatis and B. subtilis cells were disrupted by vortexing with 0.1-mm-diameter glass beads prior to incubation at 65 °C. Total RNA was extracted using a standard phenol extraction procedure (25). Residual DNA was removed by addition of 1 unit of DNase I (Fermentas) twice at 37 °C at an interval of 30 min. cDNA synthesis was performed using MultiScribe reverse transcriptase (Invitrogen) following the protocol supplied by the manufacturer. To estimate the in vivo concentration of prfA and prfB transcripts, an absolute quantitative PCR was performed using the ABI 7300 cycler (Applied Biosystems). Primers specific to different bacteria were ordered from Invitrogen (Table 1). HOT FIREPol® EvaGreen® qPCR Mix Plus (includes ROX as a passive reference) from Solis BioDyne was used. qPCR cycles were set as follows: initial denaturation at 95 °C for 15 min, denaturation at 95 °C for 15 s, annealing at 63 °C for 20 s, and elongation at 72 °C for 40 s. In total, 40 cycles were performed where the signal was detected during elongation.
TABLE 1.
Primers used for qPCR
| Target | 5′ to 3′ sequence |
|---|---|
| prfA E. coli | GCTTGTACCGTTGCGGTAAT |
| CCTGACGAGCGGAAAGTATC | |
| prfB E. coli | CCGTATGTTCTCTGGCGAAT |
| GCGCAGATACATACGCTCAA | |
| prfA M. smegmatis | ACGAGGTCGAAGAACTCACC |
| GACTTCACCTCCAGCACGAT | |
| prfB M. smegmatis | ATCCTGACCGTCAAGCAGAC |
| GCGTCCTTCTCGAGTTGTTC | |
| prfA B. subtilis | GCCGACAGGTGTTGTTGTAT |
| CATAGATTCTGGCACGGAGAAC | |
| prfB B. subtilis | GCCGTTCGGATTACTCACTT |
| CTGATACAGCTTGGCCTTCA |
Quantitative Western Blot
To estimate the in vivo protein levels of the class I release factors, quantitative Western blots were performed using rabbit polyclonal antibodies against RF1 and RF2 (kind gifts from Richard Buckingham, France). Cell lysate was prepared from E. coli BL21(DE3) and harvested at an A600 of 0.1, and a total of 1 A600 unit was applied to a 12% SDS-PAGE. Known amounts of purified RF1/RF2 (0.05–1 pmol) were loaded in the same gel for making a standard curve. The proteins were transferred to the nitrocellulose membrane with a semidry method (Bio-Rad Semi-Dry Trans-Blot Cell) using 25 mm Tris, 192 mm glycine, and 20% methanol for 50 min at 18 V. The membrane was blocked with 5% nonfat milk (in wash buffer containing 100 mm Tris, pH 7.4, 100 mm NaCl, and 0.1% Tween 20). The primary antibody against RF1 or RF2 was delivered in the same 5% milk solution at a 1:10,000 dilution and incubated for 40 min at 37 °C. The membrane was washed in washing buffer for 5 min and incubated with secondary antibody (ECL anti-rabbit IgG, horseradish peroxidase-linked whole antibody, GE Healthcare). The bound antibodies were detected using Western Bright Sirius (Advansta) horseradish substrate and the ChemiDoc MP System (Bio-Rad). Band intensities were quantified by the UN-SCAN-IT gelTM Version 6.1 (Silk Scientific, Orem, UT).
RESULTS
Distribution of Stop Codons within and across Different Bacterial Phyla
We analyzed stop codon frequencies in 4684 bacterial genomes involving all annotated ORFs according to the NCBI database using the stop codon counter. The results are sorted and grouped according to bacterial phyla (for a selection of around 600 bacterial species). Because the program analyzed DNA sequences corresponding to the stop codons, the results are presented as TAA, TAG, and TGA sequences. The distribution of stop codons within and across phyla varies dramatically (Fig. 1). Although in Actinobacteria, TGA is the predominant stop codon (Fig. 1A), in Firmicutes and Tenericutes (Fig. 1, B and C), TAA is the major stop codon. In the phyla Cyanobacteria and Proteobacteria, either TAA or TGA appears as the most frequent stop codon depending on the genomic GC content (Fig. 1, D and E). However, in all these phyla, TAG is the minor stop codon. One interesting exception from this feature was found in Mollicutes, which represents a small class of Proteobacteria with the smallest genome size (26). In this class of bacteria, the frequency of RF2-specific stop codon TGA was found to be close to zero (Fig. 1F). This unique feature and its consequences are discussed later.
FIGURE 1.

Stop codon distribution in different bacterial phyla. The frequencies of three stop codons TAA (green), TAG (black), and TGA (red) in different bacteria classified and grouped in different phyla (A–E) and within the class of Mollicutes (F) are plotted as a function of genomic GC content.
Variations in Stop Codon Usage with Genomic GC Content
It has been suggested previously that genomic GC content is one of the primary driving forces behind codon usage bias (16, 18). To generate a global view on stop codon usage in bacteria, we plotted our results as a function of genomic GC content. The lowest GC content in our data set, 13.9%, belonged to Candidatus Carsonella ruddii CE isolate Thao2000 in the phylum Proteobacteria. In contrast, Anaeromyxobacter dehalogenans 2CP-C within the phylum Proteobacteria showed the highest GC content (75%). In line with the previous reports (16, 18), the frequency of the TAA codon decreased with an increase in GC content, and that of the TGA codon increased in a reciprocal manner (Fig. 2A). Interestingly, the frequency of TAG codon, despite the same G content as the TGA, remained more or less constant (∼20%) irrespective of the GC content (Fig. 2A). Thus, our results confirmed the pattern of stop codon distribution in bacteria and called for further analysis for having TAG as the minor stop codon.
FIGURE 2.
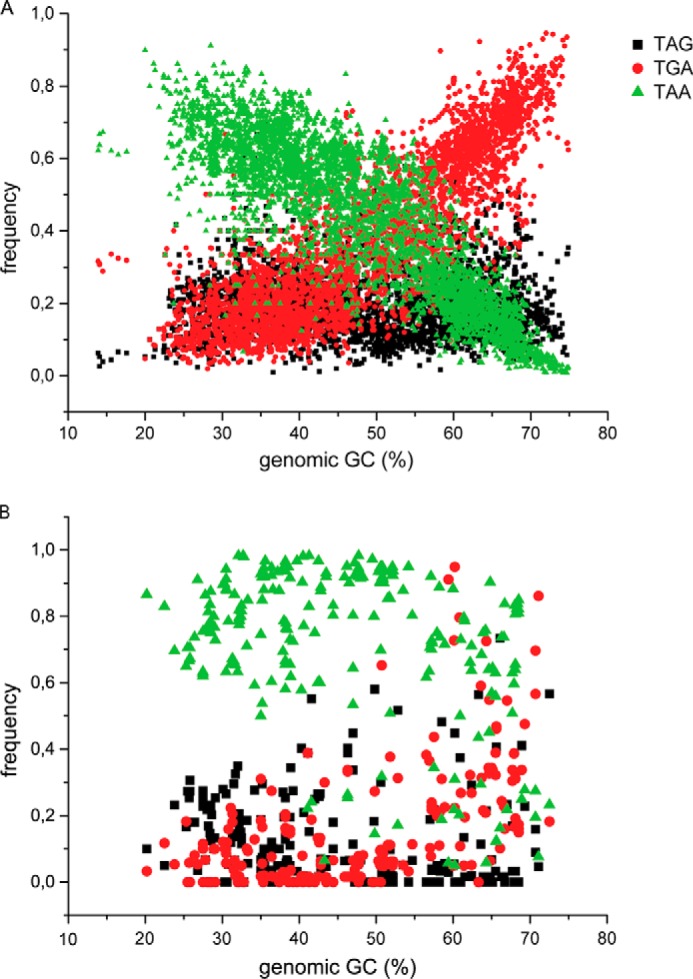
Distribution of the three canonical stop codons in bacteria. A, the frequency of the three stop codons estimated using all ORFs in 4684 bacterial sequences (chromosomes and plasmids) plotted as a function of genomic GC content. B, distribution of stop codon frequency in highly expressed genes (comprising ribosomal protein and translation factor genes) of 220 randomly selected bacteria.
Stop Codon Usage in Highly Expressed Genes
We further analyzed stop codon usage only in the highly expressed genes. For that, we extracted the coding sequences of all ribosomal proteins and some of the translation factors (IF1, IF2, IF3, EF-Tu, EF-Ts and EF-G) from 240 bacterial genomes from our original data set and analyzed them with the stop codon counter. We looked specifically at these translation-related genes as these are usually expressed at a high level during rapid growth. Interestingly, we observed a totally different distribution of the stop codons in these genes. The universal stop codon TAA was found by far to be the most preferred stop codon, and the effect of genomic GC content was much less pronounced (Fig. 2, compare B with A). At very high GC content (60–75%), some genes were found with TGA. Also, a few exceptional cases where TAG was used were also seen. Overall, our analysis showed a clear bias for TAA in the highly expressed genes.
Occurrence of Additional Stop Codons
The occurrence of additional stop codons in the downstream region from the end of the gene is often viewed as a “fail-safe mechanism” to ensure termination. It has been reported that in E. coli ∼7% of all genes possess an additional stop codon where a second stop codon is present immediately downstream of the primary stop codon (15). Because TAG is the minor stop codon, we wanted to check whether there is any bias for an additional stop codon for the genes ending with TAG. For that, we analyzed the next five in-frame codons following the primary stop codon in all genes from three different bacteria, E. coli K12, B. subtilis (substrain 168), and M. smegmatis (MC2 155). In agreement with earlier studies (15), we observed that about 8% of all genes in E. coli have a downstream stop codon (Fig. 3A). However, we observed no preference for any of the three stop codons for occurrence of additional stop codons in E. coli (Fig. 3, A–C). Alternatively, in B. subtilis and M. smegmatis, the lowest occurrence of additional downstream stop codons was found with TAG as the primary stop codon. It is also notable that the appearance of TAG as an additional stop codon is lower than the appearance of TGA and TAA in almost all cases (Fig. 3, A–C). Thus, we infer that TAG is truly a minor stop codon in all aspects.
FIGURE 3.
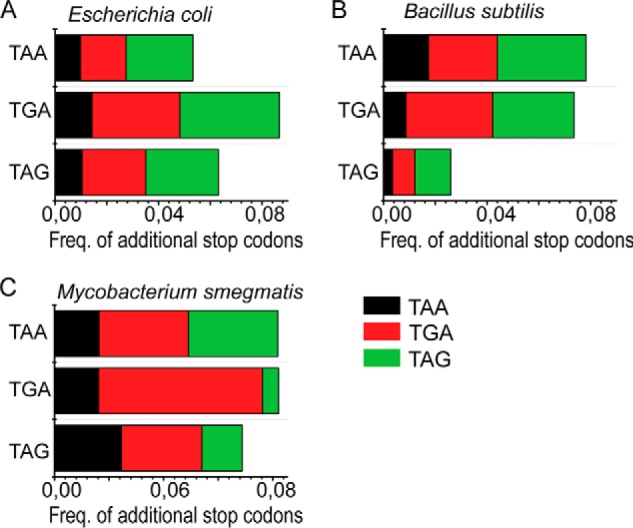
Occurrence of downstream additional stop codons. The occurrence of additional stop codons within 15 nucleotides downstream from the primary stop codons TAA, TAG, and TGA is shown. All ORFs in E. coli (A), B. subtilis (B), and M. smegmatis (C) were analyzed. The horizontal bars represent percentages of the total genes with additional stop codons TAG (black), TGA (red), and TAA (green). Freq., frequency.
Correlation of Release Factor Abundance and Frequency of TAG and TGA Codons
Because sense codon usage and tRNA abundance could be correlated in past studies (2, 3), we asked the same question in relation to stop codon usage and the abundance of the release factors RF1 and RF2. Because TAA could be read by both release factors, the analysis was restricted only to TAG and TGA codons. To follow the relative expression of the prfA and prfB genes, absolute real time qPCR was performed on total RNA extracted from exponentially growing cells. E. coli, M. smegmatis, and B. subtilis were chosen as model organisms as they show different relative frequencies of the TAG and TGA codons. In parallel, quantitative Western blotting was performed to estimate the relative amount of RF1 and RF2 proteins in E. coli. For all three systems, the prfB to prfA transcript ratio matched strikingly well with the relative frequency of TGA and TAG (Fig. 4A, compare white bars with gray bars). The prfB transcript was estimated to be 4 times in excess over prfA in E. coli, 3 times in excess in M. smegmatis, and 1.5 times in excess in B. subtilis; all of these values are highly similar to the relative frequency of TGA and TAG codons. Thus, a very good correlation was found between the frequency of TAG and TGA codons and the expression of the release factors specific to these codons. Analysis at the protein level showed RF2 expression in E. coli to be 6–7-fold higher than RF1 expression (Fig. 4A, red bars), which is in good agreement with previous studies (19, 20). Thus, the RF2:RF1 ratio matched well with the ratio of TGA to TAG.
FIGURE 4.

Expression analysis and correlation of release factors and stop codon frequency. A, correlation of the stop codon frequency (TGA/TAG) with mRNA transcripts determined with qPCR (prfB/prfA) and the amount of the release factor proteins (RF2/RF1) estimated by quantitative Western blotting using antibodies against E. coli RF1 and RF2. B, Western blotting-based determination of the amount of RFs in E. coli growing exponentially (E) and in stationary (S) phase in rich (LB) and minimal (M9) media. The bars represent the amount of the release factors in pmol estimated from the blots (presented above) done in quadruplicates. C, E. coli genes ending with TAG/TGA/TAA are grouped, and the mean expression level under fast growing condition is plotted according to the linear expression value of the microarray. TAA w/o highly are the genes ending with TAA without the highly expressed genes (yellow). D, same as C but showing expression data under biofilm-forming conditions. E, classification of the genes ending with TAG/TGA/TAA according to the biological functions.
Furthermore, we extended our analysis to quantify the amount of release factors in E. coli in different growth conditions. For that, quantitative Western blotting (Fig. 4B) was performed using equal amount of cells from the exponential and stationary phases grown in rich LB and minimal M9 media. Our result shows that RF2 is always expressed in excess over RF1 irrespective of growth condition and media. In rich LB medium, both RF1 and RF2 are expressed in higher amounts in the exponential phase compared with the stationary phase. In minimal M9 medium, this difference is less pronounced, especially for RF1. However, the ratio of RF2 to RF1 remains more or less unchanged (RF2 in 5–7 times excess over RF1) in all conditions, suggesting that RF2 is the major release factor in E. coli (Fig. 4B).
One would likely expect that the genes terminating with TAG and thereby limited by termination with only RF1 would be expressed at a low level. Indeed, when we looked into the expression level of the genes in fast growing E. coli, those terminating at TAG were found to be expressed at the lowest level compared with all others (Fig. 4C). This inference remained valid even after removal of the expression data of the highly expressed genes (a total of 62 genes) ending with TAA (Fig. 4C, TAA w/o highly). Only under biofilm conditions was the expression level of the genes ending at TAG, TGA, and TAA found to be very much equal (Fig. 4D). Interestingly, under that condition, the ratio of RF1 to RF2 in E. coli also changes to ∼1:1 as analyzed by GENEVESTIGATOR (23) by comparing microarray data from aerobically growing (21) versus biofilm-forming E. coli (22).Thus, our analysis clearly suggests a strong correlation between actual TAG and TGA frequency and the expression level of RF1 and RF2.
We further sorted the genes ending with TAG, TGA, and TAA according to their biological functions such as the genes involved in metabolic processes, biological regulation, cellular processes, localization, response to stimulus, cellular component organization, or biogenesis. The aim was to check whether a particular gene type is enriched with a particular stop codon. As shown in Fig. 4E, we did not observe clustering of TAG/TGA/TAA to any specific class of genes. In fact, the relative distribution of various classes of genes is more or less the same for all three stop codons. This implies that there is no bias for stop codon usage in the genes associated with a particular type of biological function.
DISCUSSION
By analyzing more than 8.5 million coding sequences, we have shown that stop codon usage in bacteria varies with genomic GC content in the case of the codons TAA and TGA in a reciprocal manner; however, the RF1-specific codon TAG remains more or less constant irrespective of the genomic GC content (Fig. 2A). This result fits well with the theoretical model where a bias has been applied on the TAG codon (16). Thus, our results confirm the earlier observation (16) (using only around 700 sequences) that in bacteria the usage of three stop codons is not selectively equivalent. Also, analysis of additional stop codons at the immediate downstream stop codon did not show preference for any of the three stop codons in those three species analyzed (Fig. 3).
Our analysis of the highly expressed genes shows that TAA is undoubtedly the major stop codon. The predominant occurrence of the major codon in the highly expressed genes is quite common for the sense codons (5). Thus, it is not surprising that the stop codon abundance follows the same pattern. Indeed, having TAA as the stop codon offers significant advantage over TAG or TGA as put forward in an earlier hypothesis (8–10). Most importantly, TAA can be read by both release factors and with efficiency comparable with the release factor-specific codons (13). Thus, having TAA in the highly expressed genes not only secures termination by either of the two release factors but also ensures that the termination at TAA takes place with high speed and accuracy.
Although it can be explained fairly well why TAA is the major stop codon, it remains somewhat unclear why TAG is the minor stop codon. It is interesting to note that TAG is also significantly underrepresented as the tandem stop codon when TAA and TGA are the primary stop codons (15) (Fig. 3). Earlier, the low abundance of TAG was speculated to correlate with lower efficiency of termination with RF1 (14, 19, 20). When compared for kcat/Km, which is often correlated with efficiency in an enzymatic reaction, the value reported for RF1 is 6.0 × 107 m−1 s−1, and that for RF2(Chromosomal) is 4.6 × 107 m−1 s−1, which apparently implies that RF1 has about 1.5 times higher efficiency than RF2 (27). However, the cellular concentration of RF1 and RF2 is in the μm range (19), which is significantly higher than the Km values for the cognate codons for both release factors (8.3 nm for RF1 and 66 nm for RF2 (27)). Thus, the termination efficiency in vivo is likely determined by the kcat value, which is 6 times higher for RF2(Chromosomal) (3.0 s−1) than RF1 (0.5 s−1) (27).Thus, under cellular conditions, RF1-mediated termination is certainly less efficient than that with RF2, which might be a reason why TAG evolved as a less favored stop codon.
Another explanation for TAG being the minor codon can be brought forward from the view point of affinity and recycling of the two release factors. It has been reported that the efficiency of termination by RF1 and RF2 is strongly correlated with the affinity of the release factor for the post-termination complex after hydrolysis of the peptidyl-tRNA (13). It was demonstrated that the affinity of RF1 for the post-termination complex is much higher than that of RF2. This would immediately suggest that RF1 is more difficult to recycle than RF2, which is also evident from the measurements of the spontaneous recycling time of the two release factors under the same experimental conditions (∼60 s for RF1 and 11 s for RF2) (28). In line with this argument, it has also been shown that RF1 is more dependent on RF3 for its recycling than is RF2 (13, 28). The slow dissociation of RF1 after peptide release can have a significant consequence in a polysomic scenario where multiple ribosomes are lined up on a translating mRNA. Furthermore, dissociation of the release factors is also important for turnover of the ribosomes, which is crucial for fast translation and hence for fast growth. Thus, we can infer that inefficient and strictly RF3-dependent recycling of RF1 is another reason that makes TAG a non-preferred stop codon. We suggest that the low TAG frequency can be seen as a mechanism to bypass slow recycling events, which are disadvantageous from a growth perspective.
The underrepresentation of TAG codons can also be related to the higher misreading of TAG in vivo. It was demonstrated that when an essential lysine codon in the firefly luciferase gene was replaced with other codons including the stop codons TAG but not TGA showed significantly high luciferase activity (29). This would only be possible if the protein synthesis did not stop at the TAG codon and was read by an aminoacyl-tRNA (possibly lysine) instead. Thus, TAG was shown to be a “misreading hot spot,” which could be a reason why nature has kept its frequency low.
The bias of TAG being a minor codon is not valid for chloroplasts (16). Recently it was postulated that plastidial as well as mitochondrial release factors underwent a complex evolution of reassignment of stop codons as well as reinvention of stop codons (30). In this work, the ancestor of plastidial release factors was postulated to be of Gloeobacter violaceus and/or Thermosynechococcus elongatus origin. In our database, we found that G. violaceus and T. elongatus also do not follow the bias of using TAG at a low frequency (30 and 44% TAG frequency, respectively). This may imply that not only the plastidial factors but also the frequency of the stop codons has the same origin. We are aware of a few exceptions of bacterial strains that have a high frequency of TAG in their coding sequence (listed in Table 2). Highly expressed genes in these bacteria also end frequently with TAG (see Fig. 2B). Those bacterial genomes may display examples of organisms on the verge of reassigning TGA from a stop to a sense codon (31) to expand their genetic code. This idea is supported by a very recent study showing that no reassignment of TAG is found in bacteria (32), whereas reassignment of TGA (Trp in Mycoplasma) and TAA (Tyr in Planaria) has been reported earlier (33).
TABLE 2.
Bacterial species using high frequencies of TAG
Stop codon usage of species that use unusually often the stop codon TAG is shown. The listed species span a wide range of genomic GC content. Listed are the frequencies for all coding sequences (CDS) and only for highly expressed genes. BCG str., Bacillus Calmette-Guérin strain.
| Strain | All CDS |
Highly expressed |
GC | ||||
|---|---|---|---|---|---|---|---|
| TAG | TGA | TAA | TAG | TGA | TAA | ||
| % | |||||||
| Salinibacter ruber DSM 13855 | 0.473 | 0.362 | 0.165 | 0.734 | 0.102 | 0.163 | 66.1 |
| Anaplasma marginale St. Maries | 0.507 | 0.24 | 0.253 | 0.58 | 0.274 | 0.145 | 49.8 |
| Clavibacter michiganensis subsp. michiganensis | 0.230 | 0.748 | 0.021 | 0.566 | 0.183 | 0.233 | 72.5 |
| Desulfovibrio vulgaris Hildenborough | 0.406 | 0.46 | 0.134 | 0.564 | 0 | 0.435 | 63.3 |
| Anaplasma phagocytophilum HZ | 0.401 | 0.234 | 0.365 | 0.551 | 0.189 | 0.241 | 41.6 |
| Treponema pallidum Nichols | 0.386 | 0.369 | 0.245 | 0.517 | 0.313 | 0.172 | 52.8 |
| Synechococcus sp. JA-2-3B-a(2-13) | 0.41 | 0.389 | 0.201 | 0.482 | 0.31 | 0.189 | 58.5 |
| Acaryochloris marina MBIC11017 | 0.307 | 0.226 | 0.466 | 0.448 | 0.017 | 0.534 | 47 |
| Synechococcus sp. JA-3-3Ab | 0.429 | 0.354 | 0.215 | 0.448 | 0.224 | 0.31 | 60.2 |
| Myxococcus xanthus DK 1622 | 0.249 | 0.686 | 0.064 | 0.411 | 0.338 | 0.25 | 68.9 |
| Mycobacterium bovis BCG str. Pasteur 1173P2 | 0.298 | 0.545 | 0.156 | 0.406 | 0.468 | 0.125 | 65.6 |
Our analysis of stop codon frequency draws special attention to Mollicutes, which are a class of Proteobacteria that show a frequency of TGA stop codons of zero or close to zero (Fig. 1F). This would imply that these bacteria do not contain RF2 or alternatively that RF2 recognizes some other codon. When searched in the literature, we came across a previous report that demonstrated that Mycoplasma, one representative of this class, commonly lacks the gene for release factor 2, prfB (26). Therefore, we checked the occurrence of the prfB gene in this class. It turns out that almost all Mollicutes, except Phytoplasma asteris (commonly called onion yellows), lacks the prfB gene. Thus, for this group of bacteria, the lack of TGA codon can be correlated quite well with the absence of RF2. The small percentage of TGA observed in these genomes occurred mostly in hypothetical genes and in some ribosomal protein genes (Table 3). Interestingly, it was found that TGA codes for tryptophan in these bacteria instead of “stop” (34). Moreover, in all cases, the TGA codon is found to be followed by an immediate downstream TAA or TAG stop codon (Table 3). This shows that the wrong codon annotations can still be found in available databases. It is interesting to note that prfC, the gene for the class II release factor RF3, is also frequently missing in these bacteria (35). However, no correlation of the lack of RF3 and that of RF2 could be seen.
TABLE 3.
Downstream analysis of the genes ending with TGA in Mycoplasma and Ureaplasma sp.
The bacterial strains and the locus tags for the genes in Mycoplasma and Ureaplasma that show a reannotated TGA terminating codon are listed. Interestingly, most of the genes except the hypothetical proteins have either TAA or TAG stop codons immediately following the TGA codon.
| Locus tag | Bacterial strain | Codon following TGA |
|---|---|---|
| MCAP_0107 | Mycoplasma capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0152 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0216 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0246 | M. capricolum subsp. capricolum California kid ATCC 27343 | Hypothetical protein |
| MCAP_0458 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0466 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0486 | M. capricolum subsp. capricolum California kid ATCC 27343 | Hypothetical protein |
| MCAP_0543 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0677 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0682 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAA |
| MCAP_0683 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAG |
| MCAP_0825 | M. capricolum subsp. capricolum California kid ATCC 27343 | TAG |
| MGA_0732 | Mycoplasma gallisepticum strain R | TAA |
| MGA_0733 | M. gallisepticum strain R | TAG |
| MGA_0740 | M. gallisepticum strain R | TAA |
| MGA_0928 | M. gallisepticum strain R | TAA |
| MGA_0323 | M. gallisepticum strain R | Hypothetical protein |
| MGA_0333 | M. gallisepticum strain R | Hypothetical protein |
| MG_007 | Mycoplasma genitalium G-37 | TAG |
| MG_045 | M. genitalium G-37 | Hypothetical protein |
| MG_164 | M. genitalium G-37 | TAA |
| MG_441 | M. genitalium G-37 | Hypothetical protein |
| MYPE190 | Mycoplasma penetrans HF-2 | Hypothetical protein |
| MYPE1240 | M. penetrans HF-2 | Hypothetical protein |
| MYPE1390 | M. penetrans HF-2 | TAA |
| MYPE1820 | M. penetrans HF-2 | TAA |
| MYPE2070 | M. penetrans HF-2 | TAA |
| MYPE3180 | M. penetrans HF-2 | TAG |
| MYPE4880 | M. penetrans HF-2 | Hypothetical protein |
| MYPE4910 | M. penetrans HF-2 | Hypothetical protein |
| MYPE5350 | M. penetrans HF-2 | Hypothetical protein |
| MYPE5400 | M. penetrans HF-2 | Hypothetical protein |
| MYPE5660 | M. penetrans HF-2 | Hypothetical protein |
| MYPE5730 | M. penetrans HF-2 | TAA |
| MYPE8180 | M. penetrans HF-2 | TAA |
| MYPE8700 | M. penetrans HF-2 | TAG |
| MYPE8790 | M. penetrans HF-2 | Hypothetical protein |
| MYPE9780 | M. penetrans HF-2 | Hypothetical protein |
| MYPE10030 | M. penetrans HF-2 | TAG |
| MYPE10040 | M. penetrans HF-2 | TAG |
| MPN007 | Mycoplasma pneumoniae M129 | TAA |
| MPN058 | M. pneumoniae M129 | Hypothetical protein |
| MPN178 | M. pneumoniae M129 | TAA |
| MPN286 | M. pneumoniae M129 | Hypothetical protein |
| MPN40 | M. pneumoniae M129 | TAA |
| MPN458 | M. pneumoniae M129 | Hypothetical protein |
| MPN589 | M. pneumoniae M129 | Hypothetical protein |
| MYPU_0720 | Mycoplasma pulmonis UAB CTIP | Hypothetical protein |
| MYPU_1170 | M. pulmonis UAB CTIP | TAA |
| MYPU_1190 | M. pulmonis UAB CTIP | Hypothetical protein |
| MYPU_1850 | M. pulmonis UAB CTIP | TAA |
| MYPU_2030 | M. pulmonis UAB CTIP | TAA |
| MYPU_2550 | M. pulmonis UAB CTIP | Hypothetical protein |
| MYPU_3390 | M. pulmonis UAB CTIP | TAG |
| MYPU_4290 | M. pulmonis UAB CTIP | TAA |
| MYPU_5680 | M. pulmonis UAB CTIP | TAA |
| MYPU_5730 | M. pulmonis UAB CTIP | TAG |
| MYPU_5740 | M. pulmonis UAB CTIP | TAG |
| MYPU_7200 | M. pulmonis UAB CTIP | TAG |
| UU037 | Ureaplasma urealyticum parvum biovar serovar 3 | Hypothetical protein |
| UU074 | U. urealyticum parvum biovar serovar 3 | TAA |
| UU156 | U. urealyticum parvum biovar serovar 3 | Hypothetical protein |
| UU244 | U. urealyticum parvum biovar serovar 3 | TAG |
| UU245 | U. urealyticum parvum biovar serovar 3 | TAA |
| UU250 | U. urealyticum parvum biovar serovar 3 | TAG |
| UU524 | U. urealyticum parvum biovar serovar 3 | TAA |
| UU548 | U. urealyticum parvum biovar serovar 3 | TAA |
In this study, we estimated the expression levels of RF1 and RF2 in three bacteria in parallel to stop codon frequencies using genome sequences. Our analysis showed a strong correlation between the frequency of TAG and TGA codons and the abundance of RF1 and RF2 in the cell. However, the actual frequency of the stop codons should depend on the expression level of the translating mRNAs carrying a specific stop codon, which would also vary under different physiological conditions. Our analysis using gene expression microarray data showed that the genes ending with TAG are expressed at the lowest level under fast growing condition (Fig. 4, C and D) when RF1 is also expressed at a significantly lower level than RF2. In contrast, under biofilm-forming condition, the expression levels of the genes ending with TAG or TGA become similar when RF1 and RF2 levels also change to 1:1. Thus, we conclude that the relative abundance of RF1 is tightly correlated with the frequency of actually expressed TAG codons; the cellular factors involved in the regulation of the two remain unknown. Lastly, the growth condition-dependent fluctuation of the codon usage is not specific only for the stop codons but must be applicable to all other codons. Thus, our finding from stop codon usage calls for new estimation and mathematical modeling of codon usage in bacteria, taking mRNA expression levels into account.
Acknowledgments
We thank Richard Buckingham and Valérie Heurgué-Hamard for providing antibodies against E. coli RF1 and RF2 proteins. We also thank Måns Ehrenberg and Siv Andersson for valuable discussion during revision of the manuscript.
This work was supported by Swedish Research Council Research Grants 2010-2619 Medicin, 2011-6088 (Natural Science) and 2008-6593, a Linnaeus grant to the Uppsala RNA Research Center (all to S. S.); Carl-Tryggers Foundation Grants CTS 09:341 and 10:330; the Wenner-Gren Foundation; and Knut and Alice Wallenberg Foundation Grant KAW 2011.0081 (to RiboCORE).
- RF
- release factor
- IF
- initiation factor
- EF
- elongation factor
- qPCR
- quantitative PCR.
REFERENCES
- 1. Nakamura Y., Gojobori T., Ikemura T. (2000) Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plotkin J. B., Kudla G. (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong H., Nilsson L., Kurland C. G. (1996) Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 260, 649–663 [DOI] [PubMed] [Google Scholar]
- 4. Gilchrist M. A. (2007) Combining models of protein translation and population genetics to predict protein production rates from codon usage patterns. Mol. Biol. Evol. 24, 2362–2372 [DOI] [PubMed] [Google Scholar]
- 5. Ikemura T. (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 146, 1–21 [DOI] [PubMed] [Google Scholar]
- 6. Zhang G., Ignatova Z. (2009) Generic algorithm to predict the speed of translational elongation: implications for protein biogenesis. PLoS One 4, e5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scolnick E., Tompkins R., Caskey T., Nirenberg M. (1968) Release factors differing in specificity for terminator codons. Proc. Natl. Acad. Sci. U.S.A. 61, 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharp P. M., Bulmer M. (1988) Selective differences among translation termination codons. Gene 63, 141–145 [DOI] [PubMed] [Google Scholar]
- 9. Brown C. M., Dalphin M. E., Stockwell P. A., Tate W. P. (1993) The translational termination signal database. Nucleic Acids Res. 21, 3119–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bonetti B., Fu L., Moon J., Bedwell D. M. (1995) The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol. 251, 334–345 [DOI] [PubMed] [Google Scholar]
- 11. Sun J., Chen M., Xu J., Luo J. (2005) Relationships among stop codon usage bias, its context, isochores, and gene expression level in Various eukaryotes. J. Mol. Evol. 61, 437–444 [DOI] [PubMed] [Google Scholar]
- 12. Cridge A. G., Major L. L., Mahagaonkar A. A., Poole E. S., Isaksson L. A., Tate W. P. (2006) Comparison of characteristics and function of translation termination signals between and within prokaryotic and eukaryotic organisms. Nucleic Acids Res. 34, 1959–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pavlov M. Y., Freistroffer D. V., Dincbas V., MacDougall J., Buckingham R. H., Ehrenberg M. (1998) A direct estimation of the context effect on the efficiency of termination. J. Mol. Biol. 284, 579–590 [DOI] [PubMed] [Google Scholar]
- 14. Poole E. S., Brown C. M., Tate W. P. (1995) The identity of the base following the stop codon determines the efficiency of in vivo translation termination in Escherichia coli. EMBO J. 14, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Major L. L., Edgar T. D., Yee Yip P., Isaksson L. A., Tate W. P. (2002) Tandem termination signals: myth or reality? FEBS Lett. 514, 84–89 [DOI] [PubMed] [Google Scholar]
- 16. Povolotskaya I. S., Kondrashov F. A., Ledda A., Vlasov P. K. (2012) Stop codons in bacteria are not selectively equivalent. Biol. Direct 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lind C., Sund J., Aqvist J. (2013) Codon-reading specificities of mitochondrial release factors and translation termination at non-standard stop codons. Nat. Commun. 4, 2940. [DOI] [PubMed] [Google Scholar]
- 18. Santos M. A., Moura G., Massey S. E., Tuite M. F. (2004) Driving change: the evolution of alternative genetic codes. Trends Genet. 20, 95–102 [DOI] [PubMed] [Google Scholar]
- 19. Adamski F. M., McCaughan K. K., Jørgensen F., Kurland C. G., Tate W. P. (1994) The concentration of polypeptide chain release factors 1 and 2 at different growth rates of Escherichia coli. J. Mol. Biol. 238, 302–308 [DOI] [PubMed] [Google Scholar]
- 20. Mora L., Heurgué-Hamard V., de Zamaroczy M., Kervestin S., Buckingham R. H. (2007) Methylation of bacterial release factors RF1 and RF2 is required for normal translation termination in vivo. J. Biol. Chem. 282, 35638–35645 [DOI] [PubMed] [Google Scholar]
- 21. Covert M. W., Knight E. M., Reed J. L., Herrgard M. J., Palsson B. O. (2004) Integrating high-throughput and computational data elucidates bacterial networks. Nature 429, 92–96 [DOI] [PubMed] [Google Scholar]
- 22. Yang X., Ma Q., Wood T. K. (2008) The R1 conjugative plasmid increases Escherichia coli biofilm formation through an envelope stress response. Appl. Environ. Microbiol. 74, 2690–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hruz T., Laule O., Szabo G., Wessendorp F., Bleuler S., Oertle L., Widmayer P., Gruissem W., Zimmermann P. (2008) Genevestigator V3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinformatics 2008, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mi H., Muruganujan A., Thomas P. D. (2013) PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377–D386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, Appendix E3, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26. Fraser C. M., Gocayne J. D., White O., Adams M. D., Clayton R. A., Fleischmann R. D., Bult C. J., Kerlavage A. R., Sutton G., Kelley J. M., Fritchman R. D., Weidman J. F., Small K. V., Sandusky M., Fuhrmann J., Nguyen D., Utterback T. R., Saudek D. M., Phillips C. A., Merrick J. M., Tomb J.-F., Dougherty B. A., Bott K. F., Hu P.-C., Lucier T. S., Peterson S. N., Smith H. O., Hutchison C. A., 3rd, Venter J. C. (1995) The minimal gene complement of Mycoplasma genitalium. Science 270, 397–403 [DOI] [PubMed] [Google Scholar]
- 27. Freistroffer D. V., Kwiatkowski M., Buckingham R. H., Ehrenberg M. (2000) The accuracy of codon recognition by polypeptide release factors. Proc. Natl. Acad. Sci. U.S.A. 97, 2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao H., Zhou Z., Rawat U., Huang C., Bouakaz L., Wang C., Cheng Z., Liu Y., Zavialov A., Gursky R., Sanyal S., Ehrenberg M., Frank J., Song H. (2007) RF3 induces ribosomal conformational changes responsible for dissociation of class I release factors. Cell 129, 929–941 [DOI] [PubMed] [Google Scholar]
- 29. Kramer E. B., Farabaugh P. J. (2007) The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duarte I., Nabuurs S. B., Magno R., Huynen M. (2012) Evolution and diversification of the organellar release factor family. Mol. Biol. Evol. 29, 3497–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tate W. P., Mansell J. B., Mannering S. A., Irvine J. H., Major L. L., Wilson D. N. (1999) UGA: a dual signal for “stop” and for recoding in protein synthesis. Biochemistry 64, 1342–1353 [PubMed] [Google Scholar]
- 32. Ivanova N. N., Schwientek P., Tripp H. J., Rinke C., Pati A., Huntemann M., Visel A., Woyke T., Kyrpides N. C., Rubin E. M. (2014) Stop codon reassignments in the wild. Science 344, 909–913 [DOI] [PubMed] [Google Scholar]
- 33. Osawa S., Jukes T. H., Watanabe K., Muto A. (1992) Recent evidence for evolution of the genetic code. Microbiol. Rev. 56, 229–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inamine J. M., Ho K. C., Loechel S., Hu P. C. (1990) Evidence that UGA is read as a tryptophan codon rather than as a stop codon by Mycoplasma pneumoniae, Mycoplasma genitalium, and Mycoplasma gallisepticum. J. Bacteriol. 172, 504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Margus T., Remm M., Tenson T. (2007) Phylogenetic distribution of translational GTPases in bacteria. BMC Genomics 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]