

Background: ATP binding promotes CFTR channel opening by an unknown mechanism.
Results: Introducing like charges at positions 267 and 1060 in the opposing cytosolic loops that couple the nucleotide binding domains to the pore inhibits channel opening.
Conclusion: An electrostatic interaction across the cytosolic loop interface links ATP binding to CFTR opening.
Significance: This interaction may be a target for designing CFTR drugs.
Keywords: ABC Transporter, Allosteric Regulation, Cystic Fibrosis, Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), Ion Channel, Phosphorylation
Abstract
The CFTR channel is an essential mediator of electrolyte transport across epithelial tissues. CFTR opening is promoted by ATP binding and dimerization of its two nucleotide binding domains (NBDs). Phosphorylation of its R domain (e.g. by PKA) is also required for channel activity. The CFTR structure is unsolved but homology models of the CFTR closed and open states have been produced based on the crystal structures of evolutionarily related ABC transporters. These models predict the formation of a tetrahelix bundle of intracellular loops (ICLs) during channel opening. Here we provide evidence that residues E267 in ICL2 and K1060 in ICL4 electrostatically interact at the interface of this predicted bundle to promote CFTR opening. Mutations or a thiol modifier that introduced like charges at these two positions substantially inhibited ATP-dependent channel opening. ATP-dependent activity was rescued by introducing a second site gain of function (GOF) mutation that was previously shown to promote ATP-dependent and ATP-independent opening (K978C). Conversely, the ATP-independent activity of the K978C GOF mutant was inhibited by charge- reversal mutations at positions 267 or 1060 either in the presence or absence of NBD2. The latter result indicates that this electrostatic interaction also promotes unliganded channel opening in the absence of ATP binding and NBD dimerization. Charge-reversal mutations at either position markedly reduced the PKA sensitivity of channel activation implying strong allosteric coupling between bundle formation and R domain phosphorylation. These findings support important roles of the tetrahelix bundle and the E267-K1060 electrostatic interaction in phosphorylation-dependent CFTR gating.
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR)3 anion channel is an essential mediator of electrolyte transport across the airways, gut, and exocrine glands. CFTR mutations that substantially reduce protein expression or channel activity cause cystic fibrosis, the most common recessive genetic disorder among Caucasians (1, 2). The basic factors that regulate CFTR channel opening and closing are well understood. In particular, channel opening is promoted by ATP binding to CFTR's two nucleotide binding domains (NBDs) (3). Phosphorylation of its regulatory (R) domain by a cyclic-dependent protein kinase (e.g. PKA) is also required for channel activity (4, 5). ATP-induced dimerization of the NBDs appears to link ATP binding to subsequent conformational changes of the transmembrane domains (TMs) to promote channel opening. The majority of channel closings follow ATP hydrolysis at one of the two ATP binding sites (site 2) (6). Site 1 is catalytically inactive but binds ATP tightly; thus, most cycles of channel opening and closing are temporally linked to ATP binding and subsequent hydrolysis at site 2 (6–9). R domain phosphorylation controls channel gating by unknown mechanisms that may include regulating both NBD dimerization (10) and the conformations or flexibilities of the TMs (11, 12).
CFTR is a member of the large superfamily of ABC transporters that has provided initial clues about the structural basis of CFTR channel gating. Most ABC transporters are pumps that utilize the free energy released from ATP hydrolysis to actively transport substrates across cell membranes (13, 14). CFTR is an ATP-gated ion channel for which ATP hydrolysis controls ligand occupancy, not permeation per se (15, 16). Despite this thermodynamic distinction CFTR shares substantial sequence homology with other ABC transporters especially in the NBDs and to a lesser extent in the intracellular cytosolic loops (ICLs) that couple the NBDs to the TMs that form the translocation pathway. This homology is greatest for the ABC exporter subclass for which crystal structures of bacterial exporters in the presence and absence of nucleotide are available (e.g. the Sav1866 and MsbA homodimeric exporters; 14, 17, 18). Inspection of these different structures reveals a plausible alternating access mechanism in which an exporter shifts between inward-facing and outward-facing conformations to mediate substrate binding and release, respectively. ATP binding and consequent NBD dimerization have been argued to promote this shift between inward and outward-facing conformations with subsequent ATP hydrolysis reversing the process (14, 17, 18). ATP binding and NBD dimerization may promote similar conformational changes in the CFTR channel with the distinction that one of these conformations must be open to both sides of the membrane to permit anion diffusion (19). Given the aforementioned homology between CFTR and the bacterial exporters, structural models of the open and closed states have been produced using inward-facing (MsbA) and outward-facing (Sav1866) crystal structures as templates, respectively (20–24). Certain aspects of these models have been verified experimentally; notably, the orientation of the predicted NBD dimer interface and the nature of the coupling helices that link the NBDs to the cytosolic extensions of the TMs (10, 20). Thus, the available homology models appear to be good starting points for understanding CFTR structure and function although many other features of these models remain to be tested experimentally.
A major structural feature of the outward-facing structures of the homodimeric bacterial exporters is a tetrahelix bundle along the symmetry axis that appears to link ATP-induced NBD dimerization to conformational changes in the TMs (17, 18, 25, 26). This helix bundle is created by the close apposition of the cytosolic extensions of TMs 3 and 4 across the subunit interface and extends cytosolically to where the ICLs bind to the NBDs via coupling helices. Salt bridges between E208 in ICL2 of one subunit and K212 in the other subunit stabilize the tetrahelix bundle in the outward-facing conformation of the MsbA bacterial exporter (18, 25, 26). These salt bridges appear to be functionally relevant: mutating E208 reportedly inhibited the switch between the inward- and outward-facing conformations of the MsbA exporter without affecting ATP binding (26). The available CFTR open state models exhibit an analogous tetrahelix bundle that is predicted to involve interactions between TMs 3 and 4 and their cytosolic extensions (e.g. ICL2) in the first half of the CFTR polypeptide with TMs 9 and 10 and their cytosolic extensions (e.g. ICL4) in the second half (depicted in Fig. 1). In addition, a possible salt bridge between E267 in ICL2 and K1060 in ICL4 that is located at the base of the putative bundle interface near the NBD coupling helices is a feature of at least two of these models (21, 23). The importance of the predicted tetrahelix bundle and the putative E267-K1060 interaction in particular for CFTR gating is not clear. There is a brief report that mutating E267 or K1060 inhibited macroscopic CFTR-mediated currents in transfected HEK-293 cells without apparently reducing CFTR protein expression but the mechanism for this effect was not explored (27). To explore the importance of the predicted E267-K1060 interaction and more generally the tetrahelix bundle in CFTR gating we perturbed the electrostatic interactions between these positions under a variety of conditions. Our results support an important role of the tetrahelix bundle and the E267-K1060 interaction specifically in promoting ATP-dependent channel opening as well as in facilitating spontaneous (unliganded) channel openings that occur in the absence of ATP binding or NBD dimerization. Electrostatically perturbing the bundle interface also markedly reduced the PKA sensitivity of CFTR channel activation, a result we interpret as evidence of strong allosteric coupling between R domain phosphorylation and bundle formation.
FIGURE 1.

E267 and K1060 are predicted to electrostatically interact at the CFTR tetrahelix bundle interface. A, side-by-side comparison of the outward-facing MsbA crystal structure (PDB: 3B60 and Ref. 18) and the outward-facing (open state) CFTR model from Mornon et al. (21). The four helices that comprise the homodimeric tetrahelix bundle in MsbA are colored red (r. 195–214) or blue (r. 121–133) as defined in Ref. 26. The corresponding helices in CFTR (red, r. 250–269, 1043–1062 and blue, r. 177–189, 969–981) were based on pairwise sequence alignments and structure-based visual refinement. Portions of the ICL coupling helices 2 and 4 that do not overlap the tetrahelix bundle are colored orange (r. 270–280 and 1062–1075, respectively) as defined in Ref. 46. The analogous salt bridge residues that are predicted to stabilize the tetrahelix bundle are shown in licorice representation and colored yellow (K212 in MsbA and K1060 in CFTR) and purple (E208 in MsbA and E267 in CFTR). B, bottom-up view of coupling helix in the outward-facing CFTR model (21) (left), and after 60 ns of free molecular dynamics simulations (right). An overlapping APBS electrostatic potential field (32) of the E267 (purple) and K1060 (yellow) residues is mapped as a transparent surface representation and colored by charge (red is cationic, blue is anionic). Black lines are shown to indicate the distance measurements performed in C between the K1060:NZ1 atom and E267:OE1 and E267:OE2 atoms that define the salt bridge. C, minimum distance measurement between the E267 carboxyl oxygens and the K1060 amine nitrogen during 60 ns of free molecular dynamics simulations of outward-facing CFTR model in a POPC bilayer. The original E267-K1060 distance in the Mornon et al. open-state CFTR model (4.9 Å; Ref. 21) prior to minimization is displayed as a green dot near the y axis. The salt bridge cutoff distance is shown in red at 3.4 Å.
EXPERIMENTAL PROCEDURES
Cell Culture, DNA Constructs, and Transfections
HEK 293T cells were cultured in Dulbecco's modified Eagle's Medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS) and transiently transfected with wild type (WT) or mutant CFTR cDNA as described previously (12, 28). All point mutations and deletion mutations were generated using appropriate mutagenic oligonucleotides, verified by DNA sequencing and subcloned into the pCDNA3 mammalian expression vector (Invitrogen). Cells were transfected as described previously (12, 28) and grown for 24–72 h at 37 °C in DMEM plus 10% FBS without antibiotics prior to experimentation.
Electrophysiology and Data Analysis
Macroscopic and unitary currents were recorded in the excised, inside-out configuration using previously described patch clamp techniques (12, 28). Patch pipettes were pulled from Corning 8161 glass to a tip resistance of 1.5–2.0 MΩ for macroscopic recordings and 10–12 MΩ for unitary current recordings. Pipette and bath solutions were identical and contained in mm: 140 N-methyl-d-glucamine, 3 MgCl2, 1 EGTA, and 10 TES {2-[2-hydroxy-1,1-bis(hydroxymethyl) ethyl) amino] ethanesulfonic acid}, adjusted to pH 7.3 with HCl. Stock solutions of ATP and AMP-PNP were prepared in water and adjusted to neutral pH with NaOH. Patch clamp experiments were performed at 21–23 °C. Macroscopic currents were recorded using a ramp protocol (± 80 mV) with a 10 s time period. Patches were held at +60 mV for unitary current recordings. Macroscopic currents were analog filtered at 20 Hz. Unitary current signals were analog filtered at 110 Hz and then digitally filtered at 50 Hz with Clampfit 9.2 software (Axon Instruments). The product of the number of channels in each patch (N) and the single channel open probability (Po) for that patch (NPo) was estimated using Clampfit 9.2 for records containing fewer than 8 simultaneous openings obtained prior to the addition of potentiators (see Fig. 3 for example records). Channel activity after potentiator addition was too great to resolve unitary currents. In this case NPo was estimated using the equation I = i × NPo where I is the mean steady-state macroscopic current measured after addition of potentiator, and i is the unitary current at the holding potential of +60 mV (0.45 pA). Po values estimated before potentiator addition were normalized to the maximal Po after addition of potentiator assuming that channel number (N) was constant for each excised patch. Mean burst durations (MBDs, in seconds) were estimated for multichannel records using the cycle time method described by Mathews et al. (29) where T is the length of the channel record (in seconds): MBD = [(NPo)T]/(number of openings)]. This method can be used to roughly estimate MBDs for multichannel patches with the assumption that there exists only one population of openings (29).
FIGURE 3.
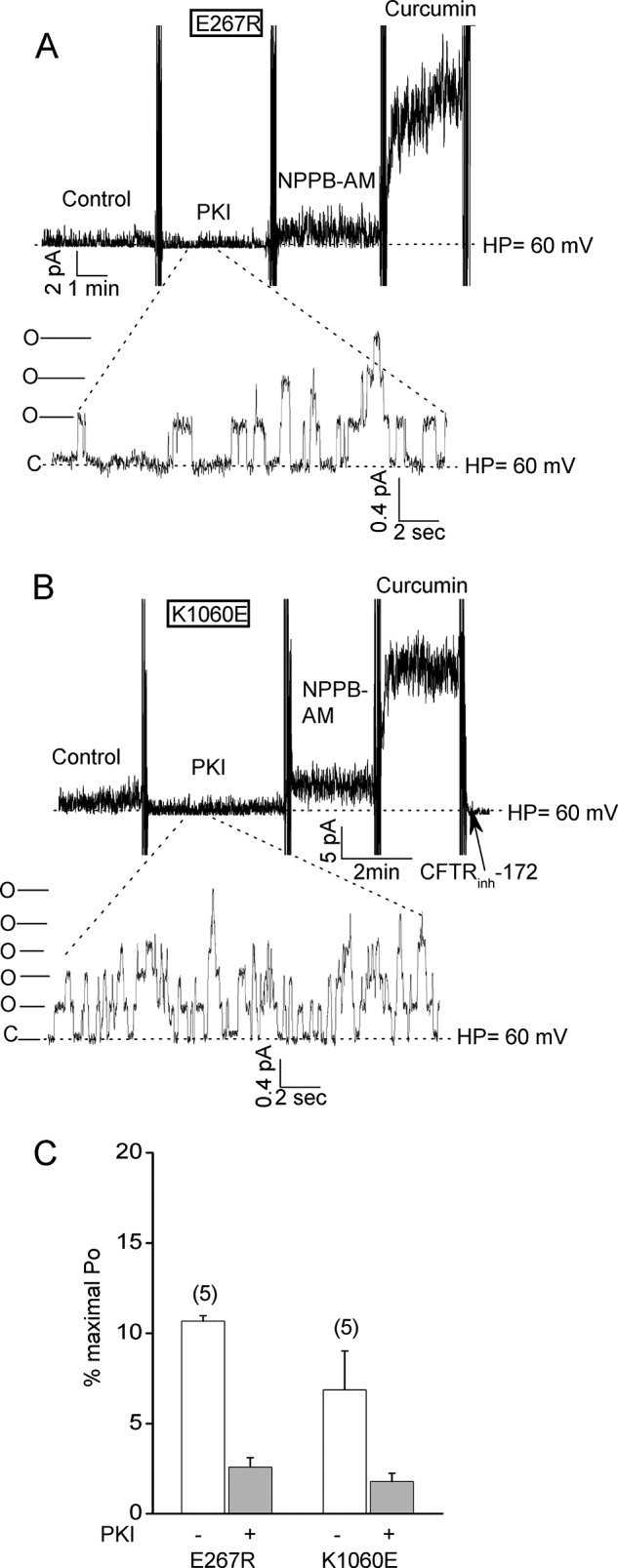
Unitary current recordings indicate that the E267R and K1060E mutations do not obviously affect single channel conductance or open channel burst duration. A and B, micropatch recordings of E267R-CFTR and K1060E-CFTR channels obtained using smaller tip pipettes in which individual channel openings and closings and unitary currents were detectable before potentiator addition. Holding potential, +60 mV. Activation conditions and reagent concentrations were the same as for Fig. 2. The magnitudes of the unitary currents at this holding potential were not different from those for WT-CFTR as follows: WT, 0.427 ± 0.003 pA (mean ± S.E.) measured for 206 openings in 2 patches; E267R, 0.419 ± 0.005 pA for 119 openings in 3 patches; K1060E, 0.431 ± 0.004 pA for 166 openings in 3 patches. The durations of channel openings (i.e. open channel bursts) before potentiator addition ranged from 0.1 to 1 s, which is also similar to that reported for WT-CFTR (29, 36). The mean open channel burst durations calculated by cycle time analysis (see “Experimental Procedures”) were 0.7 ± 0.1 and 0.6 ± 0.1 s for E267R-CFTR (n = 6 patches) and for K1060E-CFTR (n = 4), respectively. These values are not substantially different from those reported for WT-CFTR under similar activation conditions (29, 36). C, mean percent maximal Pos (± S.E.) for the indicated charge-reversal mutants before and after PKI addition. Numbers of experiments are indicated in parentheses. The Pos are normalized to the maximal Pos estimated after potentiator addition as described under “Experimental Procedures.”
Molecular Dynamics Simulations
A previously published open CFTR model based on the empirical structure of outward-facing Sav1866 was used in the construction of the simulation system (21). The CFTR model originally contained two molecules of ADP in the NBD binding pockets, which were substituted with Mg-ATP via RMSD-based superimposition. The modified CFTR model was inserted into a 180 × 180 Å pre-equilibrated patch of POPC bilayer that was generated by the Membrane Builder plugin in VMD 1.9.1 using the CHARMM36 forcefield topologies (30, 31). Surface tryptophan residues were the principal guide for depth placement. After insertion, all overlapping lipid molecules within 2.4 Å of protein were removed and the system was minimized in the absence of solvent for 15000 steps. The entire system was explicitly solvated with TIP3 water using the VMD Solvate plugin, and surface ions were subsequently neutralized with 0.15 m/liter of NaCl. The resultant system was minimized 15,000 steps and then heated from 0 to 310° K over an additional 15,000 steps. All molecular dynamics and time-independent minimization were performed in NAMD 2.9 using the NPT ensemble and CHARMM36 force fields on the Cheaha supercomputing cluster at the University of Alabama at Birmingham (31). The simulations employed Particle Mesh Ewald long-range electrostatics, Berendsen pressure control, and temperature coupling at 310 K under periodic boundary conditions. Long-range cutoffs were set at 12 Å using a switching function starting from 10 Å. The integrator utilized a 2 femtosecond time step and bonds between hydrogen atoms and heavy atoms were made rigid. Trajectory output was generated every 20 ps. Electrostatic potential analysis was performed using APBS (32), and all other analysis and rendering was performed in VMD.
RESULTS
Predicted Electrostatic Interaction between E267 and K1060 at the Presumed Tetrahelix Bundle Interface
Fig. 1A shows the tetrahelix bundle in the outward-facing MsbA crystal structure on the left and the predicted tetrahelix bundle in the CFTR outward-facing (open state) model from Mornon et al. (21) on the right. The coupling helices that are expected to link ICLs 2 and 4 to the NBDs in the CFTR model and the analogous NBD coupling helices in the MsbA structure are indicated in orange. The E208-K212 salt bridge at the base of the bundle interface in the MsbA outward-facing structure is illustrated in yellow (left). Also shown in yellow are residues E267 in ICL2 and K1060 in ICL4 in the CFTR open state model, which similarly locate to the base of the predicted helix bundle near the NBD coupling helices (right). The distance between E267 and K1060 is less than 5 Å in the CFTR open state model, as can be seen in the bottom up view in Fig. 1B (left), and is similar to the distance between these residues in a more recent CFTR open state model reported by Dalton et al. (23). Note that E267 and K1060 are far apart (> 20 Å) in the inward-facing (closed state) model of Mornon et al. (21) that lacks the tetrahelix bundle, which is a unique feature of the outer-facing conformations of the bacterial exporters (17, 18). To explore further the possibility of an electrostatic interaction between E267 and K1060 in the CFTR open conformation we performed an atomistic molecular dynamics (MD) simulation of the open state model under physiologic conditions (inserted into POPC bilayer, 150 mm NaCl, 310° K). The distance between E267 and K1060 fluctuated during the 60 ns simulation but on average shortened in comparison to the CFTR open state model to distances that were sufficient for salt bridge formation (Fig. 1, B and C).
Charge-reversal Mutations at Positions 267 and 1060 Strongly Inhibit CFTR Channel Opening
To assess the importance of E267 and K1060 in channel function we tested the effects of a series of charge-reversal and neutral substitutions at these positions on macroscopic CFTR activity in excised inside-out membrane patches (Fig. 2). Fig. 2A shows a typical macroscopic current record for an excised patch containing many wild type (WT) CFTR channels (>1000) that were activated by normally saturating concentrations of MgATP and PKA. Inhibiting the PKA in the bath by adding protein kinase A inhibitory peptide (PKI) resulted in a partial deactivation of the macroscopic current due presumably to the activity of membrane associate phosphatases in the membrane patch (12, 28). Subsequent addition of compounds that were previously shown to potentiate wild type and mutant CFTR activity (NPPB-AM and curcumin, Ref. 28) only modestly increased the current as expected given that WT-CFTR is already very active under these conditions (single channel open probability, or Po, of about 0.3–0.4 (12)). The WT-CFTR current was abolished by bath addition of a CFTR channel inhibitor (CFTR(inh)-172) as shown previously (12, 33).
FIGURE 2.

Strong inhibition of CFTR channel activity by charge- reversal mutations at positions 267 and 1060. A, macroscopic current record for excised inside-out patch containing many (> 1000) wild type (WT) CFTR channels. Ramp protocol: ± 80 mV; 10 s time period. Dotted line shows zero current level. Control condition was 110 units/ml PKA and 1.5 mm Mg-ATP. Inhibitory PKA peptide (1.4 μg/ml PKI) was added to block further phosphorylation resulting in partial current deactivation due to partial dephosphorylation by membrane-associated phosphatases (12, 28). Two CFTR potentiators, NPPB-AM (10 μm) and curcumin (30 μm), and a CFTR inhibitor (20 μm CFTR-(inh)172) were added sequentially to the bath where indicated. B, macroscopic current record for patch containing many E267R-CFTR channels showing very small control current and very large activation by potentiators. Conditions were identical to panel A. C, macroscopic record showing that E267R-CFTR channels also are strongly stimulated by the FDA-approved potentiator, VX-770 (10 μm). D, K1060E-CFTR channels also exhibit reduced control currents and strong activation by potentiators. E, mean currents (± S.E.) measured at −80 mV before and after PKI addition for the indicated constructs. Currents were normalized to the maximal currents measured after the addition of NPBB-AM and curcumin. N ranges from 5 to 17. The charge-reversal mutations at positions 267 and 1060 strongly reduced the currents before and after PKI addition. Most of the neutral substitutions had modest effects on the control currents measured before PKI addition but nearly all reduced the normalized currents measured after PKI addition compared with WT-CFTR. For all mutants except E267G the relative currents after PKI addition were significantly less than for WT (p < 0.05) by unpaired t test. F, scatter plot showing the fold current stimulation by the addition of NPPB-AM and curcumin for the indicated constructs. Each symbol represents an individual experiment. Note the break in the Y-axis. Conditions were identical to panels A–E. Potentiators were added after PKI. The charge-reversal mutants were most strongly activated because of their very low control currents but nearly all of the neutral mutants also showed stronger relative stimulation by potentiator addition compared with WT-CFTR.
In sharp contrast to this behavior of the WT-CFTR channel, mutants with charge-reversal substitutions at position 267 or 1060 exhibited markedly reduced control currents at normally saturating ATP and PKA and correspondingly much greater stimulation by NPPB-AM, curcumin or VX-770, an FDA-approved CFTR potentiator (34, 35) (Fig. 2, B–D). Fig. 2E shows mean data for a series of uncharged and charged substitutions at these positions where we normalized the currents measured before and after PKI addition to the maximal currents measured following potentiator stimulation. The charge-reversal mutations (E267R and K1060E) at these positions clearly had the most dramatic effects on CFTR activity (the E267K mutation also markedly inhibited CFTR activity; see Fig. 5). However, many of the neutral substitutions also reduced the normalized control currents measured after PKI addition because these mutations tended to be more strongly inhibited by PKI than was WT-CFTR. The greater responses of these mutants to PKI addition implies that they have altered coupling between channel gating and PKA phosphorylation, which we address more specifically in later experiments (e.g. Fig. 9). Fig. 2F is a scatter plot of the relative activation by potentiator addition for the same series of mutants. Consistent with the mean current data in Fig. 2E, the charge-reversal substitutions exhibited the biggest potentiator responses because they had the lowest currents before and after PKI addition. However, most of the neutral substitutions also exhibited greater potentiator activation compared with WT-CFTR which is consistent with their generally lower currents after PKI addition.
FIGURE 5.
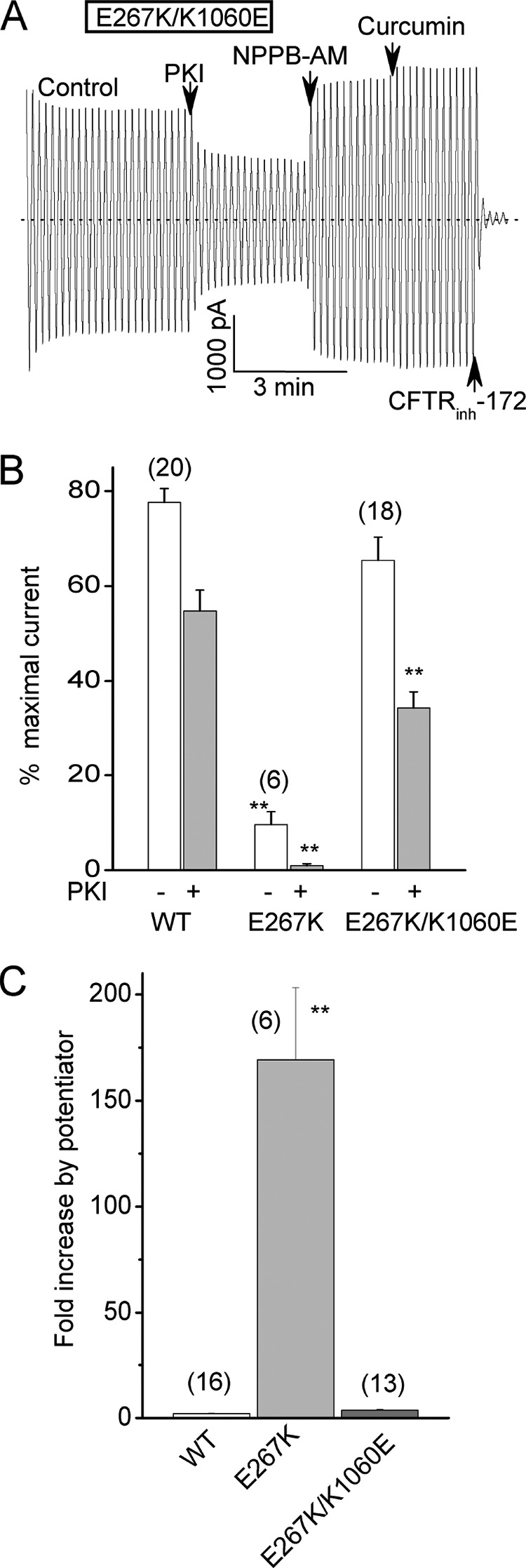
Swapping the charges between positions 267 and 1060 restores CFTR channel activity. A, macroscopic current record for a double mutant with charge-reversal substitutions at both positions (E267K/K1060E-CFTR). Conditions were identical to Fig. 2. Note the large control current and modest stimulation by potentiator addition. B, mean relative control currents (± S.E.) for the indicated constructs before and after PKI addition. Currents were normalized to the maximal currents measured after potentiator addition as for Fig. 2E. WT-CFTR data are from Fig. 2E. Note that the E267K single mutation strongly inhibited channel activity as did the E267R mutation shown in Fig. 2. See also Fig. 2E for K1060E single mutant data. **, p < 0.01 compared with WT-CFTR by unpaired t test. C, mean fold increase in current following potentiator addition for the indicated constructs. WT-CFTR data are from Fig. 2F. Error bars (S.E.) for WT and E267/K1060E are too small to be seen. **, p < 0.01 compared with both WT-CFTR and the double charge swap mutant by unpaired t test. N is indicated in parentheses.
FIGURE 9.

Charge-reversal mutations at the bundle interface strongly reduce the PKA sensitivity of CFTR activation. A–C, macroscopic current records showing that the E267R and K1060E mutants required more PKA than WT-CFTR to achieve only partial activation. Conditions other than the PKA doses were identical to Fig. 2. D, macroscopic record showing that the charge swap mutant exhibited a PKA sensitivity that was similar to that of WT-CFTR. E and F, PKA titration curves averaged over multiple experiments where the activation was normalized to the maximal currents measured after potentiator addition (panel E) or to the currents measured at the highest PKA dose tested (panel F). Symbols are means ± S.E. Ns are 5, 5, 4, and 5 for WT (black symbols), E267R (red), K1060E (blue), and E267K/K1060E (green), respectively.
Fig. 3 provides indirect but persuasive evidence that the E267R and K1060E charge-reversal mutations reduced channel activity primarily by reducing the rate of channel opening rather than by shortening the open channel bursts or decreasing the single channel conductance. Shown in Fig. 3, A and B are unitary current recordings of the E267R and K1060E mutants at a single holding potential in membrane patches excised using small tip pipettes. Unitary currents could be resolved before but not after addition of the potentiators, which robustly activated these mutants as in the macropatch experiments (mean data shown in Fig. 3C). The unitary currents for each mutant before potentiator addition were approximately the same amplitude as exhibited by WT-CFTR at this holding potential (see legend), indicating that these mutations did not obviously affect single channel conductance. In addition, the lengths of the open channel bursts exhibited by each mutant prior to potentiator addition were approximately the same as exhibited by WT-CFTR under these conditions (0.1 to 1 s; see mean burst duration data in the figure legend and compare with Refs. 29, 36). These micropatch results indicate that the E267R and K1060E charge-reversal mutants have exceptionally low Pos under control and PKI-treated conditions (10–50-fold lower than WT-CFTR) primarily because they have very low channel opening rates, not abnormally brief open channel bursts.
Thiol Modification and Charge Swap Results Support an Activity-dependent E267-K1060 Electrostatic Interaction that Promotes CFTR Opening
As another test of the importance of the E267-K1060 electrostatic interaction in channel gating we used a positively charged thiol modifier (MTSET) to acutely introduce positive charges on substituted cysteines at these two locations. We chose this reagent because in our hands MTSET has negligible effects on WT-CFTR currents under the conditions of these experiments unlike several other reagents we tested such as the negatively charged MTSCE (see WT-CFTR control data in Fig. 4C). As we showed in Fig. 2 the single E267C and K1060C mutations reduced CFTR channel activity especially after PKI addition but not to the extent of the charge-reversal substitutions. Subsequent exposure of E267C-CFTR to MTSET rapidly and markedly inhibited the post-PKI current which was reversed by bath addition of DTT (Fig. 4A). Conversely, the same positively charged reagent modestly increased the current mediated by K1060C-CFTR; an effect that also was reversed by DTT (Fig. 4B; mean data in Fig. 4C).
FIGURE 4.

A positively charged thiol modifier strongly inhibits E267C-CFTR currents in an activity-dependent manner but modestly stimulates K1060C-CFTR currents. A, E267C-CFTR macropatch record showing strong and rapid inhibition of this cysteine mutant by bath addition of 30 μm MTSET and reversal of this inhibition by 5 mm DTT. Ramp protocol and reagent concentrations were identical to Fig. 2. B, corresponding K1060C-CFTR macropatch record showing modest stimulation of this cysteine mutant by MTSET that also was reversed by subsequent DTT addition. C, mean (± S.E.) effects of MTSET addition on the currents mediated by the indicated constructs. MTSET was added after PKI addition as shown in panels A and B. N is indicated in parentheses. *, p < 0.05 compared with WT-CFTR by unpaired t test. D, E267C-CFTR macropatch record showing that MTSET inhibition was very slow when added after the current was stimulated with 2 mm AMP-PNP. Inset shows single exponential fit of MTSET inhibition time course with a time constant of almost 5 min.
If E267 and K1060 electrostatically interact at the interface of a helix bundle that is created during channel opening then the accessibility of these positions to thiol modification should be activity-dependent. To test this idea we exposed E267C-CFTR channels to the poorly hydrolyzable AMP-PNP in the continued presence of ATP prior to MTSET addition (Fig. 4D). Under these conditions AMP-PNP stimulates WT-CFTR currents by lengthening the open channel bursts because the NBD dimer is stabilized when AMP-PNP is bound to one of the two nucleotide binding sites (36–38). E267C-CFTR channels also were strongly activated by AMP-PNP but remained sensitive to subsequent treatment with MTSET followed by DTT (Fig. 4D). However, the kinetics of MTSET inhibition were markedly slower than without AMP-PNP pre-treatment (τinactivation > 4 min versus < 10 s; compare with Fig. 4A). The slow time course of MTSET inactivation of AMP-PNP- treated channels is similar to the slow time course of current deactivation following AMP-PNP washout observed for WT-CFTR (36–38). These findings support the idea that the accessibility of E267C to thiol modification is reduced in the open channel conformation(s) as would be expected if this position is buried in a helix bundle upon channel opening.
If a salt bridge between E267 and K1060 at the bundle interface is functionally important for channel opening then swapping the charges between these two positions might reverse the gating defects exhibited by the single charge reversal substitutions. Fig. 5, A--C confirm this prediction for an E267K/K1060E double mutant. This charge swap mutant behaved much like WT-CFTR both in terms of the large magnitudes of its normalized control currents (Fig. 5, A and B) and its relatively small stimulation by potentiators (Fig. 5, A and C). Taken together the results of our charge swap and thiol modification experiments support an electrostatic interaction between E267 and K1060 that promotes CFTR channel activity.
Combining E267 and K1060 Charge-reversal Mutations with a Gain of Function (GOF) Mutation Reveals the Importance of the Tetrahelix Bundle in Mediating Channel Opening in the Absence of ATP Binding or NBD Dimerization
Previously we reported a class of GOF mutation in ICL3 near the base of TM9 that increases both ATP-dependent and ATP-independent channel activity (e.g. K978C (12)). These GOF mutations also increase the ATP-free channel activity of a truncation construct that lacks NBD2 and therefore is incapable of dimerizing its NBDs (Δ1198-CFTR (12)). Here we combined one of these GOF mutations with the E267R and K1060E mutations to test: (i) if a GOF mutation could rescue the ATP-dependent channel activities of the E267 and K1060 charge-reversal mutants and (ii) if bundle formation and the predicted electrostatic interaction also facilitate channel opening in the absence of ATP binding or NBD dimerization.
Regarding the first issue, Fig. 6A shows that introducing the K978C substitution into the E267R mutant substantially restored ATP-dependent channel activity (compare with Fig. 2B). The E267R/K978C double mutant exhibited normalized control currents and potentiator responses in macropatch experiments that were similar to wild type levels (see legend for mean data). This rescue of the ATP-dependent activity of the E267 charge-reversal mutant is perhaps not surprising given that the K978C substitution reduces the free energy difference between the open and closed states both in the presence and absence of ATP (12).
FIGURE 6.

Interplay between the charge- reversal mutations at the bundle interface and a previously reported GOF mutation (K978C); evidence that the E267R and K1060E mutations also inhibit ATP-free channel activity. A, E267R/K978C double mutant behaves more like wild type channels in the presence of ATP in macropatch experiments. Conditions were identical to Fig. 2A. The mean percent control currents of this double mutant when normalized to the maximal currents measured after potentiator addition were 94 ± 5.4% and 82.8 ± 3.9% before and after PKI addition, respectively (n = 5). These values are not smaller (in fact, somewhat larger) than those for WT-CFTR (see Fig. 2E.). B–D, macroscopic records for the indicated K978C single and double mutants showing that the E267R and K1060E substitutions decreased the fractional currents remaining after ATP removal. ATP was removed by the addition of an ATP scavenger (24 U/ml hexokinase plus 10 mm glucose) followed by bath perfusion with an ATP-free solution. E267R/K978C-CFTR was activated strongly by the subsequent addition of 2 mm AMP-PNP in the absence of ATP, as reported previously for the K978C GOF mutant (panels B, C and Ref. 47). E, mean percent ATP-free currents normalized to the currents before ATP removal for the indicated constructs. N is indicated in parentheses. **, p < 0.01 when compared with the double mutants by unpaired t test.
Regarding the second issue, Fig. 6, B–E show that the E267R and K1060E mutations reduced the ATP-free activity of the K978C GOF mutant. This GOF mutant normally exhibits substantial ATP-independent currents in macropatch experiments following the addition of an ATP scavenger (hexokinase/glucose) and extensive bath perfusion with an ATP-free solution (example record in Fig. 6B; mean data in Fig. 6E). Each of the E267 and K1060 charge-reversal mutations strongly reduced these normalized ATP-free currents, which is consistent with the idea that the predicted electrostatic interaction and bundle formation also facilitate unliganded channel opening.
The latter interpretation is further supported by the results in Fig. 7, which show that the E267R mutation also reduced the ATP-free channel activity of K978C/Δ1198-CFTR, a truncation mutant lacking NBD2 but possessing the indicated GOF mutation. This construct normally exhibits substantial ATP-independent currents that are further but modestly stimulated by bath addition of a potentiator (Ref. 12 and Fig. 7A). Introducing the E267R mutation into this construct substantially reduced the basal ATP-independent currents and correspondingly increased the relative stimulation by potentiators (Fig. 7, B, D, and E). Conversely introducing a charge swap double mutation (E267K/K1060E) had no apparent effect on the basal currents and potentiator responses of this truncation construct (Fig. 7, C–E). In sum, the results of Figs. 6 and 7 support the view that the E267-K1060 interaction and tetrahelix bundle formation also promote CFTR channel opening in the absence of ATP binding or NBD dimerization.
FIGURE 7.

The charge- reversal bundle mutations also inhibit the activities of channels that lack NBD2. A, control macroscopic record showing the substantial control current and correspondingly modest stimulation by potentiator addition for the previously described K978C/Δ1198 truncation mutant lacking NBD2. Activation conditions and reagent concentrations were the same as for Fig. 2. As reported earlier (12), the channel activity of this construct is ATP-independent and is attributable to the presence of the K978C GOF mutation. B, macroscopic record showing relatively smaller control current and stronger activation by potentiator addition for a corresponding truncation construct harboring the E267R mutation. C, macroscopic record showing larger control current and a correspondingly smaller potentiator effect for a truncation construct with the charges swapped between residues 267 and 1060. D, mean relative control currents (± S.E.) for the indicated constructs before and after PKI addition. Currents were normalized to the maximal currents measured after potentiator addition as for Fig. 2E. E, mean fold increase in current following potentiator addition for the indicated constructs. Error bar (S.E.) for the charge-swap construct is too small to be seen. **, p < 0.01 compared with K978C/Δ1198 by unpaired t test. N is indicated in parentheses.
Combining a Catalytic Mutation with the E267R Mutation Rescues ATP-dependent Activity but Also Provides a Clue that R Domain Conformational Changes May Be Coupled to Bundle Formation
We next combined one of the charge-reversal mutations (E267R) with an NBD2 mutation (E1371S) that inhibits ATP hydrolysis at site 2 (Fig. 8). The E1371S mutation stabilizes the NBD dimer in the presence of ATP and greatly prolongs the open channel bursts (39, 40). Our interest was to determine if the E267R mutation inhibited the activity of the E1371S catalytic mutant and/or destabilized the NBD dimer in the absence of ATP hydrolysis. Instead, however, the E267R/E1371S-CFTR double mutant behaved similarly to the E1371S-CFTR single mutant with respect to: (i) high control currents in the presence of normally saturating ATP and PKA; (ii) negligible responses to potentiators presumably because the currents were already maximal under control conditions; and (iii) very slow deactivation upon ATP removal (τdeactivaton > 5 min), indicative of a very tight NBD dimer (Fig. 8A; compare with E1371S-CFTR data in Refs. 39, 40). We interpret these results to indicate that the E267R mutation has negligible effects on channel activity once a tight NBD dimer has been formed by ATP binding sans hydrolysis.
FIGURE 8.

Interplay between the E267R mutation and a site 2 catalytic mutation, E1371S. A, macroscopic record showing that the E267R/E1371S double mutant exhibited large control currents, very small stimulation by potentiators and very slow deactivation upon ATP removal. Inset shows single exponential fit of deactivation time course following ATP removal with a time constant of greater than 5 min. These characteristics are similar to those previously reported for the single E1371S catalytic mutant (39, 40). B and C, macroscopic records showing that the E267R substitution reduced the baseline current prior to addition of PKA (units/ml) that can be detected for the E1371S-CFTR construct. D, mean (± S.E.) baseline current measured prior to PKA addition for the indicated constructs. Baseline currents were normalized to the maximal currents measured after PKA (≥25 U/ml) and potentiator addition. *, p < 0.05 by unpaired t test. N is indicated in parentheses.
One interesting difference between the single and double E1371S mutants emerged when we assayed these constructs in the absence of bath PKA (Fig. 8, B–D). There are anecdotal reports in the literature that E1371 mutants exhibit significant channel activity in the absence of PKA stimulation (41, 42). Indeed, we observed macroscopic currents for E1371S-CFTR channels in the absence of bath PKA that averaged more than 10% of the maximal currents measured after potentiator addition (Fig. 8, C and D). WT-CFTR-mediated currents are undetectable in the absence of bath PKA under these conditions (e.g. ref. 12). Combining the E267R charge-reversal mutation with the E1371S mutation virtually abolished these PKA-independent currents (Fig. 8, B and D). This finding provides a clue that R domain conformation and bundle formation may be strongly coupled, which we follow up below.
E267R and K1060E Charge-reversal Mutations Strongly Reduce the PKA Sensitivity of Channel Gating
Fig. 9 shows that the E267R and K1060E mutations markedly impacted the PKA sensitivity of channel activation. In PKA titration experiments we observed that the activation of each charge reversal mutant required much more PKA than wild type CFTR (example records in Fig. 9, A–C; mean data in Fig. 9, E and F). The currents mediated by these mutants were not maximally active even at PKA levels that are more than 5-fold higher than required to maximally stimulate WT-CFTR. Importantly, normal PKA sensitivity was restored by introducing charge swap mutations across the putative interface (E267K/K1060E; Fig. 9, D–F). These findings indicate that PKA sensitivity is strongly coupled to tetrahelix bundle formation; more specifically, to the predicted electrostatic interaction between residues E267 and K1060 at the interface of this putative bundle. This altered PKA sensitivity occurred despite the fact that there are no consensus PKA phosphorylation sites near either of these residues in ICL2 or ICL4. The data in Figs. 8 and 9 support the emerging view that the R domain is an allosteric modulator of CFTR gating whose structure and phosphorylation state are allosterically coupled to the open channel conformation (12, 16).
DISCUSSION
The results of this study support significant roles of residues E267 and K1060 and, more generally, the predicted tetrahelix bundle in CFTR channel opening. These residues map to the interface of a tetrahelix bundle near the NBD coupling helices in homology models of the CFTR open conformation that are based on the crystal structures of bacterial exporters in their nucleotide-bound conformations. Tetrahelix bundle formation in the homodimeric MsbA exporter reportedly couples nucleotide binding at the NBDs to conformational changes in the transmembrane domains (TMDs) to mediate substrate export (26). Bundle formation in this bacterial exporter involves salt bridges at positions E208 and K212 in ICL2 that approximately correspond to the locations of E267 (ICL2) and K1060 (ICL4) in CFTR (18, 26). A mutation that disrupts these salt bridges inhibited the switch between the inward-facing and outward-facing conformations of MsbA without affecting ATP binding (26). Our data are consistent with a possibly analogous electrostatic interaction between E267 and K1060 that facilitates CFTR channel opening in the presence and absence of ATP binding. The locations of these residues at the base of the putative helix bundle near the NBD coupling helices makes them well positioned to help transmit the signals generated by ATP-induced NBD dimerization to the TMDs for CFTR channel opening. Our results also indicate strong coupling between this electrostatic interaction across the bundle interface and the PKA sensitivity of channel activation. The latter observation speaks to the mechanism (s) by which the unique R domain controls CFTR gating.
An Activity-dependent Electrostatic Interaction Across the Putative Bundle Interface that Promotes CFTR Opening
Our electrophysiologic data provide good evidence for an electrostatic interaction, possibly a salt-bridge, between E267 and K1060 that is important for channel gating. We observed very strong inhibition of CFTR activity at the macroscopic and single channel level by charge-reversal mutations at each position. This inhibition was not observed for a double mutant for which the charges were swapped between the two sites. Neutral substitutions at each position had detectable but much lesser effects on channel activity. The substantially lesser effect of an uncharged substitution is expected because of the nonlinear relationship between the single channel PO (a measure of the open-closed equilibrium constant) and the free energy difference between states (which would be increased 2-fold more by a charge-reversal mutation in the simplest case of disrupting a single salt bridge). The results of our thiol modification experiments also support an electrostatic interaction between E267 and K1060. A positively charged MTS reagent had the predicted opposite effects on channel activity for mutants with a cysteine at one or the other of these two positions. In addition, the rate of inhibition of the E267C mutant by this reagent was slowed markedly by AMP-PNP, a poorly hydrolysable ATP analog that prolongs open channel bursts and stabilizes the NBD dimer. This apparent activity dependence of the accessibility of the 267 position to modification is consistent with existing CFTR homology models and our MD simulation results which predict that E267 is buried at the bundle interface in the open channel conformation.
Our data are generally consistent with two previous reports of the impact of mutating E267 and K1060 on CFTR activity. Seibert et al. (43) provided the first evidence that the K1060 position is relevant to CFTR gating in their single channel analysis of a mutation (K1060T) that reportedly associates with mild disease (i.e. CBAVD). They observed an ∼2-fold lower single channel Po for this K1060 mutant relative to wild type CFTR in excised membrane patches under conditions that approximate our control conditions (normally saturating ATP and PKA). This moderate degree of inhibition by the uncharged K1060T substitution is consistent with our findings. In a brief report Billet et al. (27) observed that E267 and K1060 mutations reduced macroscopic (whole cell) CFTR currents in HEK-293 cells without obviously affecting protein maturation. These authors observed greater inhibition by a charge-reversal mutation than by a neutral substitution at the E267 position (E267R versus E267A), as we showed here in our more detailed mechanistic study. On the other hand, Billet et al. (27) reported that a neutral mutation at the K1060 position (A) reduced whole cell CFTR currents to about the same degree as a charge-reversal mutation (E). The apparent lack of charge-dependence for the single K1060 mutants differs from the results of our excised patch experiments. This difference may be attributable to the fact that a whole cell current measurement is a less direct readout of CFTR channel activity. Interestingly, Billet et al. (27) did report that a double mutant in which the charges were swapped between these sites (E267R/K1060E) behaved like wild type CFTR in their assay, which is consistent with an electrostatic interaction between these residues that impacts CFTR activity. In sum, the present findings and these previous reports are in general agreement that E267 and K1060 play an important role in controlling CFTR channel activity that likely involves at least in part an electrostatic interaction between these residues.
Disrupting the E267-K1060 Interaction Primarily Impacts Channel Opening, not Burst Duration or NBD Dimer Stability
The charge-reversal mutations at these two positions dramatically decreased the macroscopic currents and apparent single channel Pos in excised membrane patches without obviously affecting the durations of open channel bursts. Thus, these mutations primarily decrease the rates at which channels open rather than increase the rates at which their open bursts are terminated. This implies that the E267-K1060 interaction and by extension, formation of the tetrahelix bundle, controls entry into the open, or activated state, rather than the stability of that state. The lack of effect on the stability of the activated state is consistent with the virtually complete rescue of the ATP-dependent activity of the E267R bundle mutant that we observed when we combined this mutation with the E1371S catalytic mutation. The latter mutation abolishes ATP hydrolysis at site 2 and greatly prolongs open channel bursts by stabilizing the ATP-bound NBD dimer (39, 40). The E267R/E1371S double mutant exhibited high steady-state control currents following activation by ATP and PKA, small relative activation by potentiators and very slow deactivation upon ATP washout similar to that reported for the E1371S single mutant. We interpret these results to indicate that the E267-K1060 interaction does not strongly influence ATP binding or the stability of the ATP-induced NBD dimer. This interpretation is consistent with the report by Doshi et al. (26) that disrupting tetrahelix bundle formation in the MsbA exporter inhibited the switch between inward-facing and outward-facing conformations but had no apparent effect on nucleotide binding. Taken together, the previous MsbA results and the present CFTR data support the view that bundle formation may be a general feature of ABC transporters (pumps and channels alike) that facilitates the transition between inactive and active conformations in response to ATP binding and NBD dimerization.
The E267-K1060 Interaction Also Promotes CFTR Channel Opening in the Absence of ATP Binding or NBD Dimerization
Combining a previously reported GOF mutation (K978C) with the E267R or K1060E bundle mutations revealed that the latter mutations also inhibit ATP-free channel activity both in the presence and absence of NBD2 (i.e. for the Δ1198-CFTR truncation construct). The K978C substitution did restore nearly normal ATP-dependent activity of the E267R mutant as evidenced by high control currents in the presence of ATP and PKA and small relative activation by potentiators. The rescue of ATP-dependent gating is consistent with the previously reported effect of this GOF mutation to decrease the free energy difference between the closed and open states in the presence and absence of ATP (12). Apparently the favorable effect of the K978C mutation on the energetics of channel opening is sufficient to overcome the negative impact of the bundle mutation on gating when ATP binding and NBD dimerization can occur. Conversely, each of the E267R and K1060E mutations substantially reduced the fractional ATP-independent currents exhibited by the full-length K978C-CFTR construct and also reduced the control currents and increased the relative stimulation by potentiators for the NBD2-deletion construct bearing the GOF mutation (K978C/Δ1198-CFTR). These findings indicate that bundle formation and, more specifically, the interaction between E267 and K1060 also facilitate channel opening in the absence of ATP binding and/or NBD dimerization. Thus, channel opening appears to be conformationally coupled to the E267-K1060 interaction at the putative bundle interface independently of the nucleotide occupancies of the NBDs. This interpretation also is consistent with the MsbA results of Doshi et al. (26) who, as noted above, reported that disrupting bundle formation inhibited the switch between inward-facing and outward-facing conformations but not nucleotide binding to this bacterial exporter. According to this view, GOF mutations such as K978C enhance the channel activity of the Δ1198-CFTR construct lacking NBD2 by allosterically promoting the E267-K1060 interaction at the bundle interface.
Bundle Formation and PKA Sensitivity Appear To Be Strongly Coupled: Implications for the Role of the R Domain in Channel Gating
Phosphorylation of the large and unique R domain is normally required for CFTR channel activity. The unphosphorylated R domain inhibits channel opening by several reported mechanisms including disrupting NBD dimerization (10) and restricting the motions or flexibilities of the ICLs and TMs (11, 12). The latter mechanism is supported by our earlier observation that PKA phosphorylation of the R domain markedly increases the channel activity of the NBD2 deletion construct, K978C/Δ1198-CFTR (12). The E267R and K1060E mutations in ICL2 and ICL4 both strongly reduced the PKA sensitivity of channel activation in two ways. First, in PKA titration experiments the single charge-reversal mutants required much more PKA to achieve partial activation relative to wild type CFTR whereas the charge-swapped double mutant exhibited a nearly normal PKA sensitivity. Second, the currents mediated by the E267 and K1060 mutants including most of the neutral substitutions were more sensitive to the effect of adding PKA inhibitory peptide (PKI) to the bath, which partially reduces CFTR currents in excised HEK patches presumably because membrane associated phosphatases partially dephosphorylate CFTR channels in these patches. We interpret these differences to indicate either that the E267 and K1060 mutants are phosphorylated to a lesser degree than WT-CFTR channels in excised HEK patches before as well as after PKI addition or that these mutants require a greater degree of phosphorylation to achieve their already low activities. The former interpretation seems likely given that wild type CFTR channels are maximally active at a PKA concentration (200 units/ml) that only partially activated the single E267R and K1060E mutants. Importantly, neither ICL2 nor ICL4 has a PKA phosphorylation site in the vicinity of E267 or K1060. Thus, the large effects of these mutations on PKA sensitivity reflect either strong allosteric coupling between R domain phosphorylation and the E267-K1060 interaction at the bundle interface or a physical interaction between the R domain and these ICLs that is regulated by phosphorylation of the R domain. The former explanation is simpler in our view and is supported by other data, as discussed below. Either of these mechanisms to decrease PKA sensitivity could explain in part the large inhibition of channel opening rate by the charge-reversal mutations given that PKA phosphorylation reportedly enhances NBD dimerization (10) and NBD dimerization promotes channel opening (3).
The concept of allosteric coupling between R domain phosphorylation and the structural events that underlie channel opening (e.g. formation of the predicted tetrahelix bundle) is consistent with other findings. For example, we previously observed that the K978C GOF mutation in ICL3 near the base of TM9 increased the PKA sensitivity of channel activation even though this residue is nowhere near a PKA phosphorylation site in the polypeptide sequence (12). In addition, the increased basal activities of E1371-NBD2 catalytic mutants in the absence of added PKA that was mentioned in earlier studies (41, 42) and also observed here (Fig. 8) is consistent with an allosteric link between the channel open state and R domain conformation. These results and the present data support the view that the R domain is an allosteric modulator of CFTR gating whose structure and phosphorylation state (i.e. the accessibilities of its PKA sites to phosphorylation and dephosphorylation) are conformationally coupled to the channel open state. This view explains how, irrespective of their locations in the CFTR polypeptide, GOF or LOF mutations that bias the equilibrium toward or away from the open state can respectively increase or decrease the PKA sensitivity of channel activation (see also Ref. 16). Similar arguments have been proposed to explain the regulation of other ion channels and signaling proteins by phosphorylation (44, 45). In sum, the large effects of the E267 and K1060 mutations on the PKA sensitivity of channel activation imply strong coupling between the E267-K1060 electrostatic interaction at the predicted bundle interface and R domain phosphorylation. Such coupling can be explained most simply by viewing the R domain as an intrinsic allosteric modulator of channel gating whose structure and phosphorylation state are allosterically linked to the channel open conformation.
Acknowledgments
We thank Xiaowen Zhang and Binli Tao for preparing the CFTR mutants.
This study was supported by a National Institutes of Health Grant R01 DK056796 (to K. L. K.).
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ABC transporter
- ATP-binding cassette transporter
- GOF
- gain of function
- HEK 293
- human embryonic kidney 293
- TM
- transmembrane α-helix
- NBD
- nucleotide-binding domain
- Po
- open-channel probability
- Sav1866
- Staphylococcus aureus multidrug exporter
- MsbA
- Vibrio cholera lipid flippase
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- MBD
- mean burst duration.
REFERENCES
- 1. Zielenski J., Tsui L. C. (1995) Cystic fibrosis: genotypic and phenotypic variations. Annu. Rev. Genet. 2, 777–807 [DOI] [PubMed] [Google Scholar]
- 2. Davis P. B. (2006) Cystic fibrosis since 1938. Am. J. Crit. Care Med. 173, 475–482 [DOI] [PubMed] [Google Scholar]
- 3. Vergani P., Lockless S. W., Nairn A. C., Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng S. H., Rich D. P., Marshall J., Gregory R. J., Welsh M. J., Smith A. E. (1991) Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 66, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 5. Bozoky Z., Krzeminski M., Muhandiram R., Birtley J. R., Al-Zahrani A., Thomas P. J., Frizzell R. A., Ford R. C., Forman-Kay J. D. (2013) Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc. Natl. Acad. Sci. U.S.A. 110, E4427–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Csanády L., Vergani P., Gadsby D. C. (2010) Strict coupling between CFTR's catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proc. Natl. Acad. Sci. U.S.A. 107, 1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Basso C., Vergani P., Nairn A. C., Gadsby D. C. (2003) Prolonged nonhydrolytic interaction of nucleotide with CFTR's NH2-terminal nucleotide binding domain and its role in channel gating. J. Gen. Physiol. 122, 333–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aleksandrov L., Aleksandrov A. A., Chang X. B., Riordan J. R. (2002) The First Nucleotide Binding Domain of Cystic Fibrosis Transmembrane Conductance Regulator Is a Site of Stable Nucleotide Interaction, whereas the Second Is a Site of Rapid Turnover. J. Biol. Chem. 277, 15419–15425 [DOI] [PubMed] [Google Scholar]
- 9. Zhou Z., Wang X., Liu H. Y., Zou X., Li M., Hwang T. C. (2006) The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. J. Gen. Physiol. 128, 413–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mense M., Vergani P., White D. M., Altberg G., Nairn A. C., Gadsby D. C. (2006) In vivo phosphorylation of CFTR promotes formation of a nucleotide binding domain heterodimer. EMBO J. 25, 4728–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hegedüs T., Serohijos A. W., Dokholyan N. V., He L., Riordan J. R. (2008) Computational studies reveal phosphorylation-dependent changes in the unstructured R domain of CFTR. J. Mol. Biol. 37, 1052–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W., Wu J., Bernard K., Li G., Wang G., Bevensee M. O., Kirk K. L. (2010) ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 3888–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Higgins C. F. (1992) ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 8, 67–113 [DOI] [PubMed] [Google Scholar]
- 14. Locher K. P. (2009) Review. Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aleksandrov A. A., Aleksandrov L. A., Riordan J. R. (2007) CFTR (ABCC7) is a hydrolyzable-ligand-gated channel. Pflugers Arch. 453, 693–702 [DOI] [PubMed] [Google Scholar]
- 16. Kirk K. L., Wang W. (2011) A unified view of cystic fibrosis transmembrane conductance regulator (CFTR) gating: combining the allosterism of a ligand-gated channel with the enzymatic activity of an ATP-binding cassette (ABC) transporter. J. Biol. Chem. 286, 12813–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dawson R. J., Locher K. P. (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938 [DOI] [PubMed] [Google Scholar]
- 18. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gadsby D. C. (2009) Ion channels versus ion pumps: the principal difference, in principle. Nat. Rev. Mol. Cell Biol. 10, 344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Serohijos A. W., Hegedus T., Aleksandrov A. A., He L., Cui L., Dokholyan N. V., Riordan J. R. (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mornon J. P., Lehn P., Callebaut I. (2009) Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol. Life Sci. 66, 3469–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Norimatsu Y., Ivetac A., Alexander C., Kirkham J., O'Donnell N., Dawson D. C., Sansom M. S. (2012) Cystic fibrosis transmembrane conductance regulator: a molecular model defines the architecture of the anion conduction path and locates a “bottleneck” in the pore. Biochemistry 51, 2199–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dalton J., Kalid O., Schushan M., Ben-Tal N., Villà-Freixa J. (2012) New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J. Chem. Inf. Model. 52, 1842–1853 [DOI] [PubMed] [Google Scholar]
- 24. Rahman K. S., Cui G., Harvey S. C., McCarty N. A. (2013) Modeling the conformational changes underlying channel opening in CFTR. PLoS One 8, e74574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weng J-W., Fan K-N., Wang W-N. (2010) The conformational transition pathway of ATP binding cassette transporter MsbA revealed by atomistic simulations. J. Biol. Chem. 285, 3053–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doshi R., Ali A., Shi W., Freeman E. V., Fagg L. A., van Veen H. W. (2013) Molecular disruption of the power stroke in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 288, 6801–6813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Billet A., Mornon J-P., Jollivet M., Lehn P., Callebaut I., Becq F. (2013) CFTR: Effect of ICL2 and ICL4 amino acids in close proximity on the current properties of the channel. J. Cystic Fibrosis 12, 737–745 [DOI] [PubMed] [Google Scholar]
- 28. Wang W., Li G., Clancy J. P., Kirk K. L. (2005) Activating cystic fibrosis transmembrane conductance regulator channels with pore blocker analogs. J. Biol. Chem. 280, 23622–23630 [DOI] [PubMed] [Google Scholar]
- 29. Mathews C. J., Tabcharani J. A., Chang X. B., Jensen T. J., Riordan J. R., Hanrahan J. W. (1998) Dibasic protein kinase A sites regulate bursting rate and nucleotide sensitivity of the cystic fibrosis transmembrane conductance regulator chloride channel. J. Physiol. 508, 365–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 31. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., Mackerell A. D., Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma T., Thiagarajah J. R., Yang H., Sonawane N. D., Folli C., Galietta L. J., Verkman A. S. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 110, 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A., Zhou J., McCartney J., Arumugam V., Decker C., Yang J., Young C., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., Negulescu P. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 106, 18825–18830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramsey B. W., Davies J., McElvaney N. G., Tullis E., Bell S. C., Dřevinek P., Griese M., McKone E. F., Wainwright C. E., Konstan M. W., Moss R., Ratjen F., Sermet-Gaudelus I., Rowe S. M., Dong Q., Rodriguez S., Yen K., Ordoñez C., Elborn J. S., VX08–770-102 Study Group. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365, 1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fu J., Ji H. L., Naren A. P., Kirk K. L. (2001) A cluster of negative charges at the amino terminal tail of CFTR regulates ATP-dependent channel gating. J. Physiol. 536, 459–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hwang T. C., Nagel G., Nairn A. C., Gadsby D. C. (1994) Regulation of the gating of cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 91, 4698–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Powe A. C., Jr., Al-Nakkash L., Li M., Hwang T. C. (2002) Mutation of Walker-A lysine 464 in cystic fibrosis conductance regulator reveals functional interaction between its nucleotide-binding domains. J. Physiol. 539, 333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vergani P., Nairn A. C., Gadsby D. C. (2003) On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121, 17–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jih K. Y., Li M., Hwang T. C., Bompadre S. G. (2011) The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. J. Physiol. 589, 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou J. J., Li M. S., Qi J., Linsdell P. (2010) Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J Gen. Physiol. 135, 229–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Csanády L., Töröcsik B. (2014) Catalyst-like modulation of transition states for CFTR channel opening and closing: New stimulation strategy exploits nonequilibrium gating. J. Gen. Physiol. 143, 269–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seibert F. S., Linsdell P., Loo T. W., Hanrahan J. W., Clarke D. M., Riordan J. R. (1996) Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J. Biol. Chem. 271, 15139–15145 [DOI] [PubMed] [Google Scholar]
- 44. Volkman B. F., Lipson D., Wemmer D. E., Kern D. (2001) Two-state allosteric behavior in a single-domain signaling protein. Science 291, 2429–2433 [DOI] [PubMed] [Google Scholar]
- 45. Herlitze S., Zhong H., Scheuer T., Catterall W. A. (2001) Allosteric modulation of Ca2+ channels by G proteins, voltage-dependent facilitation, protein kinase C and Cavβ subunits. Proc. Natl. Acad. Sci. U.S.A. 98, 4699–4704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dawson J. E., Farber P. J., Forman-Kay J. D. (2013) Allosteric coupling between the intracellular coupling helix 4 and regulatory sites of the first nucleotide-binding domain of CFTR. PLoS One 8, e74347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Okeyo G., Wang W., Wei S., Kirk K. L. (2013) Converting nonhydrolyzable nucleotides to strong cystic fibrosis transmembrane conductance regulator (CFTR) agonists by gain of function (GOF) mutations. J. Biol. Chem. 288, 17122–17133 [DOI] [PMC free article] [PubMed] [Google Scholar]