

Background: Growth suppression by connexins can involve intercellular, transmembrane, or intracellular signaling.
Results: Determinants of connexin 37 (Cx37) transmembrane signaling, channel assembly, and function were identified.
Conclusion: Cx37 mutants lacking only intercellular signaling capacity fail to suppress cancer cell proliferation.
Significance: The uniquely potent growth suppression by Cx37 involves a protein conformation able to support intercellular, transmembrane, and intracellular signaling.
Keywords: Cell-Cell Interaction, Connexin, Gap Junction, Proliferation, Protein Targeting, Arterial Endothelium, Cell Cycle Control, Hemichannel, Intercellular Communication
Abstract
Connexin (Cx) 37 suppresses vascular and cancer cell proliferation. The C terminus and a channel able to function are necessary, and neither by itself is sufficient, for Cx37 to mediate growth suppression. Cx37 supports transmembrane and intercellular signaling by forming functional hemichannels (HCs) and gap junction channels (GJCs), respectively. Here we determined whether Cx37 with HC, but not GJC, functionality would suppress proliferation of rat insulinoma (Rin) cells comparably to wild-type Cx37 (Cx37-WT). We mutated extracellular loop residues hypothesized to compromise HC docking but not HC function (six cysteines mutated to alanine, C54A,C61A,C65A, C187A,C192A,C198A (designated as C6A); N55I; and Q58L). All three mutants trafficked to the plasma membrane and formed protein plaques comparably to Cx37-WT. None of the mutants formed functional GJCs, and Cx37-C6A did not form functional HCs. Cx37-N55I and -Q58L formed HCs with behavior and permeation properties similar to Cx37-WT (especially Q58L), but none of the mutants suppressed Rin cell proliferation. The data indicate that determinants of Cx37 HC function differ from other Cxs and that HC functions with associated HC-supported protein-protein interactions are not sufficient for Cx37 to suppress Rin cell proliferation. Together with previously published data, these results suggest that Cx37 suppresses Rin cell proliferation only when in a specific conformation achieved by interaction of the C terminus with a Cx37 pore-forming domain able to open as a GJC.
Introduction
Connexins (Cxs),2 the family of gene products comprising gap junction channels (GJCs), have been well documented as regulators of coordinated tissue function, including controlled growth. The 21 known members of this gene family, co-expressed in distinct combinations, work together to support cellular functions in an isoform- and tissue-specific manner (1). The Cxs share a common topology consisting of four transmembrane domains with amino and C termini located intracellularly. Six Cxs oligomerize to form a hemichannel (HC) that can support transmembrane signaling; HCs in neighboring cells can dock to form GJCs that support intercellular signaling (2, 3). Cxs thus facilitate coordinated tissue function using both these channel types (2, 3). In addition, Cxs regulate and are regulated by multiple intracellular signaling cascades through protein-protein interactions with the C terminus (4).
Cxs have been strongly linked to regulation of cell proliferation (for review, see Refs. 4–6). We have demonstrated that Cx37, but not Cx40 or Cx43, suppresses the proliferation of rat insulinoma (Rin) cells, extending the time spent in each phase of the cell cycle (7). Cx37 also contributes to controlled growth in vivo; when deleted from the genome of mice, collateral vasculogenesis and injury-induced angiogenesis are significantly enhanced (8). This growth-suppressive function requires that Cx37 be full length and able to form functional channels (9, 10); neither channel functionality nor the C-terminal domain by themselves is sufficient for Cx37 to mediate growth suppression of Rin cells. Whether the channel type required for Cx37-mediated growth suppression is the HC or GJC remains uncertain. Because Cx37 suppresses Rin cell proliferation at low plating densities where cell-cell contact and consequently GJC formation are rare (7, 11) and because Cx37-HCs are active in normal as well as low external [Ca2+] (11), consistent with their activity possibly contributing to growth suppression, we hypothesized that functional Cx37-HCs would be sufficient, and functional GJCs would be unnecessary, for Cx37-mediated suppression of Rin cell proliferation.
To test this hypothesis we needed a mutation that would prevent docking of otherwise functional HCs. Available data from Cx26 (12, 13) suggested that mutation of any one of the six highly conserved cysteines in the extracellular loops (ECLs) might be sufficient to prevent docking of HCs to form GJCs; data from Cx43 affirmed the importance of ECL cysteines in HC docking and showed that they were not necessary for HC function (14, 15). Based on these data, we tested Cx37-C61A,C65A (cysteines 61 and 65 in the first ECL mutated to alanine) for its ability to form functional HCs, but not functional GJCs. That functional HCs were not observed with two of six cysteines mutated suggested that determinants of Cx37 HC function differed from Cx26 and possibly from Cx43 as well.
Consequently, in the current study we tested the hypothesis that Cx37 would, like Cx43, form functional HCs, but not functional GJCs, when all six cysteines in the ECLs were mutated to alanine. However, recognizing that Cx37 might differ from both Cx26 and Cx43 in the ECL structural requirements for HC function, we also tested sites suggested by structural studies to be involved in HC docking but not necessarily ECL structure, Asn-55 and Gln-58 (16). These latter residues in the first ECL are highly conserved across the Cx family of proteins and were proposed to be responsible for HC docking to form GJCs. Specifically, it was suggested that Asn-55 forms a hydrogen bond with Leu-56 in the opposite protomer and that Gln-58 forms symmetrical hydrogen bonds with the Gln-58 residue in the opposite protomer (16). These hydrogen bonds were proposed to stabilize the docking of HCs in GJC formation (16–18). Thus, we hypothesized that structurally conservative substitutions for asparagine and glutamine at these sites would compromise the ability of Cx37 to form functional GJCs but preserve HC functionality. Therefore, we mutated Asn-55 to isoleucine (Cx37-N55I) and Gln-58 to leucine (Cx37-Q58L).
Thus, in the current study we created the Cx37-C6A, Cx37-N55I, and Cx37-Q58L mutants (where C6A indicates the multiple mutant C54A,C61A,C65A,C187A,C192A,C198A) to assess the structural determinants of Cx37 HC function and, with any that exhibited HC activity, to determine whether such activity would be sufficient for Cx37 to suppress the proliferation of Rin cells. Each mutant was tested for GJC-, HC-, and growth-suppressive function. We show that all three mutants were devoid of GJC function. Cx37-C6A did not exhibit HC activity comparable with Cx37-WT, suggesting that the determinants of Cx37 HC function differ from both Cx43 and Cx26. Cx37-N55I and Cx37-Q58L formed HCs with conductance, permeation, and open probability characteristics in Ca2+-containing and Ca2+-free solutions indistinguishable from Cx37-WT HCs. Despite these functional similarities, none of these ECL mutants suppressed proliferation of Rin cells. These results indicate that 1) the determinants of Cx37 HC function differ from both Cx26 and Cx43, and 2) despite the presence of the full-length C-terminal regulatory domain, Cx37 HC function is not sufficient to successfully suppress Rin cell proliferation.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Reagents were purchased from Sigma-Aldrich, except where noted. Anti-Cx37 antibody (αCx37-18264 (19)) was used with HRP-conjugated anti-rabbit secondary antibodies (Amersham Biosciences) for immunoblotting or with Cy3-conjugated anti-rabbit-IgG (Jackson ImmunoResearch; West Grove, PA) secondary antibodies for immunocytochemistry.
Mutant Connexin and Expression Vectors
Using the QuikChange site-directed mutagenesis kit (Stratagene, San Diego, CA), the C6A mutations were sequentially introduced into the pTRE2h-mCx37 plasmid (7) using the following oligonucleotide primers (and their respective reverse primers) (Operon Biotechnologies, Huntsville, AL): C54A, 5′-AGCAGTCTGATTTTGAGGCTAACACAGCCCAGCCGG-3′; C61A,C65A, 5′-GCCCAGCCGGGCGCCACCAACGTCGCCTATGACCAGGC-3′; C187A, 5′-GCCGGTGTTTGTGGCCCAGCGTGCGCCC-3′; C192A, 5′-CCAGCGTGCGCCCGCCCCCCACATCGTG-3′; C198A, 5-CCCCACATCGTGGACGCCTATGTCTCTCGACC-3′. Similarly, the N55I and Q58L mutations were introduced using the following primers (and their respective reverse primers) (Operon Biotechnologies, Huntsville, AL): N55I, 5′-GATTTTGAGTGTATCACAGCCCAGCCGG-3′; Q58L, 5′-GAGTGTAACACAGCCTTACCGGGCTGCACCAAC-3′).Sequences were confirmed by the Genomic Analysis and Technology Core at the University of Arizona.
Cell Culture and Expression Vectors
All cells were maintained at 37 °C in a humidified, 5% CO2 incubator. iRin37 cells (7) were cultured in Rin medium (RPMI 1640 medium with 10% FetalPlex (Gemini Bio-Products, Sacramento, CA), 300 μg/ml penicillin, 500 μg/ml streptomycin, 300 μg/ml G418 (Life Technologies), and 100 μg/ml hygromycin). iRin cells (7) were transfected using Lipofectamine (Life Technologies) with pTRE2h-mCx37-C6A, -N55I, or -Q58L plasmid following the manufacturer's instructions. Stably expressing cells were selected for by the addition of 100 μg/ml hygromycin and subsequently dilution-cloned.
Immunoblotting
Whole cell and Triton X-100-insoluble protein were isolated as described previously (9), and protein concentration was determined using the BCA Assay (Pierce Chemical). Protein samples were loaded onto 12% SDS-PAGE precast gels (Bio-Rad), electrophoresed, and then transferred onto nitrocellulose using the Trans-Blot Turbo transfer system (Bio-Rad). All blots were blocked with 5% nonfat dry milk followed by the addition of Cx37 primary antibody (1:5000) and HRP-conjugated secondary antibody (1:5000). Enhanced chemiluminescence strategies, with the SuperSignal West Dura system (Thermo Scientific), were used to visualize Cx37 expression (Kodak Image Station 2000). Cx37 expression level was quantified as described previously (11) by comparing sample intensity with a standard curve developed from known amounts of Cx37-GST fusion protein (residues 229–333) run in separate lanes of the same gel.
Immunofluorescence
Cellular localization of Cx37 was determined as described previously (9, 11). Briefly, iRin37-WT, iRin37-C6A, iRin37-N55I, and iRin37-Q58L cells, plated on glass coverslips and induced, or not, (2 μg/ml doxycycline) to express Cx37, were treated with 1 mm Sulfo-NHS-SS-Biotin (Thermo Scientific) to label lysine residues in the extracellular portions of membrane proteins. Cells were fixed in cold methanol and treated with 0.2% Triton X-100 for 30 min followed by 0.5 m NH4Cl for 15 min. Cells were then rinsed, blocked (in 4% fish skin gelatin, 1% normal goat serum, and 0.1% Triton X-100 in divalent cation-free PBS), and exposed to primary antibody for 2 h. After rinsing, secondary antibodies were applied (Cy3-conjugated, diluted 1:200 in blocking reagent for Cx37; Cy5-conjugated streptavidin, diluted 1:250 in blocking reagent, for detection of biotinylated surface proteins). ToPro3 (Life Technologies; 1:1000 of 1 mm stock) was added to iRin37-C6A, -N55I, and -Q58L cells to visualize nuclei following post-secondary antibody rinses. Labeled proteins were visualized with a Zeiss LSM 510 Meta-NLO confocal/multiphoton fluorescence microscope (lasers set at 514 nm for Cy3 and 633 nm for Cy5 and ToPro3 detection).
Electrophysiology
Electrophysiology studies were performed as described previously (11). Briefly, Cx37-WT, -C6A, -N55I, or -Q58L cells were plated at low density onto glass coverslips, induced with doxycycline for 24–48 h, placed in a custom-made chamber, and bathed in external solution (containing (in mmol/liter): 142.5 NaCl, 4 KCl, 1 MgCl2, 5 glucose, 2 sodium pyruvate, 10 HEPES, 15 CsCl, and 10 tetraethylammonium chloride) with normal [Ca2+]o (1 mm CaCl2) at room temperature. Patch pipettes were fabricated as described previously (7, 20) and back-filled with internal solution (in mmol/liter: 124 KCl, 14 CsCl, 9 HEPES, 9 EGTA, 0.5 CaCl2, 5 glucose, 9 tetraethylammonium chloride, 3 MgCl2, 5 disodium ATP). Cell pairs were used for evaluation of GJC conductance and single cells for HC conductance using discontinuous, single-electrode voltage clamp (NPI SEC-05LX) amplifiers (npi electronic GmbH, Tamm, Germany). Junctional conductance was evaluated using transjunctional voltages of 10–50 mV to reveal Cx37 GJC activity. HC activity was evaluated using square pulses of ±30 mV of variable durations with only short interruptions to evince the baseline. Longer recordings for HC activity were performed while exchanging the external solution with 5 mm EGTA-containing external solution (at least twice the chamber volume over 1–2 min) to reduce [Ca2+]o.
Dye Uptake Studies
Dye uptake experiments were completed as described previously (11). Briefly, Cx37-WT, -C6A, -N55I, or -Q58L cells were plated at low density and induced with doxycycline for 24–48 h (dox+) or not induced (dox−). All cells were rinsed with culture medium followed by external solution with normal [Ca2+]o (1 mm CaCl2) and then low [Ca2+]o (1 mm CaCl2, 5 mm EGTA). Dye solution was then added to each well for 15 min while plates were kept on ice and protected from light. Dye solution contained 1.25 mg/ml N,N,N-trimethyl-2-[methyl-(7-nitro-2,1,3-benzoxadiol-4-yl)amino] ethanaminium (NBD) (21) and 0.125 mg/ml tetramethylrhodamine dextran (rhodamine) (molecular weight 3,000: Molecular Probes) dissolved in external solution with no added CaCl2. After 15 min of exposure to dye, cells were rinsed with culture medium and normal [Ca2+]o external solution and then immediately imaged with an Olympus IX71 fluorescence microscope (Center Valley, PA). Differential interference contrast, NBD (41001HQ filter, Olympus), and rhodamine-dextran (U-MWIGA3 filter, Olympus) images were acquired using a CoolSNAP ES camera (Photometrics; Tucson, AZ) and V++ software (Digital Optics; Auckland, New Zealand). Each field imaged was scored for number of NBD-positive/rhodamine-dextran-negative cells and total number of cells within the visualized fields (Cell clusters of two or more were counted as one cell). Four fields for each experiment were combined, and the percentage of NBD-positive cells was calculated. t tests were performed to evaluate differences between non-induced and induced cells, with significance at p < 0.05.
Proliferation
As described previously (7) iRin37-WT, iRin37-C6A, iRin37-N55I, or iRin37-Q58L cells were seeded at 3 × 104 cells/well into 6-well plates. Cx37 expression was induced with doxycycline (dox+) or not (dox−) 24 h after initial plating. All experimental conditions were run in triplicate, and each experiment was run at least three times. Medium, with or without doxycycline, was refreshed every 48 h, and cells were harvested and counted every 3 days over a 15-day period. Doubling time was calculated using the following calculation and as described previously (7): doubling time = (t2 − t1) × (log2/log(q2/q1)), where t is time and q is the number of cells.
RESULTS
Expression (Fig. 1A) of each mutant and Cx37-WT was quantified by comparison with a standard curve generated using known amounts of Cx37-GST fusion protein (7, 9). Expression levels were comparable in the mutant- and WT-expressing cell lines (in fmol/μg of total protein: Cx37-WT, 6.57; Cx37-C6A, 8.70; Cx37-N55I, 5.80; Cx37-Q58L, 5.66). Cx37-WT and mutant proteins were found in the Triton X-insoluble protein fraction (Fig. 1A), suggestive of their ability to form protein plaques (22). Additionally, each Cx37 mutant protein colocalized with a biotinylated cell surface marker, forming apparent plaques in the surface membranes of doxycycline-induced cells (Fig. 1B). The non-induced cells did not show any evidence of Cx37 expression (Q58L dox− shown as representative). Collectively, these data suggest that all three mutant proteins and Cx37-WT were expressed at similar levels and localized to the plasma membrane where they formed apparent plaques in surface and appositional membranes (22, 23).
FIGURE 1.
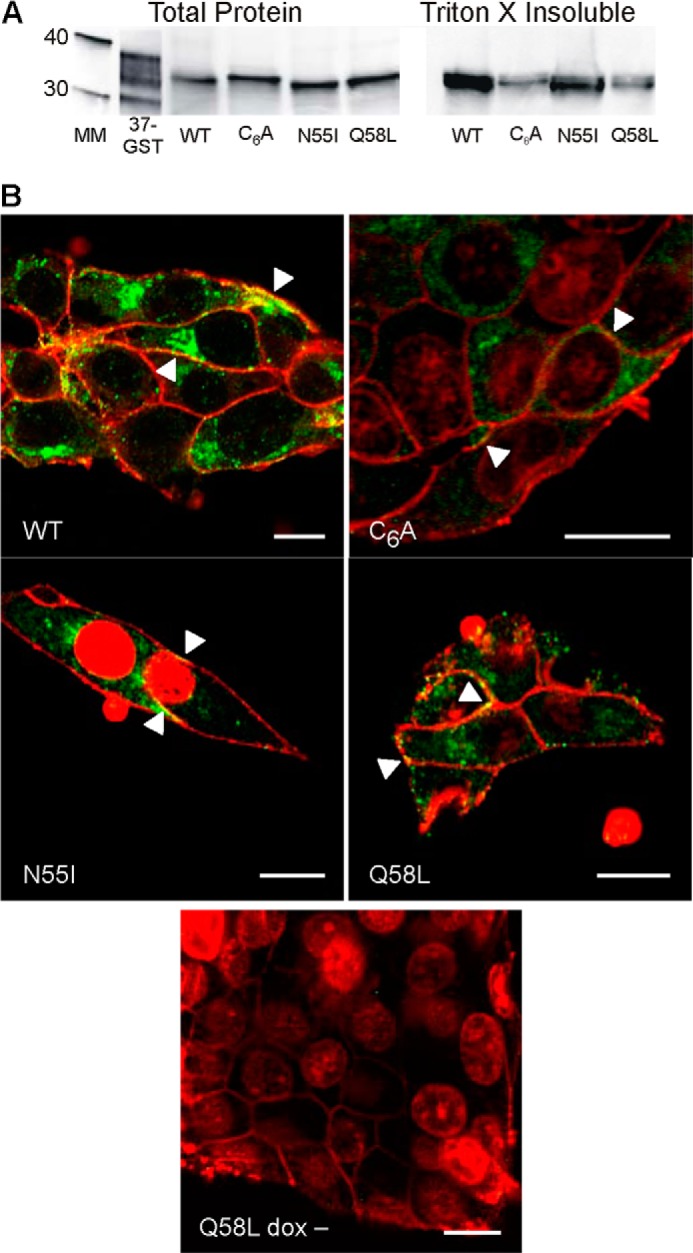
Expression and localization of Cx37 are comparable in Cx37-WT, -C6A, -N55I, and -Q58L iRin cells. A, Cx37 was detected in both the whole cell and Triton X-insoluble protein fractions of iRin cells expressing Cx37-WT, -C6A, -N55I, and -Q58L; 50 μg of total protein was loaded for each cell type. The 37-GST (GST-rCx37CT229–333, where CT indicates C terminus) lane shows positive control for antibody detection; note that the Cx37-GST fusion protein typically migrates as multiple bands (this lane was contrast-enhanced for better band visibility) (7, 9). Positions of the 30- and 40-kDa molecular mass markers are shown in the mass marker (MM) lane. B, immunocytochemistry revealed localization of WT and mutant Cx37 (green) at appositional and non-appositional membranes (arrowheads). Red staining corresponds to biotinylated proteins on the extracellular surface of the plasma membrane and to ToPro3-labeled nuclei in the C6A, N55I and Q58L images (some ToPro3-labeled nuclei are evident in unattached cells above the plane of focus). Yellow staining corresponds to Cx37 localized to the plasma membrane. Scale bars: 10 μm.
Although Cx37-WT-expressing cells were routinely electrically coupled (junctional conductance, gj = 3.7 ± 0.78 nS (n = 25)), none of the Cx37 mutant-expressing iRin cells were coupled. For each mutant, gj was not different from non-expressing cells: Cx37-C6A, 0.042 ± 0.008 nS (n = 19); Cx37-N55I, 0.053 ± 0.013 nS (n = 13); Cx37-Q58L, 0.05 ± 0.011 nS, (n = 20); non-induced Cx37-Q58L cells, 0.024 ± 0.017 nS (n = 8). This result suggests that, as expected, none of these mutants was able to form a functional GJC.
HC function was explored with voltage steps of varying duration to ± 30 mV. HC activity was observed in 11 of 24 Cx37-WT cells using this protocol. Distinct ∼500-pS events were relatively common in both normal and low [Ca2+]o conditions (Fig. 2, A and B). In addition, indistinct activity was observed, corresponding to an approximate 200-pS conductance state, which was difficult to distinguish from noise except that the amplitude of the activity diminished transiently upon switching to low [Ca2+]o (Fig. 2B). In order for HC permeation to play a role in growth suppression, these channels have to function in the presence of normal extracellular [Ca2+]. Although we previously showed such activity (11), here we examined two long duration records for differences in open probability in low versus normal [Ca2+]o. As summarized in Table 1, no obvious increase in Po was observed upon reduction of [Ca2+]o in two Cx37-WT-expressing cells.
FIGURE 2.

HC activity displayed by Cx37-WT, -N55I, and -Q58L-expressing, but not -C6A-expressing, cells is similar. A–J, distinct HC events with amplitudes of ∼500 pS were observed in normal (left column) and low [Ca2+]o (right column) in iRin cells expressing Cx37-WT (A and B), Cx37-N55I (C and D), and Cx37-Q58L (E and F), but not Cx37-C6A (G–J). Non-distinct activity centered at the ∼200-pS conductance level was evident in WT- and all mutant-expressing cells, but was diminished in low [Ca2+]o only in WT, N55I, and Q58L cells. Calibration bars (vertical/horizontal): 10 pA/1.0 s.
TABLE 1.
Open probability (Po) values for Cx37 expressing Rin cells were similar in normal and low [Ca2+]o
Po values were derived from the same cell first in normal and then low [Ca2+]o. The indicated recording times (in seconds) represent the cumulative time at +30 mV for each in each [Ca2+]o condition. NA signifies none apparent.
| Cx37 | Po in normal [Ca2+]o (time) | Po in low [Ca2+]o (time) |
|---|---|---|
| WT | 0.050 (538) | 0.051 (772) |
| 0.051 (590) | 0.193 (736) | |
| N55I | 0.020 (173) | 0.012 (765) |
| 0.017 (750) | 0.006 (1122) | |
| 0.020 (749) | 0.001 (1124) | |
| Q58L | 0.055 (758) | 0.016 (464) |
| 0.073 (624) | 0.048 (880) | |
| C6A | NA (948) | NA (2561) |
As in Cx37-WT-expressing cells, distinct 500-pS HC events as well as indistinct HC activity (∼200-pS conductance) were observed in cells expressing Cx37-N55I (Fig. 2, C and D; 5 of 19 cells) and -Q58L (Fig. 2, E and F; 14 of 40 cells). Po for Q58L was similar to Po for Cx37-WT, and for both mutants Po was unaffected by reduction of [Ca2+]o (Table 1). In contrast to the N55I and Q58L mutations, the Cx37-C6A cells lacked clearly defined ∼500-pS HC events at ± 30 mV (Fig. 2, G–J); none were observed in the 33 studied cells (11.5 h of record analyzed). In five of these cells, pulses to +30 mV lasting a total of 15.8 and 42.7 min in normal and low [Ca2+]o, respectively, revealed no distinct events (pulses to −30 mV lasting 21 and 18.7 min, respectively, were also examined with no observed events) (Table 1). Indistinct activity at ∼200 pS that did not differ between normal and low [Ca2+]o was routinely observed in these Cx37-C6A-expressing cells, consistent with instability of channel conformation under both [Ca2+]o conditions. Together, these data show that only the N55I and Q58L mutations preserved distinct transitions similar in amplitude to those displayed by Cx37-WT, and only the Q58L mutant had a similar Po.
To determine whether mutant and WT HCs were similarly permeable to large, charged molecules, we evaluated HC-mediated uptake of NBD. Fig. 3A shows representative images of HC-mediated dye uptake in each of the induced cell lines and in non-induced Cx37-WT cells. Fig. 3, B and C, summarizes data from all cell lines and experiments. The number of NBD-positive iRin37-C6A cells did not differ between expressing and non-expressing cells, indicating a lack of dye-permeable HCs. In contrast, both Cx37-N55I-expressing and Cx37-Q58L-expressing cells showed a significantly higher percentage of NBD-positive cells than their respective non-expressing counterparts, similar to Cx37-WT (Fig. 3B). Using a one-way analysis of variance with Tukey's post hoc test, we evaluated induced cell lines for possible differences; this test attributes any observed difference to the mutation, not to any combination of other differences that might exist between these cell lines (expression level, localization, regulation). There were no significant differences between the dye uptake values for Cx37-WT-, Cx37-N55I-, and Cx37-Q58L-expressing cells; however, Cx37-C6A-expressing cells were significantly less likely to take up dye than Cx37-WT-expressing cells. The range for dye-positive relative to total cells in all the individual fields counted (Fig. 3C) revealed overlapping ranges for WT-, N55I-, and Q58L-expressing cells, providing further indication of the lack of difference between these mutants and Cx37-WT.
FIGURE 3.

Cx37-WT, -N55I, and -Q58L HCs mediate NBD dye uptake; Cx37-C6A HCs do not. A, representative differential interference contrast (DIC) and NBD (NBD-M-TMA)-fluorescence images for each cell line induced to express the indicated form of Cx37 or the non-induced Cx37-WT cells. White: NBD uptake. Rhodamine images are not shown as no cells contained this dye. Scale bar, 100 μm, applies to all frames. B, NBD uptake was greater in cells induced (as compared with non-induced) to express Cx37-WT, -N55I, and -Q58L, but not Cx37-C6A. Cells counted in non-induced and induced conditions (dox−/dox+): Cx37-WT, 366/323 (n = 4); Cx37-C6A, 499/537 (n = 7); Cx37-N55I, 678/688 (n = 5); and Cx37-Q58L, 660/949 (n = 7). Error bars: S.E. * indicates significant difference between dox+ (Cx37-expressing) and dox− (non-expressing) cells within the same cell line. NS indicates no significant difference between dox+ and dox− cells. C, ratio of dye-positive to total cell number in each of the images obtained from induced Cx37-WT-, -N55I-, and -Q58L-expressing cells.
Finally, we determined whether proliferation of the iRin37-C6A, -N55I, or -Q58L cells was suppressed upon induction of Cx37 expression. Because Cx37-C6A did not form functional GJCs nor Cx37-HCs with properties similar to Cx37-WT, we expected that this mutant would not suppress proliferation of Rin cells, and it did not. However, because Cx37-N55I and -Q58L formed functional HCs whose activity could not be distinguished from Cx37-WT HCs, these mutants were expected to suppress proliferation. Surprisingly, neither of these mutants exerted an antiproliferative effect (Fig. 4) despite their similar HC properties. Indeed, the doubling time for all three of the mutants was not different from that of their respective non-expressing counterparts (Table 2). These data therefore indicate that HC function and the transmembrane signaling HCs support are not sufficient for Cx37 to suppress Rin cell proliferation.
FIGURE 4.

Expression of Cx37-C6A, -N55I, or -Q58L fails to suppress the proliferation of iRin cells. Increase in cell number over 15 days was evaluated in iRin cells induced (red filled symbols) or not (white filled symbols) to express Cx37-WT, -C6A, -N55I, or -Q58L. Proliferation was suppressed only in Cx37-WT-expressing cells. n = 3 for all groups, each experiment performed in triplicate. Error bars: S.E.
TABLE 2.
Doubling time of Rin cells induced or not to express Cx37-WT or Cx37 mutants
Only Cx37-WT altered Rin cell proliferation. Values represent the mean ± S.E.; n = 3 for each cell type.
| Cx37 | dox+ | dox− |
|---|---|---|
| WT | 8.52 ± 2.40a | 1.64 ± 0.02 |
| N55I | 2.24 ± 0.15 | 2.12 ± 0.09 |
| Q58L | 2.25 ± 0.10 | 2.43 ± 0.31 |
| C6A | 2.35 ± 0.19 | 2.42 ± 0.35 |
a Significant difference between dox + (induced) vs. dox − (non-induced) condition (t test; p < 0.05).
DISCUSSION
In previous studies exploring the mechanism of Cx37-mediated suppression of proliferation, we showed that Cx37 with mutations that rendered its channels non-functional (Cx37-T154A and Cx37-C61A,C65A) or removed the C terminus (Cx37-273tr*V5) failed to suppress proliferation of Rin cells (9–11), demonstrating that both a functional pore-forming domain and a C terminus are necessary, and neither is sufficient without the other, for Cx37 to suppress cell cycle progression and proliferation (Fig. 5). The function of both HCs and GJCs (11, 24) is prevented by the T154A and C61A,C65A mutations. In an effort to discern whether full-length Cx37 with HC function, but not GJC function, would be sufficient to mediate suppression of proliferation, we mutated highly conserved residues in the ECLs of Cxs that structural and functional data indicated would support HC function but not docking to form GJC (12–16).
FIGURE 5.
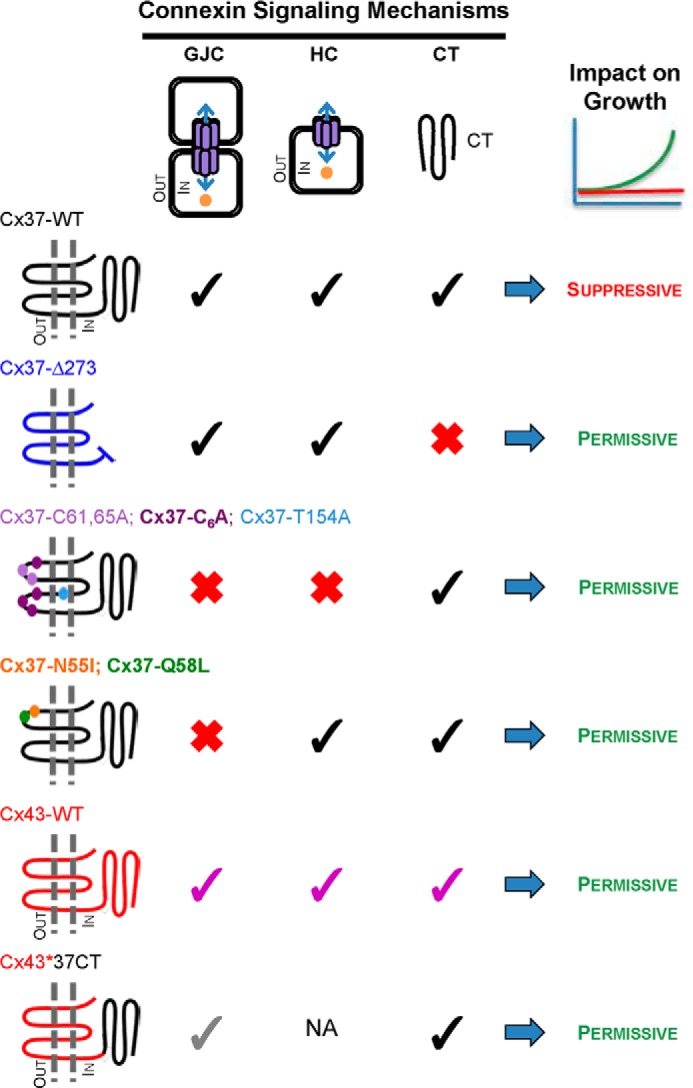
Connexin functions necessary for suppression of Rin cell proliferation. Growth-suppressive function of Cx37-WT could require functional (black checks) GJCs, HCs, or C terminus (CT), or a combination of these properties. The C terminus could influence growth through regulatory effects on channel function as well as via channel-independent mechanisms, such as through interactions with growth regulatory proteins. Truncated Cx37 (Cx37-Δ273) forms functional GJCs and HCs, but without its C terminus (red X), it fails to suppress the proliferation of Rin cells (10). Cx37-T154A (9), Cx37-C61A,C65A (11), and Cx37-C6A (present data) all retain their full-length C terminus, but none forms functional GJCs or HCs and all fail to suppress growth, indicating the necessity of channel function for growth suppression. Cx37-N55I and Cx37-Q58L fail to form functional GJCs, but retain functional HCs and their full-length C terminus, but nevertheless fail to suppress proliferation. Cx43 (purple checks) forms functional GJCs and HCs, although the properties of these channels differ from Cx37, and has a full-length C terminus with a different primary amino acid sequence from Cx37, but fails to suppress Rin cell proliferation (7). Rin cell proliferation is not suppressed by the Cx43*37CT chimera (where CT indicates C terminus) despite the presence of the Cx37 C terminus and formation of functional GJCs, albeit with unique properties (gray checks) (10, 26). NA: not available. Together, these results indicate that Cx37 exerts a growth-suppressive effect only when it is capable of all three functions, intercellular, transmembrane, and intracellular signaling, specifically those supported by the Cx37 sequence.
Three mutants were selected for study: Cx37-C6A, because Cx43 similarly mutated is unable to form functional GJCs but retains functional HCs (14, 15), and Cx37-N55I and Cx37-Q58L, because structural data stemming from Cx26 suggest these sites are involved in HC docking (16–18). We verified that all three mutants were expressed at comparable levels (7, 9, 11) and properly targeted to the plasma membrane where they formed apparent plaques; as expected, all three mutants failed to form functional GJCs. Cx37-C6A failed to form stably open HCs, but Cx37-N55I and Cx37-Q58L exhibited HC events comparable to Cx37-WT in conductivity and NBD permeability, with Cx37-Q58L demonstrating an open probability similar to Cx37-WT in both normal and low [Ca2+]o conditions. Although Cx37-N55I and Cx37-Q58L displayed WT-like HC activity, neither Cx37 mutant suppressed Rin cell proliferation comparably to Cx37-WT, thereby demonstrating that functional HCs in conjunction with a full-length C terminus are not sufficient for Cx37 to suppress Rin cell proliferation.
Cx-mediated regulation of cell proliferation can occur by channel-dependent and channel-independent mechanisms (4, 25). Previously published data indicated that channel functionality and the C-terminal domain are both necessary for growth suppression by Cx37 (7, 9, 10). These and current data indicate that neither a functional channel (HC nor combination of HC and GJC) nor the properly localized full-length C terminus is sufficient for Cx37-mediated suppression of proliferation (7, 9–11, 26). The necessity of all and sufficiency of none of the possible mechanisms for Cx-mediated growth suppression suggest that a combination of these mechanisms underlies growth suppression by Cx37. Together, the available data suggest a simple alternative model for the growth suppression by Cx37; a specific conformation of the Cx37 C terminus is required for growth suppression, a conformation that is permissive both to its interaction with cell cycle regulatory proteins as well as to the pore domain of a Cx37 protein able to form functional GJCs. This model derives strong support from Cx43*CT37 (where CT indicates C terminus). Channel data from this chimera suggest that the Cx37 C terminus does not interact with the cytoplasmic loop of Cx43 (26); subsequent NMR studies confirmed a lack of interaction between a GST-Cx37 fusion protein and the Cx43 cytoplasmic loop (10). That this chimera does not suppress Rin cell proliferation despite having a functional channel and the C terminus of Cx37 suggests that the conformation of the C terminus necessary for its interaction with cell cycle regulatory proteins is adopted only when the C terminus can interact with its own pore-forming domain. This “conformation-dependent mechanism” of growth suppression might be regarded as a hybrid of channel-independent and -dependent mechanisms in that it involves interactions of the C terminus with non-Cx regulatory proteins (widely regarded as channel-independent), but those interactions occur only when the C terminus is properly configured by a Cx37 pore-forming domain able to form a functional GJC. If this conformation-dependent mechanism were to explain Cx37-mediated growth suppression, the corollary would be that Cx37-T154A, -C6A, -C61A,C65A, -N55I, -Q58L, and Cx43*CT37 all fail to suppress proliferation because they do not support adoption of the proper conformation by the C terminus and, therefore, do not support interaction with the cell cycle regulatory proteins required for growth suppression. This is an intriguing possibility because the conformation of the C terminus and its consequent interactions with other regulatory proteins would then be expected to be regulated in a phosphorylation-dependent manner, as suggested by the work of others (27–29).
In addition to showing that Cx37 HC function is not sufficient to suppress the growth of Rin cells, the studies presented herein also demonstrate that the determinants of HC assembly and function differ between Cx26, Cx43, and Cx37. The structural stability of the ECLs has been shown to impact oligomerization (30), docking (12, 13, 16–18), and both HC and GJC function (14, 15). For Cx26, disulfide bonds formed between ECL cysteines in the endoplasmic reticulum are essential for oligomerization, subsequent HC trafficking to the plasma membrane, and HC docking to form GJCs (12). Less clear is whether these disulfide bonds are required for HC function once oligomerized as reconstituted Cx26 HCs are functional in the presence of 10 mm DTT (31), which might be expected to break those bonds. For Cx43, ECL stability in the endoplasmic reticulum is conferred by association with chaperone proteins that guide Cx monomers to the trans-Golgi network (32), where disulfide bonds between ECL cysteines form and oligomerization occurs (30). However, disulfide bond formation in Cx43 is not necessary for trafficking to the plasma membrane nor HC function, only for HC docking to form GJCs (14, 15). Cx37 appears to display properties of both these Cxs. Like Cx26, Cx37 oligomerizes in the endoplasmic reticulum (33), but unlike Cx26, disulfide bond formation between ECL cysteines is not necessary for oligomerization (14, 15); if chaperone proteins interact with Cx37 ECLs, they clearly do not interfere with oligomerization in the endoplasmic reticulum, like they do for Cx43 (32). In contrast, like Cx43, Cx37 traffics to the cell surface with and without disulfide bond formation between ECL cysteines; however, unlike Cx43, Cx37 without ECL cysteines is unable to form obviously functional HCs. That Cx37 behaves differently from either Cx26 or Cx43 is perhaps not surprising given the many functional differences between these proteins (conductance, selectivity (20, 34), HC function in normal [Ca2+]o, potent growth suppression (7)), but certainly, our data and those of others show that structure-function behaviors of Cx37 are not readily predicted by structure-function relationships determined from other Cxs, even another α-Cx, Cx43 (33).
In summary, our data show that the determinants of Cx37 HC function differ from both Cx26 and Cx43. Further, our data show that Cx37 mutants that form functional HCs with WT-like permeation and open probability characteristics and retaining a full-length C-terminal domain, but lacking functional GJCs, are not growth-suppressive when expressed in Rin cells. Taken together with previously published data (7, 9–11), these results suggest that Cx37 suppresses Rin cell proliferation only when it adopts a specific conformation achieved by interaction of the C-terminal domain with a Cx37 pore-forming domain able to function as a GJC. It is likely that this conformation uniquely supports interaction of the C terminus with cell cycle regulatory proteins that effect growth suppression. The identity of these regulatory proteins remains to be discovered.
Acknowledgment
We thank Tasha Pontifex for occasional technical assistance and insights on Cx37-mediated growth suppression.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL058732 (to J. M. B.) and T32HL007249 (to M. E. G. and J. M. B.). This work was also supported by The Finley and Florence Brown Endowed Research Award from the Sarver Heart Center, University of Arizona (to M. E. G.).
- Cx
- connexin
- GJC
- gap junction channel
- HC
- hemichannel
- Rin
- rat insulinoma
- ECL
- extracellular loop
- NBD
- N,N,N-trimethyl-2-[methyl-(7-nitro-2,1,3-benzoxadiol-4-yl)amino] ethanaminium
- dox
- doxycycline
- pS
- picosiemens
- nS
- nanosiemens
- iRin
- inducible Rin.
REFERENCES
- 1. Söhl G., Willecke K. (2004) Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232 [DOI] [PubMed] [Google Scholar]
- 2. Goodenough D. A., Paul D. L. (2003) Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 4, 285–294 [DOI] [PubMed] [Google Scholar]
- 3. Harris A. L. (2001) Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 34, 325–472 [DOI] [PubMed] [Google Scholar]
- 4. Kardami E., Dang X., Iacobas D. A., Nickel B. E., Jeyaraman M., Srisakuldee W., Makazan J., Tanguy S., Spray D. C. (2007) The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 94, 245–264 [DOI] [PubMed] [Google Scholar]
- 5. Vinken M., Decrock E., Leybaert L., Bultynck G., Himpens B., Vanhaecke T., Rogiers V. (2012) Non-channel functions of connexins in cell growth and cell death. Biochim. Biophys. Acta 1818, 2002–2008 [DOI] [PubMed] [Google Scholar]
- 6. Hervé J. C., Derangeon M. (2013) Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 352, 21–31 [DOI] [PubMed] [Google Scholar]
- 7. Burt J. M., Nelson T. K., Simon A. M., Fang J. S. (2008) Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 295, C1103–C1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fang J. S., Angelov S. N., Simon A. M., Burt J. M. (2011) Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am. J. Physiol. Heart Circ. Physiol. 301, H1872–H1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Good M. E., Nelson T. K., Simon A. M., Burt J. M. (2011) A functional channel is necessary for growth suppression by Cx37. J. Cell Sci. 124, 2448–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nelson T. K., Sorgen P. L., Burt J. M. (2013) Carboxy terminus and pore-forming domain properties specific to Cx37 are necessary for Cx37-mediated suppression of insulinoma cell proliferation. Am. J. Physiol. Cell Physiol. 305, C1246–C1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Good M. E., Ek-Vitorín J. F., Burt J. M. (2012) Extracellular loop cysteine mutant of Cx37 fails to suppress proliferation of rat insulinoma cells. J. Membr. Biol. 245, 369–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dahl G., Levine E., Rabadan-Diehl C., Werner R. (1991) Cell/cell channel formation involves disulfide exchange. Eur. J. Biochem. 197, 141–144 [DOI] [PubMed] [Google Scholar]
- 13. Foote C. I., Zhou L., Zhu X., Nicholson B. J. (1998) The pattern of disulfide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. J. Cell Biol. 140, 1187–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bao X., Chen Y., Reuss L., Altenberg G. A. (2004) Functional expression in Xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J. Biol. Chem. 279, 9689–9692 [DOI] [PubMed] [Google Scholar]
- 15. Tong D., Li T. Y., Naus K. E., Bai D., Kidder G. M. (2007) In vivo analysis of undocked connexin43 gap junction hemichannels in ovarian granulosa cells. J. Cell Sci. 120, 4016–4024 [DOI] [PubMed] [Google Scholar]
- 16. Nakagawa S., Maeda S., Tsukihara T. (2010) Structural and functional studies of gap junction channels. Curr. Opin. Struct. Biol. 20, 423–430 [DOI] [PubMed] [Google Scholar]
- 17. Maeda S., Nakagawa S., Suga M., Yamashita E. (2009) Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 458, 597–602 [DOI] [PubMed] [Google Scholar]
- 18. Gong X. Q., Nakagawa S., Tsukihara T., Bai D. (2013) A mechanism of gap junction docking revealed by functional rescue of a human-disease-linked connexin mutant. J. Cell Sci. 126, 3113–3120 [DOI] [PubMed] [Google Scholar]
- 19. Simon A. M., Chen H., Jackson C. L. (2006) Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun. Adhes. 13, 61–77 [DOI] [PubMed] [Google Scholar]
- 20. Ek-Vitorín J. F., Burt J. M. (2005) Quantification of gap junction selectivity. Am. J. Physiol. Cell Physiol. 289, C1535–C1546 [DOI] [PubMed] [Google Scholar]
- 21. Bednarczyk D., Mash E. A., Aavula B. R., Wright S. H. (2000) NBD-TMA: a novel fluorescent substrate of the peritubular organic cation transporter of renal proximal tubules. Pflugers Arch. 440, 184–192 [DOI] [PubMed] [Google Scholar]
- 22. Musil L. S., Goodenough D. A. (1991) Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol. 115, 1357–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larson D. M., Seul K. H., Berthoud V. M., Lau A. F., Sagar G. D., Beyer E. C. (2000) Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol. Cell Biol. Res. Commun. 3, 115–121 [DOI] [PubMed] [Google Scholar]
- 24. Derouette J. P., Desplantez T., Wong C. W., Roth I., Kwak B. R., Weingart R. (2009) Functional differences between human Cx37 polymorphic hemichannels. J. Mol. Cell Cardiol. 46, 499–507 [DOI] [PubMed] [Google Scholar]
- 25. Vinken M., Decrock E., De Vuyst E., Ponsaerts R., D'hondt C., Bultynck G., Ceelen L., Vanhaecke T., Leybaert L., Rogiers V. (2011) Connexins: sensors and regulators of cell cycling. Biochim. Biophys. Acta 1815, 13–25 [DOI] [PubMed] [Google Scholar]
- 26. Ek-Vitorin J. F., Burt J. M. (2013) Structural basis for the selective permeability of channels made of communicating junction proteins. Biochim. Biophys. Acta 1828, 51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larson D. M., Wrobleski M. J., Sagar G. D. V., Westphale E. M., Beyer E. C. (1997) Differential regulation of connexin43 and connexin37 in endothelial cells by cell density, growth, and TGF-β-1. Am. J. Physiol. 272, C405–C415 [DOI] [PubMed] [Google Scholar]
- 28. Morel S., Burnier L., Roatti A., Chassot A., Roth I., Sutter E., Galan K., Pfenniger A., Chanson M., Kwak B. R. (2010) Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis. 31, 1922–1931 [DOI] [PubMed] [Google Scholar]
- 29. Grosely R., Kopanic J. L., Nabors S., Kieken F., Spagnol G., Al-Mugotir M., Zach S., Sorgen P. L. (2013) Effects of phosphorylation on the structure and backbone dynamics of the intrinsically disordered connexin43 C-terminal domain. J. Biol. Chem. 288, 24857–24870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koval M., Molina S. A., Burt J. M. (2014) Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 588, 1193–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buehler L. K., Stauffer K. A., Gilula N. B., Kumar N. M. (1995) Single channel behavior of recombinant β2 gap junction connexons reconstituted into planar lipid bilayers. Biophys. J. 68, 1767–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das S., Smith T. D., Sarma J. D., Ritzenthaler J. D., Maza J., Kaplan B. E., Cunningham L. A., Suaud L., Hubbard M. J., Rubenstein R. C., Koval M. (2009) ERp29 restricts Connexin43 oligomerization in the endoplasmic reticulum. Mol. Biol. Cell 20, 2593–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith T. D., Mohankumar A., Minogue P. J., Beyer E. C., Berthoud V. M., Koval M. (2012) Cytoplasmic amino acids within the membrane interface region influence connexin oligomerization. J. Membr. Biol. 245, 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weber P. A., Chang H. C., Spaeth K. E., Nitsche J. M., Nicholson B. J. (2004) The permeability of gap junction channels to probes of different size is dependent on connexin composition and permeant-pore affinities. Biophys. J. 87, 958–973 [DOI] [PMC free article] [PubMed] [Google Scholar]