

Background: MicroRNAs have the potential to influence placental development.
Results: We have used a combination of in silico analysis and molecular techniques to identify growth-regulatory microRNAs.
Conclusion: miR-145, miR-377, and let-7a regulate placental growth.
Significance: Normal regulation of placental growth is a prerequisite for normal fetal development. Understanding the role of miRs in this process may lead to novel therapies for fetal growth complications.
Keywords: Developmental Factor, Gene Regulation, Insulin-like Growth Factor (IGF), MicroRNA (miRNA), Placenta, Proliferation, Reproduction, Trophoblast
Abstract
Placental cell growth depends on an adaptable combination of an endogenous developmental program and the exogenous influence of maternal growth factors, both of which may be influenced by microRNA (miR)-dependent effects on gene expression. We have previously shown that global miR suppression in placenta accelerates proliferation and enhances levels of growth factor signaling mediators in cytotrophoblast. This study aimed to identify miRs involved in regulating placental growth. An initial array revealed 58 miR species whose expression differs between first trimester, when cytotrophoblast proliferation is rapid, and term, by which time proliferation has slowed. In silico analysis defined potential growth-regulatory miRs; among these, hsa-miR-145, hsa-miR-377, and hsa-let-7a were predicted to target known placental growth genes and were higher at term than in the first trimester, so they were selected for further analysis. Overexpression of miR-377 and let-7a, but not miR-145, in first trimester placental explants significantly reduced basal cytotrophoblast proliferation and expression of ERK and MYC. PCR arrays, in silico analysis, Western blotting, and 3′-UTR luciferase reporter assays revealed targets of miR-145 within the insulin-like growth factor axis. Analysis of proliferation in placental explants overexpressing miR-145 demonstrated its role as a mediator of insulin-like growth factor-induced trophoblast proliferation. These findings identify miR-377 and let-7a in regulation of endogenous cell growth and miR-145 in the placental response to maternal stimulation and will aid the development of therapeutic strategies for problem pregnancies.
Introduction
Fetal growth depends on the ability of the placenta to supply nutrients adequate to meet fetal demand, which increases as gestation progresses. Placental growth, especially in early gestation, is a prerequisite of a high-capacity transport interface. The outermost syncytiotrophoblast layer of the placenta has a high turnover, with terminally differentiated and apoptotic elements shedding continuously into maternal circulation. Underlying cytotrophoblast progenitor cells divide, differentiate, and fuse with the syncytium, so their proliferation is of obvious importance for placental growth, especially during the first trimester, although turnover slows as pregnancy progresses (1). Altered rates of cytotrophoblast proliferation are associated with different pathologies; levels are enhanced with increased fetal growth (macrosomia) and diminished in fetal growth restriction (2). Factors in maternal circulation, such as the insulin-like growth factors (IGFs)2 and transforming growth factor β (TGFβ), coordinately stimulate proliferation, differentiation, and survival (3, 4) through the activation of multiple kinases (3–5) and phosphatases (5).
Gene expression can be regulated by short (18–22-nucleotide) non-coding RNAs, microRNAs (miRs), derived from long primary transcripts (pre-miRs) through sequential processing by two enzymes, Drosha and Dicer, and then incorporated into the RNA silencing complex, where they target homologous mRNAs. In mice, loss or inactivation of Dicer leads to multiple developmental defects (6, 7), and we have demonstrated that in human placenta, cytotrophoblast proliferation is increased following Dicer knockdown (8); however, the individual miRs responsible for these effects are unknown. Here we profiled placentas with different rates of trophoblast proliferation (early versus late pregnancy) to uncover differentially expressed miRs. In silico network analysis identified miRs that influence the expression of components of nodal signaling pathways, and functional studies revealed a role for three of these miRs in regulating placental growth.
EXPERIMENTAL PROCEDURES
Tissue Culture
All tissue was collected following maternal informed consent with approval from our Local Research Ethics Committee. Late first trimester (8–12-week) placentas were obtained at elective surgical or elective medical termination and dissected under sterile conditions into 5-mm3 fragments. Three pieces, selected at random, were transferred into a 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine and further dissected into 5-mm3 explants for immediate use in all experimental procedures described below.
Term (37–42-week) placentas were collected from elective Caesarean sections within 30 min of delivery. Three areas of each placenta were sampled using a transparent sheet bearing a systematic array of biopsy windows, and then chorionic, non-anchoring villi were dissected from these areas and carefully rinsed in sterile PBS for use in experiments to profile miR expression.
MicroRNA Array Profiling
Total RNA was isolated from first trimester or term tissue using a mirVANA miR isolation kit (Ambion). Quality was verified by an Agilent 2100 Bioanalyzer profile. Aliquots of RNA from all first trimester and term samples were pooled to generate a common reference sample used to normalize data across all arrays in the study. 800 ng of total RNA from individual and reference samples was labeled with Hy3TM and Hy5TM fluorescent label, respectively, using the miRCURYTM LNA Array power labeling kit (Exiqon) following the procedure described by the manufacturer. The Hy3TM-labeled samples and a Hy5TM-labeled reference RNA sample were mixed pairwise and hybridized to the miRCURYTM LNA array version 11.0 (Exiqon), which contains capture probes targeting all human miRs registered in the miRBASE version 13.0 at the Sanger Institute (9). The hybridization was performed according to the miRCURYTM LNA array manual using a Tecan HS4800 hybridization station. After hybridization, the microarray slides were scanned and stored at ozone <2.0 ppb to prevent bleaching of the fluorescent dyes. The microarray slides were scanned (Agilent G2565BA Microarray Scanner System, Agilent Technologies, Inc.), and image analysis was carried out using ImaGene version 8.0 software (BioDiscovery, Inc.). The quantified signals were background-corrected (Normexp with offset value of 10 (10)) and normalized using the global Lowess (locally weighted scatterplot smoothing) regression algorithm. The data were exported to Microsoft Excel worksheets, log2-transformed, and analyzed for differentially expressed miRs using Significance Analysis of Microarrays (SAM; version 3.01, Stanford University, Stanford, CA) in a two-class unpaired design. From SAM, the -fold change for each miR with the associated q value was determined. Only miRs with a significance of p < 0.005 between the groups (first trimester and term) were included in the heat map analysis, and these all fulfill the Bonferroni correction (a safeguard against false positives).
Quantitative RT-PCR-based Analysis of miRs
Array data were validated by assessing the expression of individual miRs using the miRCURYTM LNA Universal RT microRNA PCR system (Exiqon) following the manufacturer's instructions. Briefly, 25 ng of RNA was reverse-transcribed using Universal cDNA Synthesis kit (Exiqon) which allows polyadenylation and reverse transcription of all miRs within the sample into cDNA in a single reaction step. Individual miRs were then detected using LNA-enhanced miR-specific primer sets (Exiqon; Table 1). The relative amount of miR was normalized to 5 S rRNA, and -fold changes in miR expression from control (mean of all ΔCt values) were calculated as 2(−ΔΔCt), where ΔΔCt = ΔCt control − ΔCt treated sample and ΔCt = CtmiR − Ct5 S rRNA. PCRs were performed in duplicate using a Stratagene MX3000P real-time PCR machine. Data from first trimester and term placentas were compared using the Mann-Whitney U test.
TABLE 1.
miR-specific primers
| miR | Target sequence |
|---|---|
| Hsa-miR-29a | UAGCACCAUCUGAAAUCGGUUA |
| Hsa-miR-145 | GUCCAGUUUUCCCAGGAAUCCCU |
| Hsa-miR-675 | UGGUGCGGAGAGGGCCCACAGUG |
| Hsa-miR-377 | AUCACACAAAGGCAACUUUUGU |
| Hsa-let-7a | CUAUACAAUCUACUGUCUUUC |
| Hsa-miR-125b | ACGGGUUAGGCUCUUGGGAGCU |
| Hsa-miR-143 | UGAGAUGAAGCACUGUAGCUC |
| Hsa-miR-143-3p | GGUGCAGUGCUGCAUCUCUGGU |
Computational Analysis of miR Biological Functions and Gene Networks
A data set containing miR expression profiles of first trimester and term placental samples (generated by miR array analysis) was uploaded into Ingenuity Pathway Analysis (IPA; Ingenuity® Systems).
Functional Analysis
The functional analysis tool identified the most relevant biological functions and the right-tailed Fisher's exact test was used to calculate the probability that each of the biological functions assigned to the data set is due to chance alone. Data were expressed as inverse log (−log) of the p value. Only biological functions with −log (p value) > 1.3 (equivalent to p < 0.05) were considered for further analysis.
Network Generation
Differentially expressed miRs were overlaid in the Ingenuity® Knowledge Base onto a global molecular network to algorithmically generate a defined network (limited to 35 molecules) based on connectivity. Larger, merged networks were then generated using information contained in the Ingenuity® Knowledge Base. miRs, target genes, and gene products are represented as nodes, and the biological relationship between two nodes is represented by an edge (line). All edges are supported by at least one reference from the published literature or from canonical information stored in the Ingenuity® Knowledge Base. The intensity of the node color indicates the degree of up-regulation (red) or down-regulation (green).
Placental Cell Screen
To assess the expression of miRs in different populations of cells within the human placenta, total RNA was extracted from placental tissue homogenates that contain all of the different cell types present in the human placenta or from a panel of different placental cell lines or primary cells. The following cells were used. BeWo, JAR, and JEG cell lines were cultured as described previously and were used as models for villous trophoblast (11); the Swan-71 cell line was used to represent extravillous trophoblast cells (12). The primary cells used were cytotrophoblast and stromal cells freshly isolated from human placenta using protocols described previously (13, 14). Briefly, placental tissue was dissected, weighed, and washed thoroughly in MEM. Approximately 6 g of tissue was transferred to 20 ml of MEM containing 0.125% (v/v) trypsin, 1.3 mm EDTA (Invitrogen), and 0.4 mg/ml DNase I, grade I (Sigma), and incubated for 35 min at 37 °C, with occasional resuspension to remove trophoblast cells. Cells were harvested, and the enzymatic treatment was repeated. Remaining tissue pieces were removed by filtration (100 μm) and used for subsequent isolation of stromal cells. Trophoblast cells were loaded on a Percoll gradient, and after centrifugation at 1800 × g for 30 min, cytotrophoblast cells were collected from the 30–45% range. Cells were pelleted, resuspended in serum-free DMEM/F-12, and plated on Matrigel-coated flasks. For stromal cell isolations, tissue free from trophoblast was washed (3 × 5 min) in DMEM containing 2.5 mg/ml collagenase and 2 mg/ml hyaluronidase and incubated at 37 °C for 3 h. The suspension was allowed to settle under gravity and centrifuged for 20 min at 700 × g, and the resulting pellet was resuspended in 3 ml of MEM. The cell suspension was then loaded onto 25/60% Percoll and centrifuged for 30 min at 670 × g. The band of stromal cells and aggregates was removed from the Percoll, added to 35 ml of MEM, and centrifuged for 15 min at 100 × g. The pellet was then resuspended in DMEM, and cells were plated onto flasks coated with rat tail collagen (BD Biosciences). RNA was extracted from cells, and levels of miRs were assessed by qPCR as described previously.
Gain of Function Analysis
Overexpression of Pre-miR Mimetics in First Trimester Tissue
Pre-miR precursors for hsa-miR-377, hsa-let-7a, and hsa-miR-145 (Table 2) (Ambion) were transfected into placental explants (200–500 nm) with an Amaxa Nucleofector using basic primary mammalian epithelial cell Nucleofector buffer solution and Program U007 (Amaxa Biosystems) as described previously (15). Following transfection, first trimester tissue explants were cultured on 1% agarose-coated 24-well tissue culture plates for up to 96 h in 20% O2 at 37 °C. Overexpression of the pre-miRs was confirmed by qPCR using specific primers as described above. The effect of the three pre-miR sequences was compared with that of three controls: untreated tissue (control), tissue exposed to transfection procedure alone (mock), or tissue transfected with CyTM3-labeled pre-miR precursor negative control (Pre-C; 200–500 nm; Ambion), which has no identifiable effects on expression of known mammalian miRs. These controls formed the baselines for the evaluation of the effect of pre-miR precursors on protein expression and cell function.
TABLE 2.
miR precursor mimetic sequences
| miR | Mature miR sequence |
|---|---|
| Hsa-miR-145 | GUCCAGUUUUCCCAGGAAUCCCU |
| Hsa-miR-377 | AUCACACAAAGGCAACUUUUGU |
| Hsa-let-7a | UGAGGUAGUAGGUUGUAUAGUU |
Analysis of Cell Proliferation
Placental explants transfected with pre-miR mimetics or negative controls were maintained in culture for 48 h. BrdU (100 μm) and vehicle or IGF-I (10 nm) were added, and then explants were cultured for a further 24 h. Tissue was fixed and processed for immunohistochemistry, and levels of proliferation were assessed using monoclonal anti-Ki67 (MIB-1 clone, 1:100; DakoCytomation Ltd., Cambridgeshire, UK) or anti-BrdU (clone BU-33, 1:500; Sigma) antibodies followed by biotinylated goat anti-mouse IgG (1:200; DakoCytomation Ltd.) antibody and avidin-peroxidase. Levels of cytotrophoblast proliferation were then determined as described previously (3) and expressed as a percentage of total cytotrophoblast number. Comparisons between groups were made using one-way analysis of variance followed by planned contrasts. Data were considered significant at p < 0.05.
Immunohistochemistry
Freshly isolated placental tissue or tissue exposed to miR mimetics for up to 48 h was fixed in 4% paraformaldehyde and processed for immunohistochemistry analysis as described previously (15). Expression of key signaling molecules was examined using rabbit polyclonal ERK1/2 antibody (1:50; Cell Signaling Technologies Inc.) or mouse monoclonal c-MYC (1:100) followed by incubation with biotinylated swine anti-rabbit IgG or rabbit anti-mouse IgG (DakoCytomation, Cambridgeshire, UK). Staining was visualized using the avidin-peroxidase method with hemotoxylin counterstain as described previously (3).
Western Blotting
To confirm the effect of miR overexpression on protein expression, 48-h post-transfection lysates of placental explants were prepared in radioimmune precipitation assay buffer as described previously (5). 50 μg of protein from each sample was resolved by SDS-PAGE and transferred to nitrocellulose membranes for Western blotting with antiserum specific for ERK1/2 (rabbit polyclonal, 1:500), c-MYC (rabbit polyclonal, 1:500), p38 (rabbit polyclonal, 1:1000), and cyclin D1 (mouse monoclonal, 1:1000), all from Cell Signaling Technologies Inc., or IGF1R (1:1000, rabbit polyclonal; Santa Cruz Biotechnology, Inc. and InsightBio). Immune complexes were visualized by probing with HRP-conjugated anti-rabbit IgG or HRP-conjugated anti-mouse IgG followed by ECL. ImageJ software was used to quantitate bands. Membranes were stripped (0.1 m glycine HCl, pH 2.5) and reprobed with an antibody specific for β-actin (1:5000; Sigma clone AC-15) in order to confirm equal protein loading.
mRNA Expression Profiling
Total RNA was isolated from first trimester placental explants exposed to pre-miR-145 (500 nm) for 48 h, and 200 ng of RNA was converted to cDNA using the AffinityScript cDNA synthesis kit (Stratagene), as described above. cDNA from five individual experiments was pooled and then applied to a PI3K-AKT Signaling Pathway RT2 Profiler PCR array (SABiosciences, Qiagen, UK) and quantified by qPCR using a Stratagene MX3000P real-time PCR machine and Stratagene Brilliant SYBR Green I qPCR Mastermix, with 5-carboxy-x-rhodamine as a passive reference dye. mRNA levels were analyzed using RT2 Profiler PCR data analysis software (version 3.5; SABiosciences, Qiagen). Data were normalized against a panel of housekeeping genes (Hprt, Hsp90aba, and Gapdh) present within the array plates, and expressed relative to control (negative pre-miR control). Changes were considered significant when they varied by ≥2-fold. Western blotting on individual samples was performed to validate the array and to confirm that changes in mRNA expression were accompanied by a change in protein expression.
Assessment of miR/Target Interaction
pmiRTarget vectors containing sense or mutated MAPK1 and MYC 3′-UTR sequences cloned downstream of the firefly luciferase gene and an empty vector control were purchased from OriGene Technologies. Vectors containing 3′-UTR constructs for IGF1R were not commercially available and therefore were prepared as follows.
A 42-bp oligonucleotide and a complementary antisense strand were designed using TargetScan to correspond to the highly conserved region of IGF1R 3′-UTR to which miR-145 is predicted to bind (3804–3810 bp, ATTATTATTTGGGGGAACTGGAC). Control oligonucleotides containing a mutated miR-145 seed region within the IGF1R 3′-UTR (ATTATTATTTGGGGGAAAAGGA) were included as a control. piRGLO Dual Luciferase miRNA target expression vector was linearized with PmeI and XbaI (Roche Applied Science) restriction enzymes, and oligonucleotides (4 ng) were annealed to the linearized vector (50 ng) using T4 DNA ligase (New England Biolabs). Following ligation, the vector was transformed into XL-10 Gold Ultra competent cells (Agilent Technologies) and then purified using a Qiagen miniprep kit (Thermo Scientific). NotI digestion was performed to confirm that the sequences had been inserted correctly.
To assess interaction between miRs and their candidate target genes, 3′-UTR vectors (50 ng) and a pSV-β-galactosidase control vector (Promega) were transfected into first trimester placental explants alone or in combination with non-targeting or miR-specific mimics (100 nm). Untreated and mock-transfected samples and the empty pmiR-GLO vector were included as controls. 24 h after transfection, tissue was washed twice in PBS and lysed with passive lysis buffer (Promega). Relative amounts of Firefly luciferase were read using the Orion L microplate luminometer (Titertek) using the luciferase reporter assay (Promega) in accordance with the manufacturer's instructions. Levels of β-galactosidase were assessed using the mammalian β-galactosidase assay kit (Thermo Scientific). Levels of luciferase were normalized to β-galactosidase in each sample to control for transfection efficiency.
RESULTS
Differential Expression of miRs in First Trimester and Term Human Placenta
miRs with the potential for regulating growth were initially identified by array-profiling placentas from early and last pregnancy because the rate of trophoblast proliferation is known to be exponentially higher in first trimester than at term (16, 17). 58 miRs were differentially expressed (Fig. 1); of these, 41 were higher at term than in first trimester. Confirmatory qPCR analysis was carried out on eight miRs showing elevated expression in term tissue and one with the opposite pattern of expression (Fig. 2).
FIGURE 1.
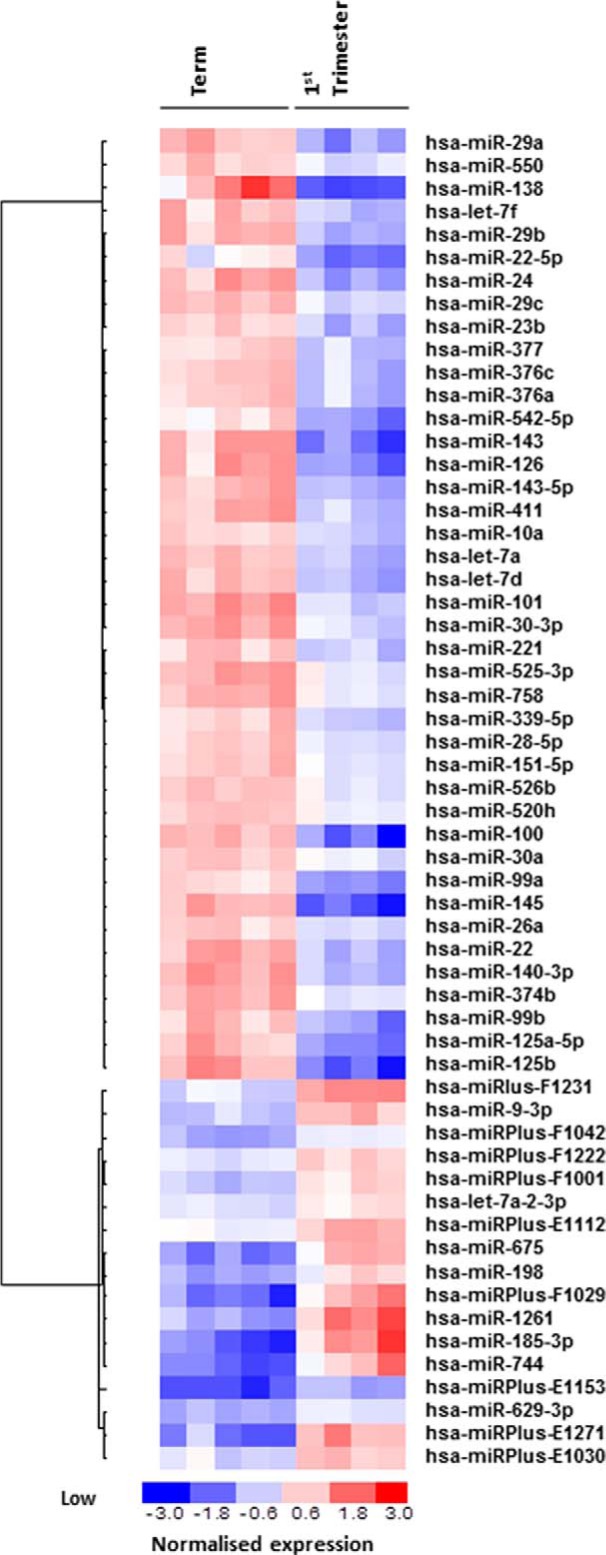
Differential expression of miRs between first trimester and term human placental tissue. Total RNA was isolated from fresh first trimester (n = 5) and term (n = 5) human placental tissue and applied to a miRCURYTM LNA array (version 11.0 hsa; Exiqon). The heat map diagram shows the result of the two-way hierarchical clustering of miRs and samples. Each row represents a miR, and each column represents a sample. The color scale shown at the bottom illustrates the relative expression level of a miR across all samples; red represents an expression level above the mean, and blue represents expression lower than the mean. The analysis was performed on log2(Hy3/Hy5) signal intensity ratios (where Hy3 represents the sample and Hy5 represents the pool control consisting of equal aliquots of RNA from each sample) that passed the filtering criteria on variation across samples; p < 0.005. miRPlus sequences are licensed (Exiqon) human sequences not yet annotated in miRBase.
FIGURE 2.

Validation of miRNA arrays. Total RNA was isolated from fresh first trimester and term human placental tissue, and qPCR analysis was performed, using individual miRNA primer sets for hsa-miR-29a (A), hsa-let-7a (B), hsa-miR-125b (C), hsa-miR-145 (D), hsa-miR-143 (E), hsa-miR-143–3p (F), hsa-miR-377 (G), and hsa-miR-675 (H). Data were normalized to 5 S rRNA expression and are presented as median and range mRNA and were analyzed by the Mann-Whitney U test. Data were considered significant if p < 0.05. Results shown are representative of at least five independent experiments.
Altered miRs Are Significantly Associated with Mitogenic Signaling Pathways
IPA was used to identify genetic networks involving differentially expressed miRs. Seven individual networks were identified (data not shown), but the majority of miRs within the functional headings of cell-cell signaling and interaction, cellular development, and cellular growth and proliferation (Fig. 3A) fell into three genetic networks, which, in turn, merged into a single larger interacting network (Fig. 3B). The large network is bridged by nodal molecules, such as mitogen-activated protein kinase (MAPK1/ERK-2), MYC, NFκB, and AKT; MAPK1/ERK-2 and MYC (highlighted by blue squares in Fig. 3B) are recognized components of promitogenic signaling pathways (18–20).
FIGURE 3.

Functional classifications and network generation of altered miRs and their potential targets. A, the data set containing the altered miRs obtained from miRCURYTM LNA array was uploaded into IPA software, and the altered miRs were classified into significantly enriched biological functions. y axis values represent an IPA network score (equal to the −log(p value), where a score of 1.3 (the threshold value; red line) is equivalent to p = 0.05) to indicate the probability that the miRs fall within these biological classifications; only cellular functions exceeding the threshold value were included. A right-tailed Fisher's exact test was used to determine the probability that each biological function identified is due to chance alone. B, networks of known (solid lines) and inferred (dashed lines) interacting molecules were created based on data within Ingenuity Knowledge Base. The color intensity of miR expression indicates the degree of up-regulated (red) or down-regulated (green) miRs. This figure depicts a large merged network composed of three individual networks bridged by two key molecules, MAPK1 (ERK2) and MYC (highlighted by blue boxes), that are known regulators of cellular proliferation. Mature miRs selected for further analyses and their precursors are circled in magenta.
Overexpression of miR-377 and let-7a Inhibits Basal Trophoblast Proliferation and Expression of Promitogenic Signaling Molecules in the First Trimester
Three miRs that were central to the large mitogenic signaling network (miR-377, let-7a, and miR-145) were selected for further analysis as candidate regulators of placental growth. Attempts to localize miRs in first trimester placental tissue using in situ hybridization were unsuccessful due to high levels of background staining in the trophoblast (data not shown). Therefore, to verify that these miRs are expressed in trophoblast, trophoblast cell lines and freshly isolated primary cytotrophoblasts were assessed by qPCR (Fig. 4). miR-377, let-7a, and miR-145 were detected in primary cytotrophoblast, and consistent with the array data, all three miRs were higher at term than in the first trimester. First trimester placental tissue explants, in which cytotrophoblast proliferation is sustained for >72 h, as indicated by Ki67 immunopositivity or BrdU incorporation, were then transfected with the three miR precursors (Fig. 5, A–C) to determine whether higher levels (as observed in term placenta) cause a reduction in cytotrophoblast proliferation. Overexpression of either hsa-miR-377 (Fig. 5A) or hsa-let-7a (Fig. 5B) but not miR-145 (Fig. 5C) significantly attenuated the proportion of Ki67-positive cytotrophoblast (Fig. 5, D and E; p < 0.05; n = 6). Similar results were obtained when BrdU incorporation was used as a marker of proliferation (Fig. 5F; p < 0.05; n = 6), suggesting that miR-377 and let-7a are negative regulators of endogenously regulated trophoblast expansion. The transfection procedure alone (Mock) or the presence of a non-targeting miR precursor (Pre C) had no effect on the expression of any of the miRs or on cytotrophoblast proliferation.
FIGURE 4.
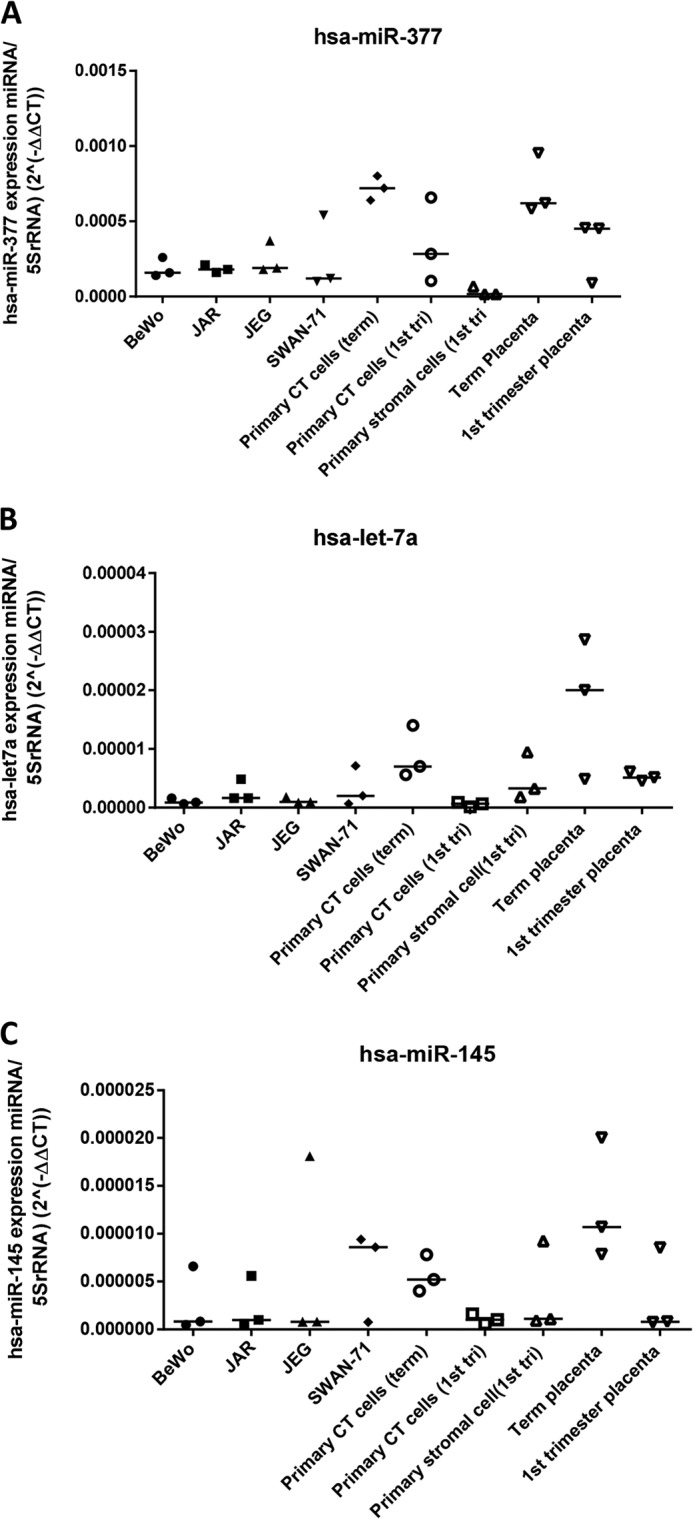
miR-377, let-7a, and miR-145 are enriched in trophoblast cells. Levels of miR-377 (A), let-7a (B), and miR-145 (C) were assessed in a panel of placental trophoblast cell lines (BeWo, JAR, JEG, and SWAN-71), primary cells (primary cytotrophoblast (CT) and primary stromal cells), and fragments of whole placental tissue from term or first trimester placenta. Levels are expressed as levels of specific miR relative to 5 S rRNA. miR-377, let-7a, and miR-145 are all expressed in both primary cytotrophoblast and trophoblast cell lines, albeit at low levels.
FIGURE 5.

miR-377 and let-7a negatively regulate trophoblast proliferation. Pre-miR precursors for miR-377, let-7a, and miR-145 or a pre-miR control (PreC) were transfected into first trimester placental explants (n = 6). The level of miR expression, quantified by qPCR, was significantly increased 24 h post-transfection (A–C). Cytotrophoblast proliferation, determined by counting the number of Ki67-positive cytotrophoblasts (D and E) or BrdU-positive cytotrophoblasts (F) in relation to total cytotrophoblast at 48 h post-transfection was reduced following overexpression of miR-377 and let-7a but not miR-145. D, arrows indicate cytotrophoblasts (CT) and syncytiotrophoblast (ST). One-way analysis of variance with planned contrasts was used to assess differences between sample groups; p < 0.05 was considered significant. Scale bars, 50 μm.
We next investigated whether miR-377 and let-7a might regulate trophoblast proliferation via modulation of two nodal promitogenic signaling molecules in the miR interaction network identified by IPA: MAPK1 and MYC (Fig. 5B). Immunohistochemistry analysis confirmed that both are expressed by first trimester cytotrophoblast (Fig. 6A). Western blots and immunohistochemistry revealed reduced expression of ERK following transfection of explants with either pre-miR-377 or pre-let-7a (Fig. 6, A–D) and demonstrated that pre-let-7a also reduced MYC expression. Immunohistochemistry analysis suggested that MYC levels in trophoblast were decreased by miR-377; however, levels in stroma were unaffected. Overexpression of miR-145 had no effect on the level of ERK or MYC. 3′-UTR luciferase reporter assays confirmed that miR-377 and let-7a, but not miR-145, directly interact with MAPK1 and MYC (Fig. 6E).
FIGURE 6.

miRs alter expression of mitogenic signaling molecules in first trimester human placenta. A–D, after treatment with miR precursors for 48 h, the expression of the nodal signaling molecules, MAPK1/ERK and MYC, were examined by immunohistochemistry (A) and Western blotting (B). Note that ERK1/2 and c-MYC are present in the villous stroma but are most abundant in cytotrophoblast in tissue transfected with the negative control pre-miR (control pre-miR). C and D, densitometry analysis confirmed that ERK1/2 (C) and c-MYC (D) protein expression are significantly reduced following miR-377 and let-7a overexpression (p < 0.05; n = 4). E, first trimester placental explants were transfected with MAPK1 or MYC sense or mutated (ΔMAPK and ΔMYC) 3′-UTR luciferase reporter constructs or empty vector in the presence of non-targeting (control mimetic) or miR-specific precursors. 24 h later, luciferase activity (normalized to that of β-galactosidase) was assessed; overexpression of miR-377 and let-7a resulted in reduced luciferase activity of MAPK (a) or MYC (b) compared with tissue transfected with control mimetic (n = 5), demonstrating that these genes are direct targets of miR-377 and let-7a. All data are representative of at least four independent experiments. Scale bars, 50 μm. Error bars, median and range.
miR-145 Regulates Multiple Components of the IGF Signaling Cascade to Influence IGF-induced Proliferation
Although in silico analysis placed miR-145 within the growth-regulatory network (Fig. 3), its overexpression did not influence basal proliferation or ERK or MYC protein expression in first trimester placenta. In other systems, miR-145 controls mitogenesis by modulating the expression of molecules within the IGF signaling pathway (21). A targeted mRNA array was used to assess the expression of 84 genes within IGF signaling cascades following transfection of first trimester explants with pre-miR-145; the level of mRNA for 29 molecules was altered by at least 2-fold compared with pre-miR control (Table 3). These data were validated by Western blot analysis of a subset of the potential miR-145 targets, including IGF1R, p38 MAPK, and cyclin D1, which confirmed reduced protein expression (Fig. 7). 3′-UTR luciferase reporter assays demonstrated that miR-145 directly interacts with IGF1R (Fig. 7C). The known roles of IGF1R and the other miR-145-regulated proteins (3, 5) supported the possibility of functional consequences of miR-145 overexpression. IPA revealed that collectively, the genes altered by miR-145 are within three functional networks: cell survival and differentiation, cell cycle and proliferation, and innate immune signaling (supplemental Figs. 1 and 2). In keeping with our prediction from experimental and in silico findings, overexpression of miR-145 in first trimester explants significantly attenuated (p < 0.05) IGF-induced cytotrophoblast proliferation (Fig. 8), as measured by Ki67 immunopositivity (Fig. 8A) and BrdU incorporation (Fig. 8B).
TABLE 3.
miR-145 regulates the expression of multiple components of the IGF axis in the first trimester placenta
Pre-miR-145 (500 nm) was transfected into first trimester placental explants. 48 h later, total RNA was extracted, and mRNA expression of multiple genes was assessed using PCR pathway arrays. Expression levels were normalized to internal controls and expressed as -fold change compared with negative control pre-miR. -Fold changes ≥2 were considered significant.
| Gene | Description | Accession number | -Fold change |
|---|---|---|---|
| RPS6KA1 | Ribosomal protein S6 kinase polypeptide 1/RSK1 | NM_009097 | −11.577 |
| TSC1 | Tuberous sclerosis 1 | NM_022887 | −8.358 |
| PRKCA | Protein kinase C, α | NM_011101 | −7.029 |
| PIK3R2 | Phosphatidylinositol 3-kinase, regulatory subunit, polypeptide2 (p85 β) | NM_008841 | −6.076 |
| CCND1 | Cyclin D1 | NM_007631 | −5.749 |
| CD14 | CD14 antigen | NM_009841 | −4.801 |
| HRAS1 | Harvey rat sarcoma virus oncogene 1 | NM_008284 | −4.238 |
| TSC2 | Tuberous sclerosis 2 | NM_011647 | −3.715 |
| HSPB1 | Heat shock protein 1 | NM_013560 | −3.638 |
| RHOA | Ras homolog gene family, member A | NM_016802 | −3.466 |
| FKBP1A | FK506 binding protein 1a | NM_008019 | −3.371 |
| MAPK8 | Mitogen-activated protein kinase 8/JNK | NM_016700 | −3.325 |
| RAF1 | V-raf-leukemia viral oncogene 1 | NM_029780 | −3.017 |
| ILK | Integrin linked kinase | NM_010562 | −2.996 |
| CDKN1B | Cyclin-dependent kinase inhibitor 1B | NM_009875 | −2.894 |
| MYD88 | Myeloid differentiation primary response gene 88 | NM_010851 | −2.854 |
| MAPK14 | Mitogen-activated protein kinase 14/p38 MAPK | NM_011951 | −2.835 |
| GUSB | Glucuronidase β | NM_010368 | −2.757 |
| PDPK1 | 3-Phosphoinositide-dependent protein kinase 1/PDK1 | NM_011062 | −2.645 |
| RBL2 | Retinoblastoma-like 2 | NM_011250 | −2.591 |
| FASl | Fas ligand (TNF superfamily, member 6) | NM_010177 | −2.400 |
| ITGB1 | Integrin β1 | NM_010578 | −2.384 |
| RASA1 | RAS p21 protein activator 1 | NM_145452 | −2.005 |
| IGF1R | Insulin-like growth factor I receptor | NM_010513 | −2.001 |
| PAK1 | P21 protein (Cdc42/Rac)-activated kinase 1 | NM_011035 | 2.726 |
| GRB2 | Growth factor receptor-bound protein 2 | NM_008163 | 2.670 |
| CASP9 | Caspase-9 | NM_015733 | 2.229 |
| IRAK1 | Interleukin-1 receptor-associated kinase 1 | NM_008363 | 2.124 |
| SRF | Serum response factor | NM_020493 | 2.009 |
FIGURE 7.

miR-145 regulates the expression of multiple components of the IGF axis in first trimester placenta. A and B, first trimester placental explants were transfected with a negative control pre-miR (Pre-C) or pre-miR-145 (500 nm) for 48 h, and then the expression of IGF1R, p38 MAPK, and cyclin D1 was assessed by Western blotting. Blots were stripped and reprobed for β-actin (as an internal control). B, densitometry revealed reduced expression of IGF1R, p38 MAPK, and cyclin D1 when miR-145 was overexpressed (p < 0.05; n = 4). C, first trimester placental explants were transfected with sense or mutated (ΔIGF1R) IGF1R 3′-UTR luciferase reporter constructs or empty vector in the presence of control or miR-145-specific mimetics. 24 h later, luciferase activity (normalized to β-galactosidase) was assessed; luciferase activity was significantly reduced in tissue with miR-145 overexpression compared with tissue transfected with control mimetic (p < 0.05; n = 5), demonstrating that IGF1R is a direct target of miR-145 in the placenta. Error bars, median and range.
FIGURE 8.
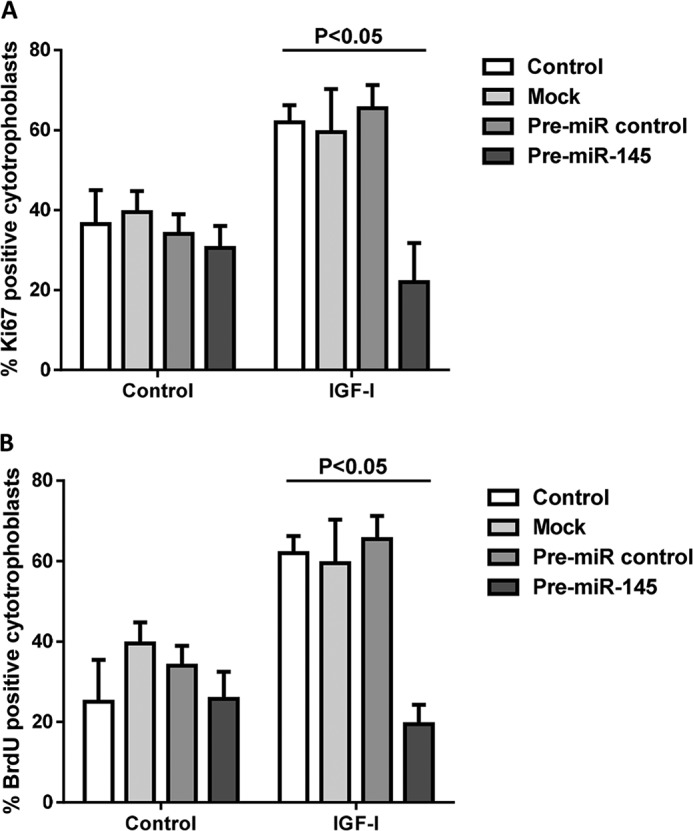
miR-145 negatively regulates IGF-induced trophoblast proliferation. First trimester placental explants (n = 6) were transfected with pre-miR-145 (500 nm) or a negative control miR. IGF-I (10 nm) or vehicle and, in some experiments, BrdU (100 μm) were added after 48 h, and then cytotrophoblast proliferation was assessed after a further 24-h period by immunohistochemical analysis of Ki67 (A) or BrdU (B) incorporation. The level of proliferating cells was determined by counting at least 200 cytotrophoblasts per treatment and expressing the number of Ki67/BrdU-positive cells as a percentage of total nuclei. One-way analysis of variance with planned contrasts was used to compare differences between groups; p < 0.05 was considered significant. Error bars, median and range.
DISCUSSION
Villous cytotrophoblast is a progenitor cell population that produces daughter cells to support the expansion of the syncytium as placental surface area increases as well as the expansion of cytotrophoblast columns, which contain the cells destined to invade maternal decidua (22). The placenta grows exponentially in the first and early second trimester, but growth has slowed down by term (23). Abundant expression of Dicer in cytotrophoblast suggests a role for miRs in the regulation of placental growth (9), and expression profiling in this study and others (16–20) has shown expression and gestational changes in miR levels that demand functional evaluation. We have previously used Dicer knockdown in explants to show that globally, miRs tend to suppress proliferation in cytotrophoblast (9). More than two-thirds of the alterations that we detected by array analysis are in the direction of increased expression at term relative to first trimester, consistent with the suppression by miRs of proteins in growth-related signaling networks.
This conclusion is supported by IPA analysis, in which the majority of miRs expressed at high levels in late pregnancy were predicted to inhibit cell growth. The predicted network contained two nodal promitogenic molecules, MAPK/ERK and MYC, both of which were localized in first trimester cytotrophoblast. The role of ERK in regulating trophoblast turnover is well documented in both human and animal systems (3, 4, 24, 25). A role for MYC in placental development is also emerging. Studies in mice show that the embryonic-lethal phenotype seen in c-MYC-null animals can be largely corrected if the expression of this gene is maintained selectively in trophoblast (26, 27). In humans, the rapid decline in MYC expression as primary term cytotrophoblast cells exit the cell cycle (28), together with the observation that JEG-3 choriocarcinoma cells have significantly reduced levels of proliferation in its absence, initially suggested a role in trophoblast regulation (29). Direct evidence is presented here; overexpression of miR-377 and let-7a reduced expression of both ERK and MYC and attenuated proliferation.
let-7 was among the first miRs to be discovered in Caenorhabditis elegans and is evolutionarily conserved (30). It is clear from numerous subsequent studies that the let-7 miR family regulates development in many species, including mammals (31). Consistent with our data, let-7a has a well documented antiproliferative role in both in animal models and in humans (32–34) involving the targeting of MYC (34–36) and ERK (37, 38). In contrast to let-7, studies on the role of miR-377 are limited. miR expression profiling has revealed that placental miR-377 expression is reduced in pre-eclampsia (39, 40); however, its targets and functions were not explored. In keeping with our data, miR-377 expression was found to be up-regulated in fibroblast cell lines under growth arrest, and its expression is reported to be negatively correlated with that of promitogenic signaling molecules (41). Both trophoblast proliferation (1) and ERK expression (42) are altered in placentas from pregnancies complicated by pre-eclampsia; thus, it is tempting to speculate that altered miR-377 expression influences these events and contributes to the pathogenesis of the disease. Given the importance of miR-377 in regulating placental growth, the expression of miR-377 in other pregnancy pathologies associated with altered placental growth (e.g. fetal growth restriction) should also be explored.
In contrast to miR-377 and let-7a, and despite its involvement in regulating c-MYC expression, miR-145 did not alter basal trophoblast proliferation. ERK levels were not affected by miR-145 overexpression. However, in non-small cell lung cancer cells, miR-145 reduced proliferation independently of ERK (37). An alternative explanation may lie in the fact that in humans, miR-145 is present on chromosome 5q33 ∼1.3 kb from miR-143, and the two are regulated by a common promoter (43). Although each of these miRs can act independently to influence cellular events, including proliferation, in some instances, a coordinated, or synergistic, role for the miR-143/145 cluster is observed (44–46). This is the case in other cells, where miR-143 and miR-145 form an integrated program of gene regulation by acting in synergy to influence the expression of common targets, including members of the MAPK/ERK pathway, while also functioning individually to alter the expression of other independent sets of genes (46). miR-143 was also altered in our miR array; thus, it is possible that miR-145 and miR-143 work in combination to inhibit basal trophoblast proliferation.
Overexpression of miR-145 alone was, however, sufficient to negatively influence IGF-induced proliferation. Multiple components of the IGF axis, including IGF1R, PDK1, Ras, Raf, and cyclin D1, were simultaneously reduced. Thus, miR-145 may fine tune IGF-induced proliferation by coordinately targeting several components within the cascade. Interestingly, although we have evidence that IGF1R, PDK1, and cyclin D1 regulate IGF-induced trophoblast proliferation (3, 5), IGF1R-mediated signaling does not appear to influence basal proliferation (3, 47). This suggests an explanation for the failure of miR-145 to influence basal trophoblast proliferation; instead, the role for miR-145 in the placenta may be to regulate the function of IGF or other growth factors. Our findings are consistent with those of other studies undertaken in hepatic and vascular cells, which demonstrate that whereas the other roles of miR-145 depend on both components of the miR-143/145 cluster, miR-145, independently of miR-143, influences IGF-induced proliferation, modulating IGF1R and IRS-1 expression (21, 48). It will be interesting to determine, in future studies, the combined role of miR-143/145 in placenta; data from the gene array performed after miR-145 overexpression suggest regulation of cell survival and/or the innate immune response.
In summary, we have used a proliferation-focused, hypothesis-generating approach, together with in silico analysis and molecular techniques, to uncover three miRs expressed by the placenta that lie within a network of mitogenic signaling molecules exerting gestational regulation on growth. Two of these, miR-145 and let-7a, have well documented roles in regulating proliferation in other systems, and we have revealed a novel role for another miR, miR-377. These data validate the approach taken to identify growth-regulatory pathways in the placenta and highlight the potential of this methodology for dissecting the function of placental miRs. Ultimately, we hope to develop miR-based approaches to therapeutic targeting for correcting the abnormal placental growth and cell turnover seen in pregnancy pathologies.
Supplementary Material
This work was supported by awards from the Biochemical Society (United Kingdom) Guildford Bench Methodology Fund, the National Institute for Health Research Manchester Biomedical Research Centre, and a Society for Endocrinology Early Career Grant. The Maternal and Fetal Health Research Centre is supported by funding from the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network.

This article contains supplemental Figs. 1 and 2.
- IGF
- insulin-like growth factor
- miR
- microRNA
- MEM
- minimum Eagle's medium
- qPCR
- quantitative PCR
- IPA
- Ingenuity Pathway Analysis.
REFERENCES
- 1. Arnholdt H., Meisel F., Fandrey K., Löhrs U. (1991) Proliferation of villous trophoblast of the human placenta in normal and abnormal pregnancies. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 60, 365–372 [DOI] [PubMed] [Google Scholar]
- 2. Jansson T., Powell T. L. (2006) Human placental transport in altered fetal growth: does the placenta function as a nutrient sensor? a review. Placenta 27, 91–97 [DOI] [PubMed] [Google Scholar]
- 3. Forbes K., Westwood M., Baker P. N., Aplin J. D. (2008) Insulin-like growth factor I and II regulate the life cycle of trophoblast in the developing human placenta. Am. J. Physiol. Cell Physiol. 294, C1313–C1322 [DOI] [PubMed] [Google Scholar]
- 4. Forbes K., Souquet B., Garside R., Aplin J. D., Westwood M. (2010) Transforming growth factor-β (TGFβ) receptors I/II differentially regulate TGFβ1 and IGF-binding protein-3 mitogenic effects in the human placenta. Endocrinology 151, 1723–1731 [DOI] [PubMed] [Google Scholar]
- 5. Forbes K., West G., Garside R., Aplin J. D., Westwood M. (2009) The protein-tyrosine phosphatase, SRC homology-2 domain containing protein tyrosine phosphatase-2, is a crucial mediator of exogenous insulin-like growth factor signaling to human trophoblast. Endocrinology 150, 4744–4754 [DOI] [PubMed] [Google Scholar]
- 6. Bernstein E., Kim S. Y., Carmell M. A., Murchison E. P., Alcorn H., Li M. Z., Mills A. A., Elledge S. J., Anderson K. V., Hannon G. J. (2003) Dicer is essential for mouse development. Nat. Genet. 35, 215–217 [DOI] [PubMed] [Google Scholar]
- 7. Yang W. J., Yang D. D., Na S., Sandusky G. E., Zhang Q., Zhao G. (2005) Dicer is required for embryonic angiogenesis during mouse development. J. Biol. Chem. 280, 9330–9335 [DOI] [PubMed] [Google Scholar]
- 8. Forbes K., Farrokhnia F., Aplin J. D., Westwood M. (2012) Dicer-dependent miRNAs provide an endogenous restraint on cytotrophoblast proliferation. Placenta 33, 581–585 [DOI] [PubMed] [Google Scholar]
- 9. Griffiths-Jones S., Grocock R. J., van Dongen S., Bateman A., Enright A. J. (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ritchie M. E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G. K. (2007) A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 [DOI] [PubMed] [Google Scholar]
- 11. Aplin J. D. (2010) Developmental cell biology of human villous trophoblast: current research problems. Int. J. Dev. Biol. 54, 323–329 [DOI] [PubMed] [Google Scholar]
- 12. Straszewski-Chavez S. L., Abrahams V. M., Alvero A. B., Aldo P. B., Ma Y., Guller S., Romero R., Mor G. (2009) The isolation and characterization of a novel telomerase immortalized first trimester trophoblast cell line, Swan 71. Placenta 30, 939–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris L. K., Keogh R. J., Wareing M., Baker P. N., Cartwright J. E., Aplin J. D., Whitley G. S. (2006) Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am. J. Pathol. 169, 1863–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ingman K., Cookson V. J., Jones C. J., Aplin J. D. (2010) Characterisation of Hofbauer cells in first and second trimester placenta: incidence, phenotype, survival in vitro and motility. Placenta 31, 535–544 [DOI] [PubMed] [Google Scholar]
- 15. Forbes K., Desforges M., Garside R., Aplin J. D., Westwood M. (2009) Methods for siRNA-mediated reduction of mRNA and protein expression in human placental explants, isolated primary cells and cell lines. Placenta 30, 124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heazell A. E., Lacey H. A., Jones C. J., Huppertz B., Baker P. N., Crocker I. P. (2008) Effects of oxygen on cell turnover and expression of regulators of apoptosis in human placental trophoblast. Placenta 29, 175–186 [DOI] [PubMed] [Google Scholar]
- 17. Kar M., Ghosh D., Sengupta J. (2007) Histochemical and morphological examination of proliferation and apoptosis in human first trimester villous trophoblast. Hum. Reprod. 22, 2814–2823 [DOI] [PubMed] [Google Scholar]
- 18. Grandori C., Cowley S. M., James L. P., Eisenman R. N. (2000) The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653–699 [DOI] [PubMed] [Google Scholar]
- 19. Goodman R. H., Smolik S. (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev. 14, 1553–1577 [PubMed] [Google Scholar]
- 20. Cobb M. H., Hepler J. E., Cheng M., Robbins D. (1994) The mitogen-activated protein kinases, ERK1 and ERK2. Semin. Cancer Biol. 5, 261–268 [PubMed] [Google Scholar]
- 21. La Rocca G., Badin M., Shi B., Xu S. Q., Deangelis T., Sepp-Lorenzinoi L., Baserga R. (2009) Mechanism of growth inhibition by MicroRNA 145: The role of the IGF-I receptor signaling pathway. J. Cell Physiol. 220, 485–491 [DOI] [PubMed] [Google Scholar]
- 22. Aplin J. D. (1991) Implantation, trophoblast differentiation and haemochorial placentation: mechanistic evidence in vivo and in vitro. J. Cell Sci. 99, 681–692 [DOI] [PubMed] [Google Scholar]
- 23. Schneider H. (1996) Ontogenic changes in the nutritive function of the placenta. Placenta 17, 15–26 [DOI] [PubMed] [Google Scholar]
- 24. Saba-El-Leil M. K., Vella F. D., Vernay B., Voisin L., Chen L., Labrecque N., Ang S. L., Meloche S. (2003) An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 4, 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fu Y. Y., Gao W. L., Chen M., Chai K. X., Wang Y. L., Chen L. M. (2010) Prostasin regulates human placental trophoblast cell proliferation via the epidermal growth factor receptor signaling pathway. Hum. Reprod. 25, 623–632 [DOI] [PubMed] [Google Scholar]
- 26. Dubois N. C., Adolphe C., Ehninger A., Wang R. A., Robertson E. J., Trumpp A. (2008) Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 135, 2455–2465 [DOI] [PubMed] [Google Scholar]
- 27. Davis A. C., Wims M., Spotts G. D., Hann S. R., Bradley A. (1993) A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7, 671–682 [DOI] [PubMed] [Google Scholar]
- 28. Morrish D. W., Dakour J., Li H., Xiao J., Miller R., Sherburne R., Berdan R. C., Guilbert L. J. (1997) In vitro cultured human term cytotrophoblast: a model for normal primary epithelial cells demonstrating a spontaneous differentiation programme that requires EGF for extensive development of syncytium. Placenta 18, 577–585 [DOI] [PubMed] [Google Scholar]
- 29. Pu H. H., Duan J., Wang Y., Fan D. X., Li D. J., Jin L. P. (2012) Thymic stromal lymphopoietin promotes the proliferation of human trophoblasts via phosphorylated STAT3-mediated c-Myc upregulation. Placenta 33, 387–391 [DOI] [PubMed] [Google Scholar]
- 30. Reinhart B. J., Slack F. J., Basson M., Pasquinelli A. E., Bettinger J. C., Rougvie A. E., Horvitz H. R., Ruvkun G. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906 [DOI] [PubMed] [Google Scholar]
- 31. Ambros V. (2011) MicroRNAs and developmental timing. Curr. Opin. Genet. Dev. 21, 511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong Q., Meng P., Wang T., Qin W., Qin W., Wang F., Yuan J., Chen Z., Yang A., Wang H. (2010) MicroRNA let-7a inhibits proliferation of human prostate cancer cells in vitro and in vivo by targeting E2F2 and CCND2. PloS One 5, e10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. He X. Y., Chen J. X., Zhang Z., Li C. L., Peng Q. L., Peng H. M. (2010) The let-7a microRNA protects from growth of lung carcinoma by suppression of k-Ras and c-Myc in nude mice. J. Cancer Res. Clin. Oncol. 136, 1023–1028 [DOI] [PubMed] [Google Scholar]
- 34. Liu Y., Yin B., Zhang C., Zhou L., Fan J. (2012) Hsa-let-7a functions as a tumor suppressor in renal cell carcinoma cell lines by targeting c-myc. Biochem. Biophys. Res. Commun. 417, 371–375 [DOI] [PubMed] [Google Scholar]
- 35. Shu J., Xia Z., Li L., Liang E. T., Slipek N., Shen D., Foo J., Subramanian S., Steer C. J. (2012) Dose-dependent differential mRNA target selection and regulation by let-7a-7f and miR-17–92 cluster microRNAs. RNA Biol. 9, 1275–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhong Z., Dong Z., Yang L., Chen X., Gong Z. (2012) Inhibition of proliferation of human lung cancer cells by green tea catechins is mediated by upregulation of let-7. Exp. Ther. Med. 4, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhong M., Ma X., Sun C., Chen L. (2010) MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem. Biol. Interact. 184, 431–438 [DOI] [PubMed] [Google Scholar]
- 38. Torrisani J., Bournet B., du Rieu M. C., Bouisson M., Souque A., Escourrou J., Buscail L., Cordelier P. (2009) let-7 microRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum. Gene Ther. 20, 831–844 [DOI] [PubMed] [Google Scholar]
- 39. Mayor-Lynn K., Toloubeydokhti T., Cruz A. C., Chegini N. (2011) Expression profile of microRNAs and mRNAs in human placentas from pregnancies complicated by preeclampsia and preterm labor. Reprod. Sci. 18, 46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu X. M., Han T., Sargent I. L., Yin G. W., Yao Y. Q. (2009) Differential expression profile of microRNAs in human placentas from preeclamptic pregnancies vs normal pregnancies. Am. J. Obstet. Gynecol. 200, 661.e1–661.e7 [DOI] [PubMed] [Google Scholar]
- 41. Maes O. C., Sarojini H., Wang E. (2009) Stepwise up-regulation of microRNA expression levels from replicating to reversible and irreversible growth arrest states in WI-38 human fibroblasts. J. Cell Physiol. 221, 109–119 [DOI] [PubMed] [Google Scholar]
- 42. Shin J. K., Jeong Y. T., Jo H. C., Kang M. Y., Chang I. S., Baek J. C., Park J. K., Lee S. A., Lee J. H., Choi W. S., Paik W. Y. (2009) Increased interaction between heat shock protein 27 and mitogen-activated protein kinase (p38 and extracellular signal-regulated kinase) in pre-eclamptic placentas. J. Obstet. Gynaecol. Res. 35, 888–894 [DOI] [PubMed] [Google Scholar]
- 43. Iio A., Nakagawa Y., Hirata I., Naoe T., Akao Y. (2010) Identification of non-coding RNAs embracing microRNA-143/145 cluster. Mol. Cancer 9, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akao Y., Nakagawa Y., Hirata I., Iio A., Itoh T., Kojima K., Nakashima R., Kitade Y., Naoe T. (2010) Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. Cancer Gene Ther. 17, 398–408 [DOI] [PubMed] [Google Scholar]
- 45. Noguchi S., Yasui Y., Iwasaki J., Kumazaki M., Yamada N., Naito S., Akao Y. (2013) Replacement treatment with microRNA-143 and -145 induces synergistic inhibition of the growth of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling pathways. Cancer Lett. 328, 353–361 [DOI] [PubMed] [Google Scholar]
- 46. Pagliuca A., Valvo C., Fabrizi E., di Martino S., Biffoni M., Runci D., Forte S., De Maria R., Ricci-Vitiani L. (2013) Analysis of the combined action of miR-143 and miR-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene 32, 4806–4813 [DOI] [PubMed] [Google Scholar]
- 47. Forbes K., Skinner L., Garside R., Aplin J. D., Westwood M. (2012) The PTP, SHP-1, acts on multiple tyrosine kinase receptors to negatively regulate human cytotrophoblast proliferation. Cell. Mol. Life Sci. 69, 4029–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Law P. T., Ching A. K., Chan A. W., Wong Q. W., Wong C. K., To K. F., Wong N. (2012) MiR-145 modulates multiple components of the insulin-like growth factor pathway in hepatocellular carcinoma. Carcinogenesis 33, 1134–1141 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.