

Background: Mycobacterial cytochrome P450 families 125 and 142 initiate metabolism of host cholesterol.
Results: Family 142 enzymes were able to oxidize cholesterol esters, whereas those in family 125 were not.
Conclusion: Structural differences impede cholesterol ester oxidation only in the 125 family.
Significance: The ability of the 142 family to oxidize cholesterol esters gives Mycobacterium tuberculosis access to additional pools of intracellular cholesterol.
Keywords: Cholesterol, Cholesterol Metabolism, Cytochrome P450, Mycobacterium smegmatis, Mycobacterium tuberculosis
Abstract
Mycobacteria share a common cholesterol degradation pathway initiated by oxidation of the alkyl side chain by enzymes of cytochrome P450 (CYP) families 125 and 142. Structural and sequence comparisons of the two enzyme families revealed two insertions into the N-terminal region of the CYP125 family (residues 58–67 and 100–109 in the CYP125A1 sequence) that could potentially sterically block the oxidation of the longer cholesterol ester molecules. Catalytic assays revealed that only CYP142 enzymes are able to oxidize cholesteryl propionate, and although CYP125 enzymes could oxidize cholesteryl sulfate, they were much less efficient at doing so than the CYP142 enzymes. The crystal structure of CYP142A2 in complex with cholesteryl sulfate revealed a substrate tightly fit into a smaller active site than was previously observed for the complex of CYP125A1 with 4-cholesten-3-one. We propose that the larger CYP125 active site allows for multiple binding modes of cholesteryl sulfate, the majority of which trigger the P450 catalytic cycle, but in an uncoupled mode rather than one that oxidizes the sterol. In contrast, the more unhindered and compact CYP142 structure enables enzymes of this family to readily oxidize cholesteryl esters, thus providing an additional source of carbon for mycobacterial growth.
Introduction
Mycobacterium tuberculosis is the causative agent of tuberculosis, a disease which has resurfaced in recent years aided by the emergence of drug resistant strains, to account for ∼2 million deaths annually (1). It is estimated that nearly one-third of the world's population is latently infected with this pathogen, which is spread in aerosolized droplets from actively infected individuals (1). Following inhalation of the bacilli, the bacteria are phagocytosed by alveolar macrophages, where they modulate the host phagosome environment by preventing fusion with the lysosome and persist as an intracellular pathogen (2). Cholesterol is an abundant intracellular molecule that M. tuberculosis has the ability to import and utilize (3). Disruption of this genetic machinery leads to attenuation in infection models and loss of the ability to grow in vitro on cholesterol as the sole carbon source, indicating the importance of cholesterol availability to the intracellular viability of the pathogen (3–6). One of the primary steps in the cholesterol degradation pathway is oxidation of the cholesterol side chain to a carboxylic acid by the cytochrome P450 (CYP)2 enzymes CYP125A1 and CYP142A1, which in turn, can be further catabolized by the β-oxidation pathway (7–9).
This cholesterol degradation pathway is conserved in the soil-dwelling relative of M. tuberculosis, Mycobacterium smegmatis, in which CYP125A3 and CYP142A2 serve as paralogous enzymes (10). Both sets of paralogs share ∼70% sequence identity, similar kinetic activities toward 4-cholesten-3-one, and overall structural similarity. Although some mycobacteria, such as the CDC1551 strain of M. tuberculosis and Mycobacterium bovis BCG, have lost a functional cyp142 gene, they still maintain an intact biochemical pathway via CYP125 (11–13). Despite efforts to obtain a substrate bound structure of CYP142A1 (14), only structures of CYP125A1 and CYP142A2 complexed with a substrate have been reported (10, 12). Structural and sequence comparisons revealed two ∼10 residue insertions in the CYP125 sequences, which are not present in the CYP142 sequences, (residues 58–67 and 100–109 in the CYP125A1 sequence; Table 1) that form a cap over the active site in the CYP125 enzymes. We hypothesized that this structural difference might create a steric barrier that prevents CYP125 from catabolizing the larger cholesterol ester molecules, compounds that have been shown to accumulate in M. tuberculosis infected macrophages (15, 16).
TABLE 1.
N-terminal sequence alignments of CYPs 142A1, 142A2, 125A1, and 125A3

Here, we show that the CYP142 subfamily, initially postulated to play a purely redundant role relative to the CYP125 enzymes in vivo (6, 10), can readily metabolize cholesteryl sulfate and cholesteryl propionate in vitro. In contrast, CYP125 enzymes cannot oxidize cholesteryl propionate and can only oxidize cholesteryl sulfate with a much lower activity than the CYP142 enzymes due to a much higher fraction of uncoupled catalytic turnover. The crystal structure of CYP142A2 in complex with cholesteryl sulfate reveals an active site that closely surrounds the substrate, as opposed to the larger binding pocket observed previously with CYP125A1 (12). We propose that the cholesteryl sulfate side chain may enter the active site in a manner that triggers the catalytic cycling of CYP125, but in which the substrate is not in position to be oxidized efficiently. Thus, CYP142 enzymes may play an important role during M. tuberculosis infection by providing access to additional reservoirs of intracellular cholesterol than would otherwise be available to the pathogen.
EXPERIMENTAL PROCEDURES
Chemicals
1,4-Cholestadiene-3-one was obtained from Research Plus (Barnegat, NJ). All other chemicals, including cholesterol, cholesteryl sulfate, cholesteryl propionate, spinach ferredoxin, spinach ferredoxin-NADP+-reductase, bovine liver catalase, glucose 6-phosphate, glucose-6-phosphate dehydrogenase, N,O-bis(trimethylsilyl)trifluoroacetamide, and methyl-β-cyclodextrin (MβCD), were purchased from Sigma-Aldrich.
Protein Expression and Purification
CYP125A1 (from M. tuberculosis), CYP125A3 (from M. smegmatis), CYP142A1 (from M. tuberculosis), and CYP142A2 (from M. smegmatis) were expressed and purified as described previously (6, 10). Briefly, Escherichia coli DH5α cells containing the pCWori expression vector with the gene of interest were grown at 37 °C and 250 rpm in TB medium containing ampicillin (100 μg ml−1) to A600 = 0.7–0.8. Expression was induced by the addition of 1 mm isopropyl β-D-1-thiogalactopyranoside and 0.5 mm δ-aminolevulinic acid, and the culture continued for 36 h at 25 °C and 180 rpm. The cultures were harvested by centrifugation and stored at −80 °C. Cell pellets were thawed on ice and resuspended with agitation in 50 mm Tris-HCl, pH 7.5, buffer containing 0.15 m NaCl and 1 mm phenylmethylsulfonyl fluoride, followed by the addition of lysozyme (0.5 mg ml−1) and DNase (0.1 mg ml−1). The cells were disrupted by sonication using a Branson sonicator (six cycles of 1 min followed by 30-s rests), the sonicates clarified by centrifugation at 100,000 × g for 45 min at 4 °C, and the proteins were purified on a nickel-nitrilotriacetic acid column washed with 10 column volumes of resuspension buffer plus 20 mm imidazole and then eluted with resuspension buffer plus 250 mm imidazole. The eluted fractions were concentrated, dialyzed against 50 mm Tris-HCl, pH 7.5, and the enzyme concentration was determined from difference spectra using the extinction coefficient 91,000 m−1 cm−1 (17). The fraction of P420 species never exceeded 5%.
UV-visible Spectroscopy
UV-visible absorption spectra were recorded on a Cary UV-visible scanning spectrophotometer (Varian) using a 1-cm path length quartz cuvette at ambient temperature in 50 mm potassium phosphate buffer, pH 7.4, containing 150 mm NaCl. Spectral titrations were performed using 2 μm P450 with the sequential addition of substrates from concentrated stocks in methanol (cholesteryl sulfate) or buffer (cholesteryl propionate), with the corresponding solution added to the reference cuvette. Cholesteryl propionate was initially dissolved in 1:10 chloroform/acetone with 0.05% Tween 20 (v/v), dried under N2 gas, and resuspended in buffer. The final concentration of methanol was always <1.5%. Difference spectra were recorded from 350 to 700 nm with a scanning rate of 120 nm/min. Data were fitted to the quadratic equation (Equation 1) or Hill equation (Equation 2) using GraphPad Prism, where Aobs is the observed absorption shift, Amax is the maximal shift, KS (or S50 for the Hill equation), is the apparent dissociation constant, [Et] is the enzyme concentration, [S] is the ligand concentration, and n is the Hill coefficient.
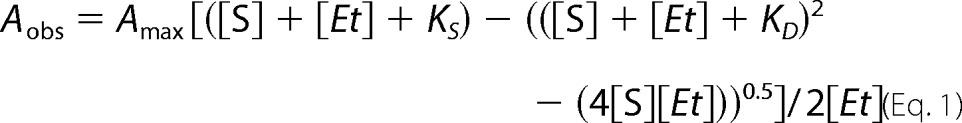 |
Cholesteryl Sulfate Oxidation
The enzymes CYP125A1, CYP125A3, CYP142A1, and CYP142A2 (0.5 μm) were incubated at ambient temperature for 5 min with cholesteryl sulfate in 50 mm potassium phosphate (pH 7.4) containing 0.05% (w/v) MβCD, 150 mm NaCl, and 10 mm MgCl2. Reactions were initiated by the addition of an NADPH regenerating system consisting of 1 mm NADPH, 1 μm spinach ferredoxin, 0.2 units ml−1 spinach ferredoxin-NADP+ reductase, 0.1 mg ml−1 bovine liver catalase, 0.4 units ml−1 glucose-6-phosphate dehydrogenase, and 5 mm glucose 6-phosphate. Aliquots of 50 μl were taken at 0, 10, 20, and 30 min and quenched with 150 μl of acetonitrile containing 5% 5 mm ammonium bicarbonate and 10 μm 1,4-cholestadiene-3-one as an internal standard. The reaction mixtures were centrifuged at 10,000 × g for 4 min. The supernatants were directly analyzed by LC-MS using a Waters Micromass ZQ coupled to a Waters Alliance HPLC system equipped with a 2695 separations module, a Waters 2487 Dual λ Absorbance detector, and a reverse phase C18 column (Waters Xterra C18 column, 3.5 μm, 2.1 × 50 mm). The column was eluted isocratically at a flow rate of 0.2 ml/min (solvent A, H2O + 5 mm ammonium bicarbonate; solvent B, CH3CN) with a gradient starting at 70% A for 2 min and then ramped to 95% B >15 min and finally maintained for 20 min. The elution was monitored at 240 nm. The mass spectrometer settings were as follows: mode, ES−; capillary voltage, 3.5 kV; cone voltage, 30 V; desolvation temperature, 250 °C. The selected ion currents of parent ions of the individual metabolites were collected and analyzed by MS using a quadrupole instrument in the negative ion mode.
To determine the Km values, the data points were fitted to the quadratic equation (Equation 3) or Hill equation (Equation 4) using GraphPad Prism where Kobs is the product forming rate determined at any ligand concentration, Kmax is the maximal rate, Km (or S50 for the Hill equation) is the substrate concentration at which the half-maximal rate is achieved, [Et] is the total enzyme concentration used, [S] is the ligand concentration, and n is the Hill coefficient.
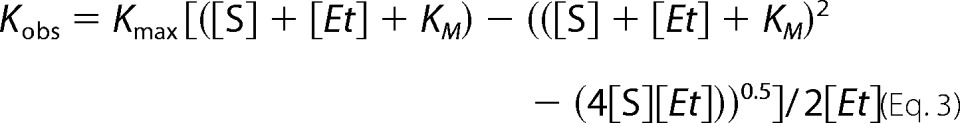 |
Cholesteryl Propionate Oxidation
CYP125A1, CYP125A3, CYP142A1, and CYP142A2 (1 μm) were incubated at ambient temperature for 5 min with cholesteryl propionate (0.1 mm) in 50 mm potassium phosphate (pH 7.4) containing 0.05% (w/v) MβCD, 150 mm NaCl, and 10 mm MgCl2. For 18O incorporation assays, the reaction mixtures were perfused with a 1:1 mixture of 16O2 and 18O2 (Sigma Aldrich) for several minutes prior to initiation. Reactions were initiated by addition of an NADPH regeneration system consisting of 1 mm NADPH, 1 μm spinach ferredoxin, 0.2 units ml−1 spinach ferredoxin-NADP+ reductase, 0.1 mg ml−1 bovine liver catalase, 0.4 units ml−1 glucose-6-phosphate dehydrogenase, and 5 mm glucose 6-phosphate. After 1 h, the reactions were quenched with an equal volume of 1 n HCl, 0.1 mm cholesterol was added as an internal standard, and the mixtures then extracted twice with 2 ml of hexanes. The combined extracts were dried under N2 gas, derivatized for 1 h at 37 °C with 75 μl of N,O-bis(trimethylsilyl)trifluoroacetamide (Sigma Aldrich), and analyzed on an Agilent 6850 gas chromatograph fitted with an HP-5MS column (30 m, 0.25 mm internal diameter, Agilent) coupled to an Agilent 5973 network mass selective detector in the flame ionization mode operating at 70 eV. Helium was the carrier gas. The temperature program was 150 °C for 2 min, 150–300 °C at 10 °C/min, and 300 °C for 18 min.
NADPH Oxidation
CYP125A1, CYP125A3, CYP142A1, and CYP142A2 (0.5 μm) were incubated at ambient temperature for 3 min with 0.1 mm substrate, 0.1 units/ml ferredoxin reductase, and 0.5 μm ferredoxin, in 50 mm potassium phosphate (pH 7.4) containing 0.05% (w/v) MβCD, 150 mm NaCl, and 10 mm MgCl2. The reactions were initiated by the addition of 200 nmol NADPH, and NADPH oxidation was monitored for 5 min and calculated from the absorption changes at 340 nm using the extinction coefficient 6.22 mm−1 cm−1.
CYP142A2 Protein Crystallization and Structure Determination
CYP142A2 was expressed and purified as described above with the following additional steps. Fractions eluted from the nickel-nitrilotriacetic acid column were further purified by flow-through chromatography on SP-Sepharose Fast-Flow (Amersham Biosciences) and subsequent binding to Q-Sepharose Fast-Flow (Amersham Biosciences), both equilibrated with 50 mm Tris-HCl, pH 7.5, buffer and washed with five column volumes of equilibration buffer. After washing, the proteins were eluted with 0.5 m NaCl in 50 mm Tris-HCl, pH 7.5. The protein was then buffer-exchanged into 50 mm potassium phosphate, pH 7.4, 10% glycerol. The heme-containing protein concentration was determined to be 900 μm by CO difference spectroscopy (17) using ϵ(450–490) = 91 mm−1 cm−1 in the presence of 50 mm potassium phosphate, 0.05% MβCD, and 50 μm 4-cholesten-3-one ligand. Protein containing only 50 mm potassium phosphate, pH 7.4, and 10% glycerol was flash-frozen in liquid nitrogen and stored at −80 °C until thawing on ice prior to setting up crystals. Upon thawing, 4 μl of a 50 mm stock dissolved in dimethyl sulfoxide was added to 100-μl aliquots, giving an approximate 2 mm cholesteryl sulfate concentration. One μl of protein solution and 1 μl of well solution, incubated in sitting drop vapor diffusion CryschemTM (Hampton) crystal trays resulted in crystals with space group P212121, containing two molecules in the asymmetric unit. The well solution contained 20–25% PEG 3350, 10–13% glycerol, and 50 mm MgCl2. After a 3–4-day incubation at 25 °C, crystals were harvested into progressively increasing concentrations of cryo-solution containing mother liquor supplemented with 20% glycerol and flash frozen in liquid nitrogen.
Data were collected at 100 K using an in-house Rigaku Rotaflex RU-200 x-ray generator with a rotating copper anode. Images were recorded with an R-AXIS IV image plate area detector using ϕ = 1° oscillations from 65–365° with 15-min exposures per frame. Data were indexed, integrated, and scaled using the HKL2000 software package (18). Intensities were converted to structure factors, and a random Rfree test set was generated from ∼5% of the total reflections using the software, truncate, as implemented in the CCP4 suite (19). Initial phases for molecular replacement using Phaser (20) were taken from a previous CYP142A2 x-ray crystal structure (PDB code 2YOO), in which the heme and 4-cholesten-3-one ligands were removed. Refinement was performed in phenix.refine (21) with individual coordinate and isotropic B-factor refinement for all atoms. The last modeled residue, His-404 of molecule A, is the lone Ramachandran outlier. This residue is part of the C-terminal His6 tag and has little effect on the overall quality of the model. To avoid model bias, cholesteryl sulfate composite omit |FO − FC| electron density maps were calculated using an iterative simulated annealing, model building, and refinement protocol implemented in the Phenix AutoBuild wizard with default parameters. Final statistics are provided in Table 2.
TABLE 2.
Data collection and refinement statistics
r.m.s.d., root mean square deviation.
| Data set | CYP142A2 C3S (PDB code 4TRI) | CYP142A2 ligand-free (PDB code 4UAX) |
|---|---|---|
| Radiation source | Rigaku RU-200 copper rotating anode | ALS synchrotron BL 8.3.1 |
| Space group | P212121 | P212121 |
| Unit cell dimensions | a = 61.45, b = 96.14, c = and 145.89 Å | a = 56.65, b = 83.52, and c = 94.50 Å |
| Resolution range (Å) (highest shell) | 50 (2.00) | 20 (1.78) |
| Wavelength (Å) | 1.54 | 1.12 |
| Total observations | 260,004 | 226,507 |
| Unique reflections (highest shell) | 56,014 | 42,943 |
| Completeness (%) (highest shell) | 94.3 (90.1) | 98.1 (89.6) |
| CC1/2 highest shell | 0.833 | 0.824 |
| Rsym/Rmeas (highest shell) | 0.086 (0.596)/0.097 (0.680) | 0.084 (0.444)/0.106 (0.558) |
| 〈I/σ〉 (highest shell) | 20.40 (2.41) | 16.05 (2.95) |
| Redundancy (highest shell) | 4.6 (3.9) | 5.3 (4.35) |
| B-factor, Wilson plot (Å2) | 27.1 | 11.4 |
| Reflections used in refinement | 55,965 | 42,890 |
| Resolution range (Å) used in refinement | 29.06 (2.00) | 19.59 (1.78) |
| No. of protein atoms fit | 6,300 | 3,145 |
| No. of heteroatoms fit | 162 | 43 |
| No. of waters fit | 720 | 597 |
| Rwork (%) | 16.3 | 16.7 |
| Rfree (%) | 21.3 | 19.6 |
| r.m.s.d. bond length (Å) | 0.007 | 0.011 |
| r.m.s.d. bond angle | 1.17° | 1.08° |
| Average B, all atoms (Å2) | 28.2 | 17.0 |
| Average B, protein atoms (Å2) | 27.6 | 15.5 |
| Average B, heteroatoms (Å2) | 28.2 | 7.9 |
| Ramachandran residues favored/allowed/outliers | 768/29/1 | 349/17/0 |
Substrate-free crystals were grown at 4 °C by the hanging drop vapor diffusion method and crystallized in space group P212121 with one molecule per asymetric unit. The well solution contained 250 μl of 1.27 m sodium citrate dibasic, 60 mm CaCl2, and 100 mm BisTris propane. The protein concentration was 250 μm in 10 mm Tris, pH 7.5, buffer with 0.25 mm cholesterol. Crystals were transferred into a cryo-solution consisting of 1.5 m sodium citrate with 0.25 mm cholesterol. Crystals were then flash frozen in liquid nitrogen. Diffraction data were collected at 100 K using the ALS synchrotron, beamline 8.3.1. The wavelength was 1.1158 Å with a detector distance of 225 mm and a starting ϕ = 102° with 1° oscillations per frame (135 frames total). Integration and scaling were perfomed in XDS (22), and intensities were converted to structure factors using SCALA from the CCP4 suite (19). Molecular replacement was performed in Phaser (20) utilizing phases from a previous substrate-free crystal structure (PDB code 3ZBY). A fraction of the data (5%) were flagged for an Rfree test set, and refinement was performed using the Phenix package (21) using an individual coordinate and and isotropic B-factor refinement strategy. There were no Ramachandran outliers. All figures were generated using molecule B. Final statistics are given in Table 2.
RESULTS
Binding of cholesteryl sulfate and cholesteryl propionate was monitored by substrate-induced spectral changes of the heme iron. As expected, both cholesterol esters bound to CYP142A1 and CYP142A2 producing a type I absorption shift, indicative of displacement of the axial water ligand from the iron atom. A fit of the spectral titrations to a single site binding model (fitting to the Hill equation did not significantly alter the results; see “Experimental Procedures”) revealed that M. tuberculosis CYP142A1 bound both substrates more tightly, with KS = 17.6 ± 3.6 versus 32.8 ± 6.6 μm for cholesteryl sulfate and KS = 0.16 ± 0.09 versus 1.5 ± 0.1 μm for cholesteryl propionate for CYP142A1 and CYP142A2, respectively, whereas M. smegmatis CYP142A2 produced a larger change in the amplitude of the spectral response (Fig. 1, A and B). Unlike canonical cytochromes P450 such as CYP142, CYP125 enzymes are predominantly high-spin in the resting state, and binding of substrates such as 4-cholesten-3-one shift the spin equilibrium further toward the high-spin state (10). However, binding of cholesteryl sulfate to M. tuberculosis CYP125A1 induced a spectral shift back to the low-spin heme iron electronic configuration, suggesting that this substrate enters the active site in a manner that reorders the binding pocket waters so that the distal water ligand is more tightly coordinated to the heme iron. This binding exhibited an apparent cooperative nature and was fit to the Hill equation (see “Experimental Procedures”), with S50 = 35.1 ± 2.6 μm and n = 1.8 ± 0.2, although there is no evidence for multiple binding events (Fig. 1A). No spectral changes were observed for the binding of cholesteryl sulfate to M. smegmatis CYP125A3, or for cholesteryl propionate to either of the CYP125 paralogs.
FIGURE 1.

A, plots of the binding of cholesteryl sulfate to CYP142A1, CYP142A2, and CYP125A1 and representative difference spectra. The binding parameters derived from the plots are indicated. B, plots of the binding of cholesteryl propionate to CYP142A1 and CYP142A2 and representative difference spectra. The binding parameters derived from the plots are indicated in arbitrary units (AU).
Cholesteryl propionate was observed to be a substrate for both CYP142, but not CYP125, enzymes. Incubations of CYP142 with cholesteryl propionate, MβCD, and an NADPH regeneration system (see “Experimental Procedures”), yielded a product peak detected by gas chromatography-mass spectrometry, with a retention time of ∼30 min, and a mass corresponding to the trimethylsilyl-derivatized alcohol product, i.e. +88 Da to 456 Da (Fig. 2A). Performing the reaction under a 1:1 mixture of 18O/16O gas shifted the mass of the product to give a doublet peak at +88/90 Da, demonstrating efficient incorporation of isotopically labeled O2, consistent with a cytochrome P450-mediated reaction (Fig. 2B). Because the propionate group is lost during the mass spectrometric ionization process, and the parent ions of neither the substrate nor the product are observed, the oxidation reaction must occur at one of the cholesterol carbons; presumably at C26 where CYP142 oxidizes both cholesterol and 4-cholesten-3-one.
FIGURE 2.

A, catalytic turnover of cholesteryl propionate by CYP142A1 and CYP142A2 in the presence and absence of the cofactor NADPH. Note the product peak at ∼30 min. B, incorporation of 18O into the product formed from cholesterol propionate by CYP142A1 and CYP142A2 in the presence of 16O2 versus a 1:1 mixture of 16O2/18O2, reported in arbitrary units (AU).
Cholesteryl sulfate was a substrate for all four of the enzymes, although the CYP142 enzymes were ∼10-fold more active toward this substrate than the CYP125 proteins (Fig. 3). Incubation of the enzymes with cholesteryl sulfate, MβCD, and an NADPH regeneration system (see “Experimental Procedures”), generated a new peak corresponding to the product, detected on the LC-MS with a retention time of ∼11.7 min and a mass of 481 Da (data not shown). The +16-Da shift from the substrate mass confirms the incorporation of a single oxygen atom and is consistent with a cytochrome P450-mediated hydroxylation reaction. Whereas the CYP142 enzymes displayed typical Michaelis-Menten kinetics, with Km = 9.2 ± 2.6 and 39.8 ± 13.7 μm for CYP142A1 and CYP142A2, respectively, both CYP125 enzymes showed sigmoidal turnover kinetics with S50 = 46.2 ± 17.4 and 49.1 ± 11.8 μm and n = 1.6 ± 0.5 and 2.7 ± 1.2, for CYP125A1 and CYP125A3, respectively.
FIGURE 3.

Catalytic oxidation of cholesteryl sulfate by CYP142A1 (A) and CYP142A2 (B). The binding parameters derived from the plots are indicated.
The crystal structure of CYP142A2 complexed with cholesteryl sulfate revealed the substrate in an orientation relative to the heme similar to that of the previously published 4-cholesten-3-one structure (10) (PDB code 2YOO), with the bulky sulfate group projecting out toward the solvent (Fig. 4A). To avoid model bias a simulated annealing composite-omit |FO − FC| electron density map was also generated (Fig. 5). We were unsuccessful in crystallizing the cholesterol bound enzyme as we initially wanted to compare cholesteryl sulfate and cholesterol binding. Instead, crystals were ligand-free with a pentacoordinate iron (PDB code 4UAX). A water molecule sits near the heme iron but is too far to be coordinated (∼2.6 Å away) and is hydrogen-bonded to the Gly-233 carbonyl oxygen atom. The crystal structure is almost identical to the previously reported ligand-free structure (PDB code 3ZBY) in which ethylene glycol sits in the active (C-α carbon root mean square deviation ∼ 0.27 ± 0.25 Å). In CYP142A2, the cholesteryl sulfate is rotated with respect to the heme compared with the position of 4-cholesten-3-one in CYP125A1 (PDB code 2X5W) (12). Oxidation of one or the other of the two terminal methyl carbons will generate either the R- or the S-stereochemistry, depending on which methyl group is oxidized. It has been shown that CYP142A1-driven 4-cholesten-3-one hydroxylation results primarily in the 26(S)-product, whereas CYP125A1 generates the opposite stereochemistry (6). The structural basis for this can be inferred from which of the ω-methyl groups is positioned closest (within 4.1 Å) to the heme iron atom. The CYP142A2 4-cholesten-3-one and cholesteryl sulfate ligands overlay perfectly near the heme, positioning the pro-(R)-methyl group nearest the iron, whereas in the CYP125A1 4-cholesten-3-one complex the pro-(S)-methyl group resides nearest to the iron.
FIGURE 4.

A, slab view of the CYP142A2 (PDB code 4TRI) active site complexed with cholesteryl sulfate (green sticks). The CYP125A1 (red, PDB code 2X5W) and CYP142A2 (white, PDB code 2YOO) 4-cholestene-3-one ligands. B, the CYP125A1 active site in complex with 4-cholestene-3-one (red sticks) overlaid with the CYP142A2 cholesteryl sulfate ligand (green). The inner lining of the binding pocket has been colored semi-transparent red. C, the CYP142A2 active site complexed with cholesteryl sulfate (green sticks) overlaid with the CYP125A1 4-cholestene-3-one ligand in red. The inner lining of the binding pocket has been colored semi-transparent green. D, CYP142A2 with the FG and BC helices colored green. The corresponding regions of CYP125A1 have been superimposed in red.
FIGURE 5.
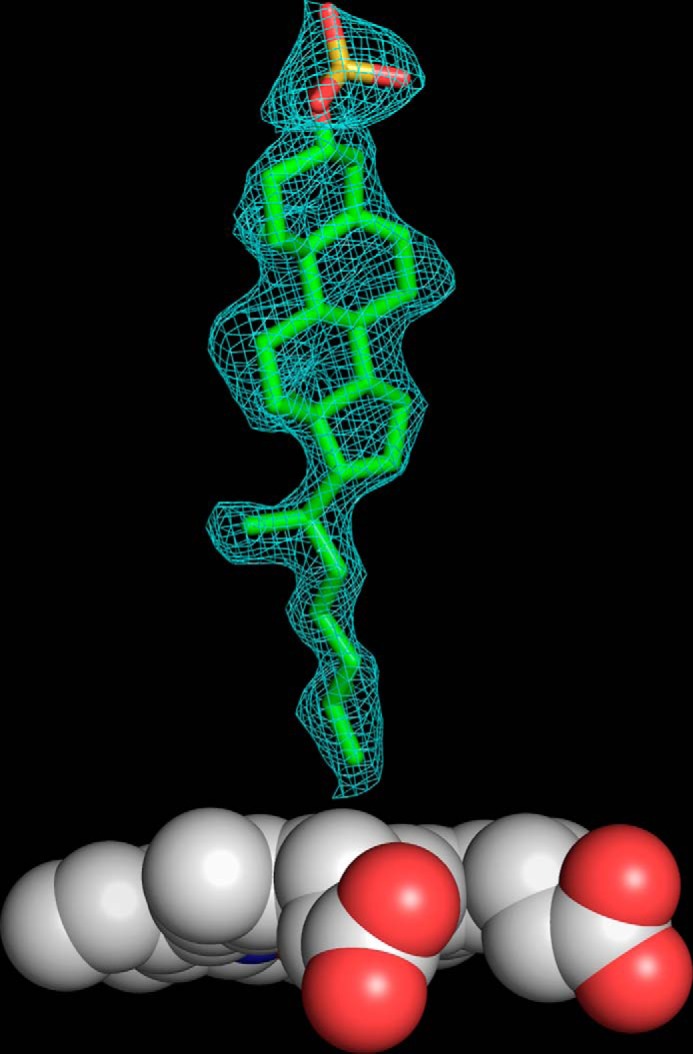
Simulated annealing composite-omit |FO − FC| electron density map contoured at 1.0σ of CYP142A2 in complex with cholesteryl sulfate (PDB code 4TRI). The electron density for the heme is not shown.
The binding cavity of CYP142A2 almost perfectly surrounds the cholesteryl sulfate ligand with a total surface area of ∼494.4 Å2 (volume ∼ 1007 Å3) calculated by F-pocket (23) and a cholesteryl sulfate ligand surface area of ∼475 Å2 (volume ∼ 676 Å3) calculated by the Volume Assessor webserver (24). Consequently, there is very little room for conformational deformation of the substrates from that seen in the crystal structure. CYP125A1, however, has a substantially larger active site with a surface area of ∼644.2 Å2 (volume ∼ 1491 Å3) (Fig. 4, B and C). It is therefore possible that CYP125A1 has the ability to bind substrates in multiple modes compared with the tighter fit seen in CYP142A2.
To investigate whether CYP125 enzymes bind cholesteryl sulfate in multiple modes, we examined the rate of NADPH oxidation by each enzyme in the absence and presence of various substrates (Table 3). The presence of substrate activates the NADPH oxidation rate between two to seven times for each of the enzymes. Although NADPH consumption is stimulated most robustly by cholesterol and 4-cholesten-3-one, cholesteryl sulfate also elevates NADPH consumption by all four enzymes. Interestingly, despite the fact that the CYP142 enzymes form the cholesteryl sulfate product at a rate approximately 10 times greater than that of their CYP125 counterparts (Fig. 3, A and B), the NADPH oxidation rate for all four enzymes differs by no more than 2–3-fold, indicating a larger degree of catalytic uncoupling for the CYP125 enzymes compared with the CYP142 enzymes in the reconstituted system. This suggests that cholesteryl sulfate can bind within the active site of CYP125 enzymes in a manner that triggers the catalytic cycle but does not position the alkyl side chain for efficient oxidation.
TABLE 3.
NADPH oxidation rate (all values min−1)
| Ligand-free | Cholesterol | 4-Cholesten-3-one | Cholesteryl sulfate | |
|---|---|---|---|---|
| CYP125A1 | 2.6 ± 0.1 | 11.3 ± 0.1 | 14.0 ± 0.1 | 4.7 ± 0. 1 |
| CYP125A3 | 1.6 ± 0.1 | 3.8 ± 0.1 | 5.7 ± 0.1 | 3.3 ± 0.1 |
| CYP142A1 | 3.9 ± 0.1 | 6.0 ± 0.1 | 8.5 ± 0.1 | 5.9 ± 0.1 |
| CYP142A2 | 2.4 ± 0.1 | 8.8 ± 0.1 | 11.2 ± 0.1 | 8.7 ± 0.1 |
DISCUSSION
Cholesterol is a critical intracellular molecule for M. tuberculosis persistence of infection (3–6). One of the primary steps in the cholesterol degradation pathway is the three-step oxidation of the cholesterol alkyl side chain to the carboxylic acid by CYP125A1 and CYP142A1, which in turn, can be catabolized via the β-oxidation pathway (7–9). This pathway is conserved in the soil dwelling relative M. smegmatis, whose genome encodes CYP125A3 and CYP142A2, enzymes that share ∼70% sequence identity with their M. tuberculosis paralogs (10). Despite the seemingly redundant catalytic behavior of these two enzymes, structural comparisons reveal two peptide insertions into the CYP125 sequences that fold to form a cap region over the substrate binding cavity. To determine the role this region may play in substrate selectivity, we examined the ability of each enzyme to bind and oxidize two model cholesterol esters, cholesteryl propionate and cholesteryl sulfate.
Both CYP142 enzymes were able to bind cholesteryl propionate, displaying a type I spin transition caused by displacement of the axial water ligand from the heme iron atom, with a consequent shift of the Soret maximum from the low-spin state with a Soret absorbance maximum at 419 nm to a high-spin configuration with the Soret maximum at 393 nm. Analysis of the oxidation reactions by gas chromatography-mass spectrometry demonstrated that both CYP142A1 and CYP142A2 oxidize cholesteryl propionate to form an alcohol product into which an 18O-atom was incorporated in the presence of 18O2. That the hydroxyl group is located on the sterol and not propionate group, the orientation of the sterol molecule in the crystal structures, and the demonstrated C-26 hydroxylation of cholesterol and 4-cholesten-3-one indicate that the product was the 26-hydroxylated derivative of cholesteryl propionate. In contrast, CYP125A1 and CYP125A3 were unable to bind or oxidize cholesteryl propionate, as we proposed from the steric hindrance imposed by the cap region and orientation of the binding pocket.
Unlike cholesteryl propionate, all four enzymes were able to oxidize cholesteryl sulfate at some level, but with important differences between the two CYP families. Both CYP142 enzymes bound cholesteryl sulfate as a canonical type I substrate and kinetic analyses of product formation were readily fit to a classic Michaelis-Menten model. However, CYP125A1, but not CYP125A3, displayed spectral changes indicative of the binding of cholesteryl sulfate. In contrast to the CYP142 enzymes, CYP125A1 exhibited a reverse type I spectral transition from the high-spin resting state, with a spectral peak at 393 nm, to a low-spin state with a maximum at 419 nm. This transition did not fit to a typical single substrate binding model, but rather to a Hill binding model with n = 1.8 ± 0.2. Somewhat unexpectedly, both CYP125 enzymes oxidized cholesteryl sulfate to the side-chain alcohol product, but at a much slower rate than the CYP142 enzymes and with kinetics that fit to a Hill equation with n = 1.6 ± 0.5 for CYP125A1 and n = 2.7 ± 1.2 for CYP125A3. The apparent cooperative behavior of the CYP125 proteins was not previously observed with smaller substrates such as cholesterol or 4-cholesten-3-one (10, 12), which makes it unlikely that multiple molecules of cholesteryl sulfate are bound simultaneously in the active site. We propose instead that cholesteryl sulfate has multiple binding modes within the larger CYP125 binding pocket (as compared with that of CYP142), which leads to the observed lower catalytic activity, complex kinetic behavior, and higher rate of uncoupling compared with CYP142.
Our proposal is consistent with structural comparison of the ligand bound forms of CYP142A2 (in complex with cholesteryl sulfate), and CYP125A1 (in complex with 4-cholesten-3-one). The two amino acid sequence insertions, combined with the movement of the FG helices, create a larger active site for CYP125A1 with two pores above the site. These pores are blocked by the FG helices in the ligand bound structure of CYP142A2 (Fig. 4D). A crystal structure of CYP125A1 in complex with the LP10 inhibitor (PDB code 2XC3), which also induces a reverse type I spectral shift upon binding to the enzyme, was reported previously (25). This inhibitor straddles the two entry channels with one of its two branches extending into each of the channels. It is conceivable that cholesteryl sulfate may occupy one of these pores as well.
Substrate binding in cytochromes P450 involves a gating mechanism in which electron transfer is promoted by substrate binding. Early studies with P450cam showed that substrate binding induces a low- to high-spin shift that is linearly correlated with a rise in redox potential of the heme, enabling electron transfer to occur (26, 27). This phenomenon prevents the possible uncoupling reaction in which redox partner-derived electrons can be channeled into superoxide, water, or hydrogen peroxide rather than productive hydroxylation. In an aqueous environment, the potential for competition between substrate and water for active site space is always present, and an active site with less non-catalytic water results in an enzyme with tighter coupling. Subtle mutations that alter the active site topology have been linked to increased water in the binding pocket and disruption of the native proton delivery network (28–30). For example, in other cytochrome P450 enzymes such as P450cam mutations that introduce bulky side chains that exclude water from the active site favor increased coupling, whereas mutations that introduce smaller side chains favor hydrogen peroxide formation (28). Subtle changes near the active site have also been shown to alter the structure of the proton relay system in iron-bound dioxygen intermediates of P450cam (31) and P450eryF (32). In the case of CYP125, binding of cholesteryl sulfate may result in a substrate that is poorly positioned for alkyl chain oxidation but is close enough to initiate the catalytic cycle of the enzyme. This would rationalize our experimental results, which show that CYP125 remains active with regards to NADPH oxidation, but is largely uncoupled when incubated with cholesteryl sulfate.
In summary, we have demonstrated that the CYP142 subfamily, initially postulated to play a purely redundant role to CYP125 enzymes in vivo, can metabolize cholesteryl sulfate and cholesteryl propionate, whereas CYP125 enzymes were only able to oxidize cholesteryl sulfate at a much slower rate and could not detectably oxidize cholesteryl propionate. The crystal structure of CYP142A2 in complex with cholesteryl sulfate reveals an active site that closely surrounds the substrate, as opposed to the larger binding pocket previously observed with CYP125A1 (12). We propose that the cholesteryl sulfate side-chain may enter the CYP125 active site in a manner that triggers catalytic cycling but places the substrate in an unsuitable position for efficient oxidation due to steric hindrance from two ∼10-amino acid insertions in the CYP125 sequence that form a cap over the active site. Most notably, the results indicate that CYP142 enzymes may play an important role during M. tuberculosis infection by providing access to additional reservoirs of esterified intracellular cholesterol that otherwise may not be available to the pathogen.
Acknowledgments
We thank Christopher Waddling for tireless work in maintaining and improving the University of California at San Francisco macromolecular structure facility and in particular, the in-house x-ray generator, where data for the cholesteryl sulfate bound CYP142A2 crystal structure was collected. This publication also includes work performed at the Advanced Light Source (ALS) in Berkeley, CA. ALS is a national user facility supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U. S. Department of Energy under Contract DE-ACO2-05CH11231. We thank Drs. Clinton Nishida, Debashree Basudhar, and other members of the Ortiz de Montellano laboratory for technical assistance and useful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants AI074824 and GM25515.
The atomic coordinates and structure factors (codes 4UAX and 4TRI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CYP
- cytochrome P450
- MβCD
- methyl-β-cyclodextrin
- PDB
- Protein Data Bank.
REFERENCES
- 1. World Health Organization (2014) World Health Organization Fact Sheets on Tuberculosis, http://www.who.int/mediacentre/factsheets/fs104/en/
- 2. Russell D. G., Cardona P. J., Kim M. J., Allain S., Altare F. (2009) Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 10, 943–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pandey A. K., Sassetti C. M. (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U.S.A. 105, 4376–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang J. C., Miner M. D., Pandey A. K., Gill W. P., Harik N. S., Sassetti C. M., Sherman D. R. (2009) igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J. Bacteriol. 191, 5232–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yam K. C., D'Angelo I., Kalscheuer R., Zhu H., Wang J. X., Snieckus V., Ly L. H., Converse P. J., Jacobs W. R., Jr., Strynadka N., Eltis L. D. (2009) Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 5, e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnston J. B., Ouellet H., Ortiz de Montellano P. R. (2010) Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J. Biol. Chem. 285, 36352–36360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ouellet H., Johnston J. B., de Montellano P. R. (2011) Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 19, 530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ouellet H., Johnston J. B., Ortiz de Montellano P. R. (2010) The Mycobacterium tuberculosis cytochrome P450 system. Arch. Biochem. Biophys. 493, 82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McLean K. J., Belcher J., Driscoll M. D., Fernandez C. C., Le Van D., Bui S., Golovanova M., Munro A. W. (2010) The Mycobacterium tuberculosis cytochromes P450: physiology, biochemistry and molecular intervention. Future Med. Chem. 2, 1339–1353 [DOI] [PubMed] [Google Scholar]
- 10. García-Fernández E., Frank D. J., Galán B., Kells P. M., Podust L. M., García J. L., Ortiz de Montellano P. R. (2013) A highly conserved mycobacterial cholesterol catabolic pathway. Environ. Microbiol. 15, 2342–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klansek J. J., Yancey P., St Clair R. W., Fischer R. T., Johnson W. J., Glick J. M. (1995) Cholesterol quantitation by GLC: artifactual formation of short-chain steryl esters. J. Lipid Res. 36, 2261–2266 [PubMed] [Google Scholar]
- 12. Ouellet H., Guan S., Johnston J. B., Chow E. D., Kells P. M., Burlingame A. L., Cox J. S., Podust L. M., de Montellano P. R. (2010) Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol. Microbiol. 77, 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Capyk J. K., Kalscheuer R., Stewart G. R., Liu J., Kwon H., Zhao R., Okamoto S., Jacobs W. R., Jr., Eltis L. D., Mohn W. W. (2009) Mycobacterial cytochrome P450 125 (CYP125) catalyzes the terminal hydroxylation of c27 steroids. J. Biol. Chem. 284, 35534–35542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Driscoll M. D., McLean K. J., Levy C., Mast N., Pikuleva I. A., Lafite P., Rigby S. E., Leys D., Munro A. W. (2010) Structural and biochemical characterization of Mycobacterium tuberculosis CYP142: evidence for multiple cholesterol 27-hydroxylase activities in a human pathogen. J. Biol. Chem. 285, 38270–38282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kondo E., Kanai K. (1976) Accumulation of cholesterol esters in macrophages incubated with mycobacteria in vitro. Jpn. J. Med. Sci. Biol. 29, 123–137 [DOI] [PubMed] [Google Scholar]
- 16. Kondo E., Kanai K. (1974) Further studies on the increase in cholesterol ester content of the lungs of tuberculous mice. Jpn. J. Med. Sci. Biol. 27, 59–65 [DOI] [PubMed] [Google Scholar]
- 17. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. II. solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 18. Otwinowski Z., Minor W., Carter C. W., Jr. (1997) Processing of X-ray diffraction data collected in oscillation mode in Methods in Enzymology, pp. 307–326, Academic Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- 19. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmidtke P., Bidon-Chanal A., Luque F. J., Barril X. (2011) MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 27, 3276–3285 [DOI] [PubMed] [Google Scholar]
- 24. Voss N. R., Gerstein M. (2010) 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 38, W555–W562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ouellet H., Kells P. M., Ortiz de Montellano P. R., Podust L. M. (2011) Reverse type I inhibitor of Mycobacterium tuberculosis CYP125A1. Bioorg. Med. Chem. Lett. 21, 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fisher M. T., Sligar S. G. (1985) Control of heme protein redox potential and reduction rate: linear free-energy relation between potential and ferric spin state equilibrium. J. Am. Chem. Soc. 107, 5018–5019 [Google Scholar]
- 27. Sligar S. G. (1976) Coupling of spin, substrate, and redox equilibria in cytochrome P450. Biochemistry 15, 5399–5406 [DOI] [PubMed] [Google Scholar]
- 28. Loida P. J., Sligar S. G. (1993) Molecular recognition in cytochrome P-450: mechanism for the control of uncoupling reactions. Biochemistry 32, 11530–11538 [DOI] [PubMed] [Google Scholar]
- 29. Madrona Y., Hollingsworth S. A., Khan B., Poulos T. L. (2013) P450cin active site water: implications for substrate binding and solvent accessibility. Biochemistry 52, 5039–5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choonkeun K., Haeyoung K., Oksoo H. (2000) The role of serine-246 in cytochrome P450eryF-catalyzed hydroxylation of 6-deoxyerythronolide B. Bioorg. Chem. 28, 306–314 [DOI] [PubMed] [Google Scholar]
- 31. Nagano S., Poulos T. L. (2005) Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J. Biol. Chem. 280, 31659–31663 [DOI] [PubMed] [Google Scholar]
- 32. Nagano S., Cupp-Vickery J. R., Poulos T. L. (2005) Crystal structures of the ferrous dioxygen complex of wild-type cytochrome P450eryF and its mutants, A245S and A245T: investigation of the proton transfer system in P450eryF. J. Biol. Chem. 280, 22102–22107 [DOI] [PubMed] [Google Scholar]