

Background: Molecular regulation of breast cancer oncogene ErbB2 is relatively unclear.
Results: An imaging-based screen identified PKC-dependent movement of ErbB2 from cell surface into an intracellular signaling compartment.
Conclusion: PKC-α and -δ are novel regulators of ErbB2 entry into a special compartment where unique oncogenic signals may originate.
Significance: PKC targeting may improve therapy against certain breast cancers.
Keywords: Breast Cancer, Confocal Microscopy, Endocytosis, Protein Kinase C (PKC), Receptor Tyrosine Kinase, ErbB2, Endocytic Traffic, High-Content Fluorescence Microscopy, Kinase Inhibitors, Small Molecule Screening
Abstract
ErbB2 overexpression drives oncogenesis in 20–30% cases of breast cancer. Oncogenic potential of ErbB2 is linked to inefficient endocytic traffic into lysosomes and preferential recycling. However, regulation of ErbB2 recycling is incompletely understood. We used a high-content immunofluorescence imaging-based kinase inhibitor screen on SKBR-3 breast cancer cells to identify kinases whose inhibition alters the clearance of cell surface ErbB2 induced by Hsp90 inhibitor 17-AAG. Less ErbB2 clearance was observed with broad-spectrum PKC inhibitor Ro 31-8220. A similar effect was observed with Go 6976, a selective inhibitor of classical Ca2+-dependent PKCs (α, β1, βII, and γ). PKC activation by PMA promoted surface ErbB2 clearance but without degradation, and ErbB2 was observed to move into a juxtanuclear compartment where it colocalized with PKC-α and PKC-δ together with the endocytic recycling regulator Arf6. PKC-α knockdown impaired the juxtanuclear localization of ErbB2. ErbB2 transit to the recycling compartment was also impaired upon PKC-δ knockdown. PMA-induced Erk phosphorylation was reduced by ErbB2 inhibitor lapatinib, as well as by knockdown of PKC-δ but not that of PKC-α. Our results suggest that activation of PKC-α and -δ mediates a novel positive feedback loop by promoting ErbB2 entry into the endocytic recycling compartment, consistent with reported positive roles for these PKCs in ErbB2-mediated tumorigenesis. As the endocytic recycling compartment/pericentrion has emerged as a PKC-dependent signaling hub for G-protein-coupled receptors, our findings raise the possibility that oncogenesis by ErbB2 involves previously unexplored PKC-dependent endosomal signaling.
Introduction
ErbB2 (HER2/neu) is a member of the epidermal growth factor receptor (EGFR)6 family of receptor tyrosine kinases (RTKs). ErbB2 overexpression, most often due to gene amplification, is causally linked to 20–30% of breast cancers and smaller percentages of other cancers (1, 2). ErbB2 overexpression in breast cancer predicts poor prognosis, a scenario significantly ameliorated by the introduction of targeted therapy with humanized monoclonal antibody Trastuzumab (Herceptin; Genentech-Roche Inc.). Frequent de novo and acquired resistance to Trastuzumab, however, are major issues and have incited efforts to elucidate the cell biology of ErbB2 receptor to improve its therapeutic targeting. For example, ErbB2 exhibits a unique dependence on Hsp90 for its stability (3–6). Accordingly, Hsp90 inhibitors, such as 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), sensitize tumor cells to traditional chemotherapeutic agents (7–9), and synergize with Trastuzumab in killing ErbB2-overxpressing breast cancer cells (10–14). These therapeutic combinations are undergoing clinical evaluation and have demonstrated substantial benefit (11). Identification of novel ErbB2 regulatory mechanisms could therefore offer new therapeutic strategies in breast cancer.
A fundamental cell biological trait of RTKs is their ligand-enhanced internalization into the endocytic pathway. Within the ErbB receptor family, endocytic traffic has been most clearly elucidated for EGFR. Ligand-activated EGFR is internalized through clathrin-dependent and multiple clathrin-independent pathways (15–18). The subsequent endocytic routing of internalized EGFR and other RTKs has emerged as a major determinant of their function. The Cbl-family of ubiquitin ligase-dependent sorting of RTKs for lysosomal degradation helps terminate signaling (19). Alternatively, EGFR can enter the endocytic recycling compartment (ERC) from where it traffics back to the cell surface for additional rounds of ligand binding (20). Furthermore, internalized RTKs continue to signal within endosomes, thereby sustaining signaling as well as generating signals distinct from those at the cell surface (15, 21, 22).
Despite critical physiological functions of ErbB2 in multiple organ systems (23, 24) and its prominent role in highly-prevalent cancers (2, 25), molecular determinants of its endocytic traffic remain unclear. A significant impediment to experimental analyses has been the lack of a ligand to directly activate ErbB2. However, the ability of ErbB2 to serve as a preferred hetero-dimerization partner for other family members, have prompted studies using indirect ErbB2 activation with ligands that bind to its heterodimeric partners (18, 26–28), or those using chimeric receptors that incorporate extracellular ligand-binding domains of EGFR (29). For example, ErbB3/4-directed ligand neuregulin 1 (NRG1)/heregulin 1 (HRG1) has been shown to induce relatively rapid endocytosis of ErbB2 (as well as ErbB3 and 4) in neurons and Schwann cells (30, 31). Genetic and pharmacological evidence suggests that NRG1-induced ErbB2 endocytosis in neural cell systems is dynamin- and clathrin-dependent (32). In general, the fates of internalized ErbB2 in Schwann cells and neurons have not been investigated in detail. However, ErbB receptor endocytosis was shown to be required for the activation of specific biochemical pathways, such as Erk and Akt phosphorylation, and clustering of nicotinic acetylcholine receptor at the presynaptic region (31, 33, 34), suggesting that ErbB2 may enter endosomal signaling compartments. EGF stimulation of mammary epithelial cells expressing low levels of endogenous ErbB2 was shown to induce substantial ErbB2 internalization (35), with subsequent degradation (35, 36), comparable to that of EGFR. However, when ErbB2 was overexpressed in these cells, it was internalized but showed reduced degradation and enhanced recycling (36). NRG1/HRG1 stimulation of ErbB2-overexpressing breast cancer cells was reported to induce rapid ErbB2 endocytosis as well as ubiquitination and degradation (27). However, the subcellular itinerary of ErbB2 in these systems has not been elucidated.
Use of chimeric receptors, utilizing EGF-binding extracellular sequences of EGFR fused to transmembrane/cytoplasmic regions of ErbB2, has shown that ErbB2 cytoplasmic sequences confer reduced efficiency of endocytosis and lysosomal down-regulation (29, 37), and accentuate recycling (38). Endogenously overexpressed ErbB2 in breast cancer cell lines or ectopically-overexpressed ErbB2 has been shown to be phosphorylated, and presumably activated, but not targeted for degradation upon activation of EGFR with EGF; in fact, ErbB2 overexpression reduces EGFR degradation and promotes its endocytic recycling (39, 40). Thus, it appears that reduced endocytosis or inefficient lysosomal targeting with enhanced recycling promote oncogenic signaling by ErbB2 (41).
A number of studies have shown that Trastuzumab or other anti-ErbB2 antibodies induce the internalization of overexpressed ErbB2 with slower kinetics than that of ligand-activated EGFR (42–44). Notably, ErbB2 removal from the cell surface correlates positively with tumor inhibitory activity of anti-ErbB2 monoclonal antibodies (44). The mechanisms of endocytosis induced by anti-ErbB2 antibodies remain unclear although clathrin-dependent endocytosis has been invoked (45). Furthermore, ErbB2 bound to Trastuzumab was shown to undergo multiple cycles of ErbB2 recycling and to slowly enter the lysosomes, leading to ErbB2 down-regulation (10, 46). Similarly, increased endocytosis and down-regulation of surface ErbB2 has been observed with artificial peptide and aptamer ligands (47, 48).
Recent work has shown that HSP90 inhibitors induce rapid down-regulation of surface ErbB2 together with its ubiquitination and degradation. A number of studies have now documented that ErbB2 is targeted to lysosomes upon HSP90 inhibition (10, 46, 49–55). It is however controversial how this takes place. Some studies have suggested that Hsp90 inhibitors enhance the clathrin-dependent or clathrin-independent endocytosis of ErbB2 (54, 55). Other studies suggest that ErbB2 is constitutively internalized, and that HSP90 inhibitors do not influence this rate but instead promote sorting of ErbB2 away from the recycling pathway and into lysosomes (46, 51).
Collectively, studies thus far indicate that overexpressed ErbB2, a driver oncogene in breast and other cancers, is inefficiently targeted for lysosomal degradation, is less efficiently endocytosed and is preferentially sorted into recycling. Importantly, ErbB2-targeted therapeutic approaches appear to promote ErbB2 sorting into lysosomes and its degradation. Despite the importance of a balance between recycling and lysosomal degradation as a determinant of ErbB2 function and therapeutic targeting, mechanisms that regulate ErbB2 recycling remain unclear.
Prior studies suggest an important regulatory role of downstream kinases in modulating the endocytic itinerary of EGFR. Multiple studies have shown that MAP kinase family member p38 mediates stress-induced EGFR endocytosis into a recycling compartment (56–59). Furthermore, PKC-α has been shown to alter the endocytic fate of activated EGFR from lysosomal degradation to recycling (60). Other studies showed that PKC-δ (61, 62), protein kinase A (61–64), Rho kinase (65), and LIM kinase (66), as well as lipid kinase diacylglyecerol kinase δ and phospholipase D modulate EGFR endocytosis (64, 67, 68, 68–70). Finally, c-Src has been shown to promote EGFR endocytosis likely into a recycling compartment (71–74). A previous study showed that PMA treatment of ErbB2-transfected fibroblasts induced the juxtamembrane threonine-686 phosphorylation of ErbB2, as well as ErbB2 internalization and intra-cytoplasmic accumulation, but no further characterization of intracellular compartments was carried out (75). In another study, assessment of cell surface levels of ErbB2 by flow cytometry or ErbB2 protein levels by Western blotting implicated PKC-α as a positive regulator of cell surface levels of ErbB2 in certain breast cancer cell lines that show high surface ErbB2 but without gene amplification (referred to as Her2 2+ based on immunohistochemistry) (76). However, no cell biological analyses were carried out and any role of PKC-α in the endocytic routing of ErbB2 was not examined. Thus, while endocytic recycling has emerged as a key biological characteristic of ErbB2, and ErbB2 is well-established to signal through a network of protein and lipid kinases, a potential role for cellular kinases in endocytic recycling of ErbB2 remains unexplored.
Here, we carried out an imaging-based screen of a subset of the cellular kinome using available kinase inhibitors to assess their impact on the removal of ErbB2 from the cell surface that was induced by the Hsp90 inhibitor 17-AAG. We identify PKC-α and PKC-δ as positive regulators of surface ErbB2 entry into the endocytic pathway and show that PKCs promote ErbB2 traffic along a recycling itinerary.
EXPERIMENTAL PROCEDURES
Reagents
The sources for reagents were as follows: Chemicals: Tocriscreen® Kinase Inhibitor Toolbox with 80 Kinase inhibitor stocks (10 mm in DMSO) (Fig. 2B for inhibitor list and specificity), Ro 31-8220 mesylate and Fasudil hydrochloride from Tocris Bioscience (Bristol, UK); 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) from Biomol International (Plymouth, PA); enzyme immunoassay assay-grade BSA, paraformaldehyde (PFA), Saponin, phorbol myristate acetate (PMA), and Go 6976 from Sigma-Aldrich.
FIGURE 2.

Small molecule kinase inhibitor library screen identifies protein kinase C as a potential regulator of ErbB2 down-regulation from the cell surface. A, SKBR-3 cells seeded and labeled with Alexa Fluor® 488-conjuagated anti-ErbB2 as in Fig. 1. Pre-warmed medium containing various inhibitors (1 μm final), with or without 17-AAG (100 nm final) was added, and cells were incubated at 37 °C for 8 h. Cell surface ErbB2 levels were computed and are presented as mean fluorescence intensity of duplicates. Inhibitors that reduced the 17-AAG-induced clearance of ErbB2 (i.e. more ErbB2 remaining at the cell surface) by >20% are indicated in red: Fasudil (no. 25), Ro-31–8220 (no. 28), and Genistein (no. 41). B, list of kinase inhibitor library (specificity per vendor is shown). C and D, to validate the selected inhibitors, SKBR-3 cells were treated with DMSO (vehicle control) or 1 μm final concentrations of Fasudil (C) or Ro 31–8220 (D), with or without 100 nm 17-AAG for 8 h, and cell surface ErbB2 levels analyzed by FACS-staining with Alexa Fluor® 488-conjugated-anti-ErbB2 antibody. E. SKBR-3 cells were seeded in 6-well plates and live cells stained with Alexa Fluor 488® conjugated anti-ErbB2 antibody for 1 h at 4 °C. Stained cells were pre-treated without or with Ro 31–8220 (1 μm) at 37 °C for 1 h followed by addition of DMSO (vehicle) or 17-AAG (25 nm) for 8 h. Cells were paraformaldehyde-fixed followed by imaging under a confocal microscope.
Antibodies: Alexa Fluor® 488- or 647-conjugated mouse anti-human ErbB2 monoclonal antibodies (mAb) (CD340; clone 24D2) (cat. 324410 and 3244412, respectively) from Biolegend Inc. (San Diego, CA), and Alexa Fluor® mouse mAb IgG1 (MOPC-21) controls (cat. 400129 and 400130) from Biolegend; mAb anti-human ErbB2 (clone Neu 24.7; cat. 554299) from BD-Pharmingen (Becton Dickinson, San Jose, CA); Goat anti-human ErbB2 polyclonal antibody (PAb) (cat. AF1129) from R&D Systems (Minneapolis, MN); rabbit anti-ErbB2-pY-1248 PAb (cat. sc-293110) from Santa Cruz Biotechnology (Santa Cruz, CA); Alexa Fluor® 594-conjugated Phalloidin (cat. A12381) and Alexa Fluor® 594-conjugated concanavalin A (cat. C11253) (Con-A) were from Invitrogen; mouse anti-Beta-actin mAb (cat. A5441) from Sigma; rabbit anti-PKC-α mAb (cat. ab32376), rabbit anti-PKC-δ Mab (cat. ab131478), and rabbit anti-ARF6 Mab (cat. ab131261) from Abcam (Cambridge, MA); and rabbit PAb anti-p44/42 MAPK (ERK1/2) (cat. 9102), and anti-phospho-MAPK (p44/42, Thr202/Tyr204) (cat. 9101) from Cell Signaling Technology Inc. (Danvers, MA); purified mouse anti-phosphotyrosine mAb (4G10) from Dr. Brian Druker (Oregon Health Science University, Portland); HRP-conjugated secondary antibodies for Western blotting from Invitrogen (Grand Island, NY).
Transfection Reagents and Plasmids: XtremeGENE 9 transfection reagent from Roche Applied Science (Indianapolis, IN); siRNA smartpools and Dharmafect I transfection reagent from Dharmacon division of Thermo-Fisher (Pittsburgh, PA); pHACE expression vectors encoding wild type (cat. 21232) and constitutively active (cat. 21234) PKC-α from Addgene; GFP-PKC-α and GFP-PKC-δ (77) from Dr. Yusuf Hannun (Stony Brook Cancer Center, Stony Brook, NY). Ds-Red tagged WT ARF6 construct was generated by PCR-amplifying human ARF6 sequences (Forward primer: CACCCTCGAGATGGGGAAGGTGCTA; Reverse primer: TCGAAAGCTTAGAT) and cloning into DsRed-Monomer-N1 vector (Clontech Laboratories Inc., Mountain View, CA). Arf6 T27N construct was generated by site-directed mutagenesis of Thr-27 in WT Arf6 construct to Asn (sequence ACA to AAC; mutagenic primers: Forward-GCGGCCGGCAAGAC; Reverse: CAACTTCTACAGGATGTTTGTCTTGCCG) and mutation confirmed by DNA sequencing.
Media
Media and supplements were from Invitrogen and fetal bovine serum from Hyclone. Other Materials: Hoechst 3342 trihydrochloride trihydrate from Invitrogen; and Vectashield mounting medium from Vector Laboratories (Burlingame, CA).
Cell Culture
SKBR-3 cells (ATCC) were cultured in Alpha Modification of Eagle's Medium containing 5% fetal bovine serum, 10 mm HEPES, 1 mm each of sodium pyruvate, nonessential amino acids and glutamine, 50 μm 2-ME, and 1% penicillin/streptomycin solution at 37 °C under 5% CO2 (10, 78).
High-Content Imaging-based Screening Assay
15,000 SKBR-3 cells were seeded/well in 100 μl medium in Thermo-Scientific® Nunc® 96-microwell black optical bottom plates, using a Thermo-Scientific® multidrop 384 automatic dispenser in the 96-well mode, and the plates were incubated for 48 h at 37 °C in a Thermo-Scientific® Cytomat 6X incubator. To stain the surface ErbB2 on live cells, ice-cold culture media containing Alexa Fluor® 488-conjugated anti-human ErbB2 (4 μg/ml) or mouse IgG1 control antibodies were added using Beckman Coulter Biomek FX, and plates incubated in the dark on ice. After 1 h, the culture medium was removed and cells were rinsed thrice with ice-cold culture media. DMSO (0.01% final), 17-AAG (100 nm final), kinase inhibitors (1 μm final), or a combination of 17-AAG and kinase inhibitors were added in pre-warmed medium and the cells incubated at 37 °C for 8 h. The medium was removed, and the cells were rinsed three times with ice-cold PBS. The cells were then fixed with 4% paraformaldehyde for 2 h at room temperature and rinsed with PBS. The nuclei were stained with Hoechst 3342, according to the manufacturer's instructions. The fluorescence signals corresponding to Hoechst 3342 and Alexa 488 were automatically collected from cell populations with the Cellomics Arrayscan VTI equipped with XF53 filters for Hoechst and FITC. Images were obtained from 300 objects (cells)/well (up to 10 fields/well). Cellomics Compartmentalization Analysis bio-application software was used to separate membrane signals from intracellular and nuclear compartments (schematic and representative image shown in Fig. 1). Surface signals from the entire imaged population were averaged to provide a quantitative measure of the mean fluorescence intensity of cell surface ErbB2. Kinase inhibitors that inhibited the 17-AAG-induced clearance of ErbB2 from the cell surface by >20% were analyzed further.
FIGURE 1.

Development of an automated high-content fluorescence imaging-based assay for the measurement of surface levels of ERBB2. A–C, SKBR-3 cells were seeded in duplicate in the inner wells of 96-well plates (B, schematic at the top), labeled with Alexa Fluor® 488-conjugated anti-ErbB2 for 1 h at 4 °C, treated with DMSO (vehicle control), or increasing concentrations (5–500 nm) of 17-AAG for 8 h and fixed in 4% paraformaldehyde. The plates were scanned in a CellomicsTM Arrayscan VTI fluorescent microscope imager (A, B) and the CellomicsTM compartmental analysis software was used to quantify the fluorescence intensity of ErbB2 at the plasma membrane (C). The Y-axis represents the mean fluorescence intensity ± S.D. The presented data are from a single experiment representative of three. D and E, SKBR-3 cells were seeded in 6-well plates for 72 h and treated with DMSO (vehicle control) or increasing concentrations (5–500 nm) of 17-AAG for 8 h. In D, cells were trypsinized, washed in FACS buffer, and live cells stained with Alexa Fluor® 488-conjugated anti-ErbB2 for 1 h on ice. Cell surface levels of ErbB2 were quantified using flow cytometry and analyzed using BD Cellquest™ software. X-axis, mean fluorescence intensity; Y-axis, cell counts. In E, Triton X-100 lysates were prepared, and 25-μg aliquots of lysate protein resolved by SDS-PAGE followed by immunoblotting with anti-ErbB2 and anti-CHIP (loading control) antibodies.
Surface ErbB2 Quantification using Fluorescence-activated Cell Sorter (FACS) Analysis
SKBR-3 cells were seeded in 6-well plates at a density of 300,000 cells/ml and grown for 48 h. Following drug treatments, cells were rinsed with ice-cold PBS and released with trypsin/EDTA (Invitrogen). Trypsinization was stopped by adding excess ice-cold culture medium. Cell suspensions were transferred to FACS tubes, washed thrice in ice-cold FACS buffer (2% fetal bovine serum/2% BSA in PBS). For live-cell surface ErbB2 staining, cells were incubated for 1 h on ice in the dark with Alexa Fluor® anti-human ErbB2 mAb or mouse IgG1 (control) diluted in FACS buffer, followed by three washes in the same buffer. Cells were fixed at room temperature in 4% PFA for 10 min, run on a BD FACScalibur flow cytometer and analyzed with CellQuest® software.
Confocal Immunofluorescence Microscopy
SKBR-3 cells were seeded at a density of 75,000 cells per well on glass coverslips inside 24-well plates and grown for 48 h. For live-cell surface ErbB2 staining, ice-cold culture medium containing Alexa Fluor® anti-human ErbB2 or mouse IgG1 (control) antibodies were added, and plates incubated in the dark for 1 h on ice. The cells were rinsed three times with ice-cold culture medium, and incubated with pre-warmed medium containing the indicated drugs. The cell culture medium was removed, and the coverslips rinsed three times with ice-cold PBS. Cells were then fixed with 4% PFA at room temperature for 10 min.
To stain for intracellular proteins, PFA was removed, and the cells on coverslips were permeabilized for 10 min in immunofluorescence (IF) buffer (2% BSA/PBS) containing 0.2% saponin, rinsed in 2% BSA/PBS, serially incubated with primary and secondary antibodies for 1 h each at room temperature with three rinses (5 min each) in 2% BSA/PBS after each antibody incubation. The coverslips were then rinsed once with PBS, and mounted on glass microscope slides with Vectashield mounting media. Images were captured using a Zeiss 710 Meta Confocal Laser Scanning Microscope at 63× magnification. Merged fluorescence pictures were generated using ZEN 2012® software from Carl Zeiss.
siRNA and Transient Transfections
Wet reverse transfection with Dharmafect 1 transfection reagent was used to introduce Dharmacon siRNA Smartpools (80 nm final) into SKBR-3 cells, and transient transfections were accomplished using Xtremegene 9, both according to the manufacturer's instructions.
Western Blotting
Following cell culture and drug treatments, SKBR-3 cells were rinsed twice with ice-cold PBS, and attached cells were lysed in ice-cold Triton X-100 lysis buffer (0.5% Triton X-100, 50 mm Tris (pH 7.5), 150 mm sodium chloride from (Fisher), 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, and 10 mm sodium fluoride) (Sigma) for 20 min. The lysates were transferred to pre-cooled Eppendorf tubes, spun at 13,000 rpm for 10 min at 4 °C, and supernatants collected and assayed for protein concentration using the Thermo-Scientific Pierce® BCA assay. The indicated amounts of protein lysates were resolved by 9% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with antibodies, as previously described (79).
RESULTS
Substantial evidence indicates that overexpressed ErbB2 traffics through the endocytic recycling pathway (15–18). As endocytic recycling of ErbB2 is thought to critically contribute to its oncogenic signaling (18, 80), and enhanced lysosomal degradation of ErbB2 is seen with ErbB2-targeted therapeutics (10, 78), a better understanding of the biochemical pathways that regulate ErbB2 recycling is of substantial interest. Analyses of the related family member EGFR suggest an important role of cellular kinases (62). However, the role of protein kinases in ErbB2 recycling is unclear. To investigate such a role, we performed a kinase inhibitor screen using high-content cell imaging.
Kinase Inhibitor Screen Using a High-content Fluorescence Microscopy Imaging Assay of ErbB2 Clearance from the Cell Surface
We reasoned that cellular kinases could positively or negatively regulate endocytic traffic of ErbB2. Therefore, we developed an immunofluorescence imaging assay in which the cell surface pool of ErbB2 on live SKBR-3 cells was labeled with an Alexa-Fluor 488-labeled anti-human ErbB2 antibody on ice and then allowed to internalize at 37 °C, in the absence or presence of 17-AAG. The level of cell surface ErbB2 at the end of the assay (8 h) was quantified on individual cells using high-content fluorescence microscopy imaging followed by compartmental analysis to specifically demarcate the membrane-associated signals (Fig. 1A). The surface ErbB2 signals in untreated cells completely colocalized with fluorescent concanavalin A used as a stable surface marker (Fig. 1B) as well as with signals obtained with concurrent indirect staining using a second anti-human ErbB2 antibody (data not shown). To validate this approach, we carried out concurrent high-content image analysis (Fig. 1C) and FACS analysis (Fig. 1D) of SKBR-3 cells that were either left untreated, or treated with DMSO (vehicle) or increasing concentrations (5–500 nm) of 17-AAG. FACS analysis of live cells with anti-ErbB2 Alexa Fluor® 488 demonstrated the expected dose-dependent reduction in the cell surface ErbB2 levels that mirrored the effects seen with high-content imaging (Fig. 1, C versus D). Under these conditions, 17-AAG induced the expected dose-dependent degradation of ErbB2 as shown by Western blotting (Fig. 1E). Thus, we concluded that the high-content imaging assay was suitable for a kinase inhibitor screen.
Kinase Inhibitor Screen Identifies PKCs as a Potential Regulator of ErbB2 Surface Down-regulation
We utilized a commercial kinase inhibitor library composed of inhibitors against a subset of tyrosine, serine/threonine and lipid kinases (Fig. 2, A and B) to assess the role of the kinome in regulating ErbB2 clearance from the cell surface of SKBR-3 cells, using the assay described above. The inhibitors were tested at 1 μm final concentration, with or without concurrent treatment with 100 nm 17-AAG. High content images were obtained 8 h post-treatment, and the levels of ErbB2 remaining at the cell surface following treatments were quantified. Among the inhibitors that reduced the 17-AAG-induced clearance of surface ErbB2, three met the 20% cutoff limit: Fasudil (no. 25) an inhibitor of Rho-dependent protein kinase; Ro 31-8220 (no. 28), a broad inhibitor of PKC family of kinases; and Genistein, a relatively nonspecific protein tyrosine kinase inhibitor (Fig. 2, A and B and associated legend). Genistein was not considered further due to its broad effects (115). FACS analysis of the remaining two hits showed that Fasudil did not affect the 17-AAG-induced ErbB2 clearance from the cell surface (Fig. 2C) while the effect of Ro 31-8220 was reproducible (Fig. 2D). Confocal imaging confirmed this result by showing that Ro 31-8220 diminished the 17AAG-induced clearance of ErbB2 from the cell surface (Fig. 2E). A second broad inhibitor of PKCs, GF109203X (Fig. 2, A and B, No. 33) also inhibited the effect of 17-AAG on ErbB2 down-regulation although its effect was below the cutoff. Therefore, the role of PKC in ErbB2 down-regulation from the cell surface was considered further. Several kinase inhibitors promoted the 17-AAG-induced ErbB2 down-regulation (Fig. 2, A and B). These candidates are being characterized independently and are not described here.
PKC Inhibition, Activation, and Genetic Manipulations Implicate PKC-α in Regulating ErbB2 Entry into Endocytic Pathway
As PKC-α has been implicated in regulating the endocytic traffic of EGFR (81), we compared Ro 31-8220, which has broad inhibitory activity for PKCs and also certain other kinases (82–85), with a more selective PKC inhibitor Go 6976 (selective for classical Ca2+-dependent PKC isozymes α, β1, βII, and γ). We also assessed the effect of PMA, an activator of classical and novel PKC isoforms (86–88). FACS analysis indicated that G0 6976, similar to Ro 31-8220, reduced the 17-AAG-induced clearance of ErbB2 from the cell surface (Fig. 3A). Furthermore, PMA by itself reduced the surface levels of ErbB2 and further increased the 17-AAG-induced clearance of ErbB2 from the cell surface (Fig. 3A, top panel). Western blot analysis revealed that PKC inhibitors modestly inhibited the 17-AAG-induced ErbB2 degradation (Fig. 3A, bottom panel). However, treatment with PMA, which reduced the surface ErbB2 levels substantially (Fig. 3A, top panel), did not induce any destabilization of ErbB2 protein nor did it enhance the 17-AAG-induced ErbB2 degradation (Fig. 3A, bottom panel). Western blotting for PKC-α levels showed the expected PMA-induced down-regulation (Fig. 3A, bottom panel), indicating that PMA was able to activate its target.
FIGURE 3.
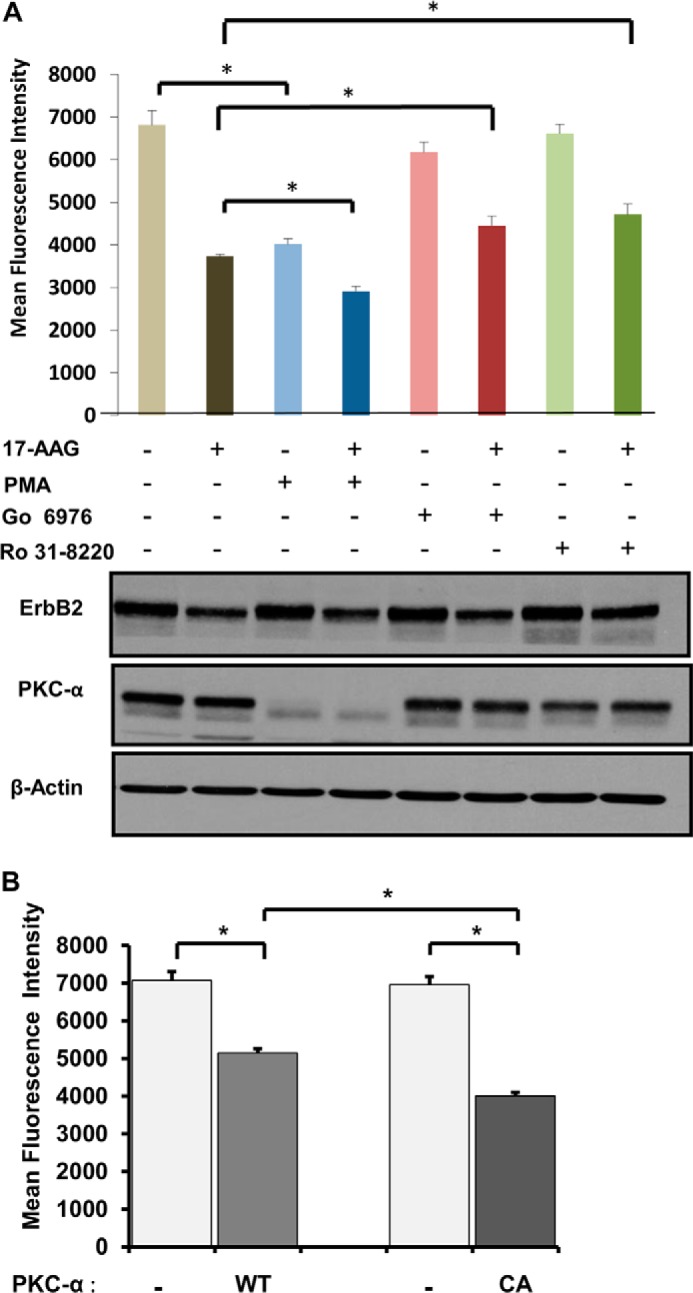
Role of PKC-α in ErbB2 clearance from the cell surface. A, SKBR-3 cells were pre-treated with DMSO, Ro 31–8220 (1 μm), Go 6976 (1 μm), or PMA (100 nm) for 1 h, and incubations continued without or with 17-AAG (25 nm) for 8 h. Cells were trypsinized, stained with Alexa Fluor® 488-anti-ErbB2 antibody, and analyzed by FACS to quantify cell surface ErbB2 levels. The bars represent mean fluorescence intensity values ± S.D. Triton X-100 lysates were prepared from parallel cultures treated as those used for FACS and 25-μg aliquots of lysate protein resolved by SDS-PAGE followed by immunoblotting with anti-ErbB2, anti-PKC-α, and anti-β-actin (loading control) antibodies. The presented data are from a single experiment representative of three. B, SKBR-3 cells were transiently co-transfected with expression constructs encoding wild-type or constitutively active (CA) PKC-α together with dsRed plasmid. Live cells were stained with Alexa Fluor® 488 conjugated-anti-ErbB2, and cell surface levels of ErbB2 on ds-Red-negative (light bars) and ds-Red-positive (dark bars) cells were quantified using FACS. The mean fluorescence intensity values ± S.D. are presented; asterisks indicate statistically significant differences (p < 0.005). The presented data are from a single experiment representative of three.
Overall, the use of an additional PKC inhibitor and PMA further established that Ca2+-dependent PKCs regulate the internalization of surface ErbB2. Previous studies of SKBR-3 cell line have shown that it expresses PKC-α but not detectable levels of PKC-β or PKC-γ (89). Therefore, to further establish the role of PKC-α, we transfected SKBR-3 cells with plasmids encoding wild type or constitutively-active PKC-α. The levels of surface ErbB2 on transfected cells were quantified using FACS analysis. Expression of constitutively-active PKC-α led to a reduction in the level of cell surface ErbB2 compared with that with WT PKC-α (Fig. 3B). Altogether, these results strongly suggest that PKC-α promotes ErbB2 entry into the endocytic pathway.
PMA-induced Down-regulation of Cell Surface ErbB2 Requires ErbB2 Kinase Activity
Results with PMA treatment and ectopic PKC-α expression (above) suggested that PKC-α activation results in the removal of ErbB2 from the cell surface. To assess whether the internalization process requires the ErbB2 kinase activity, we carried out FACS analysis of SKBR-3 cells treated with PMA in the presence or absence of ErbB2 kinase inhibitor lapatinib at concentrations that effectively reduced the ErbB2 tyrosine phosphorylation (data not shown; also see Figs. 7B and 8D). While SKBR-3 cells that were not pretreated showed a substantial PMA-induced reduction in surface ErbB2 levels (Fig. 4A), pretreatment with lapatinib essentially completely eliminated this effect (Fig. 4B). These results suggest that ErbB2 kinase activity is required for activated PKC-α-dependent ErbB2 internalization.
FIGURE 7.
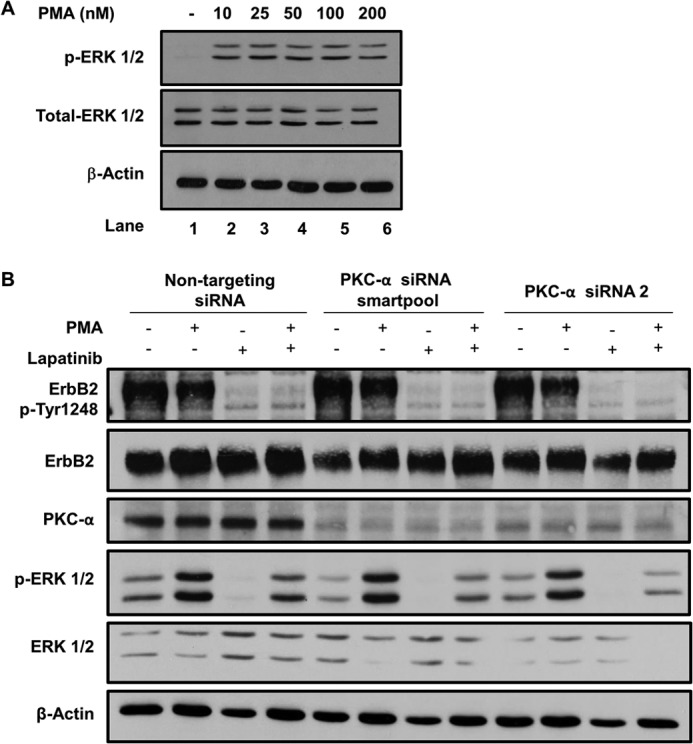
ErbB2 kinase activity but not PKC-α are required for PMA-induced ERK phosphorylation. A, SKBR-3 cells seeded in 6-well plates were treated with DMSO (vehicle) or the indicated concentrations of PMA for 8 h. 25-μg aliquots of Triton X-100 lysate proteins were subjected to immunoblotting for p-ERK 1/2, total ERK 1/2, and β-actin (loading control). B, SKBR-3 cells were transfected with a non-targeting siRNA (control) or PKC-α-specific siRNAs (a smartpool or siRNA no. 2) for 48 h, treated without or with Lapatinib (1 μm) for 1 h, and further incubated in the presence of DMSO (vehicle control) or PMA (100 nm) for 1 h. Western blotting with anti-ErbB2-pY-1248, anti-ErbB2, anti-PKC-α, anti-p-ERK 1/2, anti-ERK 1/2, and anti-β-actin (loading control) antibodies was carried out as in A.
FIGURE 8.

PKC-δ co-localizes with ErbB2 in the Arf6-positive juxtanuclear compartment and is required for PMA-induced Erk phosphorylation. A and B, SKBR-3 cells were transiently transfected to co-express GFP-tagged wild-type PKC-δ and ds-Red-tagged WT Arf6 (A) or Arf6-T27N mutant (B). The cells were stained with Alexa Fluor® 647-conjugated anti-ErbB2 antibody and treated with DMSO (vehicle) or PMA (100 nm) for 1 h. The cells were paraformaldehyde-fixed and permeabilized as in Fig. 5, and imaged under a confocal microscope. C, SKBR-3 cells were transfected with a non-targeting control siRNA or a PKC-δ-specific siRNA smartpool for 48 h. Live cells were stained with Alexa Fluor® 488-conjugated anti-ErbB2 antibody in cold and treated with PMA (100 nm) for 1 h at 37 °C. The cells were stained for PKC-δ (red) and imaged as in A. D, SKBR-3 cells were transfected with a non-targeting control siRNA or a PKC-δ-specific siRNA smartpool for 48 h, treated without or with Lapatinib (1 μm) for 1 h, and further incubated in the presence of DMSO (vehicle control) or PMA (100 nm) for 1 h. Western blotting with anti-ErbB2-pY-1248, anti-ErbB2, anti-PKC-δ, anti-p-ERK 1/2, anti-ERK 1/2, and β-actin (loading control) antibodies was carried out as in Fig. 7.
FIGURE 4.
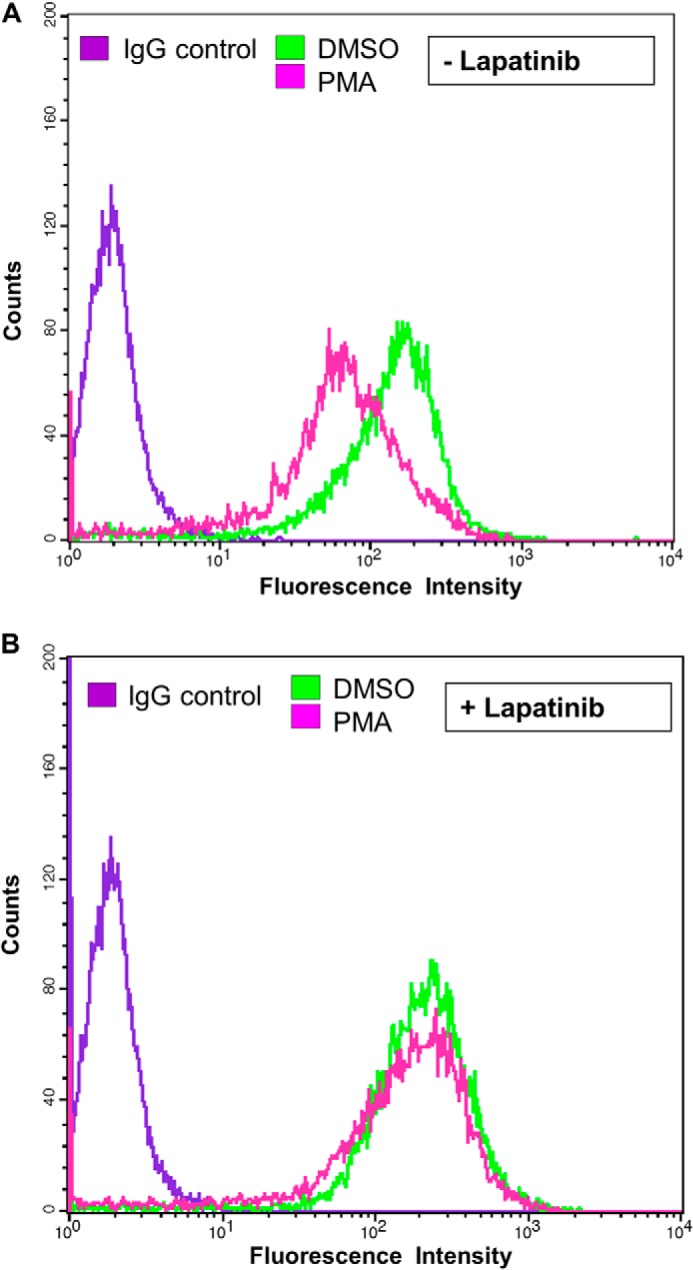
Requirement of ErbB2 kinase activity for PMA-induced down-regulation of cell surface ErbB2. SKBR-3 cells were cultured in low serum (0.5% FBS) medium without (A) or with (B) Lapatinib (100 nm) for 24h followed by further incubation in the absence (DMSO) or the presence of PMA (25 nm) for 8 h. Live cells were stained with Alexa Fluor® 488-conjugated anti-ErbB2 antibody and cell surface ErbB2 levels quantified using FACS.
PKC-α-dependent Entry of ErbB2 into a Juxtanuclear Compartment
The ability of PMA to reduce the surface levels of ErbB2 without inducing ErbB2 degradation (Fig. 3A) suggested the possibility that PKCs may regulate ErbB2 entry into a non-degradative endocytic compartment. To assess if this is the case, we carried out confocal imaging of untreated and PMA-treated SKBR-3 cells for endogenous ErbB2 and PKC-α. In untreated cells, ErbB2 expectedly showed the predominant surface localization, while PKC-α showed a predominantly diffuse cytoplasmic and some plasma membrane localization (Fig. 5A, top panel). In contrast, PMA-treated SKBR-3 cells showed a substantial accumulation of ErbB2 in a juxtanuclear compartment (Fig. 5A, lower left panel). In these cells, PKC-α became localized to the same region (lower middle panel), with substantial colocalization with ErbB2 (lower right panel). Next we transfected SKBR-3 cells with non-targeting control siRNA or PKC-α-directed siRNA, and examined the localization of ErbB2 following PMA treatment. Staining for PKC-α demonstrated the specific knockdown of PKC-α with PKC-α-targeted siRNA (Fig. 5B, compare upper versus lower middle panel). Substantial, albeit incomplete, PKC-α knockdown was confirmed by Western blotting (Fig. 5C); some variability of knockdown in individual cells was seen by immunofluorescence analysis (data not shown). Notably, while control siRNA-transfected SKBR-3 cells treated with PMA exhibited a juxtanuclear pool of ErbB2 (Fig. 5B, left upper panel) that co-localized with PKC-α (Fig. 5B, middle and right upper panels), this pool was not seen in cells with effective PKC-α knockdown (left lower panel). These results indicated that PKC-α promotes the entry of ErbB2 into a juxtanuclear endocytic compartment.
FIGURE 5.
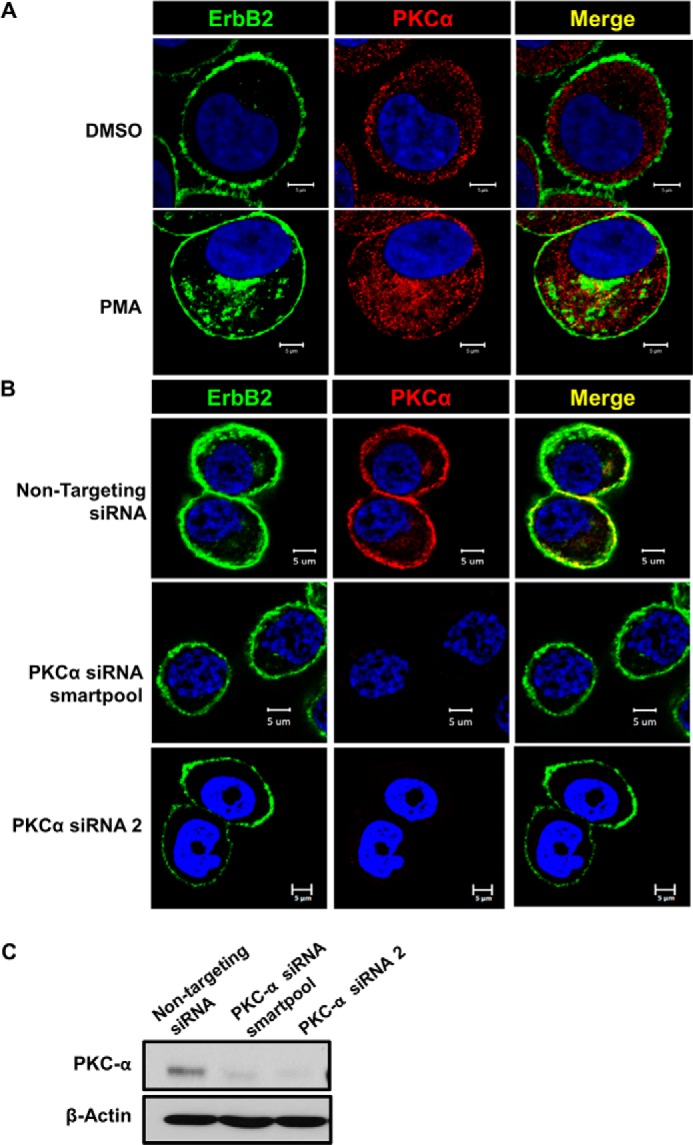
PKC-α-regulates surface ErbB2 translocation into a juxtanuclear compartment. A, live SKBR-3 cells on coverslips were stained with Alexa Fluor® 488-conjugated anti-ErbB2 antibody in cold, followed by treatment with DMSO (vehicle control) or PMA (100 nm) at 37 °C for 1 h. The cells were paraformaldehyde-fixed, permeabilized with saponin (0.2% in PBS), stained for PKC-α (red) and imaged under a confocal microscope. B, SKBR-3 cells were transfected with a non-targeting siRNA (control) or a PKC-α-specific siRNA (a smartpool or siRNA no. 2) for 48 h. Live cells were stained with Alexa Fluor® 488-conjugated anti-ErbB2 antibody in cold and treated with PMA (100 nm) for 1 h at 37 °C. The cells were stained for PKC-α (red) and imaged as in A. C, SKBR-3 cells were transfected with a non-targeting siRNA (control) or a PKC-α-specific siRNA smartpool for 48 h as in B, and 40-μg aliquots of Triton X-100 lysate protein subjected to Western blotting with anti-PKC-α and β-actin (loading control) antibodies.
The PKC-α-dependent Juxtanuclear ErbB2 Localization Represents Entry into an Arf6-regulated Endocytic Recycling Compartment
Prior studies have identified a PKC-dependent juxtanuclear compartment “pericentrion,” which was further characterized as a subset of the ERC (67, 90, 91). Arf6 has been established as a critical small GTPase regulator of endocytic recycling (92), and recent studies have shown that PKC-α and Arf6 exhibit functional cross-talk and colocalize in the ERC (67, 93, 94). Therefore, we considered the possibility that PKC-α-dependent internalization of ErbB2 into a juxtanuclear compartment represented its entry into Arf6-regulated ERC. To assess this possibility, we co-transfected SKBR-3 cells with dsRed-tagged WT or dominant-negative Arf6-T27N mutant, known to prominently localize to ERC (92), together with GFP- PKC-α. Cells were either left untreated or were PMA-treated for 1 h followed by confocal imaging. Notably, cells co-transfected with either WT (Fig. 6A) or dominant-negative mutant Arf6 (Fig. 6B) together with PKC-α (positive for red and green fluorescence) exhibited substantial juxtanuclear localization of ErbB2 even without PMA treatment (Fig. 6A, top panels). Importantly, juxtanuclear ErbB2 colocalized with transfected WT or mutant Arf6 as well as PKC-α (Fig. 6, A and B, right panels). Biochemical assessment of transfected Arf6 proteins by Western blotting confirmed their similar expression levels (Fig. 6C). To further confirm the findings above, SKBR-3 cells were transfected with GFP-PKC-α and treated with PMA, followed by confocal microscopy analysis for colocalization of endogenous ErbB2 and Arf6 with transfected GFP-PKC-α. Indeed, transfected GFP-PKC-α colocalized with endogenous Arf6 and ErbB2 in the juxtanuclear region (Fig. 6D). Together, these results demonstrate a PKC-α-dependent localization of ErbB2 in Arf6-regulated ERC.
FIGURE 6.

ErbB2 in the juxtanuclear compartment co-localizes with PKC-α and ERC marker Arf6. A and B, SKBR-3 cells were transiently transfected to co-express GFP-tagged wild-type PKC-α, ds-Red-tagged WT Arf6 (A) or Arf6-T27N mutant (B). The cells were stained with Alexa Fluor® 647-conjugated anti-ErbB2 antibody, treated with DMSO (vehicle) or PMA (100 nm) for 4 h, paraformaldehyde-fixed and permeabilized as in Fig. 5, and imaged under a confocal microscope. C, SKBR-3 cells were transiently transfected to co-express GFP-tagged wild-type PKC-α, and ds-Red-tagged WT Arf6 or Arf6-T27N mutant as in A and B, and 40-μg aliquots of Triton X-100 lysate protein were used for Western blotting with anti-Arf6 and β-actin (loading control) antibodies. D, SKBR-3 cells transiently transfected and stained as in A were treated with PMA (100 nm) for 4 h, paraformaldehyde fixed, stained with an anti-Arf6 antibody and imaged under a confocal microscope.
PMA-induced Activation of Erk in SKBR-3 Cells Is ErbB2 Kinase-dependent
Analysis of PKC-orchestrated pericentrion/ERC has demonstrated that it is a site of activation of signaling pathways downstream of a number of receptors (67, 95). The Erk pathway is prominent among those activated at the pericentrion. Since Erk pathway activation is a prominent component of overexpressed ErbB2-dependent oncogenic signaling in breast cancer cells (96), we investigated if Erk activation induced upon PKC activation exhibits ErbB2 kinase-dependence. Treatment of SKBR-3 cells with PMA at a concentration as low as 10 nm led to a substantial increase in p-Erk level while total Erk level remained unchanged (Fig. 7A). In SKBR-3 cells pre-treated with lapatinib, blotting with anti-ErbB2-pY1248 showed a nearly complete loss of ErbB2 phosphorylation (Fig. 7B, top panel), and reduced the basal p-Erk levels (Fig. 7B, fourth panel), confirming that treatment with lapatinib effectively inhibited the ErbB2 kinase activity and drive for Erk activation. Notably, lapatinib pretreatment markedly reduced the magnitude of PMA-induced increase in p-Erk levels compared with that in the absence of lapatinib pretreatment (Fig. 7B, fourth panel).
PMA-induced Activation of Erk in SKBR-3 Cells Is Mediated by PKC-δ
To test if the PMA-induced Erk activation was mediated through PKC-α, we transfected SKBR-3 cells with control siRNA, or a PKC-α-specific siRNA pool or an individual siRNA (no. 2) selected out of several tested for effective knockdown. Both PKC-α-specific siRNAs induced substantial PKC-α knockdown, as shown by Western blotting (Fig. 7B, third panel). However, this knockdown had little impact on the extent of PMA-induced p-Erk levels, suggesting that Erk pathway activation occurs through other classical or novel PKCs.
Since previous studies of SKBR-3 cell line have shown it to lack the expression of the remaining classical PKCs, PKC-β, or PKC-γ (89), it appeared more likely that PMA-induced Erk activation occurred through novel PKCs. A previous study examining the mechanism of estrogen-induced Erk activation in MCF7 breast cancer cell line implicated an autocrine loop involving HRG1 activation of ErbB2; in this system, Erk activation appeared to be mediated by PKC-δ (97). More recently, PKC-δ was shown to mediate Erk activation from ErbB2 in ErbB2-overexpressing breast cancer cell lines and PKC-δ gene knock-out delayed oncogenesis in an ErbB2 transgenic mouse model (98). These findings pointed to PKC-δ as a potential mediator of PMA-induced Erk activation in our system. To assess the role of PKC-δ, we first examined if PKC-δ also colocalized with ErbB2 in the ERC. First, we co-transfected SKBR-3 cells with GFP-PKC-δ and either WT Arf6 or Arf6-T27N. A fraction of GFP-PKC-δ colocalized with WT Arf6 (Fig. 8A) or Arf6-T27N mutant (Fig. 8B) both in DMSO-treated and PMA-treated cells. Notably, a small fraction of ErbB2 colocalized with GFP-PKC-δ and WT Arf6 in a juxtanuclear region in PMA-treated SKBR-3 cells (Fig. 8A). Also, a small fraction of ErbB2 co-localized with juxtanuclear GFP-PKC-δ and Arf6-T27N in untreated as well as PMA-treated cells (Fig. 8B). To assess if PKC-δ colocalizes with ErbB2 and the role of PKC-δ in ErbB2 localization to ERC, we carried out confocal immunofluorescence analysis of PMA-treated SKBR-3 cells transfected with a control siRNA or a PKC-δ-specific siRNA. Juxtanuclear ErbB2 was seen to colocalize with PKC-δ in control siRNA transfected cells (Fig. 8C). Juxtanuclear localization of ErbB2 was reduced in cells transfected with PKC-δ-specific siRNAs (Fig. 8C; see effective knockdown under Fig. 8D).
We therefore assessed the impact of PKC-δ siRNA on PMA-induced Erk activation in SKBR-3 cells. PKC-δ-specific siRNA effectively reduced the endogenous PKC-δ levels compared with non-targeting siRNA (Fig. 8D, third panel). PMA induced a substantially lower level of p-Erk in SKBR-3 cells with PKC-δ knockdown compared to non-targeting siRNA-transfected cells, although the reduction was somewhat less robust than that seen with lapatinib (Fig. 8D, fourth panel). These results suggest that while PKC-α and PKC-δ promote the entry of ErbB2 into ERC, PKC-δ is the predominant mediator of ErbB2 signaling into Erk pathway.
DISCUSSION
Overexpressed ErbB2 drives oncogenesis in breast and other cancers, and provides a target for clinically used targeted therapies in breast cancer. An important determinant of the oncogenicity of ErbB2 is its propensity to avoid lysosomal degradation and to instead traffic via the endocytic recycling pathway (15–18). Molecular pathways that regulate ErbB2 recycling however remain unexplored. Here, we developed and used a high-content imaging-based screen to interrogate a focused kinase inhibitor library to identify candidate protein/lipid kinases that might facilitate cell surface ErbB2 internalization in an ErbB2-overexpressing breast cancer cell line SKBR-3. Our screen revealed the ability of a broad-spectrum PKC inhibitor, Ro 31–8220, and to a lesser extent another PKC inhibitor GF 109203X, to reduce the clearance of ErbB2 from the surface of cells treated with 17-AAG. Detailed biochemical and imaging-based studies presented here provide evidence that PKC-α promotes the ErbB2 entry into the endocytic recycling compartment (ERC), where PKC-α colocalized with ErbB2. We show that ErbB2 entry into this compartment requires the kinase activities of ErbB2 and PKC-α. Furthermore, we show that PKC-δ can also localize to the same compartment and colocalize with ErbB2. Finally, we show that PKC-δ, but not PKC-α, is key to PKC-mediated Erk activation. These findings carry significant implications for our understanding of ErbB2 biology and suggest new links between ErbB2 traffic and oncogenic signaling.
The PKC family of serine-threonine kinases is well-established as a pivotal mediator as well as regulator of signaling downstream of RTKs, G-protein-coupled receptors, and other cell surface proteins, and members of this family have emerged as critical regulators of physiological processes as well as disease pathogenesis (86–88). Various PKCs have been shown to play important roles in human and experimental oncogenesis (86–88). Pertinent to our results, PKC-α has been linked to critical pro-oncogenic traits, including proliferation, migration, epithelial-mesenchymal transition, and resistance to anti-estrogens in cellular and animal-based experimental studies of breast cancer (99). Recent clinical correlative studies also suggest that overexpression of PKC-α is associated with poor prognosis in breast cancer (100–102). PKC-δ is also known to be overexpressed in a subset of breast cancers and meta-analysis of expression data revealed its specific association with poor survival in ErbB2+ breast cancer (98). The same study also established that PKC-δ gene knock-out delayed tumor development in MMTV-ErbB2 transgenic mice by reducing cellular proliferation.
While cellular, animal and clinical studies strongly support a pro-oncogenic role of PKC-α and PKC-δ in breast cancer, the mechanisms by which they play such roles in ErbB2-driven breast cancer remain less certain. Prior cellular and biochemical studies suggest an important functional role of PKC-α in ERbB2-driven oncogenic traits. HRG-induced apoptosis of SKBR-3 cells was shown to be potentiated by inhibition of PKC-α activity (89). PKC-α was shown to be activated in ErbB2-overexpressing breast cancer cell lines and to promote uPAR expression and invasive behavior in concert with c-Src (102). ErbB2 overexpression was also found to down-regulate the expression of extracellular matrix component biglycan in a PKC-dependent manner; biglycan down-regulation was demonstrated to be critical for oncogenesis (103). Furthermore, PKC-α was found to be one of the key downstream mediators of ErbB2-induced cysteine cathepsin expression and breast cancer cell invasiveness (104). Reduction of PKC-α levels upon treatment with anti-ErbB2 antibody Trastuzumab was shown to contribute to cardiomyocyte death, further supporting a role of PKC downstream of ErbB2 (105). ErbB2 was shown to induce primordial ovarian follicle growth in a PKC-dependent manner (106). While these lines of evidence support a clear role for PKC-α in ErbB2-driven oncogenesis, a role for PKC-α as a determinant of ErbB2 endocytic traffic has not been demonstrated. Given the strong collective evidence that endocytic recycling of ErbB2 is a major determinant of its oncogenic potential as an ErbB receptor family member (80, 107), our studies provide a new cell biological mechanism to link PKC-α to ErbB2-driven oncogenesis.
Previous studies have shown that PKC-dependent juxtamembrane phosphorylation regulates the endocytic traffic of EGFR by reducing the sorting of EGFR toward lysosomes while promoting its traffic into the ERC (60). Thus, our findings that PKC-α promotes the entry of ErbB2 into the ERC and that this requires the kinase activities of ErbB2 as well as PKC-α indicate that PKC activation provides a common regulatory control for the dichotomous fate of activated ErbB receptors into lysosomal degradation versus endocytic recycling. Consistent with our findings, PMA treatment of ErbB2 transfected fibroblasts has been reported to induce ErbB2 juxtamembrane threonine-686 phosphorylation, as well as ErbB2 internalization and intra-cytoplasmic accumulation, although no further characterization of intracellular compartments was carried out (75). Distinct from our conclusions, another study has shown that PKC-α is required to maintain high cell surface levels of ErbB2 in breast cancer cell lines that lack gene amplification (referred to as Her2 2+ based on immunohistochemistry) (76). Since no cell biological analyses of ErbB2 endocytic traffic were performed in that study, a direct comparison with our results is difficult at the present time, although it is quite plausible that ErbB2 traffics distinctly based on the levels of overexpression, as studies discussed under the introduction section suggest.
Our results also show that PKC-δ colocalizes with ErbB2 in SKBR-3 cells, although further analyses are needed to assess how critical a role this isozyme plays relative to PKC-α. The role of PKC-δ in ErbB2 endocytic traffic has not been clarified previously. Inhibition of PKC-δ was previously shown to relieve the block of EGFR traffic from early to recycling endosome that was imposed by a calmodulin inhibitor (61). Our initial findings, together with the key role of PKC-δ in ErbB2-driven oncogenesis recently identified using transgenic mice (98) raise the possibility that PKC-δ may also regulate endocytic recycling of ErbB2, especially under conditions of cancer-associated overexpression.
Recent studies of the G-protein coupled receptor internalization and its linkage to signaling have defined a critical role of PKCs in organizing a special juxtanuclear organelle, the pericentrion, whose further characterization has led to its identification as a subset of the ERC (90, 108). Detailed analysis of this organelle have revealed it to serve as an intracellular hub for the accumulation of internalized cell surface receptors prior to their redirection to other destinations such as recycling back to cell surface or targeting to the lysosome. Indeed, the down-regulation of PKC-α upon its sustained activation with bryostatin or PMA has also been linked to endosome-lysosome traffic (109) and this effect is enhanced by 17-AAG (110). Biochemical characterization of the pericentrion/ERC compartment revealed that it contains a number of cell surface receptors including EGFR (67). As has been observed in the PKC-dependent GPCR entry into pericentrion, we observed that endogenous ErbB2 in SKBR-3 cells colocalizes with endogenous as well as ectopically expressed PKC-α, that the colocalization requires the kinase activity of both, and that this compartment is regulated by a known ERC regulatory small GTPase Arf6 which in turn colocalizes with ErbB2 and PKC-α. Furthermore, PKC-δ also colocalizes with Arf6 and ErbB2 in this compartment. Notably, enhanced signaling downstream of PKCs, measured as PMA-induced Erk activation, was ErbB2 kinase activity-dependent. This is of interest, since PKC-dependent Erk activation in the GPCR system requires their entry into the pericentrion. Our results show that PKC-δ is more dominant in mediating PMA-induced Erk activation, while PKC-α appears to be dispensable, distinct from a requirement of PKC-α for ErbB2 localization to ERC. Further studies are needed to directly establish a potential requirement of the pericentrion/ERC co-localization of ErbB2/PKC in PKC-mediated signaling and biological responses in ErbB2-overexpressing cancer cells.
It is of note that c-Src is an essential downstream signaling component in ErbB2-driven oncogenesis (111), and Src is one of the downstream effectors of ErbB2 signaling whose activation has been shown to be linked to PKC-α or PKC-δ (98, 102). We have shown that c-Src interacts with oncogenic EGFR mutants in non-small cell lung cancer cells in the ERC, an interaction required for mutant EGFR-mediated oncogenic transformation (112, 113). Src has been shown to be a component of the pericentrion (67), suggesting that ErbB2 may signal in collaboration with Src in the ERC.
Together, our results suggest a novel positive feedback loop in ErbB2 signaling involving the PKC activation-dependent entry of ErbB2 into the ERC, establishing an endosomal signaling hub for ErbB2 signaling through ERC-localized PKCs, especially PKC-δ, and other signaling partners. This suggestion is consistent with an increasing appreciation of the critical role of endosomal signaling downstream of RTKs (114), although such a role for ErbB2 has not yet been investigated. This model is further supported by a large body of literature that has established a role for PKCs in the spatiotemporal regulation of receptor signaling (88).
Acknowledgments
We thank Dr. Jenny Black for suggestions and critical reading of the manuscript, Dr. Brian Druker for 4G10 antibody, and Dr. Yusuf Hannun for PKC constructs. This manuscript is dedicated to the fond memory of Victor Fung, Ph.D., a former Program Officer at the NCI and former Scientific Review Officer of the Cancer Etiology study section of the CSR, National Institutes of Health, for his wisdom, compassion, integrity, love of the sciences and the arts, incredible culinary skills, and above all, his contributions to the career development of so many investigators during his own distinguished career.
This work was supported by the National Institutes of Health Grants CA116552, CA105489, CA99163, CA87986, Dept. of Defense Grant W81WH-11-1-0167, NE DHHS LB506-2014-01, and Nebraska DHHS LB606-18123-Y3 (to H. B.); National Institutes of Health Grants CA96844 and CA144027, and Dept. of Defense Grants W81XWH-07-1-0351 and W81XWH-11-1-0171 (to V. B.); National Institutes of Health Grant CA127239 (to A. N.); Nebraska Center for Nanomedicine-Center for Biomedical Research Excellence (NCN-COBRE) seed grant (to S. M. R.); and the NCI Core Support Grant to the UNMC Buffett Cancer Center.
- EGFR
- epidermal growth factor receptor
- ErbB2
- epidermal growth factor receptor family 2
- RTK
- receptor tyrosine kinase
- PKC
- protein kinase C
- ERC
- endocytic recycling compartment
- FACS
- fluorescence-activated cell sorting
- PFA
- paraformaldehyde
- MAb
- monoclonal antibody
- PAb
- polyclonal antibody
- 17-AAG
- 17-N-allylamino-17-demethoxygeldanamycin
- HSP90
- heat shock protein 90.
REFERENCES
- 1. Santarius T., Shipley J., Brewer D., Stratton M. R., Cooper C. S. (2010) A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 10, 59–64 [DOI] [PubMed] [Google Scholar]
- 2. Baselga J., Swain S. M. (2009) Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 9, 463–475 [DOI] [PubMed] [Google Scholar]
- 3. Xu W., Marcu M., Yuan X., Mimnaugh E., Patterson C., Neckers L. (2002) Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. U.S.A. 99, 12847–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou P., Fernandes N., Dodge I. L., Reddi A. L., Rao N., Safran H., DiPetrillo T. A., Wazer D. E., Band V., Band H. (2003) ErbB2 degradation mediated by the co-chaperone protein CHIP. J. Biol. Chem. 278, 13829–13837 [DOI] [PubMed] [Google Scholar]
- 5. Neckers L. (2003) Development of small molecule Hsp90 inhibitors: utilizing both forward and reverse chemical genomics for drug identification. Curr. Med. Chem. 10, 733–739 [DOI] [PubMed] [Google Scholar]
- 6. Sharp S., Workman P. (2006) Inhibitors of the HSP90 molecular chaperone: current status. Adv. Cancer Res. 95, 323–348 [DOI] [PubMed] [Google Scholar]
- 7. Münster P. N., Basso A., Solit D., Norton L., Rosen N. (2001) Modulation of Hsp90 function by ansamycins sensitizes breast cancer cells to chemotherapy-induced apoptosis in an RB- and schedule-dependent manner. Clin Cancer Res. 7, 2228–2236 [PubMed] [Google Scholar]
- 8. Ohba S., Hirose Y., Yoshida K., Yazaki T., Kawase T. (2010) Inhibition of 90-kDa heat shock protein potentiates the cytotoxicity of chemotherapeutic agents in human glioma cells. J. Neurosurg. 112, 33–42 [DOI] [PubMed] [Google Scholar]
- 9. Lu X., Xiao L., Wang L., Ruden D. M. (2012) Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 83, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raja S.M., Clubb R.J., Bhattacharyya M., Dimri M., Cheng H., Pan W., Ortega-Cava C., Lakku-Reddi A., Naramura M., Band V., Band H. (2008) A combination of Trastuzumab and 17-AAG induces enhanced ubiquitinylation and lysosomal pathway-dependent ErbB2 degradation and cytotoxicity in ErbB2-overexpressing breast cancer cells. Cancer Biol. Ther. 7, 1630–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Modi S., Stopeck A., Linden H., Solit D., Chandarlapaty S., Rosen N., D'Andrea G., Dickler M., Moynahan M. E., Sugarman S., Ma W., Patil S., Norton L., Hannah A. L., Hudis C. (2011) HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 17, 5132–5139 [DOI] [PubMed] [Google Scholar]
- 12. Modi S., Stopeck A. T., Gordon M. S., Mendelson D., Solit D. B., Bagatell R., Ma W., Wheler J., Rosen N., Norton L., Cropp G. F., Johnson R. G., Hannah A. L., Hudis C. A. (2007) Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J. Clin. Oncol. 25, 5410–5417 [DOI] [PubMed] [Google Scholar]
- 13. Widakowich C., Dinh P., de Azambuja E., Awada A., Piccart-Gebhart M. (2008) HER-2 positive breast cancer: what else beyond trastuzumab-based therapy? Anticancer Agents Med. Chem. 8, 488–496 [DOI] [PubMed] [Google Scholar]
- 14. Zsebik B., Citri A., Isola J., Yarden Y., Szöllosi J., Vereb G. (2006) Hsp90 inhibitor 17-AAG reduces ErbB2 levels and inhibits proliferation of the trastuzumab resistant breast tumor cell line JIMT-1. Immunol. Lett. 104, 146–155 [DOI] [PubMed] [Google Scholar]
- 15. Sorkin A., Goh L. K. (2008) Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 314, 3093–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vaidyanath A., Hashizume T., Nagaoka T., Takeyasu N., Satoh H., Chen L., Wang J., Kasai T., Kudoh T., Satoh A., Fu L., Seno M. (2011) Enhanced internalization of ErbB2 in SK-BR-3 cells with multivalent forms of an artificial ligand. J. Cell Mol. Med. 15, 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henriksen L., Grandal M. V., Knudsen S. L., van Deurs B., Grøvdal L. M. (2013) Internalization mechanisms of the epidermal growth factor receptor after activation with different ligands. PLoS One 8, e58148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roepstorff K., Grøvdal L., Grandal M., Lerdrup M., van Deurs B. (2008) Endocytic down-regulation of ErbB receptors: mechanisms and relevance in cancer. Histochem. Cell Biol. 129, 563–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mohapatra B., Ahmad G., Nadeau S., Zutshi N., An W., Scheffe S., Dong L., Feng D., Goetz B., Arya P., Bailey T. A., Palermo N., Borgstahl G. E., Natarajan A., Raja S. M., Naramura M., Band V., Band H. (2013) Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 1833, 122–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roepstorff K., Grandal M.V., Henriksen L., Knudsen S. L., Lerdrup M., Grøvdal L., Willumsen B. M., van Deurs B. (2009) Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10, 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sigismund S., Confalonieri S., Ciliberto A., Polo S., Scita G., Di Fiore P. P. (2012) Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev. 92, 273–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waterman H., Sabanai I., Geiger B., Yarden Y. (1998) Alternative intracellular routing of ErbB receptors may determine signaling potency. J. Biol. Chem. 273, 13819–13827 [DOI] [PubMed] [Google Scholar]
- 23. Lee K. F., Simon H., Chen H., Bates B., Hung M. C., Hauser C. (1995) Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378, 394–398 [DOI] [PubMed] [Google Scholar]
- 24. Park S. K., Miller R., Krane I., Vartanian T. (2001) The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J. Cell Biol. 154, 1245–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ursini-Siegel J., Schade B., Cardiff R. D., Muller W. J. (2007) Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 7, 389–397 [DOI] [PubMed] [Google Scholar]
- 26. Sliwkowski M. X., Schaefer G., Akita R. W., Lofgren J. A., Fitzpatrick V. D., Nuijens A., Fendly B. M., Cerione R. A., Vandlen R. L., Carraway K. L., 3rd (1994) Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J. Biol. Chem. 269, 14661–14665 [PubMed] [Google Scholar]
- 27. Magnifico A., Tagliabue E., Ardini E., Casalini P., Colnaghi M. I., Ménard S. (1998) Heregulin β1 induces the down-regulation and the ubiquitin-proteasome degradation pathway of p185HER2 oncoprotein. FEBS Lett. 422, 129–131 [DOI] [PubMed] [Google Scholar]
- 28. King C. R., Borrello I., Bellot F., Comoglio P., Schlessinger J. (1988) Egf binding to its receptor triggers a rapid tyrosine phosphorylation of the erbB-2 protein in the mammary tumor cell line SK-BR-3. Embo J. 7, 1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sorkin A., Di Fiore P. P., Carpenter G. (1993) The carboxyl terminus of epidermal growth factor receptor/erbB-2 chimerae is internalization impaired. Oncogene 8, 3021–3028 [PubMed] [Google Scholar]
- 30. Vartanian T., Goodearl A., Viehöver A., Fischbach G. (1997) Axonal neuregulin signals cells of the oligodendrocyte lineage through activation of HER4 and Schwann cells through HER2 and HER3. J. Cell Biol. 137, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y., Tao Y. M., Woo R. S., Xiong W. C., Mei L. (2007) Stimulated ErbB4 internalization is necessary for neuregulin signaling in neurons. Biochem. Biophys. Res. Commun. 354, 505–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sidiropoulos P. N., Miehe M., Bock T., Tinelli E., Oertli C. I., Kuner R., Meijer D., Wollscheid B., Niemann A., Suter U. (2012) Dynamin 2 mutations in Charcot-Marie-Tooth neuropathy highlight the importance of clathrin-mediated endocytosis in myelination. Brain 135, 1395–1411 [DOI] [PubMed] [Google Scholar]
- 33. Zhong C., Du C., Hancock M., Mertz M., Talmage D. A., Role L. W. (2008) Presynaptic type III neuregulin 1 is required for sustained enhancement of hippocampal transmission by nicotine and for axonal targeting of α7 nicotinic acetylcholine receptors. J. Neurosci. 28, 9111–9116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang X. L., Huang Y. Z., Xiong W. C., Mei L. (2005) Neuregulin-induced expression of the acetylcholine receptor requires endocytosis of ErbB receptors. Mol. Cell Neurosci. 28, 335–346 [DOI] [PubMed] [Google Scholar]
- 35. Worthylake R., Wiley H. S. (1997) Structural aspects of the epidermal growth factor receptor required for transmodulation of erbB-2/neu. J. Biol. Chem. 272, 8594–8601 [DOI] [PubMed] [Google Scholar]
- 36. Worthylake R., Opresko L. K., Wiley H. S. (1999) ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 274, 8865–8874 [DOI] [PubMed] [Google Scholar]
- 37. Baulida J., Kraus M. H., Alimandi M., Di Fiore P. P., Carpenter G. (1996) All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 271, 5251–5257 [DOI] [PubMed] [Google Scholar]
- 38. Meijer I. M., van Rotterdam W., van Zoelen E. J., van Leeuwen J. E. (2012) Recycling of EGFR and ErbB2 is associated with impaired Hrs tyrosine phosphorylation and decreased deubiquitination by AMSH. Cell Signal. 24, 1981–1988 [DOI] [PubMed] [Google Scholar]
- 39. Wang Z., Zhang L., Yeung T. K., Chen X. (1999) Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell. 10, 1621–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stern D. F., Heffernan P. A., Weinberg R. A. (1986) p185, a product of the neu proto-oncogene, is a receptorlike protein associated with tyrosine kinase activity. Mol. Cell Biol. 6, 1729–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mosesson Y., Mills G. B., Yarden Y. (2008) Derailed endocytosis: an emerging feature of cancer. Nat. Rev. Cancer. 8, 835–850 [DOI] [PubMed] [Google Scholar]
- 42. Watanabe T. M., Higuchi H. (2007) Stepwise movements in vesicle transport of HER2 by motor proteins in living cells. Biophys. J. 92, 4109–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu W., Okollie B., Artemov D. (2007) Controlled internalization of Her-2/neu receptors by cross-linking for targeted delivery. Cancer Biol. Ther. 6, 1960–1966 [DOI] [PubMed] [Google Scholar]
- 44. Friedman L. M., Rinon A., Schechter B., Lyass L., Lavi S., Bacus S. S., Sela M., Yarden Y. (2005) Synergistic down-regulation of receptor tyrosine kinases by combinations of mAbs: implications for cancer immunotherapy. Proc. Natl. Acad. Sci. U. S. A. 102, 1915–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maier L. A., Xu F. J., Hester S., Boyer C. M., McKenzie S., Bruskin A. M., Argon Y., Bast R. C., Jr. (1991) Requirements for the internalization of a murine monoclonal antibody directed against the HER-2/neu gene product c-erbB-2. Cancer Res. 51, 5361–5369 [PubMed] [Google Scholar]
- 46. Austin C. D., De Mazière A. M., Pisacane P. I., van Dijk S. M., Eigenbrot C., Sliwkowski M. X., Klumperman J., Scheller R. H. (2004) Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell. 15, 5268–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hashizume T., Fukuda T., Nagaoka T., Tada H., Yamada H., Watanabe K., Salomon D. S., Seno M. (2008) Cell type dependent endocytic internalization of ErbB2 with an artificial peptide ligand that binds to ErbB2. Cell Biol. Int. 32, 814–826 [DOI] [PubMed] [Google Scholar]
- 48. Mahlknecht G., Maron R., Mancini M., Schechter B., Sela M., Yarden Y. (2013) Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc. Natl. Acad. Sci. U.S.A. 110, 8170–8175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lerdrup M., Bruun S., Grandal M. V., Roepstorff K., Kristensen M. M., Hommelgaard A. M., van Deurs B. (2007) Endocytic down-regulation of ErbB2 is stimulated by cleavage of its C terminus. Mol. Biol. Cell. 18, 3656–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lerdrup M., Hommelgaard A. M., Grandal M., van Deurs B. (2006) Geldanamycin stimulates internalization of ErbB2 in a proteasome-dependent way. J. Cell Sci. 119, 85–95 [DOI] [PubMed] [Google Scholar]
- 51. Cortese K., Howes M. T., Lundmark R., Tagliatti E., Bagnato P., Petrelli A., Bono M., McMahon H. T., Parton R. G., Tacchetti C. (2013) The HSP90 inhibitor geldanamycin perturbs endosomal structure and drives recycling ErbB2 and transferrin to modified MVBs/lysosomal compartments. Mol. Biol. Cell. 24, 129–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vuong T. T., Berger C., Bertelsen V., Rødland M. S., Stang E., Madshus I. H. (2013) Preubiquitinated chimeric ErbB2 is constitutively endocytosed and subsequently degraded in lysosomes. Exp Cell Res. 319, 32–45 [DOI] [PubMed] [Google Scholar]
- 53. Marx C., Held J. M., Gibson B. W., Benz C. C. (2010) ErbB2 trafficking and degradation associated with K48 and K63 polyubiquitination. Cancer Res. 70, 3709–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barr D. J., Ostermeyer-Fay A. G., Matundan R. A., Brown D. A. (2008) Clathrin-independent endocytosis of ErbB2 in geldanamycin-treated human breast cancer cells. J Cell Sci. 121, 3155–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pedersen N. M., Madshus I. H., Haslekås C., Stang E. (2008) Geldanamycin-induced down-regulation of ErbB2 from the plasma membrane is clathrin dependent but proteasomal activity independent. Mol. Cancer Res. 6, 491–500 [DOI] [PubMed] [Google Scholar]
- 56. Nishimura M., Shin M. S., Singhirunnusorn P., Suzuki S., Kawanishi M., Koizumi K., Saiki I., Sakurai H. (2009) TAK1-mediated serine/threonine phosphorylation of epidermal growth factor receptor via p38/extracellular signal-regulated kinase: NF-{κ}B-independent survival pathways in tumor necrosis factor α signaling. Mol. Cell Biol. 29, 5529–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tong J., Taylor P., Peterman S. M., Prakash A., Moran M. F. (2009) Epidermal growth factor receptor phosphorylation sites Ser991 and Tyr998 are implicated in the regulation of receptor endocytosis and phosphorylations at Ser1039 and Thr1041. Mol. Cell Proteomics 8, 2131–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zwang Y., Yarden Y. (2006) p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. Embo J. 25, 4195–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Winograd-Katz S. E., Levitzki A. (2006) Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 25, 7381–7390 [DOI] [PubMed] [Google Scholar]
- 60. Bao J., Alroy I., Waterman H., Schejter E. D., Brodie C., Gruenberg J., Yarden Y. (2000) Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J. Biol. Chem. 275, 26178–26186 [DOI] [PubMed] [Google Scholar]
- 61. Lladó A., Tebar F., Calvo M., Moretó J., Sorkin A., Enrich C. (2004) Protein kinase Cδ-calmodulin crosstalk regulates epidermal growth factor receptor exit from early endosomes. Mol. Biol. Cell. 15, 4877–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lladó A., Timpson P., Vilà de Muga S., Moretó J., Pol A., Grewal T., Daly R. J., Enrich C., Tebar F. (2008) Protein kinase Cδ and calmodulin regulate epidermal growth factor receptor recycling from early endosomes through Arp2/3 complex and cortactin. Mol. Biol. Cell. 19, 17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salazar G., González A. (2002) Novel mechanism for regulation of epidermal growth factor receptor endocytosis revealed by protein kinase A inhibition. Mol. Biol. Cell. 13, 1677–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Norambuena A., Metz C., Jung J. E., Silva A., Otero C., Cancino J., Retamal C., Valenzuela J. C., Soza A., González A. (2010) Phosphatidic acid induces ligand-independent epidermal growth factor receptor endocytic traffic through PDE4 activation. Mol. Biol. Cell. 21, 2916–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaneko T., Maeda A., Takefuji M., Aoyama H., Nakayama M., Kawabata S., Kawano Y., Iwamatsu A., Amano M., Kaibuchi K. (2005) Rho mediates endocytosis of epidermal growth factor receptor through phosphorylation of endophilin A1 by Rho-kinase. Genes Cells. 10, 973–987 [DOI] [PubMed] [Google Scholar]
- 66. Nishimura Y., Yoshioka K., Bernard O., Bereczky B., Itoh K. (2006) A role of LIM kinase 1/cofilin pathway in regulating endocytic trafficking of EGF receptor in human breast cancer cells. Histochem. Cell Biol. 126, 627–638 [DOI] [PubMed] [Google Scholar]
- 67. Idkowiak-Baldys J., Baldys A., Raymond J. R., Hannun Y. A. (2009) Sustained receptor stimulation leads to sequestration of recycling endosomes in a classical protein kinase C- and phospholipase D-dependent manner. J. Biol. Chem. 284, 22322–22331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shen Y., Xu L., Foster D. A. (2001) Role for phospholipase D in receptor-mediated endocytosis. Mol. Cell Biol. 21, 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Antonescu C. N., Danuser G., Schmid S. L. (2010) Phosphatidic acid plays a regulatory role in clathrin-mediated endocytosis. Mol. Biol. Cell. 21, 2944–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cai J., Crotty T. M., Reichert E., Carraway K. L., 3rd, Stafforini D. M., Topham M. K. (2010) Diacylglycerol kinase δ and protein kinase C(α) modulate epidermal growth factor receptor abundance and degradation through ubiquitin-specific protease 8. J. Biol. Chem. 285, 6952–6959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kasai A., Shima T., Okada M. (2005) Role of Src family tyrosine kinases in the down-regulation of epidermal growth factor signaling in PC12 cells. Genes Cells. 10, 1175–1187 [DOI] [PubMed] [Google Scholar]
- 72. Medts T., de Diesbach P., Cominelli A., N′Kuli F., Tyteca D., Courtoy P. J. (2010) Acute ligand-independent Src activation mimics low EGF-induced EGFR surface signaling and redistribution into recycling endosomes. Exp. Cell Res. 316, 3239–3253 [DOI] [PubMed] [Google Scholar]
- 73. Ware M. F., Tice D. A., Parsons S. J., Lauffenburger D. A. (1997) Overexpression of cellular Src in fibroblasts enhances endocytic internalization of epidermal growth factor receptor. J. Biol. Chem. 272, 30185–30190 [DOI] [PubMed] [Google Scholar]
- 74. Helikar T., Kochi N., Kowal B., Dimri M., Naramura M., Raja S. M., Band V., Band H., Rogers J. A. (2013) A comprehensive, multi-scale dynamical model of ErbB receptor signal transduction in human mammary epithelial cells. PLoS One. 8, e61757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ouyang X., Gulliford T., Epstein R. J. (1998) The duration of phorbol-inducible ErbB2 tyrosine dephosphorylation parallels that of receptor endocytosis rather than threonine-686 phosphorylation: implications for the physiological role of protein kinase C in growth factor receptor signaling. Carcinogenesis 19, 2013–2019 [DOI] [PubMed] [Google Scholar]
- 76. Magnifico A., Albano L., Campaner S., Campiglio M., Pilotti S., Ménard S., Tagliabue E. (2007) Protein kinase Cα determines HER2 fate in breast carcinoma cells with HER2 protein overexpression without gene amplification. Cancer Res. 67, 5308–5317 [DOI] [PubMed] [Google Scholar]
- 77. Feng X., Zhang J., Barak L. S., Meyer T., Caron M. G., Hannun Y. A. (1998) Visualization of dynamic trafficking of a protein kinase C βII/green fluorescent protein conjugate reveals differences in G protein-coupled receptor activation and desensitization. J. Biol. Chem. 273, 10755–10762 [DOI] [PubMed] [Google Scholar]
- 78. Raja S. M., Clubb R. J., Ortega-Cava C., Williams S. H., Bailey T. A., Duan L., Zhao X., Reddi A. L., Nyong A. M., Natarajan A., Band V., Band H. (2011) Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol. Ther. 11, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bonita D. P., Miyake S., Lupher M. L., Jr., Langdon W. Y., Band H. (1997) Phosphotyrosine binding domain-dependent up-regulation of the platelet-derived growth factor receptor α signaling cascade by transforming mutants of Cbl: implications for Cbl's function and oncogenicity. Mol. Cell. Biol. 17, 4597–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Harari D., Yarden Y. (2000) Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 19, 6102–6114 [DOI] [PubMed] [Google Scholar]
- 81. Wang X. Q., Yan Q., Sun P., Liu J. W., Go L., McDaniel S. M., Paller A. S. (2007) Suppression of epidermal growth factor receptor signaling by protein kinase C-α activation requires CD82, caveolin-1, and ganglioside. Cancer Res. 67, 9986–9995 [DOI] [PubMed] [Google Scholar]
- 82. Alessi D. R. (1997) The protein kinase C inhibitors Ro 318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1β (Rsk-2) and p70 S6 kinase. FEBS Lett. 402, 121–123 [DOI] [PubMed] [Google Scholar]
- 83. Han Z., Pantazis P., Lange T. S., Wyche J. H., Hendrickson E. A. (2000) The staurosporine analog, Ro-31–8220, induces apoptosis independently of its ability to inhibit protein kinase C. Cell Death Differ. 7, 521–530 [DOI] [PubMed] [Google Scholar]
- 84. Standaert M. L., Bandyopadhyay G., Antwi E. K., Farese R. V. (1999) RO 31–8220 activates c-Jun N-terminal kinase and glycogen synthase in rat adipocytes and L6 myotubes. Comparison to actions of insulin. Endocrinology 140, 2145–2151 [DOI] [PubMed] [Google Scholar]
- 85. Beltman J., McCormick F., Cook S. J. (1996) The selective protein kinase C inhibitor, Ro-31–8220, inhibits mitogen-activated protein kinase phosphatase-1 (MKP-1) expression, induces c-Jun expression, and activates Jun N-terminal kinase. J. Biol. Chem. 271, 27018–27024 [DOI] [PubMed] [Google Scholar]
- 86. Zeng L., Webster S. V., Newton P. M. (2012) The biology of protein kinase C. Adv. Exp. Med. Biol. 740, 639–661 [DOI] [PubMed] [Google Scholar]
- 87. Konopatskaya O., Poole A. W. (2010) Protein kinase Cα: disease regulator and therapeutic target. Trends Pharmacol. Sci. 31, 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 [DOI] [PubMed] [Google Scholar]
- 89. Le X. F., Marcelli M., McWatters A., Nan B., Mills G. B., O'Brian C. A., Bast R. C., Jr. (2001) Heregulin-induced apoptosis is mediated by down-regulation of Bcl-2 and activation of caspase-7 and is potentiated by impairment of protein kinase Cα activity. Oncogene 20, 8258–8269 [DOI] [PubMed] [Google Scholar]
- 90. Alvi F., Idkowiak-Baldys J., Baldys A., Raymond J. R., Hannun Y. A. (2007) Regulation of membrane trafficking and endocytosis by protein kinase C: emerging role of the pericentrion, a novel protein kinase C-dependent subset of recycling endosomes. Cell Mol. Life Sci. 64, 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Becker K. P., Hannun Y. A. (2003) cPKC-dependent sequestration of membrane-recycling components in a subset of recycling endosomes. J. Biol. Chem. 278, 52747–52754 [DOI] [PubMed] [Google Scholar]
- 92. Schweitzer J. K., Sedgwick A. E., D'Souza-Schorey C. (2011) ARF6-mediated endocytic recycling impacts cell movement, cell division and lipid homeostasis. Semin. Cell Dev. Biol. 22, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Padival A. K., Hawkins K. S., Huang C. (2004) High glucose-induced membrane translocation of PKC βI is associated with Arf6 in glomerular mesangial cells. Mol. Cell. Biochem. 258, 129–135 [DOI] [PubMed] [Google Scholar]
- 94. Melendez A. J., Harnett M. M., Allen J. M. (2001) Crosstalk between ARF6 and protein kinase Calpha in Fc(γ)RI-mediated activation of phospholipase D1. Curr. Biol. 11, 869–874 [DOI] [PubMed] [Google Scholar]
- 95. Gabriel L., Lvov A., Orthodoxou D., Rittenhouse A. R., Kobertz W. R., Melikian H. E. (2012) The acid-sensitive, anesthetic-activated potassium leak channel, KCNK3, is regulated by 14–3-3β-dependent, protein kinase C (PKC)-mediated endocytic trafficking. J. Biol. Chem. 287, 32354–32366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Katz M., Amit I., Yarden Y. (2007) Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta. 1773, 1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Keshamouni V. G., Mattingly R. R., Reddy K. B. (2002) Mechanism of 17-β-estradiol-induced Erk1/2 activation in breast cancer cells. A role for HER2 AND PKC-δ. J. Biol. Chem. 277, 22558–22565 [DOI] [PubMed] [Google Scholar]
- 98. Allen-Petersen B. L., Carter C. J., Ohm A. M., Reyland M. E. (2014) Protein kinase Cδ is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene 33, 1306–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Urtreger A. J., Kazanietz M. G., Bal de Kier Joffé E. D. (2012) Contribution of individual PKC isoforms to breast cancer progression. IUBMB Life. 64, 18–26 [DOI] [PubMed] [Google Scholar]
- 100. Tonetti D. A., Gao W., Escarzaga D., Walters K., Szafran A., Coon J. S. (2012) PKCα and ERβ Are Associated with Triple-Negative Breast Cancers in African American and Caucasian Patients. Int. J. Breast Cancer. 2012, 740353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lonne G. K., Cornmark L., Zahirovic I. O., Landberg G., Jirstrom K., Larsson C. (2010) PKCα expression is a marker for breast cancer aggressiveness. Mol. Cancer 9, 76–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tan M., Li P., Sun M., Yin G., Yu D. (2006) Up-regulation and activation of PKCα by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKCα and Src inhibitors. Oncogene 25, 3286–3295 [DOI] [PubMed] [Google Scholar]
- 103. Recktenwald C. V., Leisz S., Steven A., Mimura K., Müller A., Wulfänger J., Kiessling R., Seliger B. (2012) HER-2/neu-mediated down-regulation of biglycan associated with altered growth properties. J. Biol. Chem. 287, 24320–24329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rafn B., Nielsen C. F., Andersen S. H., Szyniarowski P., Corcelle-Termeau E., Valo E., Fehrenbacher N., Olsen C. J., Daugaard M., Egebjerg C., Bøttzauw T., Kohonen P., Nylandsted J., Hautaniemi S., Moreira J., Jäättelä M., Kallunki T. (2012) ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol. Cell. 45, 764–776 [DOI] [PubMed] [Google Scholar]
- 105. Gordon L. I., Burke M. A., Singh A. T., Prachand S., Lieberman E. D., Sun L., Naik T. J., Prasad S. V., Ardehali H. (2009) Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J. Biol. Chem. 284, 2080–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li-Ping Z., Da-Lei Z., Jian H., Liang-Quan X., Ai-Xia X., Xiao-Yu D., Dan-Feng T., Yue-Hui Z. (2010) Proto-oncogene c-erbB2 initiates rat primordial follicle growth via PKC and MAPK pathways. Reprod. Biol. Endocrinol. 8:66, pp1–9, 7827–7828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Citri A., Yarden Y. (2006) EGF-ERBB signaling: toward the systems level. Nat. Rev. Mol. Cell Biol. 7, 505–516 [DOI] [PubMed] [Google Scholar]
- 108. Idkowiak-Baldys J., Becker K. P., Kitatani K., Hannun Y. A. (2006) Dynamic sequestration of the recycling compartment by classical protein kinase C. J. Biol. Chem. 281, 22321–22331 [DOI] [PubMed] [Google Scholar]
- 109. Lum M. A., Pundt K. E., Paluch B. E., Black A. R., Black J. D. (2013) Agonist-induced down-regulation of endogenous protein kinase Cα through an endolysosomal mechanism. J. Biol. Chem. 288, 13093–13109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lum M. A., Balaburski G. M., Murphy M. E., Black A. R., Black J. D. (2013) Heat Shock Proteins Regulate Activation-Induced Proteasomal Degradation of the Mature Phosphorylated Form of Protein Kinase C. J. Biol. Chem. 288, 27112–27127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Marcotte R., Zhou L., Kim H., Roskelly C. D., Muller W. J. (2009) c-Src associates with ErbB2 through an interaction between catalytic domains and confers enhanced transforming potential. Mol. Cell. Biol. 29, 5858–5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chung B. M., Raja S. M., Clubb R. J., Tu C., George M., Band V., Band H. (2009) Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol. 10, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chung B. M., Dimri M., George M., Reddi A. L., Chen G., Band V., Band H. (2009) The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene 28, 1821–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lanzetti L., Di Fiore P. P. (2008) Endocytosis and cancer: an 'insider' network with dangerous liaisons. Traffic 9, 2011–2021 [DOI] [PubMed] [Google Scholar]
- 115. Rusin A., Krawczyk Z., Grynkiewicz G., Gogler A., Zawisza-Puchałka J., Szeja W. (2010) Synthetic derivatives of genistein, their properties and possible applications. Acta Biochim Pol. 57, 23–34 [PubMed] [Google Scholar]