

Background: Heat shock factor 1 is a stress-responsive transcription factor that targets diverse genomic loci.
Results: Extended and cooperatively oriented genomic binding sequences dictate HSF1 DNA binding specificity.
Conclusion: Cooperativity in HSF1 DNA binding strongly influences the constellation of bound target genes.
Significance: Differential HSF1 target gene selectivity may underlie the diverse functions for HSF1 in protein misfolding disease and cancer.
Keywords: Cancer, DNA-binding Protein, Gene Regulation, Neurodegeneration, Transcription Factor
Abstract
The heat shock transcription factor 1 (HSF1) activates expression of a variety of genes involved in cell survival, including protein chaperones, the protein degradation machinery, anti-apoptotic proteins, and transcription factors. Although HSF1 activation has been linked to amelioration of neurodegenerative disease, cancer cells exhibit a dependence on HSF1 for survival. Indeed, HSF1 drives a program of gene expression in cancer cells that is distinct from that activated in response to proteotoxic stress, and HSF1 DNA binding activity is elevated in cycling cells as compared with arrested cells. Active HSF1 homotrimerizes and binds to a DNA sequence consisting of inverted repeats of the pentameric sequence nGAAn, known as heat shock elements (HSEs). Recent comprehensive ChIP-seq experiments demonstrated that the architecture of HSEs is very diverse in the human genome, with deviations from the consensus sequence in the spacing, orientation, and extent of HSE repeats that could influence HSF1 DNA binding efficacy and the kinetics and magnitude of target gene expression. To understand the mechanisms that dictate binding specificity, HSF1 was purified as either a monomer or trimer and used to evaluate DNA-binding site preferences in vitro using fluorescence polarization and thermal denaturation profiling. These results were compared with quantitative chromatin immunoprecipitation assays in vivo. We demonstrate a role for specific orientations of extended HSE sequences in driving preferential HSF1 DNA binding to target loci in vivo. These studies provide a biochemical basis for understanding differential HSF1 target gene recognition and transcription in neurodegenerative disease and in cancer.
Introduction
All organisms encounter a diverse array of stresses that originate from external (e.g. temperature, toxins, and infection) or physiological (e.g. developmental signals or disease) stimuli. As these stresses damage the proteome through oxidation, misfolding, and/or aggregation, organisms have developed sophisticated mechanisms to repair this damage and limit accumulation of damaged proteins (1–5). For example, a major cellular response to stress serves to increase transcription of genes encoding protein chaperones, the protein degradation machinery, and anti-apoptotic proteins that prevent protein misfolding-induced cell death (6, 7). This stress response system is critical for protecting cells from proteotoxicity by accelerating the turnover of damaged proteins and favoring refolding (8, 9). Imbalances in the stress response system are associated with many diseases, including neurodegeneration and cancer, highlighting the need to gain a detailed understanding of the regulation of stress gene expression.
Heat shock transcription factor 1 (HSF1)4 is a primary mediator of stress-responsive transcription that regulates the expression of many pro-survival genes, including those encoding protein chaperones (10). HSF1 is a multidomain stress-activated transcription factor consisting of the following: an amino-terminal helix winged-loop helix DNA binding domain (DBD); three leucine zipper domains (LZ1–3) that form coiled-coil interactions to facilitate HSF1 multimerization; a central regulatory domain that is extensively modified by phosphorylation, acetylation, and sumoylation; an additional leucine zipper domain (LZ4); and a carboxyl-terminal transcription activation domain (3). Although a number of post-translational modifications are thought to either activate or repress HSF1, the role of specific post-translational modifications on HSF1 activity is not well understood. Under normal conditions, HSF1 largely exists as a repressed monomer in the cytoplasm and is thought to be bound, directly or indirectly, by the protein chaperones Hsp90, Hsp70, and Hsp40 (11). The LZ4 domain has been proposed to function as an auto-inhibitory module through intramolecular coiled-coil interactions with LZ1–3 that repress multimerization (12, 13). In response to proteotoxic stress, HSF1 trimerizes, accumulates in the nucleus, and binds to DNA sequences known as heat shock elements (HSEs), which consist of inverted repeats of the consensus sequence nGAAn (Fig. 1A) (3, 14). Importantly, although the consensus HSE sequence is well characterized, an enormous variety of HSEs exists throughout the human genome, which vary in their primary sequence, length, and orientation of nGAAn repeats (15, 16). A striking example of this diversity is the satellite III repeat region of chromosome 9 in human cells, which serves as a template for the noncoding satellite III RNA involved in the stress response. HSF1 binds to this genomic region, consisting of hundreds of GAA repeats in numerous orientations (17). Although many studies have been carried out to understand the contributions of the consensus HSE sequence for a single HSF1 trimer, the specific features that influence HSF1 binding to the variety of HSEs found in the genome, such as those containing extended sequences, are not well understood.
FIGURE 1.

Expression and purification of human HSF1 derivatives. A, schematic of the current model of HSF1 activation. Upon stress, HSF1 transitions from a monomer to a trimer and binds HSEs throughout the genome. B, diagram of the constructs used for expressing HSF1 derivatives. WT human HSF1, HSF1 with mutations M391K, L395P, and L398R (LZ4m), and HSF1 with amino acids 138–198 deleted (ΔLZ1–3) were cloned into the pET15b E. coli expression vector with an amino-terminal His6 tag. C, gel filtration chromatography of HSF1 derivatives using Sephacryl s400 media. WT hHSF1 elutes as two distinct peaks, whereas ΔLZ1–3 and LZ4m elute as single peaks corresponding to HSF1 monomer and HSF1 trimer, respectively. Each peak was isolated and concentrated to 1 mg/ml. D, SDS-PAGE analysis of 2 μg of the four species of purified HSF1.
Increasing evidence suggests that numerous stressors of environmental and physiological origin activate HSF1 (18–22). Importantly, activation of HSF1 in different cellular contexts has been shown to result in remarkably varied gene expression patterns and genomic binding fingerprints (20, 23, 24). This observation is highlighted by ChIP-seq studies in cell culture models of polyglutamine disease and cancer. Immortalized mouse striatal neurons expressing an expanded polyglutamine Huntingtin protein demonstrate a unique HSF1 binding profile when compared with cells expressing a nonpathological form of Huntingtin (24). Moreover, genome-wide binding of HSF1 is drastically altered in malignant transformed cells, compared with benign heat-shocked cells (23). This striking diversity of HSF1 target genes reinforces the importance of understanding how HSF1 recognizes a variety of HSE sequences.
Although relatively little structural information on HSF1 is available, the structure of the HSF1 DNA binding domain from the yeast Kluyveromyces lactis presented the interesting finding that HSF makes a single direct contact with the major groove of the nGAAn sequence, whereas the rest of the protein-DNA interaction occurs through water-mediated hydrogen bonds to the phosphate backbone (25). This observation supports the notion that there is substantial flexibility in HSF1 target sequences that may drive binding site preferences. Here, we characterize DNA-binding site preferences in vitro using fluorescence polarization and differential scanning fluorimetry, coupled with chromatin immunoprecipitation assays. We demonstrate a role for extended HSE sequences in specific sequence orientation and in driving preferential HSF1 DNA binding. This binding site preference is dictated by contributions from HSF1 homomultimerization and, autonomously, via the HSF1 DNA binding domain. Together, these studies provide a biochemical basis for understanding differential HSF1 target gene recognition and transcription that is observed in neurodegenerative disease and cancer.
EXPERIMENTAL PROCEDURES
Expression and Purification of Human HSF1 Derivatives
A DNA cassette encoding codon-optimized human HSF1 (Genscript) was cloned into the pET15b expression vector containing an amino-terminal His6 tag using NdeI and XhoI to generate hHSF1-pET15b. HSF1-LZ4m was generated using mutagenic PCR on the hHSF1-pET15b plasmid to introduce the L391M, L395P, and L398P mutations in the codon-optimized gene (13). ΔLZ1–3 was generated using overlapping PCR to join fragments encoding amino acids 1–137 and 199–529 of human HSF1 and subsequent cloning into pET15b with NdeI and XhoI. The HSF1 DBD was generated using PCR to amplify the sequence encoding amino acids 1–123. The resulting plasmids were transformed into Escherichia coli strain BL21(DE3). Overnight cultures were diluted 1:100 and grown to A600 = 0.6 at 37 °C. Cultures were then transferred to 15 °C, induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, and grown for 16 h. Cell pellets were lysed in nickel-nitrilotriacetic acid buffer (NB: 50 mm HEPES, pH 7.5, 300 mm NaCl) supplemented with 20 mm imidazole HCl (Im-HCl), using sonication three times with 30-s bursts. Lysates were cleared by centrifugation at 20,000 × g for 30 min. The cleared lysate was incubated with 2 ml (bed volume) of nickel-nitrilotriacetic acid-agarose beads (Qiagen) per liter of culture. Beads were washed twice with NB + 40 mm Im-HCl, twice with NB supplemented with 40 mm Im-HCl, 5 mm ATP, and 20 mm MgCl2, and then once with NB + 40 mm Im-HCl. Bound protein was eluted with NB + 250 mm Im-HCl. Eluted proteins were then separated on a Sephacryl s400 (GE Healthcare) gel filtration column using an Akta FPLC (GE Healthcare) at a flow rate of 1.3 ml/min in 25 mm HEPES, pH 7.5, and 150 mm NaCl. Fractions were collected, pooled, concentrated, and aliquoted at 10 μm (∼0.6 mg/ml), flash-frozen in N2, and stored at −80 °C.
Fluorescence Polarization (FP) Assays
Fluorescently labeled HSE and mutant HSE (mHSE) oligonucleotides with a 5′-fluorescein amide modification (IDT) were used for FP. The labeled oligonucleotides were annealed to complementary sequences by heating to 95 °C and slow cooling to room temperature. FP experiments were performed in 25 mm HEPES, pH 7.5, 75 mm NaCl. 1 nm labeled oligonucleotide was added to FP Buffer, and a baseline milli-polarization value was taken. Human HSF1 derivatives were titrated into the reaction, and milli-polarization values were taken at each concentration of HSF1. Curves shown are representative, and Kd values are calculated from three independent experiments using a one-site binding fit of the curves in GraphPad Prism 5.
Thermal Denaturation Profiling
Differential scanning fluorimetry experiments were performed in a CFX384 RT-PCR thermocycler (Bio-Rad). A temperature gradient that started at 25 and increased to 95 °C at a rate of 0.5 °C per 30 s was used to generate melting curves. Fluorescence readings were taken using the FRET channel. Each reaction was composed of 25 mm HEPES, pH 7.5, 75 mm NaCl, 5 μm HSF1, 5× SYPRO Orange Dye (Invitrogen), 1 mm tris(2-carboxyethyl)phosphine, and DNA to a final volume of 10 μl. The reaction was plated into Hard-Shell PCR Plates 384-Well CLR/WHT (Bio-Rad). This solution was equilibrated for at least 5 min prior to initiation of the differential scanning fluorimetry experiment. DNA concentration was used at a 3-fold molar excess over HSF1, as calculated using three HSF1 molecules binding to a single HSE. This ratio was chosen because we found that a 3-fold molar excess of DNA saturated HSF1 without significant nonspecific binding to mHSE. For clarity, the resulting data sets were trimmed to 25–75 °C and then normalized as a percentage of the highest and lowest relative fluorescence unit values within individual wells. The Tm was then calculated from the maximum of the first derivative plot of the normalized relative fluorescence unit melting curves. Finally, ΔTm was calculated by subtracting the Tm for a control mHSE from the Tm of each individual HSE, The final data are presented as the mean ΔTm and S.E. Statistical analyses of WT and mutant genomic HSEs were performed by one-way analysis of variance followed by Newman-Keuls Multiple Comparison Test using Graph Pad Prism 5.
Quantitative Chromatin Immunoprecipitation
HEK293T cells were grown in 15-cm dishes to 75% confluency and maintained at 37 °C or heat-shocked at 42 °C for 20 min. Cells were cross-linked with 1% formaldehyde on ice for 5 min, quenched with 125 mm glycine for 5 min, washed twice with cold PBS, and then lysed in 2 ml of Lysis Buffer (25 mm HEPES, 150 mm NaCl, 100 mm KCl, 5 mm MgCl2, 1% Triton X-100, 0.05% SDS, 0.03% Nonidet P-40, 1 mm EDTA, pH 7.4). Lysates were then sonicated three times with 30-s bursts and cleared by centrifugation at 14,000 × g for 15 min. Lysates were then split twice into 1-ml aliquots (30 μl saved for input) as follows: 1 ml incubated with 5 μl of affinity-purified rabbit anti-HSF1 antibody overnight and 1 ml incubated without antibody. The next day, 50 μl of Dynabeads Protein G (Invitrogen) were added for 4 h at 4 °C. Beads were washed twice with Lysis Buffer, twice with Lysis Buffer + 0.5 m NaCl, and then twice with TE, pH 7.5. Proteins were eluted with TE + 1% SDS for 10 min at 65 °C. Beads were spun at 14,000 rpm for 10 min, and the supernatant was incubated overnight at 65 °C to reverse cross-links. The next day samples were treated with proteinase K for 1.5 h and purified using Qiagen PCR-cleanup kit prior to quantitative PCR analysis.
RESULTS
Expression and Purification of Human HSF1 Derivatives from E. coli
To examine the DNA-binding site characteristics for human HSF1 in vitro, three derivatives of a synthetic His6-tagged human HSF1 gene were codon-optimized for expression in E. coli and purified as follows: 1) wild type human HSF1 (WT); 2) trimeric HSF1 harboring three point mutations (L391M, L395P, L398P) in the LZ4 domain that disrupt the proposed auto-inhibitory intramolecular coiled-coil interaction (LZ4m); and 3) monomeric HSF1 with the LZ1–3 deleted (ΔLZ1–3) (Fig. 1B). The three HSF1 variants were purified using nickel-nitrilotriacetic acid affinity chromatography followed by Sephacryl s400 size exclusion chromatography. As shown in Fig. 1C, wild type HSF1 elutes as two distinct peaks, indicative of an equilibrium between monomer and trimer. The LZ4m HSF1 derivative, known to be constitutively multimerized when expressed in mammalian cells, elutes as a single peak corresponding to the HSF1 homotrimer, and the HSF1 ΔLZ1–3 derivative elutes as a single peak corresponding to the monomeric protein. These results confirm the hypothesis that the LZ4 domain represses trimerization by demonstrating that the HSF1 LZ4m protein is constitutively trimerized in the absence of eukaryotic regulation (13, 26). Colloidal blue staining of the purified proteins demonstrates that the HSF1 monomer, trimer, ΔLZ1–3, and LZ4m proteins are highly purified (Fig. 1D).
HSF1 DNA Binding in Vitro Correlates with Multimeric State
Fluorescence polarization assays were used to quantitatively characterize the DNA binding features of the HSF1 derivatives in vitro, using a fluorescein-labeled double-stranded oligonucleotide containing a canonical HSE sequence (HSE) and a derivative containing a mutated HSE sequence (mHSE), to which HSF1 fails to bind, as negative control (Fig. 2A). As shown in Fig. 2B, specific DNA binding was detected for all HSF1 derivatives, with a range of affinity for the specific HSE sequence. Both HSF1 homotrimeric species demonstrated low nanomolar affinity for the specific HSE whereas the HSF1 monomer exhibited an ∼10-fold lower affinity than the HSF1 trimer (Fig. 2C). However, the constitutive monomer (ΔLZ1–3) bound DNA with ∼70-fold lower affinity than the HSF1 trimer. The affinity difference between the constitutive monomer and monomeric WT HSF1 may reflect HSE-binding inducing trimerization. Collectively, these results demonstrate that the purified HSF1 derivatives specifically bind to an HSE sequence in vitro.
FIGURE 2.

Fluorescence polarization analysis of HSF1-HSE binding correlates with multimeric state of HSF1 derivatives. A, sequences used for FP analysis. dsDNA containing the inverted nGAAn repeats (HSE) was used for specific DNA binding. A dsDNA with specific mutations disrupting the nGAAn repeat (mHSE) was used as a negative control. B, representative binding curves generated from FP experiments for the four HSF1 derivatives. An increase in millipolarization units (MP) is indicative of direct binding to the fluorescently labeled HSE probe. The HSF1 variants saturate the HSE probe with different binding kinetics. C, Kd values calculated from a hyperboloidal curve fit for the four HSF1 derivatives.
HSE Sequence Element Enhances HSF1 Thermal Stability
To quantitatively evaluate HSF1 binding to distinct HSEs, thermal denaturation profiling was used in which a fluorescent dye, SYPRO Orange, reported on the melting temperature of HSF1 in the absence or presence of cognate HSE sites. Thermal denaturation profiling has been used to characterize changes in the stability of proteins in different buffer conditions or in the presence of ligands. SYPRO Orange is an environmentally sensitive dye that fluoresces when bound to hydrophobic residues of proteins as they unfold during a thermal stimulus. Changes in protein stability are then identified by an apparent shift in the melting curve generated by SYPRO Orange fluorescence throughout the temperature gradient (27–29). We utilized the thermal denaturation profiling technique to investigate interactions between HSF1 and its natural ligand, DNA. As shown in Fig. 3A, a DNA oligonucleotide containing a functional HSE consensus sequence significantly stabilized all of the HSF1 derivatives tested, the increase in relative fluorescence occurs at a higher temperature in the presence of a specific HSE. The concentration of HSE oligonucleotide-binding sites was optimized in the presence of 5 μm HSF1 to achieve the largest increase in melting temperature. Under these conditions, the mutated HSE did not evoke a shift in HSF1 thermal denaturation, indicating that the HSF1-DNA binding interaction, rather than a change in the ionic strength of the reaction, drives the shift in thermal denaturation profile. Using these conditions we determined that the HSF1 monomer, trimer, and LZ4m species all experience a 20 °C shift in thermal stability in the presence of an HSE sequence, although the thermal stability of the ΔLZ1–3 monomeric variant was shifted by 5 °C (Fig. 3B). The large thermal shifts observed provide indirect evidence that HSF1-HSE interactions are stable, consistent with the idea that dissociation of HSF1 from DNA in vivo may require acetylation of HSF1 on lysine 80 within the DNA binding domain (30) or other active processes.
FIGURE 3.

Thermal denaturation profiling of HSF1 derivatives. A, representative melting curves for all derivatives in the presence of nonspecific (mHSE) or specific (HSE) DNA. WT monomer, WT trimer, and LZ4m demonstrate enhanced thermal stability in the presence of HSE DNA. ΔLZ1–3 is also stabilized but to a much lesser extent than the other HSF1 derivatives. B, thermal shifts calculated from the melting curves. The first derivative of the melting curve was calculated, and the highest derivative value was taken as the melting temperature. Melting temperatures of HSF1 in the presence of the mHSE were subtracted from melting temperatures for the HSE curves for each HSF1 derivative to provide a thermal shift (ΔTm). WT monomer, WT trimer, and LZ4m are stabilized by ∼20 °C, whereas ΔLZ1–3 stability is shifted by 4 °C.
Extended HSEs in Tandem Orientations Facilitate Cooperative HSF1 Binding
Chromatin immunoprecipitation-sequencing experiments have demonstrated that HSEs throughout the mammalian genome exhibit a variety of lengths, orientations, and sequences (15, 16, 23, 24). To understand the influence of HSE variation on the HSF1-binding site selection, the thermal shifts caused by a panel of oligonucleotides containing distinct binding sites that are representative of genomic HSF1 binding sequences were measured. It has been previously shown that HSF1 makes critical polar contacts with the guanine of the 5′-nGAAn-3′ sequence within the major groove (25). To understand the importance of the orientation of this critical guanine, an oligonucleotide consisting of tandem repeats of the nGAAn sequence with no inversion was used in thermal denaturation experiments (head to tail oligonucleotide; Fig. 4A). This HSE sequence maintains the contact site for the HSF1 interaction, but alters the orientation of the bound HSF1 molecules. DNA sequences containing three HSEs in tandem, with different orientations between the HSEs, were then analyzed. These extended HSEs can theoretically accommodate three HSF1 trimers, but their orientations will dictate the possible interactions between trimers. A parallel oriented HSE (II HSE) contains three HSE-binding sites, but the orientation of sequence between trimers forces a head to tail interaction between adjacently bound trimers similar to the orientation in the H2T oligonucleotide (Fig. 4A). Finally, a triple HSE (triHSE) contains three HSEs but allows for head-to-head and tail-to-tail interactions between trimers.
FIGURE 4.

Thermal denaturation profiling of HSE variants identifies a role for extended HSEs. A, diagram of the HSE variants used. HSE is the same as used in Fig. 3. Head to tail (H2T) HSE is generated by inverting the middle nGAAn repeat of the HSE sequence. This disrupts the typical head to head interface of the DNA binding domain while bound to DNA. The II HSE is constructed by placing three HSE sites in tandem with an orientation that provides a head to tail interaction between trimers (red boxes). The triple HSE (triHSE) is three HSEs oriented so that head to head or tail to tail interactions (red boxes) are present between trimers. B, ΔTm values in the presence of HSE variants. C, triHSE variants with one HSE disrupted by an mHSE. A triHSE with two trimer binding sites oriented in a cooperative fashion imparts a thermal shift between a single HSE and triHSE, whereas a triHSE with two trimer binding sites in noncooperative orientation or separated by a mHSE do not demonstrate an increased thermal stability.
Using this collection of HSE-binding site variants, HSF1 binding was evaluated by thermal stability profiling. As shown in Fig. 4B, inverting the middle nGAAn (H2T HSE) resulted in a significant reduction in HSF1 binding (−12 °C for WT monomer, WT trimer, and LZ4m and −3 °C for ΔLZ1–3) suggesting that the interface present in the nGAAnnTTCn HSE DNA-binding site contributes to binding affinity and furthermore suggesting the importance of sequence orientation in driving stable HSF1-HSE interactions. The II HSE, containing binding sites fostering head to tail interactions between trimers, does not differ from a single HSE in driving differences in HSF1 thermal stability. This suggests that although multiple trimers can be bound adjacently, they do not bind cooperatively under these conditions. However, when HSF1 binds the triHSE that contains continuous inversions of the nGAAn sequence that would facilitate head-to-head and tail-to-tail interactions between HSF1 trimers, the thermal shift is increased by 7–10 °C. This additional thermal shift may be due to cooperative interactions between HSF1 trimers, which would enhance the thermal stability of the HSF1-HSE complex.
The importance of HSE orientation in thermal stabilization of HSF1 is further supported by data shown in Fig. 4C, with DNA molecules containing two HSE-binding sites in which the third HSE is mutated to a nonspecific site. Using the WT HSF1 homotrimer, we demonstrate that when one cooperative site is available between trimers, an additional thermal shift is observed that is greater than a single HSE but less than the triHSE that contains two cooperative interfaces. The increased thermal shift is not observed when the two adjacent HSEs do not contain a cooperative orientation or if the HSEs are separated by a nonfunctional HSE. Together, these results support the hypothesis that specific HSE sequences and orientations facilitate cooperative HSF1 binding as measured by an increase in HSF1 thermal stability.
HSF1 DNA-binding Domain Facilitates Cooperative HSE Binding
It was unexpected that an increase in thermal stability would be observed with HSF1 ΔLZ1–3 binding to the cooperatively oriented triHSE because this form of HSF1 cannot trimerize. To test whether HSF1 domains in addition to the trimerization domain mediate this cooperative binding effect, potential cooperative interactions between DNA binding domains were evaluated. This possibility was considered because DBDs of adjacent HSF1 monomers might be predicted to assemble in an orientation resembling that found within a trimer.
To determine whether the HSF1 DBD contributes to the increase in thermal stability, the HSF1 DBD was purified, and thermal shifts imparted by HSEs were analyzed. Fig. 5A shows that the HSF1 DBD is purified to homogeneity using a strategy similar to that of the other HSF1 derivatives. As shown in Fig. 5B, the thermal shifts imparted by the HSEs have a similar relationship for the HSF1 DBD as compared with the full-length HSF1 homotrimer. Specifically, the DBD exhibits an increase in melting temperature when the HSE contains continuously inverted nGAAn repeats present in the triHSE, as compared with the single HSE, H2T HSE, or II HSE site.
FIGURE 5.

HSF1 DNA-binding domain participates in cooperative HSE binding. A, SDS-PAGE analyses of the purified HSF1 DBD with molecular weight indicated. B, DBD and WT trimer thermal shifts in the presence of HSE variants. Although the magnitude of the thermal shift is different between the two HSF1 derivatives, a similar relationship is observed between the different HSEs for the two derivatives. C, representation of the K. lactis DBD crystal structure (25) modeled in PyMOL. The dimer interface (green) observed in the crystal structure is predicted to be present between trimers in cooperative HSEs but not in noncooperative HSEs, suggesting that this interface may contribute to cooperative binding to extended HSEs in the proper orientation.
The observation that the HSF1 DNA binding domain cooperatively binds the triHSE is supported by the structural characterization of the K. lactis HSF1 DBD in the HSF-DNA co-crystal structure (Fig. 5C). The crystal structure was solved with two adjacent DBDs in a tail-to-tail orientation. Importantly, this orientation is present between DBDs within a trimer when bound to an HSE but also between adjacently bound trimers with properly oriented extended HSEs, such as that found in the triHSE sequence. The observed inter-DBD interaction interface described in this structure is quite extensive, with numerous hydrogen bonds between the “wing” and “turn” of the adjacent DNA binding domains. This interface may be important not only for the high affinity HSE DNA binding by an individual trimer but also for cooperative binding of multiple HSF1 trimers. Although the report on the HSF1-DNA co-crystal modeled and speculated about cooperative interactions between trimers, data shown here represent the first quantitative measurement of cooperativity between human HSF1 trimers for HSEs oriented in such a manner as to permit cooperative binding (31). Moreover, we propose that this predicted HSF1 inter-DBD interaction surface may provide an additional level of stability and regulation for HSF1-DNA interactions when present in extended genomic HSE sequences.
Cooperative Orientations in Genomic HSEs Influence HSF1 Binding in Vitro and in Vivo
It is important to ascertain whether the cooperative HSF1-DNA interactions observed in vitro have consequences for HSF1 DNA binding in vivo. To address this question, a set of genomic HSEs recently identified in human K562 cells by comprehensive ChIP-seq experiments was evaluated for HSF1 binding in vitro (15). In this study, some genomic HSE sequences were bound by HSF1, although others were bound only by the related transcription factor HSF2. Interestingly, sequences that were bound by HSF1 in vivo generally contain extended and/or cooperative HSE sequences. Table 1 illustrates a panel of genomic sequences and highlights the diversity of HSEs in length and orientation of the critical guanine (red). A panel of DNA oligonucleotides containing these HSE sequences was evaluated in HSF1 thermal shift assays. Interestingly, a range of HSF1 thermal shifts was elicited by the distinct genomic HSEs (Fig. 6A). The sequence and orientation of the MLL HSE resembled the H2T HSE (Fig. 4) and elicited little increase in HSF1 thermal stability. In contrast, the prototypical HSE from the HSPA1A (Hsp70) promoter or that from the UBB ubiquitin gene promoter drove thermal shifts similar to a “perfect” HSE. Importantly, the genomic HSEs associated with the greatest shift in HSF1 thermal stability have orientations that resemble the cooperative HSEs shown in Fig. 4.
TABLE 1.
Genomic HSE sequences
Sequences taken from eight genomic loci demonstrate that the orientation of the guanine required for critical HSF1 major groove contacts is varied in genomic HSEs. Cooperative sequences contain guanines (shown in red) that are specifically spaced on alternating strands.
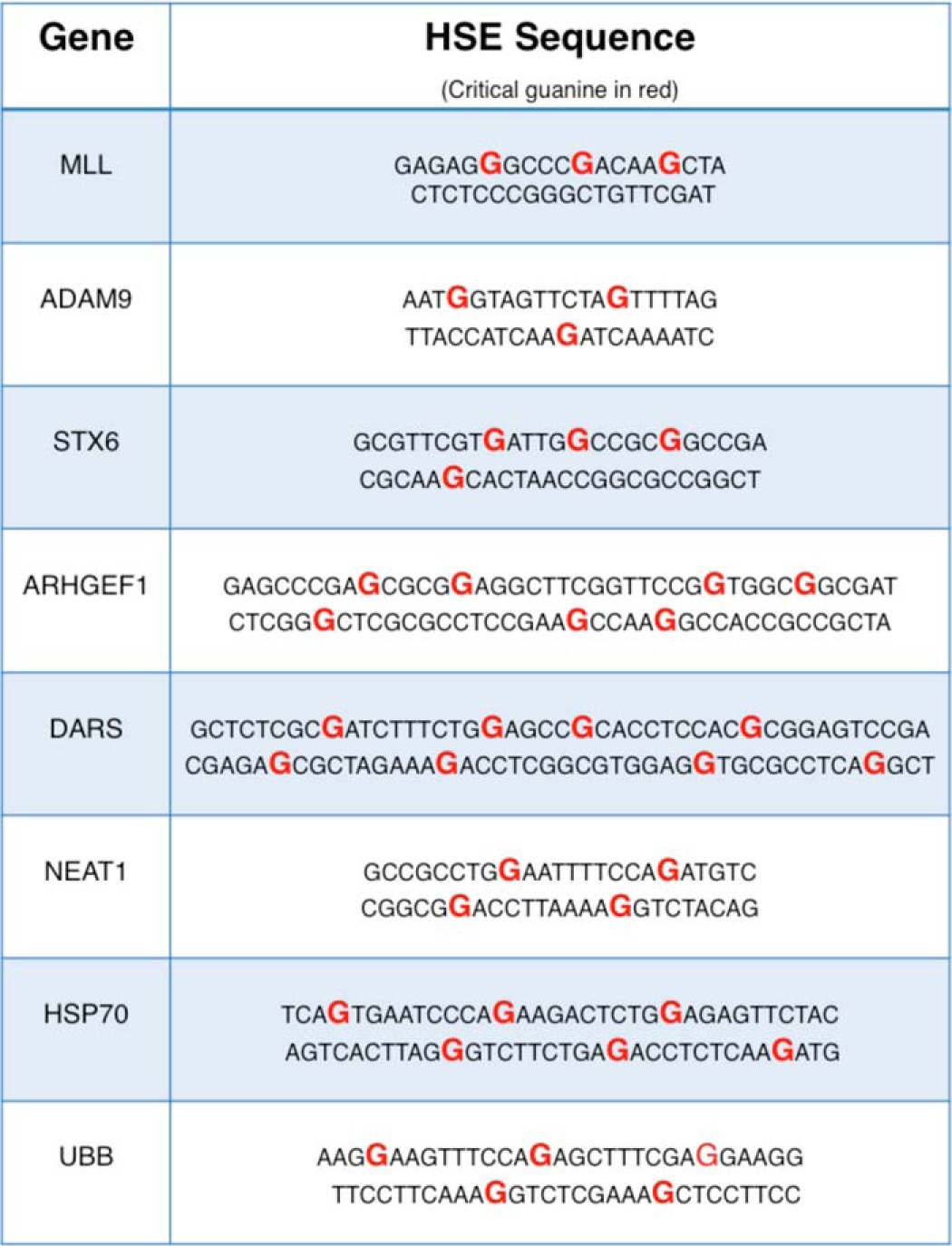
FIGURE 6.

HSF1 binding to genomic HSEs in vitro by thermal denaturation correlates with binding in vivo. A, HSF1 thermal shifts elicited by genomic HSEs. The magnitude of thermal shift correlates with the cooperative nature of the HSE. B, chromatin immunoprecipitation analysis of endogenous human HEK293T cell HSF1 binding to genomic sequences tested in B. A strong correlation can be drawn between in vitro HSE binding as assessed by thermal shift and in vivo binding measured by quantitative ChIP.
To evaluate whether the HSF1 thermal shifts caused by specific HSEs in vitro correlate with the strength of HSF1 DNA binding in vivo, HSF1 DNA binding was evaluated by quantitative chromatin immunoprecipitation experiments in human HEK293T cells grown at control temperature (37 °C) or heat shocked for 1 h at 42 °C. As shown in Fig. 6B, a strong correlation exists between HSE sequences driving strong HSF1 thermal shifts in vitro and HSF1 binding to HSEs in human cells. Furthermore, the strongest binding observed in vivo was seen in HSEs with cooperative orientations. Together, these results support the hypothesis that HSF1 prefers extended HSE sequences with specifically oriented GAA repeats.
To further test the importance of cooperativity in genomic HSE binding, a panel of mutated genomic HSEs was generated (Fig. 7A). To test the influence of cooperative binding to the MLL HSE, a noncooperative guanine was substituted with thymine to allow binding to a potentially cooperative guanine located on the opposing strand. Similarly, an ARHGEF1 HSE mutant predicted to accommodate cooperative binding was created by inverting an internal TTC sequence to facilitate cooperative binding throughout the HSE. In both cases, HSF1-HSE binding was enhanced (+1 °C for MLL and +5 °C for ARHGEF1) as measured by thermal shifts (Fig. 7B). To ascertain whether cooperative sites can be disrupted in genomic sequences, a variant was constructed in the HSPA1A HSE that is predicted to lack cooperative sequences between trimer binding sites without disrupting either trimer binding site individually (Fig. 7A). This sequence imparts a significantly lower thermal shift, −8 °C compared with WT, suggesting that inter-trimer interactions are important for binding at the Hsp70 promoter. Moreover, a UBB HSE lacking the signatures of a predicted cooperative orientation results in significantly reduced HSF1 binding affinity, −7 °C compared with WT. Collectively, these data demonstrate that the orientation of extended HSE sequences dictates the DNA binding affinity of HSF1 to genomic loci in human cells.
FIGURE 7.
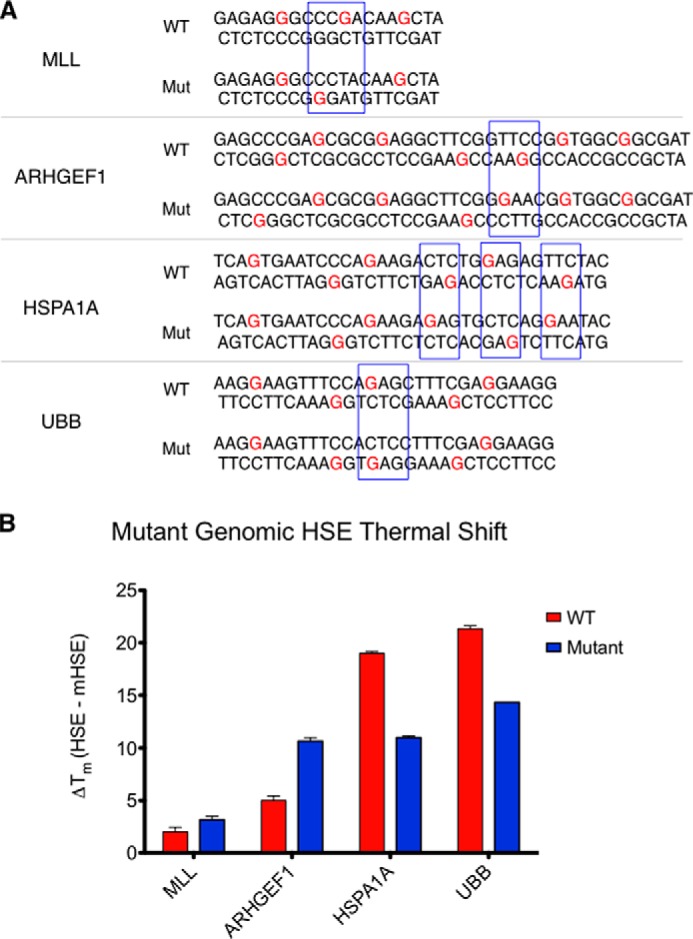
Directed mutation of cooperativity within genomic HSE sequences results in changes in HSF1 DNA binding affinity. A, sequences of “wild type” (WT) and mutant (Mut) genomic HSEs within the MLL, ARHGEF1, HSPA1A, and UBB genes. Subtle mutations within the MLL and ARHGEF1 HSEs foster increased cooperativity, whereas mutations within the HSPA1A and UBB sequence disrupt cooperative interactions between trimers. B, thermal shifts elicited by the genomic HSE variants. MLL and ARHGEF1 HSE mutants exhibit higher affinity binding, whereas the HSPA1A and UBB HSE mutants exhibit lower binding affinity for HSF1. All mutants demonstrated significantly different melting profiles as measured by one-way analysis of variance followed by Newman-Keuls multiple comparison test.
DISCUSSION
Increasing evidence demonstrates that the human heat shock transcription factor 1 targets a diverse array of genomic loci for gene regulation, which has important implications for normal cellular physiology, during stress and in disease. Elucidating the mechanism by which HSF1 selects different genomic loci is important for understanding the consequences of HSF1 activation in the diverse chromatin environment of different physiological and disease states.
The purification of HSF1 using tandem affinity and gel filtration revealed that recombinant human HSF1 exists in an equilibrium between monomer and trimer in vitro and that the two species can be purified by size exclusion chromatography. In this work, we also developed a method for purifying HSF1 LZ4m and ΔLZ1–3, derivatives of HSF1 that are important tools for investigating HSF1 DNA binding. For example, the results obtained with purified HSF1 LZ4m lend biochemical support to the suggestion that the carboxyl-terminal coiled-coil, LZ4, auto-inhibits trimer formation, as we found this derivative to be exclusively multimeric (13, 26).
One limitation to our detailed knowledge of HSF1-DNA interactions has been the low throughput assays commonly used, such as electrophoretic mobility shift assays (EMSA), that are also difficult to quantitate. Initial analysis of recombinant human HSF1 DNA binding with fluorescence polarization, a facile and quantitative assay, revealed differences in the affinity of HSF1 derivatives for a consensus HSE sequence. Moreover, we found that thermal denaturation profiling was amenable to high throughput, enabling studies of DNA binding to a wide array of putative interaction sites such as the many variants found to be bound by HSF1 in the human genome.
Using these platforms, the binding of HSF1 variants to genomic HSE sequences was analyzed, and the results were compared with binding in the context of chromatin in human HEK293T cells. Importantly, the thermal shift profiling method accurately reported on the relative affinity of the genomic interactions, further validating this experimental platform. Furthermore, the results suggest that there are HSF1 binding preferences to extended, cooperative HSEs, supporting the notion that this mechanism for DNA binding contributes to selectivity. It is important to note that the correlation between in vitro and in vivo binding is not perfect, particularly for HSEs found in the promoters of genes such as DARS and HSPA1A. This observation suggests that another layer of complexity exists in HSF1 binding in vivo, which could be attributed to the previously reported heteromultimerization of HSF1 with HSF2, chromatin environments, post-translational modifications, and other factors (18, 32–34). However, our results suggest that when HSF1 is presented with a given array of accessible HSEs, it will prefer cooperatively oriented HSEs. These distinct binding preferences are likely to impact the kinetics and amplitude of HSF1 transcription activation at different loci and dictate the function of HSF1 under diverse physiological and disease contexts.
Acknowledgments
We thank Drs. Maria Schumacher, Nam Tonthat, Charles Pemble IV, and Nathan Nicely for providing valuable insight into this work and reagents. We thank members of the Thiele laboratory for critical comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-NS065890.
- HSF1
- heat shock transcription factor 1
- HSE
- heat shock element
- DBD
- DNA binding domain
- FP
- fluorescence polarization
- mHSE
- mutant HSE
- Im-HCl
- imidazole HCl
- triHSE
- triple HSE
- H2T
- head to tail
- II HSE
- parallel HSE.
REFERENCES
- 1. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 2. Neef D. W., Jaeger A. M., Thiele D. J. (2011) Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 10, 930–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akerfelt M., Morimoto R. I., Sistonen L. (2010) Heat shock factors: integrators of cell stress, development, and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glover-Cutter K. M., Lin S., Blackwell T. K. (2013) Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 9, e1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morimoto R. I. (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morimoto R. I., Kline M. P., Bimston D. N., Cotto J. J. (1997) The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 32, 17–29 [PubMed] [Google Scholar]
- 7. Broadley S. A., Hartl F. U. (2009) The role of molecular chaperones in human misfolding diseases. FEBS Lett. 583, 2647–2653 [DOI] [PubMed] [Google Scholar]
- 8. Bailey C. K., Andriola I. F., Kampinga H. H., Merry D. E. (2002) Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 11, 515–523 [DOI] [PubMed] [Google Scholar]
- 9. Okiyoneda T., Barrière H., Bagdány M., Rabeh W. M., Du K., Höhfeld J., Young J. C., Lukacs G. L. (2010) Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hahn J. S., Hu Z., Thiele D. J., Iyer V. R. (2004) Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 24, 5249–5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo Y., Guettouche T., Fenna M., Boellmann F., Pratt W. B., Toft D. O., Smith D. F., Voellmy R. (2001) Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 276, 45791–45799 [DOI] [PubMed] [Google Scholar]
- 12. Farkas T., Kutskova Y. A., Zimarino V. (1998) Intramolecular repression of mouse heat shock factor 1. Mol. Cell. Biol. 18, 906–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rabindran S. K., Haroun R. I., Clos J., Wisniewski J., Wu C. (1993) Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science 259, 230–234 [DOI] [PubMed] [Google Scholar]
- 14. Pelham H. R. (1982) A regulatory upstream promoter element in the Drosophila hsp 70 heat-shock gene. Cell 30, 517–528 [DOI] [PubMed] [Google Scholar]
- 15. Vihervaara A., Sergelius C., Vasara J., Blom M. A., Elsing A. N., Roos-Mattjus P., Sistonen L. (2013) Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc. Natl. Acad. Sci. U.S.A. 110, E3388–E3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guertin M. J., Martins A. L., Siepel A., Lis J. T. (2012) Accurate prediction of inducible transcription factor binding intensities in vivo. PLoS Genet. 8, e1002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jolly C., Konecny L., Grady D. L., Kutskova Y. A., Cotto J. J., Morimoto R. I., Vourc'h C. (2002) In vivo binding of active heat shock transcription factor 1 to human chromosome 9 heterochromatin during stress. J. Cell Biol. 156, 775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandqvist A., Björk J. K., Akerfelt M., Chitikova Z., Grichine A., Vourc'h C., Jolly C., Salminen T. A., Nymalm Y., Sistonen L. (2009) Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol. Biol. Cell 20, 1340–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inouye S., Fujimoto M., Nakamura T., Takaki E., Hayashida N., Hai T., Nakai A. (2007) Heat shock transcription factor 1 opens chromatin structure of interleukin-6 promoter to facilitate binding of an activator or a repressor. J. Biol. Chem. 282, 33210–33217 [DOI] [PubMed] [Google Scholar]
- 20. Guisbert E., Czyz D. M., Richter K., McMullen P. D., Morimoto R. I. (2013) Identification of a tissue-selective heat shock response regulatory network. PLoS Genet. 9, e1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fawcett T. W., Sylvester S. L., Sarge K. D., Morimoto R. I., Holbrook N. J. (1994) Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J. Biol. Chem. 269, 32272–32278 [PubMed] [Google Scholar]
- 22. Mosser D. D., Kotzbauer P. T., Sarge K. D., Morimoto R. I. (1990) In vitro activation of heat shock transcription factor DNA-binding by calcium and biochemical conditions that affect protein conformation. Proc. Natl. Acad. Sci. U.S.A. 87, 3748–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mendillo M. L., Santagata S., Koeva M., Bell G. W., Hu R., Tamimi R. M., Fraenkel E., Ince T. A., Whitesell L., Lindquist S. (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riva L., Koeva M., Yildirim F., Pirhaji L., Dinesh D., Mazor T., Duennwald M. L., Fraenkel E. (2012) Poly-glutamine expanded huntingtin dramatically alters the genome wide binding of HSF1. J. Huntingtons Dis. 1, 33–45 [PMC free article] [PubMed] [Google Scholar]
- 25. Littlefield O., Nelson H. C. (1999) A new use for the ‘wing’ of the ‘winged’ helix-turn-helix motif in the HSF-DNA cocrystal. Nat. Struct. Biol. 6, 464–470 [DOI] [PubMed] [Google Scholar]
- 26. Zuo J., Baler R., Dahl G., Voellmy R. (1994) Activation of the DNA-binding ability of human heat shock transcription factor 1 may involve the transition from an intramolecular to an intermolecular triple-stranded coiled-coil structure. Mol. Cell. Biol. 14, 7557–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clemente J. C., Nulton E., Nelen M., Todd M. J., Maguire D., Schalk-Hihi C., Kuo L. C., Zhang S. P., Flores C. M., Kranz J. K. (2012) Screening and characterization of human monoglyceride lipase active site inhibitors using orthogonal binding and functional assays. J. Biomol. Screen. 17, 629–640 [DOI] [PubMed] [Google Scholar]
- 28. Nettleship J. E., Brown J., Groves M. R., Geerlof A. (2008) Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol. Biol. 426, 299–318 [DOI] [PubMed] [Google Scholar]
- 29. Cummings M. D., Farnum M. A., Nelen M. I. (2006) Universal screening methods and applications of ThermoFluor. J. Biomol. Screen. 11, 854–863 [DOI] [PubMed] [Google Scholar]
- 30. Westerheide S. D., Anckar J., Stevens S. M., Jr., Sistonen L., Morimoto R. I. (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323, 1063–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao H., Perisic O., Lis J. T. (1991) Cooperative binding of Drosophila heat shock factor to arrays of a conserved 5 bp unit. Cell 64, 585–593 [DOI] [PubMed] [Google Scholar]
- 32. Labbadia J., Cunliffe H., Weiss A., Katsyuba E., Sathasivam K., Seredenina T., Woodman B., Moussaoui S., Frentzel S., Luthi-Carter R., Paganetti P., Bates G. P. (2011) Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 121, 3306–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anckar J., Hietakangas V., Denessiouk K., Thiele D. J., Johnson M. S., Sistonen L. (2006) Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol. Cell. Biol. 26, 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holmberg C. I., Tran S. E., Eriksson J. E., Sistonen L. (2002) Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem. Sci. 27, 619–627 [DOI] [PubMed] [Google Scholar]