

Background: Translational elongation factors are extensively methylated, but the roles of these modifications are not established.
Results: Loss of methylation on elongation factor 2 in Saccharomyces cerevisiae by deletion of EFM3/YJR129C or EFM2 results in translational defects.
Conclusion: Elongation factor methylation is required for normal translational function.
Significance: Protein lysine methylation fine tunes the translational apparatus.
Keywords: Protein Methylation, Protein Synthesis, Ribosome Function, S-Adenosylmethionine (SAM), Translation Elongation Factor, Lysine Methylation
Abstract
Methylation of various components of the translational machinery has been shown to globally affect protein synthesis. Little is currently known about the role of lysine methylation on elongation factors. Here we show that in Saccharomyces cerevisiae, the product of the EFM3/YJR129C gene is responsible for the trimethylation of lysine 509 on elongation factor 2. Deletion of EFM3 or of the previously described EFM2 increases sensitivity to antibiotics that target translation and decreases translational fidelity. Furthermore, the amino acid sequences of Efm3 and Efm2, as well as their respective methylation sites on EF2, are conserved in other eukaryotes. These results suggest the importance of lysine methylation modification of EF2 in fine tuning the translational apparatus.
Introduction
Methylation of translational components has been shown to have a broad spectrum of functional consequences (1–6). Methylation of rRNA plays a role in ribosome biogenesis (6), and modifications to tRNAs increase their stability or affect translational fidelity (2). Protein modifications are also important and are found on various components, including ribosomal proteins, release factors, and elongation factors (1, 5, 7). In a few cases, the functional consequences of these methylations have been established. For example, a 3-methyl histidine on ribosomal protein Rpl3 was recently shown to be involved in large ribosomal subunit biogenesis and translational fidelity (8). In prokaryotes and eukaryotes, release factor 1 is methylated on the conserved GGQ motif that enters the peptidyl transfer center. Loss of the methyltransferase in bacteria results in termination defects (1); in yeast, the loss of the release factor 1 methylation site increases resistance to zymocin (9). However, in most cases, the functional relevance of protein methylation in translation is not known.
Methylation of elongation factors has been well established in Saccharomyces cerevisiae, and many of the modification sites are conserved in higher eukaryotes (10, 11). There are three protein elongation factors in budding yeast: the evolutionarily conserved EF1A and EF2 and the fungal-specific EF3. These three proteins guide tRNAs through the various active sites of the ribosome (12–15). EF1A ensures that correct codon matches occur between the aminoacyl-tRNA and the mRNA, whereas EF2 and EF3 help facilitate the timely translocation of peptidyl-tRNAs and removal of deacylated tRNAs. These three proteins together contain 10 methylated lysine residues (10, 11). The methyltransferases responsible for catalyzing the modification of only three of these residues have been identified (11, 16). Furthermore, the functional relevance of these modifications has been largely unexplored.
In yeast, EF1A is the most heavily methylated of the elongation factors, containing two monomethyllysines (Lys-30 and Lys-390), one dimethyllysine (Lys-316), one trimethyllysine (Lys-79), and a C-terminal lysine α-carboxyl methyl ester (10, 17). EF2 contains trimethyl Lys-509 and dimethyl Lys-613, whereas EF3 has three trimethyllysines: Lys-187, Lys-196, and Lys-789 (11). With the exception of the C-terminal methyl ester, there is no evidence that these modifications are reversible. Although the functional role of these methylations during translation elongation is unclear, the locations of these modifications hint at their importance. Structural studies of EF2 and the 40 S ribosome subunit indicate that the Lys-509 site is in close contact with ribosomal protein Rps23b (18). Lys-613 is in proximity to helix 33 of the 18 S rRNA (18) and is on the same domain as the diphthamide modification at His-699 that aids in maintaining proper transcript frame (19). The potential for enhancing contact with ribosomal components suggests that these methylation sites could be crucial to maintain proper communication with the ribosome during translocation.
Three elongation factor methyltransferases (EFMs)3 have been identified in yeast. Efm1 monomethylates Lys-30 of EF1A (11, 16). See1, which we now refer to as Efm4, dimethylates Lys-316 of EF1A (11, 16). Efm2 has been shown to dimethylate Lys-613 on EF2, and indirect evidence suggests that it may trimethylate Lys-196 of EF3 (11). This leaves five methylation events with no known responsible enzyme. Because the majority of those sites are trimethylated, we sought to search for these enzymes through trimethyllysine immunoblot-based screens.
In this study, we identified Yjr129c as the enzyme responsible for the trimethylation at lysine 509 on elongation factor 2. While this work was being prepared for publication, this finding was reported by another group, and the protein was designated Efm3 (20). In addition to mass spectrometric and immunoblot identification, we directly confirm the identity of this modification as a trimethyllysine by amino acid analysis. We then tested possible functions of elongation factor methylation, including Efm2-catalyzed modification of EF2. Deletion of EFM2 or EFM3 increased sensitivity to translation inhibitors, indicating changes in the ability for EF2 to interact and communicate with ribosomal components. Additionally, we found that translational fidelity is reduced in efm2Δ, indicating possible termination defects.
EXPERIMENTAL PROCEDURES
Strains and Growth Conditions
S. cerevisiae strains used in this study are listed in Table 1. Growth media in this study include YPD (BD Difco 242810, 1% (w/v) yeast extract, 2% (w/v) peptone, and 2% (w/v) dextrose), SD −Ura (minimal synthetic defined medium lacking uracil; 0.07% (w/v) CSM-Ura powder (MP Biomedicals, 114511212), 0.17% (w/v) yeast nitrogen base without amino acids or ammonium sulfate, 0.5% (w/v) ammonium sulfate, 2% (w/v) dextrose), SC (synthetic complete; 0.07% (w/v) CSM (MP Biomedicals, 114500012), 0.17% (w/v) yeast nitrogen base without amino acids or ammonium sulfate, 0.5% (w/v) ammonium sulfate, with or without 2% (w/v) glucose).
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Biological function | Source |
|---|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Wild type | Open Biosystems |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Wild type | Open Biosystems |
| efm3Δ a | BY4741 background | Putative/elongation factor methyltransferase | Open Biosystems |
| efm3Δ α | BY4742 background | Putative/elongation factor methyltransferase | Open Biosystems |
| efm2Δ | BY4741 background | Elongation factor methyltransferase | Open Biosystems |
| efm4Δ(see1Δ) | BY4741 background | Elongation factor methyltransferase | Open Biosystems |
| efm1Δ | BY4741 background | Elongation factor methyltransferase | Open Biosystems |
| rkm1Δ | BY4741 background | Ribosomal protein lysine methyltransferase | Open Biosystems |
| rkm2Δ | BY4741 background | Ribosomal protein lysine methyltransferase | Open Biosystems |
| yjr093cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| ykl162cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| ynl024cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| ymr209cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| yor021cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| ylr063cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| ymr310cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
| ygr283cΔ | BY4741 background | Putative methyltransferase | Open Biosystems |
Overnight 5-ml cultures were used to inoculate cultures to an A600 of 0.1 or 0.15 and grown to values needed for the specific experiment. Cultures were grown in flasks on a rotary shaker (250 rpm) at 30 °C.
Immunoblotting
Strains of interest were grown in YPD to an A600 of 0.7, and cells from 14 ml of the culture were harvested and washed twice with water. Lysis was performed using 0.2 g of glass beads (Biospec Products, 11079105) and 50 μl of lysis buffer (1% SDS, 0.7 mm PMSF). Samples were vortexed for 1 min and then incubated on ice for 1 min, repeated 10 times. Crude lysates were extracted, and beads were washed once with 50 μl of lysis buffer. Unbroken cells and membranes were pelleted by centrifugation at 12,000 × g for 15 min at 4 °C. Protein concentrations were determined using the Lowry method (21). 50 μg of protein from each sample was loaded onto a 4–12% BisTris gel (Invitrogen, NuPAGE Novex) and run at 200 V for 1 h with MOPS buffer. Rainbow full range molecular weight markers (GE Healthcare, RPN800E) were used as standards. Proteins were transferred to PVDF membrane (Hybond-P) at 30 V for 1 h. Membranes were blocked overnight at 4 °C in 5% dried nonfat milk in PBST (phosphate-buffered saline with 0.1% Tween 20 (v/v)). Membranes were washed in PBST and incubated with primary antibodies diluted into 1% dried nonfat milk in PBST for 1.5 h at room temperature and then with secondary antibodies diluted in the same solution for 1 h at room temperature. ECL was used to visualize bands (Amersham Biosciences ECL Prime Western blotting, GE Healthcare, RPN2232). After probing, membranes were stained with Ponceau (1% Ponceau S (w/v), 0.1% acetic acid (v/v)) to determine loading equality.
Antibodies in this study include anti-trimethyllysine-HRP (1:5000; Immunechem, ICP0602), anti-di-/trimethyllysine (1:10,000; Upstate Biotechnology, Inc., 07-756), anti-pan-methyllysine (1:10,000; Abcam, ab7315), and anti-rabbit IgG-HRP (1:6666; Cell Signaling, 7074). The Immunechem and Upstate Biotechnology antibodies were kind gifts from Joanna Goldberg (Emory University). The Abcam “anti-pan methyllysine” antibody was prepared against calf histone H1 containing dimethyllysine residues. The Upstate Biotechnology antibody was raised against a synthetic peptide containing dimethyllysine at position 9 of human histone H3 and is listed by the manufacturer as an anti-di-/trimethyllysine antibody. In the figures, we have emphasized which modified form the antibody prefers in the yeast elongation factors by using boldface type and underlining the respective degree of lysine methylation. Thus, we describe the Abcam antibody as α-M/D/TMK and the Upstate Biotechnology antibody as α-D/TMK. Mouse tissue cytosolic extracts were a kind gift of Dr. Jonathan Lowenson from UCLA.
In Vivo Radiolabeling and Amino Acid Analysis
Cultures of wild type and knock-out cells were grown in YPD to an A600 of 0.7, and cells from 14 ml of culture were harvested by centrifugation at 5000 × g for 5 min), resuspended in 1 ml of water, and transferred to a microcentrifuge tube. After centrifugation, cells were resuspended in 900 μl of YPD and 100 μl of S-adenosyl-l-[methyl-3H]methionine ([3H]AdoMet; 83.3 Ci/mmol; 0.55 mCi/ml in 10 mm H2SO4-ethanol (9:1); PerkinElmer Life Sciences). Cells were incubated for 30 min at 30 °C on a rotary shaker. Radiolabeled cells were washed twice with water and lysed using the glass bead method described above.
Lysates from each strain were loaded onto a 4% stacking, 12% resolving SDS/Tris-glycine polyacrylamide gel (15 × 17 × 0.2 cm) and run at 35 mA through the stacking and 45 mA through the resolving gels. Gels were Coomassie-stained (50% methanol, 10% acetic acid, 40% water, 0.2% Brilliant Blue R-250 (w/v)) and destained overnight (10% methanol, 10% acetic acid, 80% water). The protein band running just above the 97 kDa marker was excised and placed into a 6 × 50-mm glass test tube. 100 μl of 6 n HCl was added to each slice, and tubes were placed in a reaction chamber (Eldex Laboratories, 1163) containing 500 μl of 6 n HCl. Chambers were heated for 20 h in vacuo at 109 °C in a Pico-Tag vapor phase apparatus (Waters). Residual HCl was removed by vacuum centrifugation.
Dried gel slices were resuspended in 400 μl of cation exchange loading buffer (sodium citrate, 0.2 m Na+, pH 2.2). 2 μmol of each methyllysine standard was added to the sample (Sigma; Nϵ-methyl-l-lysine hydrochloride 04685, Nϵ,Nϵ-dimethyl-l-lysine monohydrochloride 19773, and Nϵ,Nϵ,Nϵ-trimethyllysine hydrochloride T1660) and loaded onto a cation exchange column (Beckman AA-15 sulfonated polystyrene resin, 0.9-cm inner diameter by 12-cm height) equilibrated with running buffer (sodium citrate, 0.3 m Na+) at 55 °C. For full separation of mono-, di-, and trimethyllysine, low pH buffer (pH 3.8) was used. To reduce run times when separation of mono- and dimethyllysine was not required, a pH 4.5 buffer was used. Buffer at pH 5.5 was used to analyze when the separation of mono-, di-, and trimethyllysine species was not needed. Amino acids were eluted in the equilibration buffer at 1 ml/min while collecting 1-min fractions at the expected elution position of the methyllysine standards. 50 μl of each fraction was added to a flat-bottom 96-well plate to detect standards by the ninhydrin method. Each well was mixed with 100 μl of ninhydrin reagent (2% ninhydrin (w/v), 0.3% hydrindantin (w/v), 75% dimethyl sulfoxide (v/v), 25% 4 m lithium acetate, pH 4.2 (v/v)), and the plate was heated at 100 °C for 15 min. Standards were detected by measuring absorbance at 570 nm using a SpectraMax M5 microplate reader. The remainder of each fraction was added to 5 ml of scintillation fluor (Safety Solve, Research Products International) in a 20-ml scintillation vial and counted for three 5-min cycles using a Beckman LS6500 instrument to detect 3H-methylated amino acids.
In-gel Trypsin Digests and Mass Spectrometry
Coomassie-stained gel slices from the 100-kDa region of fractionated polypeptides of yeast cell lysates were washed with 50 mm ammonium bicarbonate and destained by incubating in a solution of 50% 50 mm ammonium bicarbonate, 50% acetonitrile for 2–4 h until the gel slice became transparent. Slices were incubated in 100% acetonitrile and dried by vacuum centrifugation for 10 min. After incubating the dried slice in a minimal volume of 10 mm DTT in 50 mm ammonium bicarbonate for 1 h at 60 °C to reduce the disulfide bond, proteins were alkylated by treatment in 50 mm iodoacetamide in 50 mm ammonium bicarbonate for 45 min at 45 °C. Gel slices were washed by alternating 10-min incubations in 50 mm ammonium bicarbonate and 100% acetonitrile. Slices swelled on ice in a working stock solution of 20 ng/μl sequencing grade trypsin (Promega, V5111) for 45 min. Digests were performed for 16 h at 37 °C, and peptides were eluted using 50% acetonitrile, 1% trifluoroacetic acid in water. Peptides were dried by vacuum centrifugation and resuspended in 200 μl of 0.1% TFA in water.
Tryptic peptides from the 100 kDa SDS-gel band of wild type, efm2Δ, and efm3Δ lysates were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an EASY-nLC 1000 system (Thermo Scientific, Waltham, MA) coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific) and an EASY-Spray nano-electrospray ionization source. Peptides were injected onto a 75 μm × 15-cm, 3μ, 100-Å PepMap C18 reversed-phase LC column and separated using a linear gradient from 5% solvent B (0.1% formic acid in acetonitrile), 95% solvent A (0.1% formic acid in water) to 50% solvent B in 45 min at a constant flow of 300 nl/min. Eluted peptides were analyzed with a top 10 data-dependent acquisition method and identified using Proteome Discoverer (version 1.4; Thermo Scientific) coupled with MASCOT (version 2.4.1; Matrix Science, London, UK). Orbitrap MS resolving power was set to 70,000 at m/z 200 for MS1 and 17,500 at m/z 200 for MS2. Tryptic peptides with up to one missed cleavage were searched against the SwissProt S. cerevisiae database (2013; 7798 sequences) with dynamic modifications for carbamidomethyl (C), oxidation (M), deamidation (N, Q) monomethyl (K), dimethyl (K), and trimethyl (K). Precursor and product ion mass tolerances were set to 10 ppm and 0.005 Da, respectively. Methylated EF2 peptides identified by MASCOT were manually examined and confirmed from the corresponding MS/MS spectra.
Manually confirmed EF2 peptides with methylated lysine residues were further examined by targeted parallel reaction-monitoring mass spectrometry (PRM-MS) to explore the effects of EFM2 and EFM3 deletions on EF2 methylation. Samples (described above) were reanalyzed by a targeted MS/MS acquisition method using an inclusion list containing the doubly and triply charged mass-to-charge (m/z) values of the manually confirmed EF2 peptides from wild type lysate. Peaks corresponding to methylated EF2 peptides were visualized in Xcalibur Qual Browser software (Thermo Scientific) using precursor → fragment transitions extracted within 10 ppm mass accuracy. The following transitions (m/z) were used to identify methylated and unmethylated peptides of interest: 286.1920 → 372.2423 (LVEGLKTMK509R); 281.5201 → 251.1790 (LVEGLKDMK509R); 390.2060 → 402.2823 (DDFKDMK613AR); 383.1981 → 268.1712 (DDFKMMK613AR); 329.7103 → 446.2609 (LVEGLK); 376.1903 → 521.3194 (DDFKAR).
Bioinformatic Alignments and Phylogenetic Tree Construction
Whole protein sequences for translocase (EF2 or EF-G) were aligned using Clustal Omega. A protein-protein BLAST search was performed using S. cerevisiae Efm3 (UniProt P47163) or Efm2 (UniProt P32324) as the query. Sequences were aligned using MUSCLE (multiple-sequence comparison by log-expectation) in MEGA 6. The evolutionary history was inferred using the neighbor-joining method to create phylogenetic trees. The evolutionary distances were computed using the p-distance method. All positions containing gaps and missing data were partially eliminated. In instances where poor homology was found, organisms were eliminated from the alignment or replaced by a different representative from the same kingdom.
Translation Inhibitor Assays
Changes in sensitivity to various translation inhibitors were determined using serial dilution spot test growth assays. Briefly, cells were grown at 30 °C in YPD medium to an A600 of ∼0.5. 1 ml of each culture was centrifuged down at 5000 × g for 5 min. Cells were washed with water, and the pellets were diluted to an A600 of 0.5. The cells were then diluted in a 5-fold series in water under sterile conditions. 3 μl of each dilution was spotted onto a 10-cm 2% agar plate containing YPD or YPD + antibiotic and incubated at 30 °C for 2–5 days. Antibiotics used were cycloheximide (Sigma, C7698), puromycin (VWR, 97064-280), paromomycin (Sigma, P9297), anisomycin (Sigma, A9789), tunicamycin (Sigma, T7765), and verrucarin A (Sigma, V4877). Levels of the drug transporter Pdr5 were measured by Northern blot as described previously (8).
Dual Luciferase Translational Fidelity and Frameshift Assays
The dual luciferase systems were used as described previously (8, 22–24). Stop codon read-through and amino acid misincorporation reporters and control vectors were generously provided by Dr. David Bedwell and Ming Du (University of Alabama, Birmingham, AL). Frameshift reporter plasmids were generously provided by Dr. Jonathan Dinman (University of Maryland). All vectors (Table 2) were transformed into wild type, efm2Δ, and efm3Δ cells by the LiOAc-ssDNA-PEG method. The assay was performed as described with the Dual-Luciferase reporter assay system (Promega) using a SpectraMax M5 microplate reader.
TABLE 2.
Vectors used for Dual-Luciferase assays
| Experimental purpose | Control vector | Experimental vector | Source |
|---|---|---|---|
| Amino acid misincorporation | CTY775/luc CAAA | CTY775/luc CAAA FF K529 (AAA to AAT, K → N) | Gift from Dr. David Bedwell (University of Alabama) |
| Stop codon read-through, UAA | CTY775/luc CAAC | CTY775/luc UAAC | |
| Stop codon read-through, UAG | CTY775/luc CAGC | CTY775/luc UAGC | |
| Stop codon read-through, UGA | CTY775/luc CGAC | CTY775/luc UGAC | |
| −1 frameshift | pJD375, 0-frame control | pJD376 (L-A virus element) | Gift from Dr. Jonathan Dinman (University of Maryland) |
| +1 frameshift | pJD375, 0-frame control | pJD377 (Ty1) |
Structural Analysis of Efm3 and Related Methyltransferases
The structures of Efm3, Efm2, and human FAM86A were modeled using the Protein Homology/analogY Recognition Engine version 2.0 (Phyre2). Efm3 modeling was performed using one-to-one threading with METTL21D (VCP-KMT) Chain B from Homo sapiens (PDB entry 4LG1) using the global alignment method with default settings for secondary structure scoring and weight. Efm2 and FAM86A modeling used intensive mode. Structural figures of the catalytic region of these models and crystal structures of other related enzymes were generated in MacPyMOL (DeLano Scientific).
RESULTS
Deletion of the EFM3 Gene in S. cerevisiae Results in the Loss of Trimethyllysine in One or More 100-kDa Proteins
An immunoblot-based screen against trimethyllysine residues on polypeptides from whole cell lysates of putative methyltransferase knock-out strains was performed (Fig. 1A). When using the Upstate Biotechnology antibody nominally specific for di- and trimethyllysine residues, distinct bands were detected near 100 and 50 kDa, corresponding to the approximate molecular masses of EF2/EF3 and EF1A, respectively. The patterns were similar in each case with the exception that there was no detectable signal in the 100 kDa band from the extract of the strain with a deletion of the EFM3 (YJR129C) gene (Fig. 1A).
FIGURE 1.

Deletion of EFM3/YJR129C results in loss of trimethylated lysines in 100-kDa polypeptides in S. cerevisiae. Immunoblots were performed on whole cell lysates as described under “Experimental Procedures” with various antibodies directed against methylated lysine residues. The positions of molecular weight markers are shown on the left. A, polypeptides from lysates of strains with deletions of candidate methyltransferase genes were probed with anti-di-/trimethyllysine (Upstate Biotechnology, 07-756). Arrows indicate the distinct signals at 100 and 50 kDa, correlating to EF2/EF3 and EF1A, respectively. B, polypeptides from known and putative EFM knock-out strains were immunoblotted with multiple methyllysine antibodies. The same membrane was probed, stripped, and reprobed with anti-di-/trimethyllysine (left; Upstate Biotechnology, 07-756), anti-dimethyllysine (middle left; Abcam, Ab7315), or anti-trimethyllysine (middle right; Immunechem, ICP0602). The Ponceau-stained membrane is shown on the right as a protein loading control. The type of methyllysine recognized by each antibody is indicated below each blot with the boldface and underlined letter representing the strongest specificity. C, comparison of wild type and EFM knock-out strains, including efm3Δ, in both mating type backgrounds. All samples were run on the same gel; spaces indicate where non-relevant lanes were removed.
In a separate experiment using deletion strains of known EFMs, we confirmed the loss of 100 kDa immunoreactivity in the efm3Δ (yjr129cΔ) strain (Fig. 1B). We then stripped and reprobed the membrane with two additional antibodies to methylated lysine residues. A complete loss of immunoreactivity in the 100 kDa region was seen with the Upstate Biotechnology di-/trimethyllysine antibody but not with the Abcam dimethyllysine antibody (Fig. 1B). In the latter case, we detected a slight reduction of immunoreactivity in the 100 kDa band. This same reduction was noted in the efm2Δ lysate, which suggested that Efm3 might be acting on the same substrates for Efm2, EF2, and/or EF3. A significant loss of signal is also detected at the 50 kDa position in efm4Δ (see1Δ), which dimethylates lysine 316 on EF1A, confirming the identity of this band.
To confirm that the loss of signal was not due to secondary mutations, the immunoblots were repeated again with gene deletions of the EFM3/YJR129C gene in a- and α mating type backgrounds (Fig. 1C). The same loss of signal was observed in both strains.
Due to the potential nonspecific binding of the antibodies and varied preference of the different antibody preparations between di- and trimethyllysine species, amino acid analysis was utilized to detect the specific type of methylation lost in the efm3Δ strain. S. cerevisiae is capable of taking up exogenous methyl donor, AdoMet, from the media. By supplementing cultures with [3H]AdoMet, substrates methylated during incubation incorporate tritiated methyl groups. After in vivo labeling, cells were lysed, and the resulting lysates were resolved by SDS-PAGE. The Coomassie-stained bands in the 100 kDa region were excised and acid-hydrolyzed into amino acids. The resulting hydrolysate was loaded onto a high resolution cation exchange column capable of separating methyl derivatives of the amino acids. Analysis of wild type 3H-hydrolysates showed the presence of [3H]trimethyllysine and [3H]dimethyllysine but no [3H]monomethyllysine (Fig. 2A, left). Hydrolysates from EFM2 deletion strains, a known EF2-dimethylating enzyme (11), were used as a positive control to ensure that the excised gel slice contained EF2. 3H-Hydrolysates from efm2Δ maintained the presence of trimethyllysine, but no detectable dimethyllysine was seen (Fig. 2A, right).
FIGURE 2.

Deletion of EFM2 results in loss of dimethylated lysine, and deletion of EFM3 results in loss of trimethylated lysine in 100-kDa polypeptides. Lysates from in vivo radiolabeled cells were fractionated by SDS-PAGE, and the 100 kDa gel region was excised. Gel slices were acid-hydrolyzed as described under “Experimental Procedures.” The resulting hydrolysates were loaded onto a high resolution cation exchange column with standards of methylated lysine derivatives. A, using a pH 3.8 elution buffer, radiolabeled methylated lysine derivatives were separated. The position of the standards, detected by ninhydrin reactivity, is shown by the dashed line. Due to a tritium isotope effect, the radiolabeled derivatives elute slightly before the non-labeled standards (58). Each trace is representative of three independent experiments. B, separation of hydrolysates from radiolabeled wild type and efm3Δ cells in both mating type backgrounds. A pH 4.5 elution buffer was used. The ninhydrin profiles are shown for methylated standards run in a separate experiment due to interference from the large amounts of ammonium ion present in the gel hydrolysates. Each trace is representative of two independent experiments. C, the amount of di- and trimethyllysine radioactivity as a percentage of the total radioactivity in the hydrolysate is shown, with error bars reflecting the S.D. p values from Student's t test are shown when less than 0.05.
Wild type and efm3Δ cells from both mating type backgrounds were then in vivo labeled, and the 100-kDa regions were acid-hydrolyzed. Analysis of the wild type 3H-hydrolysates demonstrated the presence of tri- and dimethyllysine in both mating types (Fig. 2B, top panels). By contrast, 3H-hydrolysates from efm3Δ showed an almost complete loss of the trimethyllysine peak (Fig. 2B, bottom panels). To account for variations in the amount of radioactivity loaded onto the columns, one-fifth of the volume of the load (25 μl) was counted for normalization. The fractions of radioactivity for both di- and trimethyllysine relative to the total radioactivity were calculated (Fig. 2C). No significant differences in the relative amount of dimethyllysine were detected between mating types and knockouts, indicating that Efm3 is specifically responsible for the trimethylated lysine on the protein in this region.
Taken together, these results suggest that EFM3/YJR129C encodes a protein lysine methyltransferase that catalyzes the trimethylation of at least one polypeptide of about 100 kDa. Because this is the approximate size of the polypeptides of EF2 and EF3, we investigated whether EF2/EF3 methylation is altered in the absence of this methyltransferase.
Lysine 509 on EF2 Is Unmethylated in the Absence of Efm3
To confirm that Efm3 is acting on an elongation factor, the 100 kDa region on a gel containing non-radioactive lysates was subjected to in-gel trypsin digestion. The resulting peptides were loaded onto a C18 reversed phase column and analyzed by LC-MS/MS on a Q-Exactive Orbitrap mass spectrometer. A protein sequence search reported EF2 (UniProt P32324) as the main component of the 100 kDa protein band. Known methylations on EF2 peptides LVEGLKR and DDFKAR at Lys-509 (di- and trimethyl) and Lys-613 (mono- and dimethyl) were identified, respectively, with additional manual MS/MS spectra confirmation in wild type lysate samples (Fig. 3, A and B). The effect of efm2Δ and efm3Δ on the methylation status of Lys-509 and Lys-613 was examined using a sensitive targeted mass spectrometry method that scans specifically for EF2 methylated peptides observed in wild type samples and their corresponding unmethylated variants. Extracted precursor → fragment ion chromatograms specific to each peptide showed a loss of di- and trimethylation at Lys-509 on the LVEGLKR peptide but no effect on Lys-613 methylation in efm3Δ samples (Fig. 3C). Deletion of EFM2 resulted in a loss of mono- and dimethylation at Lys-613 on peptide DDFKAR without affecting methylation at Lys-509. Further evidence of the effect of Efm2 and Efm3 on EF2 methylation was confirmed by the presence of unmethylated LVEGLK and DDFKAR peptides (not observed in fully methylated wild type samples) in efm3Δ and efm2Δ samples, respectively. A similar approach for analyzing the impact of Efm3 also showed the loss of Lys-509 on EF2 (20).
FIGURE 3.

Deletion of EFM3 results in loss of trimethyllysine 509 on elongation factor 2. The 100-kDa protein bands from wild type and knock-out lysates were excised, in-gel trypsin-digested, and analyzed by LC-MS/MS. A Mascot search identified EF2 as the top hit (score = 13,763.99) for the 100 kDa band. Known trimethylation at Lys-509 (TMK509) (A) and dimethylation at Lys-613 (DMK613) (B) were identified in EF2 peptides LVEGLKR and DDFKAR, respectively, in wild type lysate. Dimethylation at Lys-509 (DMK509) and monomethylation at Lys-613 (MMK613) were also observed in EF2 from wild type lysate. C, the effect of efm2Δ and efm3Δ on the methylation of Lys-509 and Lys-613 was examined by comparison of EF2 from wild-type lysate with that from knock-out lysates. PRM-MS with extracted precursor → product ion transitions (within 10 ppm) was used to identify methylated peptides in each sample condition (WT, efm2Δ, and efm3Δ).
In our experiments, we detected peptides from EF3, but sequence coverage did not include the known methylated sites. Thus, it is possible that Efm2 and/or Efm3 methylates one or more of the three sites on EF3.
Evolutionary Conservation of Efm3 and Efm2 Correlates with the Conservation of Their Respective EF2 Methylation Sites
A BLAST search was performed to identify potential homologs of Efm2 and Efm3. The sequences were aligned using MUSCLE, and a phylogenetic tree was constructed to visually demonstrate the conservation (Fig. 4, A and C). The Lys-509 and Lys-613 lysine residues and surrounding sequence are highly conserved in Animalia, Plantae, and Fungi but less so in Bacteria, Archaea, and Protista.
FIGURE 4.
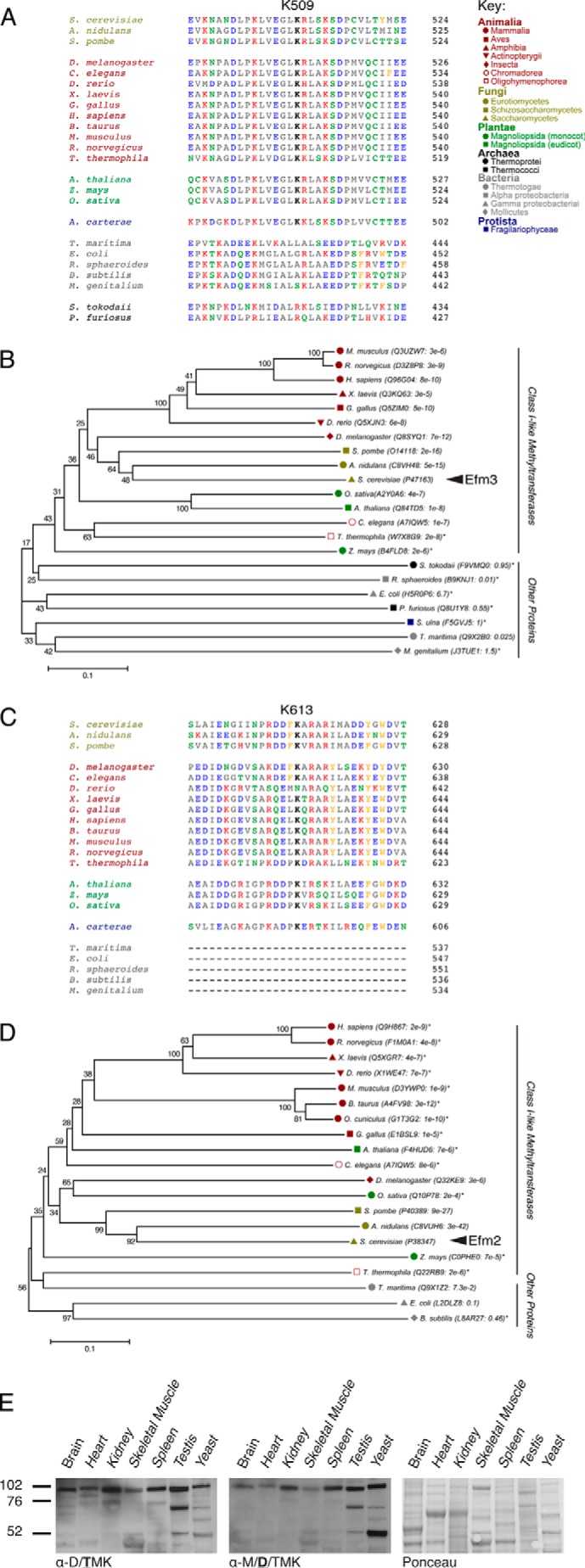
Conservation of EF2 methylation sites and methyltransferases among six kingdoms of life. The region corresponding to the Lys-509 trimethylation site (A) and the Lys-613 dimethylation site (C) in S. cerevisiae is shown for representative organisms. The methylated lysine residue is shown in boldface type. Aliphatic residues are shown in gray, acidic residues in blue, basic residues in red, polar residues in green, and aromatic residues in orange. In C, Archaea were removed from alignment due to significant sequence differences. B and D, phylogenetic trees depicting evolutionary conservation of Efm3 (B) and Efm2 (D). The UniProt ID of the top ranking alignment for each organism is indicated, along with their respective E values. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown beside the branches. The evolutionary distances are in units of the number of amino acid differences per site. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. All proteins were mutual best hits except for those indicated with an asterisk. In B, Amphidinium carterae was removed because no significant homolog to Efm3 was found. Synedra ulna was used as a representative Protista but had no homolog for EF2. In D, Protista, Archaea, Rhodobacter sphaeroides, and Mycoplasma genitalium were completely removed due to no significant homology to Efm2. E, polypeptides from cytosolic extracts from various mouse tissues were probed by immunoblotting with anti-di-/trimethyllysine antibodies (left; Upstate Biotechnology, 07-756). The membrane was then stripped and reprobed with anti-dimethyllysine (middle; Abcam, ab7315). The membrane was Ponceau-stained to ensure equal loading (right).
The conservation of these enzymes correlated well with the conservation of the methylation sites (Fig. 4, B and D); organisms that have homologous enzymes also have the corresponding methylation sites, whereas the Protista, Bacteria, and Archaea do not seem to have either the methylation sites or the enzymes. To confirm the similarity of the methylation reactions in yeast and mammals, cytosolic extracts from mouse tissues were immunoblotted with methyllysine antibodies. Although the protein and methylation patterns varied between tissues, each sample tested showed a distinct trimethyllysine band just below the 102 kDa marker (Fig. 4E). This corresponds to the molecular mass of mouse EF2 (95 kDa) and suggests that EF2 in higher eukaryotes may also be similarly methylated.
Deletion of Efm2 or Efm3 Results in Altered Sensitivity to Translational Inhibitors
To elucidate possible roles of EF2 methylation, wild type and methyltransferase knock-out cells were exposed to a variety of translational inhibitors to look for changes in sensitivity (Fig. 5A). Inhibitors were selected to affect different stages of translation, including those blocking translocation (cycloheximide (25)), inhibiting peptidyl transfer (verrucarin A (26, 27)), or inducing premature termination (puromycin, (28, 29)). Additional inhibitors were chosen that act as tRNA structural mimics (anisomycin (30) and paromomycin (31)) or that block initiation via the unfolded protein response (tunicamycin (32, 33)).
FIGURE 5.

efm2Δ and efm3Δ cells have altered sensitivity to translational inhibitors. A, wild type, efm2Δ and efm3Δ cells were cultured to an A600 of 0.5 and 5-fold serial diluted in water. 3 μl from each dilution was spotted onto a YPD agar plate with or without inhibitors at the indicated concentrations. Images shown are representative of at least three individual replicates. Cells shown are on the same antibiotic plate with other tested strains; only strains relevant to this study are displayed. YPD without inhibitor ensures equal cell loading between strains. The number of days each plate was incubated is indicated below each panel. B, a Northern blot against the multidrug transporter Pdr5 transcript was performed as described (8). The 25 and 18 S ribosomal RNAs are shown as loading controls.
Both efm2Δ and efm3Δ cells demonstrated increased sensitivity to verrucarin A, cycloheximide, and tunicamycin. No differences in sensitivity as compared with wild type were seen for paromomycin, puromycin, and anisomycin. In each case, similar results were found for efm2Δ and efm3Δ, indicating that both methylation sites are required for resistance to these antibiotics. Given the role of EF2 in translocation, it is not surprising that sensitivity of cells is altered when exposed to inhibitors that block translocation either directly or indirectly.
Pdr5 is a multidrug transporter that can export several translation inhibitors, and changes in its expression have been shown to affect drug sensitivity of various mutant strains (34, 35). To confirm that the observed growth phenotypes were not due to variations in PDR5 mRNA expression, the transcript levels of PDR5 were examined by Northern blot. No significant differences were detected between the various strains (Fig. 5B), indicating that the increased sensitivity of the efm2Δ and efm3Δ strains to verrucarin A, cycloheximide, and tunicamycin is unlikely to be due to variations in drug export efficiencies.
Deletion of EFM2, but Not EFM3, Results in Increased Stop Codon Read-through
Wild type and knock-out strains were transformed with dual luciferase reporter vectors (22–24). These vectors contain two luciferase genes, Renilla and firefly, with one of three types of modifications: an inserted stop codon, an inactivation mutation in the firefly gene, or a frameshift signal between the two genes. Changes in stop codon read-through, amino acid misincorporation, and programmed frameshift can be measured by the amount of active firefly produced. No significant changes were observed between wild type and efm3Δ cells for any of these assays (Fig. 6). However, efm2Δ cells showed a 2- and 3-fold higher stop codon read-through for UAG and UAA, respectively, although no differences were seen in misincorporation or frameshifting (Fig. 6).
FIGURE 6.

efm2Δ but not efm3Δ cells have increased stop codon read-through. Dual luciferase assays were utilized to measure the percentage of stop codon read-through, amino acid misincorporation, and frameshifting. Titles of each panel indicate the stop codon analyzed, the misincorporation of lysine, or the direction of programmed ribosomal frameshift (PRF). Values for three or four replicates with S.D. are shown as error bars. p values are displayed where differences were less than 0.05.
Lysine Methyltransferase Structures Share Similarities in the Catalytic Core
The identity of the methyltransferases responsible for four of the methylated lysine residues on the yeast elongation factors is still unknown. In an attempt to narrow down the search for these enzymes, we turned to enzyme structural analysis for potential similarities in known lysine methyltransferases. Efm2 and Efm3 are members of Group J, methyltransferases predicted to have similar substrate types (36). Group J proteins share homology with human Family 16 enzymes, most of which are protein lysine methyltransferases (37–39). Family 16 includes FAM86A, the homolog of Efm3 (20). Here we specifically compared the catalytic centers of modeled Phyre2 structures of Efm2, Efm3, and FAM86A with crystal structures of methyltransferases from the Family 16 and members of the SET domain protein lysine methyltransferases (Fig. 7).
FIGURE 7.

Similarity in the modeled catalytic centers of Efm2 and Efm3 with SET domain lysine methyltransferases and human Family 16 Class I methyltransferases. Top, the SET domain enzymes, SETD6 (PDB entry 3QXY), LSMT (PDB entry 2H2J), and SET7/9 (PDB entries 3M53 (wild type) and 4J71 (Y335F)) have a catalytically required tyrosine residue (blue) that is positioned between the methyl group and adenine ring of AdoMet (green). Alignment of SET7/9 Y335F showed that the phenylalanine (orange) is positioned almost identically to the wild type tyrosine. Residues implicated in substrate binding are shown in pink (41, 47). The substrate-binding tyrosine (pink) and a nearby side chain carbonyl help balance the full negative charge of the substrate lysine (yellow). The aromatic plane of the tyrosine residue is ∼4.4, 4.2, and 3.5 Å away (in SETD6, LSMT, and SET7/9, respectively) from the substrate lysine ϵ-nitrogen atom, suggesting a cation-π interaction (49). Bottom panels, the catalytic sites on crystal structures of the Class I human METTL21A (PDB entry 4LEC), VCP-KMT (PDB entry 4LG1), and CaM-KMT (PDB entry 4PWY) methyltransferases as well as those of the Phyre2 models of S. cerevisiae Efm2 and Efm3 and human FAM86A are displayed in a similar orientation as the three SET domain enzymes shown above. The Class I enzymes have a tyrosine or phenylalanine residue (blue) oriented similarly as the catalytic tyrosine in the SET enzymes. The aspartate residue (light pink), previously shown to be catalytically required in VCP-KMT (38), could serve a similar purpose as the tyrosine-carbonyl pairings in the SET domain enzymes in stabilizing the substrate lysine. A conserved tryptophan residue (pink) in the Class I enzymes is well suited to form cation-π interactions in a manner similar to the phenylalanine residue in the SET domain methyltransferases.
SET domain enzymes differ substantially in sequence and in the overall structure of the AdoMet binding site from Class I methyltransferases (40). Interestingly, these enzymes appear to share a structurally analogous tyrosine or phenylalanine residue in the active site (Fig. 7). In the SETD6 methyltransferase, a Y285A mutation resulted in the loss of catalytic activity, whereas in the SET7/9 enzyme, a corresponding Y335F mutation reduced AdoMet binding affinity without a large change in the catalytic turnover rate (41, 42). Additionally, mutation of the SET7/9 tyrosine to p-aminophenylalanine hindered AdoMet binding by 10,000-fold but only reduced kcat by 35-fold (43). These results suggest two distinct roles of this tyrosine, one of CH···O hydrogen bonding to the AdoMet methyl group and one of a possible cation-π interaction with the substrate lysine. Importantly, a tyrosine or phenylalanine, part of the previously noted DXX(Y/F) motif of the Family 16 and Group J enzymes, is present and similarly positioned in the catalytic site (Fig. 7) (38). Although there does not appear to be CH···O hydrogen bonding between the tyrosine residue and AdoMet, the possibility of cation-π interactions between the aromatic ring and substrate lysine remains. Such cation-π interactions have been previously noted in methyllysine recognition proteins (44–46).
In the SET domain enzymes shown in Fig. 7, the positive charge on the substrate lysine is balanced by the partial negative charge on a hydroxyl group of a different tyrosine residue and a backbone carbonyl group (47). A similar balance has been noted in human SET8 and SUV4-20H2 (48). In the Class I enzymes described here, the aspartate residue of the DXX(Y/F) motif, shown to be catalytically required in VCP-KMT, appears well positioned for similar charge stabilization of a substrate lysine (Fig. 7) (38). Finally, a substrate binding phenylalanine in SETD6 and LSMT (Rubisco large subunit methyltransferase) is positioned similarly to a tryptophan in these Class I enzymes. Here, the aromatic residues could also provide substrate stabilization through cation-π interactions (49).
The clear conservation of the DXX(Y/F) motif in Family 16 and Group J led us to ask if other yeast Class I protein lysine methyltransferases evolved a similar substrate binding mechanism. Surprisingly, no analogous motif is found in other types of protein lysine methyltransferases. For example, both Dot1 and Efm4 lack the DXX(Y/F) motif. Thus, the similarity in catalytic structure between SET domain and Group J enzymes may be of limited use in searching for new protein lysine methyltransferases. However, these observations suggest a convergent evolution of lysine recognition from distinct classes of enzymes.
DISCUSSION
With the identification of both EF2 methyltransferases, it is now possible to examine how these methylation events influence protein synthesis. We assayed cells lacking these methyltransferases for their sensitivity to antibiotics, misincorporation, frameshifting, and stop codon read-through. Cells lacking either Efm2 or Efm3 exhibited increased sensitivity to several antibiotics affecting translation. Whereas efm2Δ and efm3Δ cells showed no difference in misincorporation and frameshifting compared with wild type, efm2Δ displayed increased stop codon read-through.
EF2 is responsible for coordinating the complex translocation step and maintaining the correct reading frame of the mRNA, yet no effect was observed in frameshifting in the methyltransferase-deficient cells. This is perhaps surprising, given that loss of the diphthamide modification in EF2 has been shown to affect frameshifting (19). The close structural proximity of the diphthamide residue to the methylated Lys-613 residue (50) suggested a similar role. Overall, our results indicate that the methylation of EF2 may help to correctly establish connections and contact with the ribosome that, when no longer there, make it more difficult to overcome structural alterations in the presence of translational inhibitors. This is emphasized by the locations of these modifications and the results of this study.
Interestingly, the Efm2 modified Lys-613 residue interacts with helix 33 of the small subunit rRNA, a yeast-specific contact, due to an insertion in domain IV (18). This domain inserts directly into the tRNA binding pockets of the ribosome (51) and is thought to mimic the anticodon domain of the A site tRNA. Loss of this methylation by Efm2 could result in loss of contact points and ultimately alter the ability of EF2 to recognize when termination should occur. This is supported by our translational fidelity data, which demonstrated possible defects in termination and not necessarily elongation. efm2Δ cells demonstrated increased stop codon read-through but no changes in amino acid misincorporation or frameshifting. This is generally an indicator of termination defects (22) and suggests that EF2 may also have a role in termination. In bacteria, EF2 has already been shown to interplay with release and termination factors to stimulate the dissociation of peptidyl-tRNA from the P site (52), and a similar association with termination could be present in yeast. Additionally, the E values for fungal Efm2 homologs are significantly better than values across species for Efm2 or Efm3 (Fig. 4D). It is possible that this is a fungus-specific modification necessary to modulate contacts with the ribosome.
It now appears that all of the methyltransferases modifying EF2 have been identified. However, enzymes that modify two of the sites on EF1A and those that modify the three sites of EF3 have not been found. To fully understand why the elongation factors are methylated, it is essential to uncover these enzymes. Recent work has been done to predict substrate category (protein versus nucleotide versus small molecule) and has correctly predicted Efm3 as a protein methyltransferase (53). However, the specifics of residue type and extent of modification are a much more complex question. Protein lysine methyltransferases are found in both the SET domain and the seven-β-strand (Class I) families. The SET domain family has been extensively characterized in terms of substrate recognition (48). The Class I methyltransferases, although representing the largest family, have a wide variety of substrates, making substrate definition more complex. In protein arginine methyltransferases, the recognition of substrates is relatively well understood, with their characteristic post-motif-II double-E loop and THW motif (54). For some Class I protein lysine methyltransferases, a similarly positioned post-motif-II DXX(Y/F) sequence has been noted (Fig. 7). Our structural analysis comparing two distinct classes of methyltransferases may provide clues regarding the catalytic role of this motif.
Non-histone protein methylation is a largely unexplored yet critical aspect of a multitude of cellular processes, including metabolism and cell signaling (7). In the budding yeast, S. cerevisiae, 86 known and putative methyltransferases have been identified, with 16 having no known substrates (36, 55); of these latter enzymes, six are predicted to have protein substrates (53). Additionally, 40 yeast methyltransferases have human homologs, suggesting that the modifications may be important for a properly functioning cell (56). About two-thirds of the known yeast enzymes are involved with methylating various components of the translational apparatus (Table 3). Because methylation can regulate, enhance, block, or fine tune interactions, this extensive set of methylation events indicates an intricate system for producing an efficient and accurate protein-making machine as well as suggesting potential mechanisms for its regulation (57). A properly functioning cell is highly dependent on correctly synthesized proteins, giving credence to the idea that fine tuning the translational apparatus with methylation is essential for optimal cell fitness.
TABLE 3.
S. cerevisiae methyltransferases involved in modifying translation components, grouped by substrate type
| rRNA | tRNA | mRNA | Translation factors | Ribosomal proteins |
|---|---|---|---|---|
| Spb1 | Trm1 | Abd1 | Mtq1 | Hmt1 |
| Nop1 | Trm2 | Mtq2 | Rmt2 | |
| Nop2 | Trm3 | Efm1 | Ntm1 | |
| Rrp8 | Trm5 | Efm2 | Hpm1 | |
| Mrm2 | Trm7 | Efm3 | Rkm1 | |
| Dim1 | Trm8 | Efm4 | Rkm2 | |
| Mrm1 | Trm9 | Rkm3 | ||
| Emg1 | Trm10 | Rkm4 | ||
| Bmt2 | Trm11 | Rkm5 | ||
| Rcm1 | Trm13 | Sfm1 | ||
| Bmt5 | Trm44 | |||
| Bmt6 | Ppm2 | |||
| Bud23 | Gcd14 | |||
| Tyw3 | ||||
| Trm140 | ||||
| Ncl1 |
Acknowledgments
We thank Dr. Jonathan Lowenson for providing mouse extracts and Dr. Alexander Patananan and Kanishk Jain for assistance in the bioinformatics work. We also thank the following for generous reagent gifts: Dr. Joanna Goldberg (Emory University School of Medicine), Dr. Jonathan Dinman (University of Maryland), and Dr. David Bedwell (University of Alabama). Finally, we thank Dr. Raymond Trievel (University of Michigan) for helpful insights into the methyltransferase structural analyses.
This work was supported, in whole or in part, by National Institutes of Health Grants GM026020 (to S. G. C.) and GM007185, a Ruth L. Kirschstein National Research Service Award (to M. C. D.).
- EFM
- elongation factor methyltransferase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- AdoMet
- S-adenosyl-l-methionine
- [3H]AdoMet
- S-adenosyl-l-[methyl-3H]methionine
- MMK
- monomethyllysine
- DMK
- dimethyllysine
- TMK
- trimethyllysine
- PRM-MS
- parallel reaction-monitoring mass spectrometry
- PDB
- Protein Data Bank.
REFERENCES
- 1. Graille M., Figaro S., Kervestin S., Buckingham R. H., Liger D., Heurgué-Hamard V. (2012) Methylation of class I translation termination factors: structural and functional aspects. Biochimie 94, 1533–1543 [DOI] [PubMed] [Google Scholar]
- 2. Jackman J. E., Alfonzo J. D. (2013) Transfer RNA modifications: nature's combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 4, 35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu J., Jia G. (2014) Methylation modifications in eukaryotic messenger RNA. J. Genet. Genomics 41, 21–33 [DOI] [PubMed] [Google Scholar]
- 4. Motorin Y., Helm M. (2011) RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2, 611–631 [DOI] [PubMed] [Google Scholar]
- 5. Polevoda B., Sherman F. (2007) Methylation of proteins involved in translation. Mol. Microbiol. 65, 590–606 [DOI] [PubMed] [Google Scholar]
- 6. Sharma S., Yang J., Watzinger P., Kötter P., Entian K. D. (2013) Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 41, 9062–9076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clarke S. G. (2013) Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem. Sci. 38, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Al-Hadid Q., Roy K., Munroe W., Dzialo M. C., Chanfreau G. F., Clarke S. G. (2014) Histidine methylation of yeast ribosomal protein Rpl3p is required for proper 60S subunit assembly. Mol. Cell Biol. 34, 2903–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Studte P., Zink S., Jablonowski D., Bär C., von der Haar T., Tuite M. F., Schaffrath R. (2008) tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol. Microbiol. 69, 1266–1277 [DOI] [PubMed] [Google Scholar]
- 10. Cavallius J., Zoll W., Chakraburtty K., Merrick W. C. (1993) Characterization of yeast EF-1 α: non-conservation of post-translational modifications. Biochim. Biophys. Acta 1163, 75–80 [DOI] [PubMed] [Google Scholar]
- 11. Couttas T. A., Raftery M. J., Padula M. P., Herbert B. R., Wilkins M. R. (2012) Methylation of translation-associated proteins in Saccharomyces cerevisiae: identification of methylated lysines and their methyltransferases. Proteomics 12, 960–972 [DOI] [PubMed] [Google Scholar]
- 12. Belfield G. P., Tuite M. F. (1993) Translation elongation factor 3: a fungus-specific translation factor? Mol. Microbiol. 9, 411–418 [DOI] [PubMed] [Google Scholar]
- 13. Justice M. C., Hsu M. J., Tse B., Ku T., Balkovec J., Schmatz D., Nielsen J. (1998) Elongation factor 2 as a novel target for selective inhibition of fungal protein synthesis. J. Biol. Chem. 273, 3148–3151 [DOI] [PubMed] [Google Scholar]
- 14. Kurata S., Shen B., Liu J. O., Takeuchi N., Kaji A., Kaji H. (2013) Possible steps of complete disassembly of post-termination complex by yeast eEF3 deduced from inhibition by translocation inhibitors. Nucleic Acids Res. 41, 264–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mateyak M. K., Kinzy T. G. (2010) eEF1A: thinking outside the ribosome. J. Biol. Chem. 285, 21209–21213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lipson R. S., Webb K. J., Clarke S. G. (2010) Two novel methyltransferases acting upon eukaryotic elongation factor 1A in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 500, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zobel-Thropp P., Yang M. C., Machado L., Clarke S. (2000) A novel post-translational modification of yeast elongation factor 1A: methylesterification at the C terminus. J. Biol. Chem. 275, 37150–37158 [DOI] [PubMed] [Google Scholar]
- 18. Spahn C. M., Gomez-Lorenzo M. G., Grassucci R. A., Jørgensen R., Andersen G. R., Beckmann R., Penczek P. A., Ballesta J. P., Frank J. (2004) Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 23, 1008–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ortiz P. A., Ulloque R., Kihara G. K., Zheng H., Kinzy T. G. (2006) Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J. Biol. Chem. 281, 32639–32648 [DOI] [PubMed] [Google Scholar]
- 20. Zhang L., Hamey J. J., Hart-Smith G., Erce M. A., Wilkins M. R. (2014) Elongation factor methyltransferase 3: a novel eukaryotic lysine methyltransferase. Biochem. Biophys. Res. Commun. 451, 229–234 [DOI] [PubMed] [Google Scholar]
- 21. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 22. Salas-Marco J., Bedwell D. M. (2005) Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 348, 801–815 [DOI] [PubMed] [Google Scholar]
- 23. Plant E. P., Nguyen P., Russ J. R., Pittman Y. R., Nguyen T., Quesinberry J. T., Kinzy T. G., Dinman J. D. (2007) Differentiating between near- and non-cognate codons in Saccharomyces cerevisiae. PLoS One 2, e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harger J. W., Meskauskas A., Dinman J. D. (2002) An “integrated model” of programmed ribosomal frameshifting. Trends Biochem. Sci. 27, 448–454 [DOI] [PubMed] [Google Scholar]
- 25. Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., Liu J. O. (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carrasco L., Barbacid M., Vazquez D. (1973) The trichodermin group of antibiotics, inhibitors of peptide bond formation by eukaryotic ribosomes. Biochim. Biophys. Acta 312, 368–376 [DOI] [PubMed] [Google Scholar]
- 27. Cundliffe E., Cannon M., Davies J. (1974) Mechanism of inhibition of eukaryotic protein synthesis by trichothecene fungal toxins. Proc. Natl. Acad. Sci. U.S.A. 71, 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allen D. W., Zamecnik P. C. (1962) The effect of puromycin on rabbit reticulocyte ribosomes. Biochim. Biophys. Acta 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 29. Yarmolinsky M. B., Haba G. L. (1959) Inhibition by puromycin of amino acid incorporation into protein. Proc. Natl. Acad. Sci. U.S.A. 45, 1721–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kirillov S., Porse B. T., Vester B., Woolley P., Garrett R. A. (1997) Movement of the 3′-end of tRNA through the peptidyl transferase centre and its inhibition by antibiotics. FEBS Lett. 406, 223–233 [DOI] [PubMed] [Google Scholar]
- 31. Vicens Q., Westhof E. (2001) Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure 9, 647–658 [DOI] [PubMed] [Google Scholar]
- 32. Chan S. W., Egan P. A. (2005) Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 19, 1510–1512 [DOI] [PubMed] [Google Scholar]
- 33. Hiramatsu N., Joseph V. T., Lin J. H. (2011) Monitoring and manipulating mammalian unfolded protein response. Methods Enzymol. 491, 183–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gulshan K., Schmidt J. A., Shahi P., Moye-Rowley W. S. (2008) Evidence for the bifunctional nature of mitochondrial phosphatidylserine decarboxylase: role in Pdr3-dependent retrograde regulation of PDR5 expression. Mol. Cell Biol. 28, 5851–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang X., Moye-Rowley W. S. (2001) Saccharomyces cerevisiae multidrug resistance gene expression inversely correlates with the status of the F(0) component of the mitochondrial ATPase. J. Biol. Chem. 276, 47844–47852 [DOI] [PubMed] [Google Scholar]
- 36. Petrossian T. C., Clarke S. G. (2009) Multiple motif scanning to identify methyltransferases from the yeast proteome. Mol. Cell Proteomics 8, 1516–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jakobsson M. E., Moen A., Bousset L., Egge-Jacobsen W., Kernstock S., Melki R., Falnes P. Ø. (2013) Identification and characterization of a novel human methyltransferase modulating Hsp70 protein function through lysine methylation. J. Biol. Chem. 288, 27752–27763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kernstock S., Davydova E., Jakobsson M., Moen A., Pettersen S., Mælandsmo G. M., Egge-Jacobsen W., Falnes P. Ø. (2012) Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat. Commun. 3, 1038. [DOI] [PubMed] [Google Scholar]
- 39. Magnani R., Dirk L. M., Trievel R. C., Houtz R. L. (2010) Calmodulin methyltransferase is an evolutionarily conserved enzyme that trimethylates Lys-115 in calmodulin. Nat. Commun. 1, 43. [DOI] [PubMed] [Google Scholar]
- 40. Schubert H. L., Blumenthal R. M., Cheng X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Horowitz S., Dirk L. M., Yesselman J. D., Nimtz J. S., Adhikari U., Mehl R. A., Scheiner S., Houtz R. L., Al-Hashimi H. M., Trievel R. C. (2013) Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J. Am. Chem. Soc. 135, 15536–15548 [DOI] [PubMed] [Google Scholar]
- 42. Levy D., Kuo A. J., Chang Y., Schaefer U., Kitson C., Cheung P., Espejo A., Zee B. M., Liu C. L., Tangsombatvisit S., Tennen R. I., Kuo A. Y., Tanjing S., Cheung R., Chua K. F., Utz P. J., Shi X., Prinjha R. K., Lee K., Garcia B. A., Bedford M. T., Tarakhovsky A., Cheng X., Gozani O. (2011) Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat. Immunol. 12, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horowitz S., Adhikari U., Dirk L. M., Del Rizzo P. A., Mehl R. A., Houtz R. L., Al-Hashimi H. M., Scheiner S., Trievel R. C. (2014) Manipulating unconventional CH-based hydrogen bonding in a methyltransferase via noncanonical amino acid mutagenesis. ACS Chem. Biol. 9, 1692–1697 [DOI] [PubMed] [Google Scholar]
- 44. van Ingen H., van Schaik F. M., Wienk H., Ballering J., Rehmann H., Dechesne A. C., Kruijzer J. A., Liskamp R. M., Timmers H. T., Boelens R. (2008) Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure 16, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 45. Schalch T., Job G., Noffsinger V. J., Shanker S., Kuscu C., Joshua-Tor L., Partridge J. F. (2009) High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell 34, 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roy S., Musselman C. A., Kachirskaia I., Hayashi R., Glass K. C., Nix J. C., Gozani O., Appella E., Kutateladze T. G. (2010) Structural insight into p53 recognition by the 53BP1 tandem Tudor domain. J. Mol. Biol. 398, 489–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang Y., Levy D., Horton J. R., Peng J., Zhang X., Gozani O., Cheng X. (2011) Structural basis of SETD6-mediated regulation of the NF-κB network via methyl-lysine signaling. Nucleic Acids Res. 39, 6380–6389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Del Rizzo P. A., Trievel R. C. (2014) Molecular basis for substrate recognition by lysine methyltransferases and demethylases. Biochim. Biophys. Acta 10.1016/j.bbagrm.2014.06.008 [DOI] [PubMed] [Google Scholar]
- 49. Burley S. K., Petsko G. A. (1986) Amino-aromatic interactions in proteins. FEBS Lett. 203, 139–143 [DOI] [PubMed] [Google Scholar]
- 50. Jørgensen R., Yates S. P., Teal D. J., Nilsson J., Prentice G. A., Merrill A. R., Andersen G. R. (2004) Crystal structure of ADP-ribosylated ribosomal translocase from Saccharomyces cerevisiae. J. Biol. Chem. 279, 45919–45925 [DOI] [PubMed] [Google Scholar]
- 51. Tourigny D. S., Fernández I. S., Kelley A. C., Ramakrishnan V. (2013) Elongation factor G bound to the ribosome in an intermediate state of translocation. Science 340, 1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heurgué-Hamard V., Karimi R., Mora L., MacDougall J., Leboeuf C., Grentzmann G., Ehrenberg M., Buckingham R. H. (1998) Ribosome release factor RF4 and termination factor RF3 are involved in dissociation of peptidyl-tRNA from the ribosome. EMBO J. 17, 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Szczepińska T., Kutner J., Kopczyński M., Pawłowski K., Dziembowski A., Kudlicki A., Ginalski K., Rowicka M. (2014) Probabilistic approach to predicting substrate specificity of methyltransferases. PLoS Comput. Biol. 10, e1003514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang X., Zhou L., Cheng X. (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 19, 3509–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wlodarski T., Kutner J., Towpik J., Knizewski L., Rychlewski L., Kudlicki A., Rowicka M., Dziembowski A., Ginalski K. (2011) Comprehensive structural and substrate specificity classification of the Saccharomyces cerevisiae methyltransferome. PLoS One 6, e23168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petrossian T. C., Clarke S. G. (2011) Uncovering the human methyltransferasome. Mol. Cell Proteomics 10.1074/mcp.M110.000976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Erce M. A., Pang C. N., Hart-Smith G., Wilkins M. R. (2012) The methylproteome and the intracellular methylation network. Proteomics 12, 564–586 [DOI] [PubMed] [Google Scholar]
- 58. Zurita-Lopez C. I., Sandberg T., Kelly R., Clarke S. G. (2012) Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming ω-NG-monomethylated arginine residues. J. Biol. Chem. 287, 7859–7870 [DOI] [PMC free article] [PubMed] [Google Scholar]