

Background: Human APOE effects on RXR agonist activity are unclear.
Results: In RXR agonist-treated EFAD mice, beneficial effects in APOE4 hippocampus include ABCA1/ABCG1-induced apoE lipoprotein association/lipidation. Detrimental effects in APOE3 and APOE4 cortex might be from systemic hepatomegaly.
Conclusion: Aβ pathology and APOE genotype modulate RXR agonist effects.
Significance: Although promising for later stage Alzheimer disease, detrimental side effects limit translational application of RXR agonists.
Keywords: Alzheimer Disease, Amyloid-β (AB), Apolipoprotein E (ApoE), Drug Action, Drug Delivery, ABCA1/ABCG1 Lipid Transporters, RXR Agonists, ApoE/Aβ Complex, Hepatomegaly, Oligomeric Aβ
Abstract
Previous data demonstrate that bexarotene (Bex), retinoid X receptor (RXR) agonist, reduces soluble and insoluble amyloid-β (Aβ) in Alzheimer disease (AD)-transgenic mice either by increasing the levels of mouse apolipoprotein E (apoE) or increasing ABCA1/ABCG1-induced apoE lipoprotein association/lipidation. However, although the mechanism of action of RXR agonists remains unclear, a major concern for their use is human (h)-APOE4, the greatest AD genetic risk factor. If APOE4 imparts a toxic gain-of-function, then increasing apoE4 may increase soluble Aβ, likely the proximal AD neurotoxin. If the APOE4 loss-of-function is lipidation of apoE4, then induction of ABCA1/ABCG1 may be beneficial. In novel EFAD-Tg mice (overexpressing h-Aβ42 with h-APOE), levels of soluble Aβ (Aβ42 and oligomeric Aβ) are highest in E4FAD hippocampus (HP) > E3FAD-HP > E4FAD cortex (CX) > E3FAD-CX, whereas levels of lipoprotein-associated/lipidated apoE have the opposite pattern (6 months). In E4FAD-HP, short-term RXR agonist treatment (Bex or LG100268; 5.75–6 months) increased ABCA1, apoE4 lipoprotein-association/lipidation, and apoE4/Aβ complex, decreased soluble Aβ, and increased PSD95. In addition, hydrogel delivery, which mimics low sustained release, was equally effective as gavage for Bex and LG100268. RXR agonists induced no beneficial effects in the E4FAD-HP in a prevention protocol (5–6 months) and actually increased soluble Aβ levels in E3FAD-CX and E4FAD-CX with the short-term protocol, possibly the result of systemic hepatomegaly. Thus, RXR agonists address the loss-of-function associated with APOE4 and exacerbated by Aβ pathology, i.e. low levels of apoE4 lipoprotein association/lipidation. Further studies are vital to address whether RXR agonists are an APOE4-specific AD therapeutic and the systemic side effects that limit translational application.
Introduction
Alzheimer disease (AD),2 the most common form of dementia, has reached epidemic proportions and is a serious economic and social burden worldwide (1). Soluble forms of the amyloid-β peptide (Aβ42) (2–4) are considered a proximal cause of impaired synaptic function in AD, particularly soluble oligomeric Aβ (oAβ) (5–8). Unfortunately, therapeutic strategies targeting Aβ42 production or clearance have not yet proved to be viable due to both efficacy and safety issues (9, 10). Thus, there is a drive to develop novel therapeutic approaches targeting Aβ. Apolipoprotein E (apoE)-containing lipoproteins, produced primarily by glia, represent a promising physiological target for modulating Aβ levels in the CNS. Cramer et al. (11) identified bexarotene (Bex), a retinoid X receptor (RXR) agonist developed as an anti-cancer drug, as a potential AD therapeutic candidate to increase apoE levels. Indeed, Bex increased mouse (m)-apoE levels, decreased soluble Aβ within hours, and reduced insoluble Aβ after 3 days in transgenic mouse models expressing familial AD mutations (FAD-Tg) (11). However, recent data in multiple FAD-Tg models, although variable (12), demonstrate that Bex has no effect on insoluble Aβ, but may lower soluble Aβ42 and soluble oAβ levels (13–16). In addition, these recent studies indicate that rather than increasing APOE (gene coding for apolipoprotein E) expression, Bex may lower soluble Aβ through increasing the levels of other RXR target proteins, specifically, the ATP-binding cassette transporters (ABC) ABCA1 and ABCG1 (13–16). As ABCA1 and ABCG1 function to transport lipids to apoE-containing lipoproteins (17), increased apoE lipoprotein association/lipidation may be the mechanism for reducing soluble Aβ levels (18–24). Thus, there is a critical need to address confounding factors that may interact with the activity and mechanism of action of RXR agonists leading to inconsistent data in FAD-Tg models, particularly as Bex is currently under consideration for clinical trials.
The major concern for the use of RXR agonists in AD is human (h)-APOE4. APOE4 is the greatest genetic risk factor for sporadic AD, increasing risk up to 15-fold compared with APOE3 (25, 26). Despite representing more than half of all AD patients, APOE4 carriers are often omitted from clinical trials because they fail to respond or respond anomalously (27–30). Although the impact of h-APOE on CNS function is multifactorial, compared with APOE3, APOE4 increases insoluble and soluble Aβ levels in FAD-Tg mice (31) and in AD patients (32–34). However, the effects of RXR agonists on Aβ levels in the presence of h-APOE are unclear. The critical unresolved question is whether RXR agonists act via increasing apoE levels or increasing the ABCA1/G1-dependent lipidation of apoE. Overall, concern centers on whether APOE4 imparts a toxic gain-of-function, and thus Bex-induced increased apoE4 levels may actually increase Aβ levels or, if the APOE4 loss-of-function is lipidation of apoE4, induction of ABCA1/ABCG1 may then be beneficial. However, the majority of published data tests Bex in FAD-Tg mice expressing m-apoE (11, 12, 14–16), which is structurally and functionally distinct from any of the human isoforms. In the context of h-APOE, limited data in one FAD-Tg model (APOE/APP/PS1) demonstrate that Bex lowers soluble Aβ levels for APOE3 and APOE4 as assessed in the interstitial fluid by microdialysis, but not when measured using biochemical extractions (13). Furthermore, APOE/APP/PS1 mice display minimal Aβ accumulation (13), and the levels of Aβ pathology at the time of Bex treatment may determine the overall response. In APOE-TR mice, which do not express h-Aβ, Bex reverses the lipidation deficiency of APOE4 (35). Given the conflicting data on Bex treatment in FAD-Tg mice expressing m-apoE (12), it is vital, from a translational perspective, to further dissect the interactive effects of h-APOE and RXR agonists on Aβ accumulation in an in vivo model that expresses h-APOE.
We recently developed novel EFAD mice, which express 5×FAD mutations and h-APOE3 or h-APOE4 and are a tractable model of APOE genotype-specific Aβ accumulation (31). In EFAD mice, total brain apoE levels are lower with APOE4. However, using a three-step sequential protein extraction protocol, TBS, TBSX (TBS containing Triton X-100) and formic acid (FA), the reduced levels of apoE4 are seen only in the TBSX fraction, indicating that apoE4 is less lipoprotein-associated and/or less lipidated (31). Additional data from our group and others support the pathway that the reduced lipoprotein association/lipidation of apoE4 (31, 35) results in lower levels of apoE4/Aβ complex and higher levels of oAβ (31, 33), compromising synaptic viability. Thus, EFAD mice allow the preclinical analysis of RXR agonists in the presence of h-APOE.
There are further issues related to RXR agonists such as Bex as an AD therapeutic. Bex is a synthetic retinoic acid analogue, classed as a pan-RXR agonist, and is currently approved for treatment of cutaneous T-cell lymphoma. Adverse events, including severe but reversible hypertriglyceridemia (36–39), have prompted the design of improved Bex analogues, and LG100268 (LG) was first reported in 1995 as delivering improved potency and selectivity for RXR over other nuclear receptors (NR), particularly the retinoic acid receptors (RAR) (40), although further improvement is needed (41). In the livers of rodents and humans, Bex modulates metabolic enzymes associated with drug-drug interactions and induces hepatomegaly (14, 42), which may outweigh any beneficial effects in the CNS, particularly in an elderly population. Comparison of Bex and LG is essential to confirm that any actions of Bex represent an effect of the RXR agonist drug class, rather than any off-target effect; furthermore, LG and later generation RXR agonists may prove safer and potentially more effective than Bex for AD therapy.
In this study, EFAD mice (31) were treated with the RXR agonists Bex and LG and indirect target engagement, mechanistic pharmacodynamic (PD), PD, and efficacy markers were measured. Overall, the data demonstrate that Aβ levels and APOE genotype determine the effect of RXR agonists on PD and efficacy readouts. In untreated EFAD mice, levels of soluble Aβ (Aβ42 and oAβ) are highest in hippocampus (HP) of E4FAD mice and follow the pattern E4FAD-HP > E3FAD-HP > E4FAD-cortex (CX) > E3FAD-CX, although the levels of lipoprotein-associated/lipidated apoE have the opposite pattern. In the E4FAD-HP, short-term (5.75–6M) Bex and LG treatment support the hypothesis that RXR agonists increase ABCA1/G1 levels, apoE4 lipoprotein association/lipidation, and apoE/Aβ complex levels, which results in decreased soluble Aβ and increased PSD95 levels. In addition, hydrogel delivery, which mimics low sustained release, was equally effective as gavage for Bex and LG in lowering soluble Aβ with APOE4. However, in the 30-day prevention protocol, no beneficial effects were observed in the E4FAD-HP (5–6M). Furthermore, in E3FAD-CX and E4FAD-CX, brain regions with the lowest Aβ levels at the time of short-term treatment, RXR agonists actually increased soluble Aβ levels in the absence of any changes in ABCA1 levels or apoE lipoprotein association/lipidation. The lack of a response with RXR agonists in E4FAD-HP during prevention and the induction of detrimental CNS effects in the CX in short-term treatment may be the result of systemic side effects, particularly hepatomegaly, which have been observed in models of acute and chronic liver disease (43–46). Collectively, these data suggest that Bex and LG can address a loss-of-function associated with APOE4 and exacerbated by Aβ, i.e. lower apoE lipoprotein association/lipidation. However, future studies are vital to determine whether this pathway is relevant for APOE3 carriers or whether RXR agonists are an APOE4-specific AD therapeutic. In addition, addressing the systemic side effects of RXR agonists is important for their translational application in longer term AD prevention and treatment trials.
EXPERIMENTAL PROCEDURES
Chemicals
LG was synthesized and characterized as described previously (40, 47). Purity of Bex (LC Laboratories, Woburn, MA) and LG were >98% by RP-HPLC.
RXR Agonist Treatment (Table 1)
TABLE 1.
RXR agonist treatment protocols
| Study | Age and length of treatment | Drug delivery | Treatment |
|---|---|---|---|
| T1 | 5.75 to 6 months | Gavage | VC, sesame oil |
| (7 days) | Bex, 20 mg/ml sesame oil | ||
| LG, 20.86 mg/ml in sesame oil | |||
| Treatment, 5 μl/g (mouse weight) | |||
| T2 | 5.75 to 6 months | Hydrogel | VC, 0.4% DMSO in hydrogel |
| (7 days) | Bex, 3 mg/4.5 ml in hydrogel | ||
| LG, 3.13 mg/4.5 ml in hydrogel | |||
| Treatment, ∼4.5 ml hydrogel consumed per mouse/day (for a 30-g mouse) | |||
| T3 | 5 to 6 months | Hydrogel | As above, hydrogel replaced every 2–3 days |
| (30-day prevention) |
All experiments follow the Institutional Animal Care and Use Committee protocols of the University of Illinois at Chicago. EFAD mice (31) were generated by crossing 5×FAD mice (48) and APOE-TR mice (49). 5×FAD mice express APP K670N/M671L + I716V + V717I and PS1 M146L + L286V under the control of the neuron-specific mouse Thy-1 promoter, and in APOE-TR mice, the coding region of the h-APOE gene replaces that of the m-APOE gene. Thus, EFAD mice are APOE-TR+/+/5×FAD+/−. It should be noted, however, that the introduction of the h-APOE into FAD-Tg mouse models delays Aβ accumulation (34, 50, 51). Although the 5×FAD mice produce very high levels of primarily Aβ42 and exhibit significant amyloid plaque deposition by 6–8 weeks, introduction of APOE4 (E4FAD) delays Aβ accumulation ∼4 months, and APOE3 (E3FAD) and APOE2 (E2FAD) delay deposition ∼6 months (31, 52).
Male E3FAD (APOE3) and E4FAD (APOE4) mice were treated with Bex (100 mg/kg/day), LG100268 (LG, 104.3 mg/kg/day equimolar to Bex), or vehicle control (VC) using three treatment protocols as follows: T1, 7-day oral gavage (5.75–6M); T2, 7-day hydrogel (5.75–6M); and T3, 30-day hydrogel (5–6M). For gavage treatment, Bex (20 mg/ml) or LG (20.86 mg/ml) were suspended in sesame oil (Sigma), and mice were treated with 5 μl/g. For hydrogel treatment, Bex or LG were dissolved in DMSO and mixed in hydrogel (0.4% DMSO in hydrogel), so that a mouse (30 g) consumed 4.5 ml of hydrogel per day, containing 3 mg of Bex or 3.13 mg of LG. Mice were acclimatized to consume hydrogel (Clear H20 Inc., Portland, ME) in place of drinking water for 3 days, and hydrogel consumption was routinely monitored before and after drug treatment (53). Note: sample processing and analysis were conducted by investigators blinded to APOE genotype and drug treatment.
Pharmacokinetics
Drug delivery was identical to that used for EFAD mouse studies. 3-Month-old male C57BL/6 mice (Charles River, Wilmington, MA) were treated with Bex (100 mg/kg) or equimolar LG (104.3 mg/kg) by gavage or hydrogel. At various time points between 0 and 960 min for gavage (Fig. 1, A and B) and at 1, 4, and 24 h for hydrogel treatment (Fig. 1C), mice were sacrificed using CO2; plasma was immediately drawn from the dorsal aorta, and mice were intracardially perfused, decapitated, and brain hemispheres flash-frozen. Levels of Bex and LG in the plasma and brain tissues of were measured by LC-MS/MS analysis. Working internal standard solutions were prepared by serially diluting DMSO stock solutions of Bex or LG (10 mm) in MeCN to 10–2000 nm. Working internal standard solutions were freshly prepared in MeCN from DMSO stocks to a working concentration of 50 nm Bex and 12.5 nm LG. Bex was used as the internal standard for LG and vice versa. Analysis was carried out on commercial C57BL/6 blank plasma and perfused brain (BioreclamationIVT, New York) for calibration and quality control. Blank plasma samples (45 μl) were transferred into low retention Eppendorf tubes stored on ice, mixed with 5 μl of standard solution, and 200 μl of ice-cold internal standard solution in MeCN. Blank perfused brains were homogenized in 3 volumes of 30% MeOH using a disposable pestle. Homogenized samples (145 μl) were mixed with 5 μl of standard solution and treated with ice-cold internal standard solution in MeCN (600 μl). All samples were centrifuged at 13,000 × g for 10 min at 4 °C. The supernatant was removed, dried with N2, and reconstituted with 30% MeOH (100 μl). After centrifuging at 13,000 × g for 10 min at 4 °C, an aliquot of 25 μl was analyzed by LC-MS/MS. Analysis of plasma (50 μl) and brain samples (150 μl) from treated animals followed the same procedures. 30-day PK hydrogel treatment measurements were made on E3FAD and E4FAD mice using plasma samples obtained at sacrifice. Samples were analyzed on a QTRAP 5500 system equipped with an Agilent 1260 UHPLC, using an Acquity BEH C18 column (3.0 × 100 mm, 1.7 μm, at 40 °C) with a flow rate of 0.45 ml/min (mobile phase 50% B to 100% B over 8.5 min: A = 30% MeOH containing 5 mm NH4HCO2; B = 95% MeCN containing 5 mm NH4HCO2). Measurements were made in the negative ion multiple reaction-monitoring mode at m/z 348.48 for Bex and 363.49 for LG.
FIGURE 1.
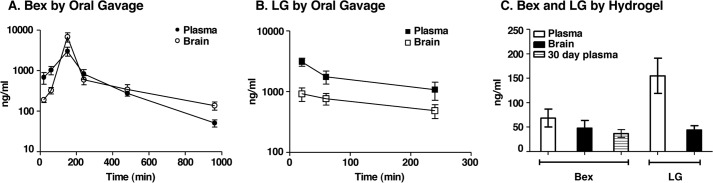
PK analysis of oral Bex and LG. Brain and plasma levels of Bex (100 mg/kg/day) or equimolar LG (104 mg/kg/day) delivered orally to mice were measured by LC-MS/MS. A, concentrations of Bex in brain and plasma at various time points after delivery by gavage to C57BL/6 mice. B, concentrations of LG in brain and plasma at various time points after delivery by gavage to C57BL/6 mice. C, averaged concentrations of Bex and LG in brain and plasma at 1, 4, and 24 h during a 24-h delivery by hydrogel to C57BL/6 mice (for 300day plasma samples) or to E3FAD and E4FAD mice. Data are expressed as mean ± S.E. (n = 4–11).
Tissue Harvest
Brain and liver tissue was harvested as described previously (31). EFAD mice were anesthetized with sodium pentobarbital (50 mg/kg) and transcardially perfused with ice-cold PBS containing protease inhibitors (Calbiochem, set 3). Right hemi-brains were dissected on ice into CX, HP, and cerebellum, snap-frozen in liquid nitrogen, and stored at −80 °C until use.
Protein Extraction
Serial extraction of soluble detergent and insoluble proteins was performed as described previously (31, 52). Briefly, the dissected brains were homogenized in 15 volumes (w/v) of Tris-buffered saline (TBS) and centrifuged (100,000 × g, 1 h at 4 °C), and the TBS-soluble extract was frozen in liquid nitrogen and stored at −80 °C. The pellet was washed in TBS, resuspended in 15 volumes of TBS containing 1% Triton X-100 (TBSX), mixed by rotation at 4 °C for 30 min, and centrifuged (100,000 × g, 1 h at 4 °C), and the TBSX-soluble fraction was stored as for TBS. Pellets were washed with TBSX, weighed, resuspended to 150 mg/ml in FA, and rotated end over end for 2 h at room temperature. Finally, samples were centrifuged (100,000 × g, 1 h at 4 °C), and the supernatant was neutralized using 20 volumes of Tris base and stored as for TBS/TBSX. Total protein was measured in the TBS and FA (QuickStartTM Bradford assay, Bio-Rad) and TBSX (Micro BCA protein assay kit, Pierce) fractions.
ELISAs
ApoE and Aβ42 levels were measured in the TBS, TBSX, and FA extracts, and oAβ and apoE/Aβ complex in the TBS extracts by ELISA. ApoE ELISA utilizes α-apoE (1:2000, Millipore) capture and α-apoE biotin-gt (1:5000, Meridian) detection antibodies as described previously (52). Aβ42 levels were measured using ELISA kits from Invitrogen (KHB3441). oAβ levels were measured using MOAB-2 based ELISA kits from Biosensis (BEK-2215-1P). ApoE/Aβ complex levels were measured based on a modified protocol described in (52). The new protocol followed the oAβ ELISA kit protocol (Biosensis) but used the α-apoE biotin-gt (1:5000, Meridian) detection antibody in place of the biotin-MOAB-2 antibody, with standards as prepared in Ref. 52. All data were normalized to total protein in each extract.
mRNA Analysis
1 ml of RNAlater solution (Ambion) was added to ∼20 mg of brain tissue, incubated at 4 °C overnight, and stored at −80 °C to ensure minimal mRNA degradation. Quantitative analysis of mRNAs was performed by the QuantiGene 2.0 Plex assay system (Affymetrix) for LXRα/β and PPARγ expression. The raw output is “mean fluorescence intensity” for each assay. Mean fluorescence intensity values are adjusted by blank value subtraction, and standardized with appropriate endogenous control gene expression value. Data are analyzed with MasterPlex®EX software (Hitachi-Solution America).
Western Blot
Gel electrophoresis and Western blot analyses were performed on 30 μg of TBSX-extracted proteins according to the manufacturer's protocols (Invitrogen). Briefly, samples in lithium dodecyl sulfate sample buffer (Invitrogen) were heated to 90 °C for 5 min and loaded onto 4–12% BisTris NuPAGE gels, electrophoresed using MOPS running buffer, and transferred to 0.2 μm PVDF membranes (Invitrogen). Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline containing 0.0625% Tween 20 (TBST) and incubated in primary antibodies followed by HRP-conjugated secondary antibodies (1:5000 dilution of 0.8 mg/ml stocks, Jackson ImmunoResearch). The following primary antibodies were used: ABCA1 (1:500, Novus Biologicals); ABCG1 (1:500, Novus Biologicals); PSD95 (1:500, Antibodies Inc.); and β-actin (1:2000, Sigma). Immunoreactivity was detected using enhanced chemiluminescence (ECL; Amersham Biosciences) and imaged on a Kodak Image Station 4000R. Protein expression was quantified by absorbance of protein/absorbance of actin.
All samples from control and RXR-treated mice within a brain region, for example E4FAD-HP, were run on the same Western blot to allow for appropriate comparison. In addition, to enable a comparison across brain regions, all the samples for the VC groups within a treatment protocol were run on a single blot, for example the VC groups for E3FAD-CX, E4FAD-CX, E3FAD-HP, and E4FAD-HP of T1. For the representative blots presented in the figures, all the lanes originate from the same gel but from different areas, i.e. the lanes were not immediately adjacent to each other. This re-organization corresponds to the specific results of a given figure.
Statistical Analysis
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey's post hoc analysis using GraphPad Prism version 5 for Macintosh. p < 0.05 was considered significant. Correlation analysis was conducted using Pearson's correlation.
RESULTS
Study Design and Pharmacokinetics
RXR Agonist Treatment Protocols of EFAD Mice
To determine the interactions between h-APOE and RXR agonists, we used three treatment protocols (see Table 1). In all studies, male E3FAD and E4FAD mice (31) were orally delivered Bex (100 mg/kg/day) or the RXR agonist LG (104.3 mg/kg/day, equimolar to Bex). T1 short-term (7 day) treatment was from 5.75 to 6 months by oral gavage. For T1, the dosing, method of oral delivery, and 7-day treatment protocol match that previously reported for Bex in FAD-Tg mice (13–16). Because Bex is almost insoluble in water, gavage must be delivered as a sesame oil slurry. For this reason and the reported toxicity of Bex in C57BL/6 mice (maximum tolerated dose ∼80 mg/kg/day) (54), we also chose an alternative mode of oral drug delivery. T2 short-term (7 days) treatment was from 5.75 to 6 months by hydrogel. For water-insoluble drugs, dissolution in DMSO and delivery in hydrogel is a viable alternative, with hydrogel replacing drinking water (53, 55). This short-term study was first completed to establish comparable results to drug delivery by gavage, thus informing protocol three. T3 medium-term (30 days) prevention protocol was from 5 to 6 months by hydrogel. The goal of T3 was to dissect the effect of RXR agonists in a prevention protocol.
As described under “Experimental Procedures,” proteins were sequentially extracted into soluble (TBS), detergent-soluble (TBS containing 1% Triton X-100 (TBSX)), and insoluble (FA) fractions (31, 52) from the dissected cortex (CX) and hippocampus (HP) of EFAD mice. Markers of indirect target engagement (ABCA1/G1 and total apoE levels), mechanistic PD (soluble apoE/Aβ complex levels, TBSX-apoE for lipoprotein association/lipidation), PD for Aβ (total Aβ42, soluble Aβ42, and soluble oAβ), and efficacy (synaptic viability) were assessed.
Pharmacokinetics (PK)
Bex and LG have brain bioavailability. Initially, the plasma PK of Bex and LG was determined. In male C57BL/6 mice, after gavage delivery of Bex (100 mg/kg), the plasma values we observed were Cmax ∼3000 ng/ml (8.6 μm) and Tmax ∼2 h (Fig. 1A). Similar values were reported for rats after bolus oral delivery of Bex, specifically Tmax ∼2 h at 100 mg/kg/day p.o. (56). In a female Tg mouse strain that is susceptible to mammary cancer (C3(1)-SV40 Tag mice), gavage delivery in sesame oil, similar to our procedure, gave reported values for plasma of Cmax ∼20,000 ng/ml (58 μm) and Tmax ∼0.5 h (57). After equimolar gavage delivery, the half-life for LG in plasma was shorter compared with Bex but with a similar Cmax (Fig. 1, A and B). Importantly, both were brain bioavailable with an area under the curve (brain/plasma) of 1.46 and 0.41 and a brain Cmax of ∼6700 ng/ml (19.4 μm) and 920 ng/ml (2.53 μm) for Bex and LG, respectively. For calibration of PK with PD, the reported cellular EC50 values for Bex and LG for RXR activation are 20–55 and 15–25 nm, respectively (40, 41).
Bex delivery to rodents has been primarily via feed an intermittent, continuous drug delivery similar to hydrogel. To estimate PK after hydrogel delivery, drug levels in plasma and brain were measured at 1, 4, and 24 h, and brain concentrations for Bex and LG were ∼50 ng/ml (140 nm) and ∼45 ng/ml (125 nm), respectively (Fig. 1C). After a 30-day delivery of Bex to EFAD mice by hydrogel, plasma concentrations were not significantly different by 7-day hydrogel (Fig. 1C). Thus, with both gavage and hydrogel, the concentrations of Bex and LG in the brain were at least 3-fold higher than the in vitro IC50 (40, 41), suggesting full agonism.
Short-term RXR Agonist Treatment Induces Beneficial Effects in E4FAD-HP (T1 and T2)
Initially, the effect of short-term (5.75–6M) gavage (T1) or hydrogel (T2) Bex and LG treatment was determined in the E4FAD-HP, the brain region with the highest Aβ pathology at the time of treatment compared with E3FAD/E4FAD-CX or E3FAD-HP (31). Indeed, the E4FAD-HP has the highest levels of insoluble Aβ42 and soluble Aβ (Aβ42 and oAβ), the lowest levels of apoE/Aβ complex, and the lowest lipoprotein-associated/lipidated apoE compared with other brain regions in E4FAD and E3FAD mice at 6 months (see other brain regions under “Results” and Refs. 31, 52). Focusing on the E4FAD-HP allowed testing of the hypothesis that RXR agonists will address a loss-of-function associated with apoE4 in treatment protocols (Fig. 2).
FIGURE 2.

Short-term RXR agonist treatment induces beneficial effects in E4FAD-HP. E4FAD mice were treated with Bex (100 mg/kg/day), LG (104 mg/kg/day, equimolar to Bex (B)) or vehicle control (VC) from 5.75 to 6 months by daily gavage (T1) or 7-day hydrogel (T2). Proteins were sequentially extracted from the E4FAD-HP using TBS, TBSX, and FA. Markers of indirect target engagement including the following: A, ABCA1 (WB and TBSX); B, ABCG1 (WB and TBSX); and C, total apoE levels (ELISA, TBS + TBSX + FA). The mechanistic pharmacodynamics include the following: D and E, apoE extraction profile (ELISA); F, soluble apoE/Aβ complex (ELISA, TBS, and VC levels = 73.4 ± 6.8 nm Aβ42/mg protein). The pharmacodynamics for Aβ include the following: G, total Aβ42 (ELISA, TBS + TBSX + FA, and VC levels = 3.6 ± 0.3 μg/mg protein); H, soluble Aβ42 (ELISA, TBS, and VC levels = 365.1 ± 51.3 pg/mg protein); and I, soluble oAβ (ELISA, TBS, and VC levels = 4.58 ± 0.5 ng/mg protein), and for efficacy: J, PSD95 (WB and, TBSX) was measured. Data are expressed as the mean ± S.E. and were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. n = 6. *, p < 0.05 versus VC.
RXR Agonists Increase ABCA1/ABCG1 but Not Total ApoE Levels in E4FAD-HP
Although Bex and LG were brain-penetrant with gavage and hydrogel delivery, it is important to demonstrate target engagement to interpret PD and efficacy data. Data are conflicted whether RXR agonists function through increasing the levels of ABCA1/ABCG1 or total apoE (13–16). Therefore, ABCA1 and ABCG1 and total apoE levels were assessed as markers of indirect target engagement after RXR agonist treatment. Both RXR agonists increased ABCA1 levels by gavage (T1) and hydrogel (T2) treatment (Fig. 2A). In T1, compared with VC, Bex and LG increased ABCA1 levels by ∼200 and 300%, respectively, as assessed by Western blot (WB) analysis (TBSX extract). Similar increases were also observed in T2 with increases in ABCA1 levels of ∼300% with Bex and 100% with LG compared with VC. Both RXR agonists also increased ABCG1 levels in T1, but not in T2 (Fig. 2B). There were no changes in total apoE levels for T1 or T2 by Bex or LG (Fig. 2C) when assessed by ELISA (total apoE = sum of apoE levels in the TBS, TBSX, and FA extracts divided by total protein).
RXR Agonists Increase Detergent-soluble/Lipoprotein-associated ApoE in E4FAD-HP
A previously developed three-step sequential protein extraction protocol (TBS, TBSX, and FA) was used to determine the effect of APOE genotype on the levels of lipoprotein-associated apoE (31, 52) and was used to homogenize the EFAD mice tissue in this study. TBSX employs a nonionic detergent to release apoE from lipoproteins without inducing the formation of new micelles, as can occur with ionic detergents such as SDS (31, 58), and thus it indicates the levels of lipoprotein-associated apoE. In EFAD mouse brain homogenates, total brain apoE levels are lower with APOE4, but this difference is seen only in the TBSX fraction, indicating that apoE4-containing lipoproteins are less lipoprotein-associated and/or less lipidated (31). As RXR agonists increased ABCA1 and ABCG1 levels, apoE levels were plotted as extraction profiles (Fig. 2, D and E). Bex and LG increased TBSX-apoE4 levels in the E4FAD-HP by ∼80% by gavage (T1) (Fig. 2D) and by ∼140% using hydrogel (T2) (Fig. 2E) compared with the VC. In contrast, TBS-soluble apoE4 levels were decreased by RXR treatment by ∼40% in both T1 and T2. Thus, although total apoE levels were not affected by RXR agonist treatment, soluble (TBS) apoE levels were decreased, and detergent-extracted apoE4 levels were increased. These data support that RXR agonists increase the lipoprotein association of apoE4.
RXR Agonists Increase ApoE/Aβ Complex Levels in E4FAD-HP
A potential consequence of RXR agonist-induced increased apoE4 lipoprotein association is increased apoE/Aβ complex levels (33, 52). To determine the effect of APOE genotype on apoE/Aβ complex levels, we previously developed an apoE/Aβ complex ELISA and demonstrated that apoE/Aβ complex levels are lower with AD and APOE4 in human CSF and with APOE4 in the TBS extracts of EFAD mice (33, 52). In this study, both RXR agonists increased apoE/Aβ complex levels by gavage (T1) and hydrogel (T2) by ∼70 and 100%, respectively, compared with VC when assessed by ELISA (Fig. 2F). Thus, short-term RXR agonist treatment increased ABCA1 levels, apoE4 lipoprotein association, and apoE/Aβ complex levels in the E4FAD-HP.
RXR Agonists Do Not Change Total Aβ42 Levels but Decrease Soluble Aβ (Aβ42 and oAβ) in E4FAD-HP
Currently, there is debate on the effect of RXR agonists on the levels of total Aβ42, measured by immunohistochemistry or biochemically. Initial data demonstrated that Bex reduced total Aβ by ∼60% in APP/PS1 mice after a 7-day gavage treatment (11), although subsequent studies failed to replicate these changes in a number of mouse models (13–16). In this study, total Aβ42 (TBS + TBSX + FA) levels were unchanged by RXR agonist treatment in both short-term treatment protocols (Fig. 2G), consistent with the latter publications with Bex in FAD-Tg mice (13–16).
Accumulating evidence over the last decade suggests that soluble Aβ42 (2–4) and soluble oAβ (5–8), rather than insoluble Aβ, may be the proximal neurotoxin in AD. By ELISA, oAβ levels are increased with APOE4 and AD in human CSF, and both soluble Aβ42 and soluble oAβ levels are higher in E4FAD mice compared with E3FAD mice (31, 33). Furthermore, although data are varied, Bex has previously been demonstrated to lower soluble Aβ in some FAD-Tg models (13–16) and in one case oAβ (13). Thus, soluble Aβ (Aβ42 and oAβ) represents an important PD read-out after RXR agonist treatment. In this study, soluble (TBS) Aβ42 levels were reduced in E4FAD-HP with RXR agonist treatment. Specifically, compared with the VC, Bex and LG lowered soluble Aβ42 levels by ∼52% by oral gavage (T1) and although not significantly by ∼23% by hydrogel (T2) (Fig. 2H). Importantly, soluble oAβ levels were also reduced with RXR agonist treatment. In the T1 study, Bex and LG decreased soluble oAβ by ∼35 and ∼49%, respectively, and in T2 both RXR agonists reduced oAβ levels by ∼32% by hydrogel (Fig. 2I).
RXR Agonists Increase PSD95 Levels in E4FAD-HP
An important read-out for the preclinical analysis of a potential AD therapeutic is a measure of neuronal viability. Increased Aβ levels are linked to reduced postsynaptic density protein 95 (PSD95) levels in humans and FAD transgenic mouse models (59–62). Thus, in this study we utilized PSD95 levels as a marker of drug efficacy. RXR agonist treatment increased PSD95 levels in E4FAD-HP by ∼160% with oral gavage (T1) and ∼60% by hydrogel treatment (T2) (Fig. 2J).
Therefore, both RXR agonists in short-term treatment protocols exhibit beneficial effects in the E4FAD-HP through increasing ABCA1/G1 levels, apoE4 lipoprotein-association, and apoE/Aβ complex levels, which result in decreased soluble Aβ (Aβ42 and oAβ) and increased PSD95 levels.
Medium-term RXR Agonist Prevention Protocol Induces No Effects on ApoE or Aβ in E4FAD-HP (T3)
To determine whether RXR agonists could prevent increases in Aβ pathology and decreases in PSD95 levels in the E4FAD-HP, a medium-term prevention study was conducted ending at 6 months (Fig. 3). E4FAD mice were treated with both Bex and LG from 5 to 6 months by hydrogel rather than gavage, as hydrogel represents less stress to the animal than repeated daily gavage and induced comparable beneficial PD and efficacy effects in short-term treatment in the E4FAD-HP. Bex and LG both increased ABCA1 (but not ABCG1) levels by 70% (Fig. 3A). However, despite demonstrating indirect target engagement, neither Bex nor LG exerted any changes on markers for mechanistic PD (TBSX-apoE and apoE/Aβ complex), PD (insoluble and soluble Aβ), or efficacy (PSD95) (Fig. 3, B–H). Thus, the beneficial effects of RXR agonists observed in E4FAD-HP in short-term treatment protocols (from 5.75 to 6 months) are not replicated in a prevention protocol when E4FAD mice are treated at an earlier stage of Aβ pathology, but still ending at 6 months.
FIGURE 3.

Medium-term RXR agonist prevention protocol induces no effects on apoE or Aβ in E4FAD-HP. E4FAD mice were treated with Bex (100 mg/kg/day), LG (104 mg/kg/day), or VC from 5 to 6 months by hydrogel (T3). In the E4FAD-HP, markers of indirect target engagement include the following: A, ABCA1 (WB and TBSX), ABCG1 (WB and TBSX); B, total apoE levels (ELISA, TBS + TBSX + FA). The mechanistic pharmacodynamics include: C, TBSX-apoE (ELISA); D, soluble apoE/Aβ complex (ELISA, TBS). Pharmacodynamics for Aβ include the following: E, total Aβ42 (ELISA, TBS + TBSX + FA); F, soluble Aβ42 (ELISA and TBS); and G, soluble oAβ (ELISA and TBS), and for efficacy, H, PSD95 (WB and TBSX) was measured. Data were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. n = 6. *, p < 0.05 versus VC.
Short-term RXR Agonist Treatment Increases Soluble Aβ (Aβ42 and oAβ) Levels in E3FAD-CX and E4FAD-CX (T1 and T2)
As data were conflicted between the treatment and prevention protocols in E4FAD-HP, the effect of short-term RXR agonist treatment in brain regions with lower levels of Aβ pathology at the time of treatment was dissected (i.e. E3FAD-CX, E4FAD-CX, E3FAD-HP). Readouts of indirect target engagement, mechanistic PD, PD for Aβ, and efficacy in the VC group are presented first, as these are an indication of the levels at the time of treatment. The effects of RXR agonists are plotted for a comparison (Figs. 4 and 5 represent T1 and, as data were similar, T2 data is not shown).
FIGURE 4.

Vehicle control (left) and short-term RXR agonist treatment (right) effects on ABCA1 levels, apoE lipoprotein association, and apoE/Aβ complex levels in E3FAD-CX, E4FAD-CX, and E3FAD-HP. EFAD mice were treated with Bex (100 mg/kg/day), LG (104 mg/kg/day, equimolar to Bex), or vehicle control (VC) from 5.75 to 6 months by daily gavage (T1). In E3FAD-CX, E4FAD-CX, and E3FAD-HP, markers of indirect target engagement include the following: A and B, ABCA1 (WB and TBSX). Mechanistic pharmacodynamics include the following: C and D, TBSX-apoE (ELISA); and E and F, soluble apoE/Aβ complex (ELISA, TBS) were measured. Note: RXR agonists induce no beneficial effects in E3FAD-CX or E4FAD-CX. Data were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. n = 6. *, p < 0.05 (as shown, left); *, p < 0.05 versus VC (right); #, p < 0.05 Bex versus LG (right).
FIGURE 5.

Vehicle control (left) and short-term RXR agonist treatment (right) effects on total Aβ42, soluble Aβ (Aβ42 and oAβ), and PSD95 levels in E3FAD-CX, E4FAD-CX, and E3FAD-HP. EFAD mice were treated with Bex (100 mg/kg/day), LG (104 mg/kg/day, equimolar to Bex), or VC from 5.75 to 6 months by daily gavage (T1). In E3FAD-CX, E4FAD-CX, and E3FAD-HP markers of pharmacodynamics for Aβ include the following: A and B, total Aβ42 (ELISA, TBS + TBSX + FA); C and D, soluble Aβ42 (ELISA, TBS); and E and F, soluble oAβ (ELISA, TBS); and for efficacy, G and H, PSD95 (WB and TBSX) was measured. Note: RXR agonists induce an increase in soluble Aβ (Aβ42 and oAβ) in E3FAD-CX and E4FAD-CX. Data were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. n = 6. *, p < 0.05 (as shown, left); *, p < 0.05 versus VC (right); #, p < 0.05 Bex (B) versus LG (right).
RXR Agonists Elicit No Effects on ABCA1 Levels, ApoE Lipoprotein Association, or ApoE/Aβ Complex Levels in E3FAD-CX and E4FAD-CX
ABCA1
Generally, in the VC groups, ABCA1 (Fig. 4A) and ABCG1 (data not shown) levels are lower in E4FAD-HP compared with other brain regions. For example, in the VC group gavage-treated mice (T1), ABCA1 levels were significantly different for E4FAD-HP versus E3FAD-CX and E4FAD-CX. RXR agonists induced no effect on ABCA1 or ABCG1 levels in the E3FAD-CX or E4FAD-CX of T1 (Fig. 4B) or by hydrogel (T2) (except increase with LG in E3FAD-CX, data not shown). However, both agonists increased ABCA1 levels by 100–200% in E3FAD-HP.
TBSX-ApoE
TBSX-apoE levels were lowest in E4FAD-HP (Fig. 4C). RXR agonists did not significantly modulate the levels of TBSX-apoE in the E3FAD-CX, E4FAD-CX, or E3FAD-HP (Fig. 4D). The only exceptions were an increase in TBSX-apoE levels in LG-treated E4FAD-CX (Fig. 4D) and a decrease in Bex-treated E3FAD-HP in T2 (data not shown).
ApoE/Aβ Complex
Consistent with published data, apoE/Aβ complex levels in the VC groups of both treatments were lower in E4FAD compared with E3FAD mice in both the CX and HP (Fig. 4E) (52). LG and Bex did not change apoE/Aβ complex levels in the E3FAD-CX or E4FAD-CX. In E3FAD-HP (Fig. 4F), LG decreased apoE/Aβ complex levels in T1, and Bex decreased apoE/Aβ complex levels in T2 (data not shown).
RXR Agonists Induce an Increase in Soluble Aβ (Aβ42 and oAβ) in the E3FAD-CX and E4FAD-CX
Total Aβ42
Consistent with previous data, total Aβ42 levels in the VC followed the order E4FAD-HP > E3FAD-HP > E4FAD-CX ≥ E3FAD-CX (Fig. 5A). Gavage (T1) and hydrogel (T2) treatment with Bex did not affect total Aβ42 levels in the CX or HP of EFAD mice (Fig. 5B). LG increased total Aβ42 levels in the E3FAD-HP of T1-treated mice and in E3FAD-CX of T2-treated mice.
Soluble Aβ42
As demonstrated previously in the VC groups, soluble Aβ42 levels followed the order E4FAD-HP > E3FAD-HP > E4FAD-CX ≥ E3FAD-CX (Fig. 5C). RXR agonist treatment increased soluble Aβ42 levels by ∼50% in the E3FAD-CX and E4FAD-CX (Fig. 5D). These data were significant for Bex and LG in the E4FAD-CX in T1, and LG in E3FAD-CX for T1 and T2. In E3FAD-HP, Bex increased soluble Aβ42 by 100% following both short-term treatments.
Soluble oAβ
oAβ levels in the VC group followed the order E4FAD-HP > E3FAD-HP > E4FAD-CX > E3FAD-CX (Fig. 5E). Both RXR agonists increased oAβ levels in E3FAD-CX and E4FAD-CX (Fig. 5F). For example, in T1, Bex increased oAβ levels by 100% in E3FAD-CX and 150% in E4FAD-CX compared with VC. In the E3FAD-HP, there were no effects on oAβ levels.
Although Nonsignificant, RXR Agonist Treatment Trends to Lower PSD95 Levels in E4FAD-CX
In the VC groups, PSD95 levels followed the pattern E3FAD-CX > E4FAD-CX > E3FAD-HP > E4FAD-HP (Fig. 5G). A nonsignificant trend was observed in E4FAD-CX for decreased PSD95 levels with RXR agonist treatment. Indeed, PSD95 levels were decreased by ∼27% by both RXR agonists in T1 (p = 0.07, Fig. 5H).
Collectively, these data demonstrate that short-term RXR agonist treatment in the CX of E3FAD and E4FAD mice does not significantly change ABCA1, TBSX-apoE, or apoE/Aβ complex levels, and in the E3FAD-HP it increases ABCA1 levels without significant increases in apoE lipoprotein association. Importantly, in the E3FAD-CX and E4FAD-CX, short-term RXR agonist treatment increases soluble Aβ levels (Aβ42 and oAβ), which tends to be associated with decreased PSD95 levels in the E4FAD-CX.
Medium-term RXR Agonist Prevention Protocol Induces No Effects on ApoE or Aβ in E3FAD-CX, E4FAD-CX, and E3FAD-HP (T3)
As RXR agonists increased oAβ levels in E3FAD-CX and E4FAD-CX and had no effect in the E3FAD-HP, as for the E4FAD-HP, the effect of a medium-term prevention protocol was dissected in these regions (5–6 months, Fig. 6). LG and Bex increased levels of ABCA1 (Fig. 6A), but not ABCG1 (Fig. 6B), in all brain regions (HP and CX) for all genotypes (APOE3 and APOE4) in EFAD mice (with exception of Bex treated E4FAD-CX). However, in general, there were no changes in TBSX-apoE levels or apoE/Aβ complex levels for any of the brain regions (Fig. 6, C and D, exceptions include Bex-induced decrease in TBSX-apoE in E3FAD-CX and LG-dependent decreased apoE/Aβ complex in E3FAD-HP). In addition, there were no effects of RXR agonist treatment on total Aβ or soluble Aβ (Aβ42 and oAβ) levels in the medium-term prevention protocol (Fig. 6, E–G). For PSD95, RXR agonists decreased levels by ∼15% in the E4FAD-CX but elicited no other effects (Fig. 6H). Thus, in a medium-term prevention study, in which treatment was initiated in brain regions with the lowest Aβ pathology at the time of treatment (lower than the short-term studies described above), despite increasing ABCA1 levels, no beneficial effects on any readouts for PD or efficacy were observed.
FIGURE 6.

Medium-term RXR agonist prevention protocol induces no effects on apoE or Aβ in E3FAD-CX, E4FAD-CX, and E3FAD-HP. EFAD mice were treated with Bex (B) (100 mg/kg/day), LG (104 mg/kg/day), or VC from 5 to 6 months by hydrogel (T3). In E3FAD-CX, E4FAD-CX, and E3FAD-HP, markers of indirect target engagement include the following: A, ABCA1 (WB and TBSX) and B, ABCG1 (WB and TBSX). The mechanistic pharmacodynamics include the following: C, TBSX-apoE (ELISA) and D, soluble apoE/Aβ complex (ELISA, TBS). The pharmacodynamics for Aβ include the following: E, total Aβ42 (ELISA, TBS + TBSX + FA); F, soluble Aβ42 (ELISA and TBS), and G, soluble oAβ (ELISA and TBS); and the efficacy, H, PSD95 (WB was TBSX) was measured. Data were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. n = 6. *, p < 0.05 versus VC.
Collective Data Analysis and Interpretation, Beneficial Effects of RXR Agonists
Increasing Aβ Levels Are Associated with Decreased Levels of ApoE Lipidation-related Genes and Proteins, Potentiated by APOE4
Our data raised the following important question. Why did Bex and LG induce beneficial effects on apoE lipoprotein association only in short-term studies in the E4FAD-HP? Critical to understanding this issue is that in 6-month EFAD mice, Aβ accumulation, including insoluble Aβ, soluble Aβ42, and soluble oAβ, are from highest to lowest as follows: E4FAD-HP > E3FAD-HP > E4FAD-CX > E3FAD-CX (Fig. 5) (31, 52). In contrast, ABCA1, ABCG1 levels, and mRNA levels of the permissive nuclear receptor heterodimer targets of RXR, namely LXRα,β and PPARγ, followed the order E3FAD-CX > E4FAD-CX > E3FAD-HP > E4FAD-HP (Fig. 7A, data from VC groups T1 and T2). Thus, the levels of apoE lipidation-related pathway genes and proteins are all decreased with increased Aβ pathology, an effect that is potentiated by APOE4. These effects may underlie the progressive decrease in lipoprotein-associated apoE with increased Aβ pathology, which is therefore the lowest in the E4FAD-HP at 6 months (Fig. 7A).
FIGURE 7.

Collective analysis, beneficial effects of RXR agonist treatment. A, protein levels of ABCA1, ABCG1, and TBSX-apoE, as well as the expression (mRNA) of LXRα, LXRβ, and PPARγ plotted across brain region as % E3FAD-CX for the VC control groups of T1 and T2. B, levels of ABCA1 and TBSX-apoE in the VC and RXR agonist treatment groups, Bex and LG combined (RXR), plotted across brain region as % E3FAD-CX. C, levels of oAβ (ng/mg protein) in the VC and RXR agonist treatment groups, Bex and LG combined (RXR), plotted across brain region. n = 12. *, p < 0.05 versus VC.
RXR Agonists May Increase Lipoprotein-associated ApoE Only at a Critically Low Threshold
Through analyzing the effect of RXR agonists in the context of the continuum of Aβ accumulation, ABCA1 levels are increased in the E3FAD-HP and E4FAD-HP; however, TBSX-apoE levels are only increased in the E4FAD-HP (Fig. 7B). These data are important as follows: 1) ABCA1 levels may only be increased once the RXR targets that increase ABCA1 levels reach a critical low threshold, equivalent to E3FAD-HP; and 2) a similar “critically low threshold” of apoE lipoprotein association may also exist, equivalent to the E4FAD-HP. Once this critically low threshold is attained, RXR agonists can increase ABCA1-dependent apoE lipoprotein association and lower levels of soluble Aβ. Indeed, it is striking that RXR agonist treatment lowers soluble oAβ levels in the E4FAD-HP to those of E3FAD-HP (Fig. 7C).
Collective Data Analysis and Interpretation, Detrimental Effects of RXR Agonists
RXR Agonist Increased oAβ Levels in the Absence of Changes in ABCA1 Levels
A further issue raised by the data is that in the brain regions with the lowest Aβ pathology at the time of short-treatment (E3FAD-CX and E4FAD-CX), RXR agonists increased oAβ levels (Fig. 7C). Additional analysis demonstrated no significant correlation between ABCA1 and oAβ (Fig. 8A, Pearson's r = +0.05) in the CX of both E3FAD and E4FAD mice (T1 and T2), indicating that the increased oAβ levels in the CX of EFAD mice are not a result of changes in ABCA1 levels and, more broadly, CNS indirect target engagement. Thus, there is likely an alternative mechanism of action for the RXR agonist-induced increases in oAβ levels in the CX of EFAD mice.
FIGURE 8.

RXR agonists induce hepatomegaly in EFAD mice. A, correlation analysis between ABCA1 and oAβ levels in EFAD-CX. Liver weights were measured in E3FAD and E4FAD mice treated with Bex, LG, or VC by 7-day gavage (5.75–6 months) (B), 7-day hydrogel (5.75–6 months) (C), and 30-day hydrogel (5–6 months) (D). Data are expressed as the mean ± S.E. and were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. n = 6. *, p < 0.05 versus VC.
RXR Agonists Induced Hepatomegaly in EFAD Mice
Evidence indicates that Bex can increase liver weight (hepatomegaly), potentially via hepatic steatosis (14). Indeed, hepatomegaly is touted as an indirect indication of bioavailability and target engagement for Bex (14). Furthermore, it is unclear whether agonists that are more selective than Bex for RXR compared with RAR (i.e. LG) will negate the hepatomegaly. In this study, short-term treatment by gavage (T1) with Bex and LG increased liver weight in E3FAD and E4FAD mice by ∼80% (Fig. 8B). The 7-day RXR agonist treatment with hydrogel (T2) also increased liver weight in E3FAD mice; LG (∼140%) induced a greater increase than Bex (60%) (Fig. 8C). Interestingly, liver weight was not affected by RXR agonist hydrogel treatment in E4FAD mice (although LG increased liver weight nonsignificantly by ∼25%). However, medium-term prevention (T3) with either RXR agonist resulted in an ∼140 and ∼70% increase in liver weight for E3FAD and E4FAD mice, respectively (Fig. 8D). Thus, both short-term treatment and medium-term prevention protocols with RXR agonists result in an enlarged liver (hepatomegaly), a potentially negative systemic side effect. One potential interpretation is that for RXR agonists there is balance between the beneficial effects for increasing apoE lipoprotein association in the CNS versus negative effects in the liver, which may also impact CNS function (see “Discussion” below). In the absence of beneficial affects in the CNS (i.e. no critical low threshold of apoE lipoprotein association), systemic liver effects may induce a stress in the CNS that is observed here as increased oAβ levels, and in some cases with decreased PSD95 levels (e.g. E4FAD-CX in T1/2 and T3). These systemic effects may also confound interpretation of longer term RXR agonist treatment strategies, such as the lack of beneficial effects in the E4FAD-HP observed in our medium-term prevention study.
DISCUSSION
Developing therapeutics targeting processes that underlie AD progression is a major challenge of the 21st century. As APOE4 is the greatest AD genetic risk factor, and either decreased apoE4 levels or apoE4 lipoprotein association may result in increased Aβ levels, targeting apoE-containing lipoproteins, particularly apoE4, is an exciting therapeutic prospect. Furthermore, the repurposing of drugs to target AD-relevant pathways is considered an important approach to accelerate drug development in AD. Thus, the provocative paper by Cramer et al. (11) raised great hope in the field, as the anti-cancer drug Bex (an RXR agonist) was demonstrated to lower Aβ levels and improve behavior through m-apoE-dependent effects in multiple FAD-Tg mouse models. However, there is concern about the effects of Bex, and therefore RXR agonists, in the presence of h-APOE4, with subsequent reports producing conflicting results on both the activity and mechanism of action of Bex (11, 12, 14–16). Thus, key factors that may determine the translatability of RXR agonists for AD, including the effects of h-APOE and the level of Aβ pathology at time of treatment, were assessed in vivo.
The first important finding of this study is that both RXR agonists induced beneficial effects in short-term treatment protocols in the E4FAD-HP, the brain region with the highest Aβ levels and lowest apoE lipoprotein association at the time of treatment. In the E4FAD-HP, Bex and LG increased ABCA1/G1 levels, apoE4 lipoprotein association, and apoE4/Aβ complex levels, decreased soluble oAβ, and improved synaptic viability (Fig. 9). These data begin to directly address important issues for the use of RXR agonists as AD therapeutics.
FIGURE 9.

Summary hypothesis, RXR agonists induce beneficial effects in the E4FAD-HP. A, data from this study support the hypothesis that in the E4FAD-HP, the short-term RXR agonist treatment (T1 and T2) is as follows: 1, increases ABCA1/G1 levels; 2, increases apoE4 lipoprotein association/lipidation (as ABCA1 and TBSX-apoE levels are positively correlated); 3, increases apoE4/Aβ complex levels (as TBSX-apoE and apoE4/Aβ complex levels are positively correlated); 4, decreases soluble Aβ levels (as oAβ and apoE4/Aβ complex levels are inversely correlated); and 5, improves synaptic viability (as oAβ and PSD95 levels are inversely correlated). Thus, RXR agonists address a loss-of-function associated with APOE4 and exacerbated by Aβ, i.e. lower apoE4 lipoprotein association/lipidation. B, schematic representation of the proposed hypothesis.
RXR Agonist Mechanism of Action, ABCA1 Versus ApoE
Post-RXR agonist treatment, ABCA1 levels are inversely correlated with oAβ levels in the E4FAD-HP (data not shown, Pearson's r value = −0.51, p < 0.01). This ABCA1-associated reduction in soluble Aβ levels is likely by increased apoE4 lipoprotein association/lipidation and not increased total apoE levels. Indeed, ABCA1 and TBSX-apoE are positively correlated (Fig. 9A, parts 1 and 2, Pearson's r value = +0.67, p < 0.0001). These data are in agreement with the increased or reduced amyloid deposition with ABCA1-KO or overexpression in FAD-TG mice, respectively (18–24). Furthermore, in the context of m-apoE, NR agonists, including GW3965 (63) and Bex (13–16), appear to function via increasing the ABCA1-dependent lipidation of apoE (11).
ApoE Lipoprotein Association/Lipidation, ApoE/Aβ Complex Levels, and Soluble Aβ Clearance
Through increasing apoE lipoprotein association, RXR agonists increased apoE/Aβ complex levels, as they are positively correlated in the E4FAD-HP (Fig. 9A, parts 2 and 3, Pearson's r value = +0.42, p < 0.05). Our novel data begin to address the mechanism by which RXR agonist-dependent increased apoE4 lipoprotein association/lipidation may increase soluble Aβ clearance, i.e. the apoE/Aβ complex. In vivo, apoE isoform specifically modulates Aβ clearance (63–69), with soluble Aβ clearance lower with apoE4 compared with apoE3. Furthermore, NR agonists increase Aβ degradation in vitro in an ABCA1 and apoE-dependent manner (63). However, the exact process of how apoE4 decreases and how NR increases Aβ clearance is unclear. The levels of the soluble apoE/Aβ complex are lower and oAβ higher with AD and APOE4 in EFAD mice and human tissue (32). Furthermore, in this study, the apoE/Aβ complex and oAβ levels are inversely correlated in the E4FAD-HP (Fig. 9A, parts 3 and 4, Pearson's r value = −0.53, p < 0.001). Thus, increased levels of the apoE/Aβ complex may contribute to the increased clearance rate of soluble Aβ after RXR agonist treatment.
Soluble Versus Insoluble Aβ
The RXR agonist-induced beneficial effects on synaptic viability, as measured by increased PSD95 levels, are likely the result of decreased levels of soluble Aβ42 and oAβ levels, rather than total/insoluble Aβ. An important caution is that using PSD95 as a measure of efficacy for synaptic viability requires future validation that RXR agonist treatment correlates with improvements in functional and cognitive readouts. Further analysis of the E4FAD-HP demonstrated an inverse correlation between levels of oAβ and levels of PSD95 (Fig. 9A, parts 4 and 5, Pearson's r value = −0.61, p < 0.001). This is in agreement with the correlation of soluble Aβ and oAβ with cognitive decline and disease severity in humans (70), and the association of oAβ levels with memory decline in FAD-Tg mice (70). Furthermore, recent data demonstrate that transient soluble oAβ levels may underlie neuronal deficits in FAD-Tg mice using a genetic approach (71). However, as a dynamic equilibrium likely exists between soluble and insoluble Aβ levels (i.e. soluble Aβ may derive from insoluble Aβ and vice versa (72, 73)), further research is required to rule out the contribution of a particular Aβ form impairing neuronal viability.
A second important finding is that the levels of soluble Aβ (Aβ42 and oAβ) at the time of treatment influence the effect of RXR agonists on PD and efficacy readouts. In EFAD mice, increasing Aβ levels in the presence of APOE4 leads to decreased levels of apoE lipidation-related pathway genes and proteins, with the lowest levels in E4FAD-HP, the only brain region examined where RXR agonists improved PD and efficacy markers. Thus, RXR agonists may elicit beneficial effects only when apoE lipoprotein association/lipidation falls below some critical pathological threshold. The mechanism through which Aβ down-regulates the expression of NRs may involve decreased production of oxysterol ligands, positive modulators of NR expression. Overall, activation of the sterol regulatory element-binding protein 2 (SREBP2) results in the production of oxysterol ligands from cholesterol, activating NRs, including LXR and RXR, to increase ABCA1 expression (74). Recent data demonstrate that Aβ down-regulates SREBP-2 cleavage (75), supporting disruptions in lipid metabolism as a potential pathogenic process in AD (76). Thus, Aβ may lower RXR expression through modulation of lipid homeostasis.
As the proposed critically low pathological threshold of apoE lipoprotein association/lipidation is not reached in the E3FAD-CX or E4FAD-CX, RXR agonists would be expected to exert no effects on soluble Aβ. However, soluble Aβ levels actually increased with both Bex and LG in the CX of E3FAD and E4FAD mice, the brain regions with the lowest Aβ levels at the time of treatment, and the effects that may be the result of hepatomegaly, as described below. These results may facilitate interpretation of the variable results previously reported for Bex (11, 12, 14–16). Several FAD-Tg mouse models expressing m-apoE were used in previous studies, with widely variable Aβ levels at the time of treatment, and the level of m-apoE associated with lipoprotein-association was not measured and was difficult to predict.
Additional AD risk factors may also modulate the activity of the RXR agonists. Recent data demonstrate that in APOE-TR mice, Bex increases apoE4 lipidation to apoE3 levels (35). These data raise the intriguing possibility that apoE4-containing lipoproteins may be less able to modulate their lipoprotein-associated/lipidated state either at baseline or in response to stressors both Aβ-dependent and Aβ-independent, specifically during aging. An additional interaction that modifies AD risk is sex and APOE genotype. Clinical data indicate that the APOE4-induced risk for AD is significantly greater, perhaps exclusive to, females compared with males (77–79). Mechanistically, APOE4 may increase AD risk in women via multiple pathways involving the cardiovascular system (80), cellular aging (81), and importantly, Aβ accumulation. Indeed, we previously demonstrated that insoluble Aβ is increased in female E4FAD mice compared with female E3FAD mice (82). Thus, although male mice were the focus of this study, one could speculate that RXR agonists may be particularly efficacious in E4FAD female mice.
Although repurposing drugs for AD is an attractive alternative, there are important considerations when adapting drugs for use in the CNS that were initially developed for peripheral indications, particularly anti-cancer drugs and specifically Bex. The major side effects associated with Bex include severe but reversible hypertriglyceridemia, central hypothyroidism, pancreatitis, and importantly hepatomegaly (83–85). In this study, only 1-week Bex treatment induced an increase in liver size up to 150%, a side effect that may have impacted CNS function, as measured by increased levels of soluble oAβ. Adverse CNS effects are observed in models of both acute and chronic liver disease (43–46, 86, 87). Indeed, hepatic encephalopathy, alcoholic liver disease and nonalcoholic fatty liver disease are associated with significant impairments in cognitive function (43–46, 86, 87). In the liver, hepatomegaly, including steatosis (fatty liver), induces neuroinflammation, vascular dysfunction, lipid peroxidation, a decreased capacity for plasma detoxification, and insulin resistance (43–46, 86, 87). Evidence indicates that these peripheral pathways, particularly inflammation, modulate neuronal function either directly or by the induction of a toxic neuroinflammatory response (43–46, 86, 87). The potential for the liver to modulate cognitive function is highlighted by encephalopathy induced by liver failure, characterized by increased plasma toxin levels, brain edema, astrocyte swelling, and impaired neuronal function (45).
As LG and Bex both showed similar effects on apoE-Aβ biomarkers and induced hepatomegaly, these actions would appear common to RXR agonists. There are at least three alternative approaches that could be pursued. The first approach is to identify an optimal dose of Bex or a similar RXR agonist to induce beneficial effects in the CNS without detrimental effects in the periphery. However, our data do not support this approach, as the lower concentrations of Bex and LG delivered via hydrogel did not attenuate hepatomegaly. The second approach would focus on identifying an RXR/NR heterodimer optimal for AD symptoms. Indeed, novel RXR agonists selective for specific NR heterodimers, or tissues, have been described (83) that may minimize liver toxicity and other unwanted side effects. The third approach would determine whether agonists for alternative NRs, including LXR, PPAR, or RAR, are more beneficial than RXR.
In conclusion, our data demonstrate that in the E4FAD-HP, RXR agonists increase apoE4 lipoprotein association/lipidation and apoE4/Aβ complex levels, which result in decreased oAβ levels and improved synaptic viability. Thus, RXR agonists address a loss-of-function associated with apoE4 and exacerbated by Aβ, i.e. lower apoE4 lipoprotein association/lipidation. However, identifying whether this pathway is relevant for APOE3 carriers and addressing the detrimental effects of RXR agonists, including hepatomegaly, may be critical for providing viable long term prevention and treatment options for AD patients.
Acknowledgments
We thank Isaac Schiefer, Zhihui Qin, Bhargava Karumudi, Rui Xiong, and Shuai Wang for technical assistance with compound preparation and analysis and Stephan Green and Vaiva Laikaite at the University of Illinois at Chicago DNA Services facility for conducting the mRNA analysis described in this work.
This work was supported, in whole or in part, by National Institutes of Health Grant P01AG030128 from NIA (to M. J. L.). This work was also supported by Alzheimer Drug Discovery Foundation grant (to M. J. L. and L. M. T.) and University of Illinois at Chicago Center for Clinical and Translational Science Grant UL1RR029879 (to M. J. L.). In collaboration with Skip Binder, M. J. L. developed the antibody MOAB-2 used to develop the apoE/Aβ complex and oAβ ELISAs. MOAB-2 is licensed as a research tool to multiple companies. Also, for research purposes, Biosensis Pty. Ltd. has licensed the rights to ELISA kits with MOAB-2 for detection of oAβ and apoE/Aβ complex.
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- ABCA1
- ATP-binding cassette subfamily A
- ABCG1
- ATP-binding cassette subfamily G
- apoE
- apolipoprotein E
- apoE/Aβ complex
- apoE in complex with Aβ
- Bex
- bexarotene
- CX
- cortex
- EFAD mice
- Tg-mice expressing 5×FAD mutations and human APOE3 or APOE4
- FAD
- familial AD
- FAD-Tg
- transgenic mice expressing one or more FAD mutations
- FA
- formic acid
- LG
- h
- human
- HP
- hippocampus
- m-apoE
- mouse apoE
- NR
- nuclear receptor
- oAβ
- oligomeric Aβ
- PD
- pharmacodynamics
- Tg
- transgenic
- RXR
- retinoid X receptor
- VC
- vehicle control
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- RAR
- retinoic acid receptor
- PPAR
- peroxisome proliferator-activated receptor
- LXR
- liver X receptor
- WB
- Western blot.
REFERENCES
- 1. Thies W., Bleiler L. (2013) 2013 Alzheimer's disease facts and figures. Alzheimers Dement. 9, 208–245 [DOI] [PubMed] [Google Scholar]
- 2. Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., Rogers J. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 4. Wang J., Dickson D. W., Trojanowski J. Q., Lee V. M. (1999) The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 158, 328–337 [DOI] [PubMed] [Google Scholar]
- 5. Tomic J. L., Pensalfini A., Head E., Glabe C. G. (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin M., Shepardson N., Yang T., Chen G., Walsh D., Selkoe D. J. (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuo Y. M., Emmerling M. R., Vigo-Pelfrey C., Kasunic T. C., Kirkpatrick J. B., Murdoch G. H., Ball M. J., Roher A. E. (1996) Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 271, 4077–4081 [DOI] [PubMed] [Google Scholar]
- 8. Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060–1065 [DOI] [PubMed] [Google Scholar]
- 9. Karran E., Hardy J. (2014) Antiamyloid therapy for Alzheimer's disease–are we on the right road? N. Engl. J. Med. 370, 377–378 [DOI] [PubMed] [Google Scholar]
- 10. Mikulca J. A., Nguyen V., Gajdosik D. A., Teklu S. G., Giunta E. A., Lessa E. A., Tran C. H., Terak E. C., Raffa R. B. (2014) Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J. Clin. Pharm. Ther. 39, 25–37 [DOI] [PubMed] [Google Scholar]
- 11. Cramer P. E., Cirrito J. R., Wesson D. W., Lee C. Y., Karlo J. C., Zinn A. E., Casali B. T., Restivo J. L., Goebel W. D., James M. J., Brunden K. R., Wilson D. A., Landreth G. E. (2012) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. LaClair K. D., Manaye K. F., Lee D. L., Allard J. S., Savonenko A. V., Troncoso J. C., Wong P. C. (2013) Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol. Neurodegener. 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fitz N. F., Cronican A. A., Lefterov I., Koldamova R. (2013) Comment on apoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 340, 924-c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Price A. R., Xu G., Siemienski Z. B., Smithson L. A., Borchelt D. R., Golde T. E., Felsenstein K. M. (2013) Comment on apoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 340, 924-d [DOI] [PubMed] [Google Scholar]
- 15. Tesseur I., Lo A. C., Roberfroid A., Dietvorst S., Van Broeck B., Borgers M., Gijsen H., Moechars D., Mercken M., Kemp J., D'Hooge R., De Strooper B. (2013) Comment on apoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 340, 924-e [DOI] [PubMed] [Google Scholar]
- 16. Veeraraghavalu K., Zhang C., Miller S., Hefendehl J. K., Rajapaksha T. W., Ulrich J., Jucker M., Holtzman D. M., Tanzi R. E., Vassar R., Sisodia S. S. (2013) Comment on apoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 340, 924-f [DOI] [PubMed] [Google Scholar]
- 17. Yu C., Youmans K. L., LaDu M. J. (2010) Proposed mechanism for lipoprotein remodelling in the brain. Biochim. Biophys. Acta 1801, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirsch-Reinshagen V., Zhou S., Burgess B. L., Bernier L., McIsaac S. A., Chan J. Y., Tansley G. H., Cohn J. S., Hayden M. R., Wellington C. L. (2004) Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207 [DOI] [PubMed] [Google Scholar]
- 19. Wahrle S. E., Jiang H., Parsadanian M., Legleiter J., Han X., Fryer J. D., Kowalewski T., Holtzman D. M. (2004) ABCA1 is required for normal central nervous system apoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993 [DOI] [PubMed] [Google Scholar]
- 20. Hirsch-Reinshagen V., Maia L. F., Burgess B. L., Blain J. F., Naus K. E., McIsaac S. A., Parkinson P. F., Chan J. Y., Tansley G. H., Hayden M. R., Poirier J., Van Nostrand W., Wellington C. L. (2005) The absence of ABCA1 decreases soluble apoE levels but does not diminish amyloid deposition in two murine models of Alzheimer's disease. J. Biol. Chem. 280, 43243–43256 [DOI] [PubMed] [Google Scholar]
- 21. Koldamova R., Staufenbiel M., Lefterov I. (2005) Lack of ABCA1 considerably decreases brain apoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235 [DOI] [PubMed] [Google Scholar]
- 22. Wahrle S. E., Jiang H., Parsadanian M., Hartman R. E., Bales K. R., Paul S. M., Holtzman D. M. (2005) Deletion of Abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242 [DOI] [PubMed] [Google Scholar]
- 23. Zelcer N., Khanlou N., Clare R., Jiang Q., Reed-Geaghan E. G., Landreth G. E., Vinters H. V., Tontonoz P. (2007) Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 10601–10606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wahrle S. E., Jiang H., Parsadanian M., Kim J., Li A., Knoten A., Jain S., Hirsch-Reinshagen V., Wellington C. L., Bales K. R., Paul S. M., Holtzman D. M. (2008) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reitz C., Mayeux R. (2010) Use of genetic variation as biomarkers for mild cognitive impairment and progression of mild cognitive impairment to dementia. J. Alzheimers Dis. 19, 229–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leoni V. (2011) The effect of apolipoprotein E (apoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer's disease. Clin. Chem. Lab. Med. 49, 375–383 [DOI] [PubMed] [Google Scholar]
- 27. Qiu W. Q., Mwamburi M., Besser L. M., Zhu H., Li H., Wallack M., Phillips L., Qiao L., Budson A. E., Stern R., Kowall N. (2013) Angiotensin converting enzyme inhibitors and the reduced risk of Alzheimer's disease in the absence of apolipoprotein E4 allele. J. Alzheimers Dis. 37, 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sperling R., Salloway S., Brooks D. J., Tampieri D., Barakos J., Fox N. C., Raskind M., Sabbagh M., Honig L. S., Porsteinsson A. P., Lieberburg I., Arrighi H. M., Morris K. A., Lu Y., Liu E., Gregg K. M., Brashear H. R., Kinney G. G., Black R., Grundman M. (2012) Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 11, 241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farlow M. R., Lahiri D. K., Poirier J., Davignon J., Schneider L., Hui S. L. (1998) Treatment outcome of tacrine therapy depends on apolipoprotein genotype and gender of the subjects with Alzheimer's disease. Neurology 50, 669–677 [DOI] [PubMed] [Google Scholar]
- 30. Risner M. E., Saunders A. M., Altman J. F., Ormandy G. C., Craft S., Foley I. M., Zvartau-Hind M. E., Hosford D. A., Roses A. D. (2006) Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 6, 246–254 [DOI] [PubMed] [Google Scholar]
- 31. Youmans K. L., Tai L. M., Nwabuisi-Heath E., Jungbauer L., Kanekiyo T., Gan M., Kim J., Eimer W. A., Estus S., Rebeck G. W., Weeber E. J., Bu G., Yu C., Ladu M. J. (2012) APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 287, 41774–41786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verghese P. B., Castellano J. M., Holtzman D. M. (2011) Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 10, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tai L. M., Mehra S., Shete V., Estus S., Rebeck G. W., Bu G., LaDu M. J. (2014) Soluble apoE/Aβ complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tai L. M., Youmans K. L., Jungbauer L., Yu C., Ladu M. J. (2011) Introducing human APOE into Aβ transgenic mouse models. Int. J. Alzheimers Dis. 2011, 810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boehm-Cagan A., Michaelson D. M. (2014) Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J. Neurosci. 34, 7293–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Torino F., Barnabei A., Paragliola R., Baldelli R., Appetecchia M., Corsello S. M. (2013) Thyroid dysfunction as an unintended side effect of anticancer drugs. Thyroid 23, 1345–1366 [DOI] [PubMed] [Google Scholar]
- 37. Sherman S. I. (2003) Etiology, diagnosis, and treatment recommendations for central hypothyroidism associated with bexarotene therapy for cutaneous T-cell lymphoma. Clin. Lymphoma 3, 249–252 [DOI] [PubMed] [Google Scholar]
- 38. Liu S., Ogilvie K. M., Klausing K., Lawson M. A., Jolley D., Li D., Bilakovics J., Pascual B., Hein N., Urcan M., Leibowitz M. D. (2002) Mechanism of selective retinoid X receptor agonist-induced hypothyroidism in the rat. Endocrinology 143, 2880–2885 [DOI] [PubMed] [Google Scholar]
- 39. de Vries-van der Weij J., de Haan W., Hu L., Kuif M., Oei H. L., van der Hoorn J. W., Havekes L. M., Princen H. M., Romijn J. A., Smit J. W., Rensen P. C. (2009) Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein. Endocrinology 150, 2368–2375 [DOI] [PubMed] [Google Scholar]
- 40. Boehm M. F., Zhang L., Zhi L., McClurg M. R., Berger E., Wagoner M., Mais D. E., Suto C. M., Davies J. A., Heyman R. A. (1995) Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J. Med. Chem. 38, 3146–3155 [DOI] [PubMed] [Google Scholar]
- 41. Jurutka P. W., Kaneko I., Yang J., Bhogal J. S., Swierski J. C., Tabacaru C. R., Montano L. A., Huynh C. C., Jama R. A., Mahelona R. D., Sarnowski J. T., Marcus L. M., Quezada A., Lemming B., Tedesco M. A., Fischer A. J., Mohamed S. A., Ziller J. W., Ma N., Gray G. M., van der Vaart A., Marshall P. A., Wagner C. E. (2013) Modeling, synthesis, and biological evaluation of potential retinoid X receptor (RXR) selective agonists: novel analogues of 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (bexarotene) and (E)-3-(3-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)-4-hydroxyphenyl)acrylic acid (CD3254). J. Med. Chem. 56, 8432–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y., Yao R., Maciag A., Grubbs C. J., Lubet R. A., You M. (2006) Organ-specific expression profiles of rat mammary gland, liver, and lung tissues treated with targretin, 9-cis retinoic acid, and 4-hydroxyphenylretinamide. Mol. Cancer Ther. 5, 1060–1072 [DOI] [PubMed] [Google Scholar]
- 43. de la Monte S. M. (2014) Relationships between diabetes and cognitive impairment. Endocrinol. Metab. Clin. North Am. 43, 245–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weissenborn K., Bokemeyer M., Krause J., Ennen J., Ahl B. (2005) Neurological and neuropsychiatric syndromes associated with liver disease. AIDS 19, S93–S98 [DOI] [PubMed] [Google Scholar]
- 45. Lewis M., Howdle P. D. (2003) The neurology of liver failure. QJM 96, 623–633 [DOI] [PubMed] [Google Scholar]
- 46. Sonneville R., Verdonk F., Rauturier C., Klein I. F., Wolff M., Annane D., Chretien F., Sharshar T. (2013) Understanding brain dysfunction in sepsis. Ann. Inten. Care 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Faul M. M., Ratz A. M., Sullivan K. A., Trankle W. G., Winneroski L. L. (2001) Synthesis of novel retinoid X receptor-selective retinoids. J. Org. Chem. 66, 5772–5782 [DOI] [PubMed] [Google Scholar]
- 48. Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R. (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sullivan P. M., Mezdour H., Aratani Y., Knouff C., Najib J., Reddick R. L., Quarfordt S. H., Maeda N. (1997) Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272, 17972–17980 [DOI] [PubMed] [Google Scholar]
- 50. Fryer J. D., Simmons K., Parsadanian M., Bales K. R., Paul S. M., Sullivan P. M., Holtzman D. M. (2005) Human apolipoprotein E4 alters the amyloid-β 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. 25, 2803–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bales K. R., Liu F., Wu S., Lin S., Koger D., DeLong C., Hansen J. C., Sullivan P. M., Paul S. M. (2009) Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tai L. M., Bilousova T., Jungbauer L., Roeske S. K., Youmans K. L., Yu C., Poon W. W., Cornwell L. B., Miller C. A., Vinters H. V., Van Eldik L. J., Fardo D. W., Estus S., Bu G., Gylys K. H., Ladu M. J. (2013) Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 288, 5914–5926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Overk C. R., Borgia J. A., Mufson E. J. (2011) A novel approach for long term oral drug administration in animal research. J. Neurosci. Methods 195, 194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janakiram N. B., Mohammed A., Qian L., Choi C. I., Steele V. E., Rao C. V. (2012) Chemopreventive effects of RXR-selective rexinoid bexarotene on intestinal neoplasia of Apc(Min/+) mice. Neoplasia 14, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Overk C. R., Lu P. Y., Wang Y. T., Choi J., Shaw J. W., Thatcher G. R., Mufson E. J. (2012) Effects of aromatase inhibition versus gonadectomy on hippocampal complex amyloid pathology in triple transgenic mice. Neurobiol. Dis. 45, 479–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Howell S. R., Shirley M. A., Grese T. A., Neel D. A., Wells K. E., Ulm E. H. (2001) Bexarotene metabolism in rat, dog, and human, synthesis of oxidative metabolites, and in vitro activity at retinoid receptors. Drug Metab. Dispos. 29, 990–998 [PubMed] [Google Scholar]
- 57. Wu K., Kim H. T., Rodriquez J. L., Hilsenbeck S. G., Mohsin S. K., Xu X. C., Lamph W. W., Kuhn J. G., Green J. E., Brown P. H. (2002) Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol. Biomarkers Prev. 11, 467–474 [PubMed] [Google Scholar]
- 58. Youmans K. L., Leung S., Zhang J., Maus E., Baysac K., Bu G., Vassar R., Yu C., LaDu M. J. (2011) Amyloid-β42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J. Neurosci. Methods 196, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sultana R., Banks W. A., Butterfield D. A. (2010) Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer's disease. J. Neurosci. Res. 88, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koffie R. M., Meyer-Luehmann M., Hashimoto T., Adams K. W., Mielke M. L., Garcia-Alloza M., Micheva K. D., Smith S. J., Kim M. L., Lee V. M., Hyman B. T., Spires-Jones T. L. (2009) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Almeida C. G., Tampellini D., Takahashi R. H., Greengard P., Lin M. T., Snyder E. M., Gouras G. K. (2005) β-Amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 20, 187–198 [DOI] [PubMed] [Google Scholar]
- 62. Bachstetter A. D., Watterson D. M., Van Eldik L. J. (2014) Target engagement analysis and link to pharmacodynamic endpoint for a novel class of CNS-penetrant and efficacious p38α MAPK inhibitors. J. Neuroimmune Pharmacol. 9, 454–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jiang Q., Lee C. Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T. M., Collins J. L., Richardson J. C., Smith J. D., Comery T. A., Riddell D., Holtzman D. M., Tontonoz P., Landreth G. E. (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li J., Kanekiyo T., Shinohara M., Zhang Y., LaDu M. J., Xu H., Bu G. (2012) Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 287, 44593–44601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Deane R., Sagare A., Hamm K., Parisi M., Lane S., Finn M. B., Holtzman D. M., Zlokovic B. V. (2008) ApoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Verghese P. B., Castellano J. M., Garai K., Wang Y., Jiang H., Shah A., Bu G., Frieden C., Holtzman D. M. (2013) ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110, E1807–E1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ly S., Altman R., Petrlova J., Lin Y., Hilt S., Huser T., Laurence T. A., Voss J. C. (2013) Binding of apolipoprotein E inhibits the oligomer growth of amyloid-β peptide in solution as determined by fluorescence cross-correlation spectroscopy. J. Biol. Chem. 288, 11628–11635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hashimoto T., Serrano-Pozo A., Hori Y., Adams K. W., Takeda S., Banerji A. O., Mitani A., Joyner D., Thyssen D. H., Bacskai B. J., Frosch M. P., Spires-Jones T. L., Finn M. B., Holtzman D. M., Hyman B. T. (2012) Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 32, 15181–15192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Larson M. E., Lesne S. E. (2012) Soluble Aβ oligomer production and toxicity. J. Neurochem. 120, 125–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fowler S. W., Chiang A. C., Savjani R. R., Larson M. E., Sherman M. A., Schuler D. R., Cirrito J. R., Lesné S. E., Jankowsky J. L. (2014) Genetic modulation of soluble Aβ rescues cognitive and synaptic impairment in a mouse model of Alzheimer's disease. J. Neurosci. 34, 7871–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hong S., Quintero-Monzon O., Ostaszewski B. L., Podlisny D. R., Cavanaugh W. T., Yang T., Holtzman D. M., Cirrito J. R., Selkoe D. J. (2011) Dynamic analysis of amyloid β-protein in behaving mice reveals opposing changes in ISF versus parenchymal Aβ during age-related plaque formation. J. Neurosci. 31, 15861–15869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hudry E., Dashkoff J., Roe A. D., Takeda S., Koffie R. M., Hashimoto T., Scheel M., Spires-Jones T., Arbel-Ornath M., Betensky R., Davidson B. L., Hyman B. T. (2013) Gene transfer of human apoE isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 5, 212ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wong J., Quinn C. M., Brown A. J. (2006) SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem. J. 400, 485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mohamed A., Saavedra L., Di Pardo A., Sipione S., Posse de Chaves E. (2012) β-Amyloid inhibits protein prenylation and induces cholesterol sequestration by impairing SREBP-2 cleavage. J. Neurosci. 32, 6490–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lim W. L., Martins I. J., Martins R. N. (2014) The involvement of lipids in Alzheimer's disease. J. Genet. Genomics 41, 261–274 [DOI] [PubMed] [Google Scholar]
- 77. Altmann A., Tian L., Henderson V. W., Greicius M. D. (2014) Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75, 563–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bretsky P. M., Buckwalter J. G., Seeman T. E., Miller C. A., Poirier J., Schellenberg G. D., Finch C. E., Henderson V. W. (1999) Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 13, 216–221 [DOI] [PubMed] [Google Scholar]
- 79. Payami H., Zareparsi S., Montee K. R., Sexton G. J., Kaye J. A., Bird T. D., Yu C. E., Wijsman E. M., Heston L. L., Litt M., Schellenberg G. D. (1996) Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: a possible clue to the higher incidence of Alzheimer disease in women. Am. J. Hum. Genet. 58, 803–811 [PMC free article] [PubMed] [Google Scholar]
- 80. Yaffe K., Haan M., Byers A., Tangen C., Kuller L. (2000) Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology 54, 1949–1954 [DOI] [PubMed] [Google Scholar]
- 81. Jacobs E. G., Kroenke C., Lin J., Epel E. S., Kenna H. A., Blackburn E. H., Rasgon N. L. (2013) Accelerated cell aging in female APOE-ϵ4 carriers: implications for hormone therapy use. PLoS One 8, e54713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kunzler J., Youmans K. L., Yu C., Ladu M. J., Tai L. (2014) APOE modulates the effect of estrogen therapy on Aβ accumulation EFAD-Tg mice. Neurosci. Lett. 560, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pérez E., Bourguet W., Gronemeyer H., de Lera A. R. (2012) Modulation of RXR function through ligand design. Biochim. Biophys. Acta 1821, 57–69 [DOI] [PubMed] [Google Scholar]
- 84. Scarisbrick J. J., Morris S., Azurdia R., Illidge T., Parry E., Graham-Brown R., Cowan R., Gallop-Evans E., Wachsmuth R., Eagle M., Wierzbicki A. S., Soran H., Whittaker S., Wain E. M. (2013) U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br. J. Dermatol. 168, 192–200 [DOI] [PubMed] [Google Scholar]
- 85. Väkevä L., Ranki A., Hahtola S. (2012) Ten-year experience of bexarotene therapy for cutaneous T-cell lymphoma in Finland. Acta Derm. Venereol. 92, 258–263 [DOI] [PubMed] [Google Scholar]
- 86. Erbas O., Sarac F., Aktug H., Peker G. (2014) Detection of impaired cognitive function in rat with hepatosteatosis model and improving effect of GLP-1 analogues (exenatide) on cognitive function in hepatosteatosis. Scientific World J. 2014, 946265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Elliott C., Frith J., Day C. P., Jones D. E., Newton J. L. (2013) Functional impairment in alcoholic liver disease and nonalcoholic fatty liver disease is significant and persists over 3 years of follow-up. Digest. Dis. Sci. 58, 2383–2391 [DOI] [PubMed] [Google Scholar]