

Background: No available inhibitors target isoforms of the biologically and medically relevant enzyme that initiates mucin-type O-glycosylation.
Results: Protein-based, fluorescent sensors were developed for T2 and T3 isoforms using modified GalNAc-transferase target sites and these were specifically activated in HEK cells lacking their corresponding GalNAc-transferases.
Conclusion: The fluorescence sensors are isoform specific.
Significance: The designed biosensors may enable high-throughput screening for specific regulators with therapeutic potential.
Keywords: Biosensor, Fluorescence, Glycosylation, Glycosyltransferase, Golgi, Protein Processing
Abstract
Humans express up to 20 isoforms of GalNAc-transferase (herein T1–T20) that localize to the Golgi apparatus and initiate O-glycosylation. Regulation of this enzyme family affects a vast array of proteins transiting the secretory pathway and diseases arise upon misregulation of specific isoforms. Surprisingly, molecular probes to monitor GalNAc-transferase activity are lacking and there exist no effective global or isoform-specific inhibitors. Here we describe the development of T2- and T3-isoform specific fluorescence sensors that traffic in the secretory pathway. Each sensor yielded little signal when glycosylated but was strongly activated in the absence of its glycosylation. Specificity of each sensor was assessed in HEK cells with either the T2 or T3 enzymes deleted. Although the sensors are based on specific substrates of the T2 and T3 enzymes, elements in or near the enzyme recognition sequence influenced their activity and required modification, which we carried out based on previous in vitro work. Significantly, the modified T2 and T3 sensors were activated only in cells lacking their corresponding isozymes. Thus, we have developed T2- and T3-specific sensors that will be valuable in both the study of GalNAc-transferase regulation and in high-throughput screening for potential therapeutic regulators of specific GalNAc-transferases.
Introduction
Mucin-type O-glycosylation, characterized by the initial addition of N-acetylgalactosamine (GalNAc) to the hydroxyl group of serine or threonine residues (and possibly tyrosine), is a large and important subgroup of protein O-glycosylation. The appended carbohydrate chains dramatically alter protein surface characteristics affecting solubility, stability, and interactions. Moreover, it is becoming increasingly apparent that proximal site-specific O-glycosylation performed by individual GalNAc-transferases may modulate regulated proteolytic processing events to control protein activity, ectodomain shedding, and cell signaling (1–3).
The O-glycan chain is built in the Golgi apparatus by the stepwise addition of individual monosaccharides (3). Although one or two isoforms exist for each of the enzymes mediating chain extension, there are up to 20 distinct isoforms of the initiating enzyme UDP-N-acetyl-α-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase (herein termed T1–T20) in humans (4). These isoforms have distinct yet overlapping substrate specificities and show distinct temporal and spatial expression patterns (4). Much remains to be determined regarding the purpose of this large number of isozymes covering a single reaction.
The GalNAc-transferases are type II transmembrane proteins. All share a domain organization in which a short cytoplasmic tail and single transmembrane domain is followed by a variable length lumenal stem region and then catalytic and lectin domains except for T20, which lacks the lectin domain (4, 5). Although binding of the catalytic domain to the substrate largely determines the substrate specificity of each isoform (6), the lectin domain influences this activity by binding GalNAc already linked to the substrate at a nearby site (7–9). Binding by the lectin domain presumably increases enzyme/substrate interaction but it also directly activates the catalytic domain (10).
Because of the large number of proteins modified by O-glycosylation in the Golgi and the potential of O-glycosylation to profoundly affect functionality of the modified proteins, it should not be surprising that abnormal GalNAc-transferase activity perturbs cell function and causes disease (10). Examples of the former include cell adhesion, migration, and tissue lubrication. An important example of the latter is the up-regulation of GalNAc-transferase activity in cancer, which is believed to cause increased invasiveness among other possible effects (11–16). Another example is a loss of function mutation in T3 that results in increased processing of fibroblast growth factor 23 (FGF23) leading to familial tumoral calcinosis and hyperphosphatemia-hyperstosis (17–19). Similarly, T2 and T11 deficiencies are connected to high serum levels of triglyceride and high-density lipoprotein cholesterol (20, 21) and heterotaxy disorder (22), respectively. There are already at least 25 well defined medical syndromes linked to defects in O-glycosylation and this number is likely to grow (1).
Despite the biological and medical significance of the GalNAc-transferase family, GalNAc-transferase isoform-specific assays are lacking that monitor their activity in living cells and there are no known inhibitors. To address these shortcomings, we initiated the development of cell-based fluorescence sensors whose fluorescence is a readout of GalNAc-transferase activity (23). The sensors are designed to traffic in the secretory pathway becoming activated if their glycosylation is inhibited. Fluorescence is dependent on inhibition because glycosylation prevents furin protease from removing a blocking domain, which occludes binding of the dye malachite green to a fluorescence activating protein domain (Fig. 1A). Thus, when glycosylation is inactive (e.g. in the presence of an active drug) the peptide sequence is cleaved by furin-like proprotein convertase processing and the sensor is activated. The biosensor operates in a “turn-on” mode and it is ratiometric because it also contains a fluorescent protein domain.
FIGURE 1.

Sensor design and evaluation of the ANGPTL3-based sensor for T2. A, the schematic shows an intact sensor on the left with its blocking domain (BD), linker domain with a threonine (T) site for GalNAc-transferase and an arginine (R) site for furin, fluorescent activating protein domain (FAP), transmembrane domain, and green fluorescent protein domain (GFP). Glycan attachment masks the furin site but if an inhibitor (?) blocks the glycan attachment then furin cleaves the linker releasing the BD. As shown on the right, the FAP then dimerizes and binds and activates the dye (malachite green (MG)). B–M, representative images are shown for HEK cell lines expressing wild type (WT, B–D), T225G (E–G), T226G (H–J), or T225G/T226G (K–M) versions of the ANGPTL3-based T2 sensor. Images are GFP, MG, and a merge of GFP (green) and MG (red). Image acquisition was identical within each channel and all but K–M were acquired on the same day. Bar = 50 μm. N, the ratio of the average MG fluorescence per image field to the average GFP fluorescence for the same fields is plotted for the indicated constructs with fold-differences shown (**, p < 0.001, n.s. = not significant, 8 fields per condition per experiment, n = 4 ± S.E.).
Our previously characterized first generation sensor (23) contained a target substrate sequence with a non-restrictive GalNAc-transferase isoform consensus sequence for O-glycosylation. Because important diseases and regulatory changes involve single GalNAc-transferase isoforms and therapeutics should ideally target specific isoforms, our intention in the present study was to convert the biosensor to an isoform-specific modality, thereby addressing whether distinct isoforms exhibit true substrate specificity. Focusing on T2 and T3 we were able to design and generate isoform-specific detection but it required optimization of known sequences targeted by these enzymes. The strategy we used to develop T2- and T3-specific sensors can now be used to develop other isoform-specific sensors. Significantly, with two or more isoform-specific sensors at hand, parallel screening can identify compounds selectively activating a single sensor thereby greatly reducing the likelihood of off-target effects.
EXPERIMENTAL PROCEDURES
Constructs
To improve recovery and expression of stable cell lines, the sequence encoding the previously described sensor (23) was excised using SpeI and NotI-HF and cloned into NheI and NotI sites of the pIRES vector. Primers were then designed encoding the target isoform-specific substrate sequences of ANGPTL3,2 FGF23, and an artificial sequence (see Table 1) and these were either cloned into the linker region by a loop-in procedure using QuikChange (Stratagene, La Jolla, CA) or by direct insertion into XhoI and BamHI sites. Subsequent point mutation of the base linker sequences was by QuikChange. All constructs were confirmed by sequencing.
TABLE 1.
Sensor linker domain sequences
Note: residues comprising furin recognition are bold and underlined and residues that are mutated from WT are in bold.
| Sensor | Version | Sequence |
|---|---|---|
| ANGPTL3-based T2 | WT | 219KPRAPRTTPF228 |
| T225G | -KPRAPRGTPF- | |
| T226G (acceptor site) | -KPRAPRTGPF- | |
| T225G/T226G | -KPRAPRGGPF- | |
| FGF23-based T3 | WT | 168HFNTPIPRRHTRSAEDDG185 |
| T171G (upstream site) | -HFNGPIPRRHTRSAEDDG- | |
| T178G (acceptor site) | -HFNTPIPRRHGRSAEDDG- | |
| H177V | -HFNTPIPRRVTRSAEDDG- | |
| H177G/T178G | -HFNTPIPRRVGRSAEDDG- | |
| H177V/S180G/A181P | -HFNTPIPRRVTRGPEDDG- | |
| H177V/T178G/S180G/A181P | -HFNTPIPRRVGRGPEDDG- | |
| Artificial sequence | WT | 1RRAYRVTPGP10 |
| T7G (acceptor site) | -RRAYRVGPGP- |
Cell Culture
HEK cells, including HEK293 controls and HEK cells deleted for T2 or T33 produced by zinc finger nuclease-mediated knock-out similarly to previously described (24, 25), were cultured in minimal essential medium (Thermo Scientific, Waltham, MA) with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) and 100 IU/ml of penicillin/streptomycin (Sigma). The cells were transfected with the constructs using the JetPEI transfection reagent (VWR International, Radnor, PA) exactly as described by the manufacture's instructions. Selection with 2 μm puromycin (Sigma) was initiated 24–48 h post-transfection. After about 2 weeks of culturing, the cells were collected using EDTA and sorted using a FacsVantage fluorescence cell sorter (BD Biosciences). Cells (5,000 per construct) expressing GFP at 80–90% of the maximum observed were collected and propagated for use.
Imaging
The cells were passed onto coverslips at 50% confluence 24 h before imaging. After transfer to an imaging chamber containing 110 nm of the dye MG11p (MG) (a generous gift from the Molecular Biosensor and Imaging Center, Carnegie Mellon University) they were imaged using an LSM 510 Meta DuoScan Spectral Confocal Microscope equipped with a ×40 objective (Zeiss, Thornwood, NY). Single optical sections were acquired and quantified using ImageJ (National Institutes of Health, Bethesda, MD) as described previously (23). Briefly, background was taken as the highest non-cell pixel value and uniformly subtracted. The GFP channel was then used as a mask to select an area of measurement. The average pixel intensity in this area was then determined for the GFP and MG channels and expressed as a ratio. For every condition, 8 fields were analyzed per experiment and each experiment was repeated 4 times. For each channel (GFP or MG), acquisition parameters were identical for every experiment. Where necessary for presentation purposes, grayscale projection of the MG channel was adjusted equally for all images in an experiment.
Flow Cytometry
Control cells or cells stably expressing the sensors were passed into 96-well plates (50,000 cells/well) and returned to the incubator for 24 h. Where indicated, a further incubation for 0–9 h was carried out in medium containing 0–10 mm benzyl-N-acetyl-α-galactosaminide (BAG, Sigma) added from a stock dissolved in dimethyl sulfoxide. All samples contained equivalent amounts of dimethyl sulfoxide (1%). Just prior to analysis the medium was replaced with 0.1 ml of PBS solution containing 110 nm MG dye and 5 mm EDTA. After 5 min at 37 °C the released cells were collected and GFP and MG fluorescence was measured by AccuriTM C6 flow cytometry (BD Biosciences) at 488 and 640 nm, respectively. Data presentation and quantification were carried out using FlowJo software. For histograms, the background subtracted MG and GFP fluorescence measurements for each cell were expressed as a ratio and binned. Background was subtracted separately in each channel using the geometric mean of all values determined for the MG and GFP channels using untransfected HEK cells. To compare average values among multiple experiments, the individual ratios calculated were collected and expressed as geometric means and these means were then compared using standard arithmetic mean calculations and statistics. Three independent experiments were performed for each.
RESULTS
T2 Sensor Based on ANGPTL3
To develop a sensor specifically sensitive to the activity of T2 we used a sequence from angiopoietin-like 3 (ANGPTL3) as the linker in our previously described fluorescent glycosylation sensor (23). All linker sequences used in this study are listed in Table 1. The chosen 219KPRAPRTTPF228 sequence of ANGPTL3 is an in vitro target of T2 and to a lesser extent T3 (26). In liver cells, which naturally express ANGPTL3 and lack T3, the sequence is a specific target of T2 (25) indicating that other family members expressed in these cells do not recognize it. Glycosylation of the sequence on Thr226 blocks furin-like proprotein processing cleavage at Arg224 (1, 26). If this glycan masking was recapitulated in the context of the sensor, the sensor should be dark under normal conditions and activated upon inhibition of glycosylation at Thr226. That is, absence of glycosylation should allow furin to cleave the linker, removing the inhibitory domain thereby allowing activation of the MG dye (Fig. 1A). Because there are no available inhibitors against GalNAc-transferases, we used mutation of the glycan acceptor site in the sequence to test sensor functioning. Human embryonic kidney (HEK) cells were used in our experiments and they express all isoforms except T5, T9, T15, T17, T19, and T20 (Proteinatlas). In anticipation of the likelihood that T3 would also glycosylate Thr226 we simultaneously tested a glycine substituted at position −1 (T225G) relative to the glycosylation site because glycine substitution at this position interferes with T3 but not T2-mediated glycosylation in vitro (27).
HEK cells were generated that stably expressed one of four versions of the ANGPTL3-based sensor: wild type (WT), T225G, T226G, or T225G/T226G. GFP fluorescence present in the sensor was used to verify expression and quantify sensor activation as a ratio of MG fluorescence per unit GFP fluorescence. As expected, all cells expressed the sensors on the cell surface as indicated by GFP fluorescence (Fig. 1, B–M). Significantly, MG fluorescence was not apparent for both the WT version (Fig. 1, B–D) and the version with a glycine at position −1 (Fig. 1, E–G), whereas there was strong activation apparent for both versions lacking the Thr226 glycan acceptor site (Fig. 1, H–M). Compared with WT, fluorescence was 267- and 558-fold greater for T226G and T225G/T226G, respectively (Fig. 1N). Each increase was well over 500 standard deviations (S.D.) indicating a remarkable signal to noise ratio. Interestingly, the T225G/T226G signal was about twice that of T226G, indicating that glycine at −1 unexpectedly enhanced cleavage of the sensor. Note that, were glycosylation to take place at Thr225 it could account for this result but an observation described in the next paragraph counters this possibility.
To test the GalNAc-transferase isoform-specific control of sensor activation, we used zinc finger-nuclease gene engineered HEK cells lacking either T2 or T3 enzymes (double knock-out was not available). If the ANGPTL3-based sensor was T2 isoform specific in HEK cells, we expected activation of the sensor in ΔT2 cells. However, whereas GFP fluorescence indicated strong expression of the WT sensor, no activation of MG fluorescence was observed (Fig. 2, A–C). Importantly though, the T225G version of the sensor was strongly activated in ΔT2 cells (Fig. 2, D–F). The specific and strong activation of the T225G version of the sensor in ΔT2 cells (≈170 S.D. over control HEK cells expressing the same construct) meets the key criterion for isoform-specific sensing. We speculate that glycine at position −1 of the acceptor site selectively interferes with recognition of Thr226 by the T3 isoform leaving T2 as the sole enzyme targeting Thr226. The reason we favor this explanation rather than T3-mediated glycosylation of Thr225 is that glycine substitution of the Thr226 glycan acceptor site led to robust activation (Fig. 2, G–I). This activation argues that Thr225, which remains present in this construct, is not glycosylated.
FIGURE 2.

Activation of the ANGPTL3-based T2 sensor in ΔT2 and ΔT3 HEK cells. A–O, representative images are shown for HEK cell lines deleted for either GalNAc T2 (ΔT2, A–I) or T3 (ΔT3, J–O) and expressing wild type (WT, A–C and J–L), T225G (D–F and M–O), or T226G (G–I) versions of the ANGPTL3-based T2 sensor. Images are GFP, MG, and a merge of GFP (green) and MG (red). Image acquisition was identical within each channel with J–O acquired on a separate day. Bar = 50 μm. P, the ratio of the average MG fluorescence per image field to the average GFP fluorescence for the same fields is plotted for the indicated constructs with fold-differences shown (**, p < 0.001, n.d. = not determined, n = 4 ± S.E.). Control HEK cell data are re-plotted from Fig. 1.
We also tested the WT and T225G sensors in ΔT3 cells. Although well expressed, neither WT (Fig. 2, J–L) nor T225G (Fig. 2, M–O) was activated, consistent with T2 being sufficient to carry out effective glycan masking of either sensor version. Quantification of all the results for ΔT2 and ΔT3 cells confirmed these conclusions (Fig. 2P). Due to the lack of availability of other cell lines, we were limited to the ΔT3 cell line in controlling for participation of other isoforms. Nevertheless, such tests were arguably unimportant (even for the ΔT3 cell line) because the level of activation of the T225G in ΔT2 cells was near maximal. This implies that T2 was the sole GalNAc-transferase in HEK capable of using the modified site (i.e. glycine at position −1) as a substrate. In summary, the T225G-modified ANGPTL3-based sensor provides a specific readout of T2 activity in HEK cells.
T3 Sensor Based on FGF23
A similar strategy was adopted to develop a sensor specific to the activity of T3. The fibroblast growth factor 23 (FGF23) sequence 168HFNTPIPRRHTRSAEDDG185 is a T3 specific target and its glycosylation on Thr178 blocks furin cleavage at Arg179 (19). This sequence was used as the linker in the sensor and mutation of the glycan acceptor site was used to test sensor functioning. In this case, there was an additional site (Thr171) to consider, which is a known target of glycosylation (19).
For the FGF23-based sensor of T3 activity, HEK cells were generated stably expressing WT, T171G, or T178G versions of the sensor. As before, GFP fluorescence confirmed sensor expression. Whereas relatively low levels of MG fluorescence were observed for WT (Fig. 3, A–C), activation was evident for both T171G (Fig. 3, D–F) and T178G (Fig. 3, G–I). Although the effect of the T171G mutation may seem surprising, in other work we observed that T3 requires glycosylation at Thr171 to glycosylate Thr178 in a process dependent on the T3 lectin domain3 (9), similar to the situation for several other GalNAc-transferase isoforms and their substrates (28, 29). More relevant to the present work, the fold-increase for these constructs (Fig. 3J) was markedly less than that above and yielded little more than 8 S.D. over background. Interestingly, this was not due to low activation. The activated constructs yielded MG/GFP fluorescence ratios that were comparable with those of the ANGPTL3-based sensor (1.0 for FGF23-based T171G versus 1.1 for ANGPTL3-based T225G/T226G). Rather, it was due to a higher level of background activation evident in the WT sensor. This is consistent with previous work showing that furin cleavage of FGF23 is highly efficient relative to its cleavage of ANGPTL3 (18) but it could also be that the FGF23-based sensor was less efficiently glycosylated.
FIGURE 3.

Evaluation of the T3 sensor based on FGF23. A–I, GFP, MG, and merged channels are shown for HEK cell lines expressing the WT (A–C), T171G (D–F), and T178G (G–I) versions of the FGF23-based T3 sensor. Bar = 50 μm. J, the MG/GFP ratio is shown for each (**,p < 0.001, n = 4 ± S.E.). K–P, HEK cell lines deleted for either T3 (K–M) or T2 (N–P) and expressing the wild type FGF23-based T3 sensor are shown. Bar = 50 μm. Q, the MG/GFP ratio is shown for the wild type T3 sensor expressed in control HEK cells or HEK cells deleted for T2 or T3 (*, p < 0.05, n.s. = not significant, n = 4 ± S.E.).
Before attempting to improve the FGF23-based sensor we tested whether its activation depended on T3. Indeed, its expression in ΔT3 cells led to greater activation of MG fluorescence (Fig. 3, K–M). The increase was small but significant (Fig. 3Q). The sensor was also tested in ΔT2 cells. For an unknown reason, we were only able to recover cells stably expressing low levels, but, as expected, absence of T2 did not activate the sensor (Fig. 3, N–Q). Thus, whereas the sensor needed improvement, it showed promise in being isoform specific.
Our intention was to maximize the signal to noise ratio where feasible and ideally achieve a maximum signal at least 20 S.D. above background so that, in the context of a screen, a signal that is 10% of the maximum will be statistically meaningful (i.e. ≥2 S.D. above background). To improve the signal to noise ratio we decided to modify the FGF23 insert sequence with the goal of making it a better substrate of T3 glycosylation. Fortunately, extensive in vitro assays were previously used to assess glycosylation of various peptide sequences (27) and recent work identified a phosphorylation site (Ser180) that negatively regulates glycosylation (31). Based on these results, we tested the single mutation H177V and the triple mutation H177V/S180G/A181P. Compared with the level of “background” activation of WT (Fig. 3, A–C), both H177V (Fig. 4, A–C) and H177V/S180G/A181P (Fig. 4, D–F) substantially reduced the amount of activation. To test whether the constructs could still be activated by the absence of their glycosylation, we generated corresponding versions lacking the T3 acceptor site. Similar to mutation of the acceptor site in the WT to generate T178G (Fig. 3, G–I), MG fluorescence was activated in H177V/T178G (Fig. 4, G–I). For an unknown reason, H177V/T178G/S180G/A181P proved less useful as it did not yield comparable levels of MG fluorescence (Fig. 4, J–L). Quantification confirmed the significant improvement using the H177V version of the sensor (Fig. 4M). Furthermore, because of the reduced noise, activation was >1000 S.D. over background.
FIGURE 4.

Optimization of the FGF23-based T3 sensor. A–L, GFP, MG, and merged channels are shown for HEK cell lines expressing H177V (A–C), H177V/S180G/A181P (D–F), H177V/T178G (G–I), and H177V/T178G/S180G/A181P (J–L) versions of the FGF23-based T3 sensor. Note that those containing the T178G substitution (G–L) lack the glycan acceptor site. Bar = 50 μm. M, quantified MG/GFP ratio for the indicated constructs without (Ø) or with (+T178G) the glycine substitution at the glycan acceptor site (**, p < 0.001, n = 4 ± S.E.).
Based on the improvement evident for the H177V version, we tested its isoform specificity using ΔT2 and ΔT3 cells. For comparison, the WT and H177V/S180G/A181P constructs were also tested. Both WT (Fig. 5, A–C) and H177V (Fig. 5, D–F) were active in ΔT3 cells indicating that these sensors depend on glycosylation by T3. Neither was activated in ΔT2 cells showing the specificity of this effect (Fig. 5, J–O). The H177V/S180G/A181P mutation failed to show any activation (Fig. 5, G–I) and quantification confirmed that the H177V modified sensor was significantly improved in both signal to noise (≈900 S.D.) and isoform specificity (Fig. 5P). These results support the utility of the H177V modified FG23-based sensor for specific determination of T3 activity.
FIGURE 5.
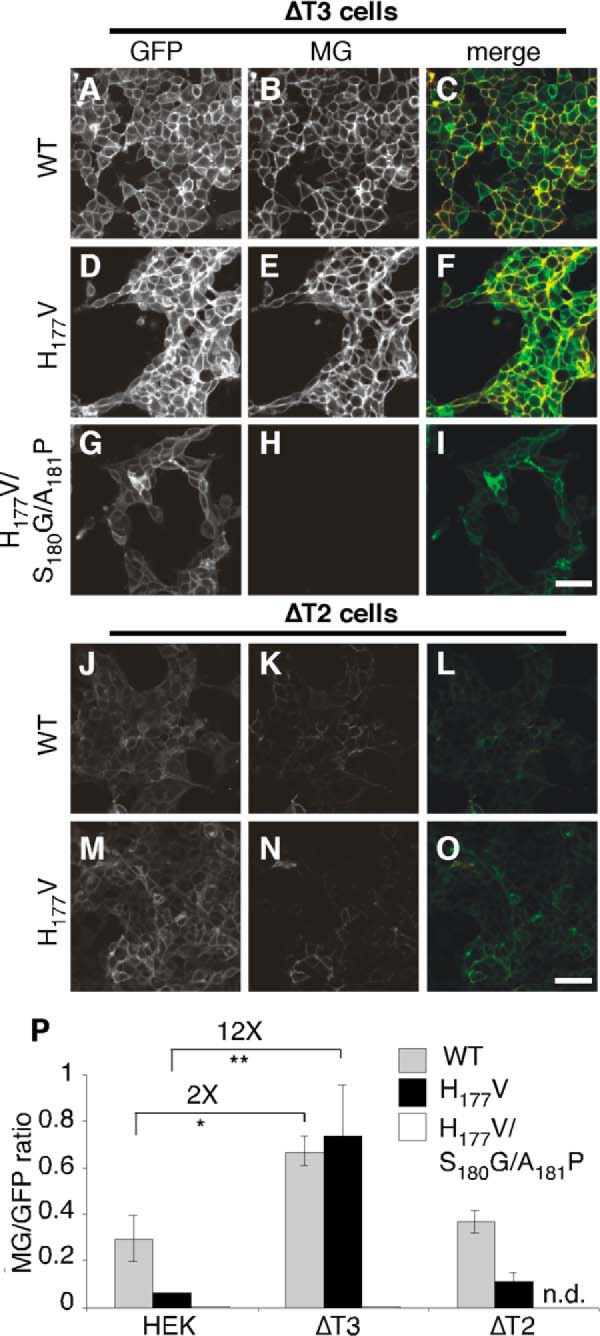
Activation of the FGF23-based T3 sensor in ΔT3 and ΔT2 HEK cells. A–O, representative images are shown for HEK cell lines deleted for either GalNAc T3 (ΔT3, A–I) or T2 (ΔT2, J–O) and expressing wild type (WT, A–C and J–L), H177V (D–F and M–O), or H177V/S180G/A181P (G–I) versions of the FGF23-based T3 sensor. Bar = 50 μm. P, the MG/GFP ratio is shown for the indicated constructs expressed in control HEK cells or HEK cells deleted for T3 or T2 (**, p < 0.001, n.d. = not determined, n = 4 ± S.E.).
T3 Sensor Based on Artificial Sequence
The previous sections demonstrate how natural target sites for O-glycosylation and proprotein convertase processing can be tuned to improve signal to noise levels and isoform specificity of the sensors. Therefore, we wanted to test a linker sequence that was entirely designed de novo. The design was based on previous in vitro analysis of the substrate specificity of T3 (27). The sequence, 1RRAYRVTPGP10, contained a single potential glycan acceptor, Thr7, placed two residues C-terminal of an Arg5 furin cleavage site in an attempt to maximize masking and thus decrease background sensor fluorescence. The sequence surrounding Thr7 was chosen to optimize it as a substrate of T3. In HEK cells stably expressing the artificial sequence-based T3 sensor, GFP fluorescence indicated good expression and MG fluorescence indicated a reasonably low level of background activation (Fig. 6, A–C). To test its ability to function, we generated cells expressing the same sensor harboring a T7G substitution. This version showed dramatically increased MG fluorescence (Fig. 6, D–F). Overall, the level of activation was 9-fold and 30 S.D. over background (Fig. 6G).
FIGURE 6.

Evaluation of a T3 sensor based on an artificial sequence. A–F, GFP, MG, and merged channels are shown for HEK cell lines expressing either an unmodified version of the T3 sensor containing an artificial sequence (A–C) or a version with a glycine substitution of the glycan acceptor site (T7G, D–F). Bar = 50 μm. G, the MG/GFP ratio is shown for each (**, p < 0.001, n = 4 ± S.E.). H–M, images for the artificial sequence-based T3 sensor expressed in control (H–J) or ΔT3 (K–M) HEK cells. Bar = 50 μm. N, the MG/GFP ratio is shown for the artificial sequence-based T3 sensor expressed in control HEK cells or HEK cells deleted for T3 (*, p < 0.05, n = 4 ± S.E.).
To test whether the artificial sequence-based sensor activity was controlled by T3, the sensor was next expressed in the ΔT3 cell line. Significantly, compared with control HEK cells (Fig. 6, H–J), the sensor was activated in ΔT3 cells (Fig. 6, K–M) and this was confirmed by quantification (Fig. 6N). Although future modification of this sequence may lead to further optimization, these findings indicate that we achieved de novo design of a T3 sensor, which, unlike the FGF23-based version, functioned in the absence of an upstream glycosylation site.
Sensor Tests in 96-well Format
As a test of the utility of the sensors in high throughput screening for glycosylation inhibitors we adopted our assays to flow cytometry in a 96-well format. Cells expressing either the ANGPTL3-based T2 sensor or the FG23-based T3 sensor were released from 96-well dishes and collected in the presence of the MG dye. The GFP and MG fluorescence per cell for 10,000 cells per well was then determined. To assess whether sensor activation could be detected, the fluorescence distribution resulting from the wild type sensors was compared with that of their corresponding glycine-substituted versions. These experiments were performed prior to our optimization of the FGF23-based T3 sensor, which served the secondary purpose of determining whether significant results were achievable with a low level of fold-activation. Indeed, both the ANGPTL3-based T2 sensor (Fig. 7A) and the FGF23-based T3 sensor (Fig. 7B) showed clear activation using this methodology. Quantification indicated an activation of over 600-fold (7816 S.D.) for the T2 sensor and 2-fold (38 S.D.) for the T3 sensor (Fig. 7C) essentially matching the values determined for these versions using microscopy (Figs. 1N and 3J). Indeed, both the fluorescence histograms and the calculated averages confirm that the difference in fold-activation between the two sensors is primarily in the high background of the T3 sensor supporting the need for optimization of this sensor to improve its glycan masking.
FIGURE 7.

Sensor tests in 96-well format using inhibitor of glycan extension. A and B, flow cytometry-based histogram distribution of MG/GFP ratios is shown for HEK cell lines expressing the indicated versions of either the ANGPTL3-based T2 sensor (A) or the FGF23-based T3 sensor (B). Note that lines in red correspond to wild type (WT) and lines in blue are for sensors lacking glycan acceptor sites. C, averages of the MG/GFP ratios are compared for each sensor version (*, p < 0.05; **, p < 0.001, n = 3 ± S.E.). D and E, histogram distribution of MG/GFP ratios is shown for the wild type T2 (D) and T3 (E) sensors either without or with incubation (6 h) in 5 mm benzyl-N-acetyl-α-galactosaminide (BAG) to inhibit glycan extension. F, averages of the MG/GFP ratios are compared for each sensor with or without BAG treatment (**, p < 0.001, n.s. = not significant, n = 3 ± S.E.). G and H, averages of the MG/GFP ratios are compared for cells expressing the T3 sensor and treated with 5 mm BAG for the times indicated or treated with BAG for 6 h at the concentrations indicated (n = 3 ± S.E.).
Next, to mimic the addition of an unknown drug that might score in a high throughput screen, we repeated the flow cytometry assay on cells expressing either of the wild type sensors after their treatment with benzyl-N-acetyl-α-galactosaminide (BAG), which is an inhibitor of chain extension. If glycan masking in either sensor required a fully extended glycan chain, we expected BAG to activate the sensor. Interestingly, this was the case for only one of the sensors. Whereas the ANGPTL3-based T2 sensor was slightly, if at all, affected by BAG (Fig. 7D), the FGF23-based T3 sensor showed activation (Fig. 7E). Quantification of the effect indicated a statistically significant activation of 1.7-fold and 17 S.D. over untreated (Fig. 7F). This compares well with its 2-fold maximum activation suggesting that this sensor strongly depends on chain extension for its masking. On one hand, the differential effect is surprising in that the glycan site flanks the furin site in ANGPTL3 but resides directly in the FGF23 site (Table 1). Also, unlike ANGPTL3, our minimal first generation sensor was activated by BAG (23) even though the relative positions of its glycan and furin sites are the same as ANGPTL3. On the other hand, the differential effect observed here is in agreement with previous work showing inhibition of processing by a single GalNAc in ANGPTL3 (26) but not FGF23 (19). As confirmation of the effect, we determined its time course (Fig. 7G) and concentration dependence (Fig. 7H). Inhibition was detected within 3 h and with as little as 0.5 mm BAG. In summary, the experiments in this section provide a “proof of principle” for the utility of the sensors in high-throughput screening and also indicate that the importance of glycan chain length for glycan masking can vary depending the target sequence and enzyme involved.
DISCUSSION
Despite the biological and medical importance of the GalNAc-transferases that initiate mucin-type O-glycosylation in the Golgi, effective inhibitors are lacking. Central to the study of the GalNAc-transferases is their presence as a large family of isoforms that show distinct substrate specificities and expression patterns. Diseases arising from GalNAc-transferase misregulation typically involve single isoforms. Also, pan-specific or general inhibitors, whereas desirable as experimental tools, will block all mucin-type O-glycosylation, which is a critically important post-translational modification for normal cell function. Thus, potential therapies will ideally target specific isoforms. With this consideration in mind, we sought to create an isoform-specific, cell-based, fluorescence biosensor to be used for high-throughput screening of potential inhibitors. Toward this goal, isoform-specific GalNAc-transferase target sequences for the T2 and T3 enzymes were used to replace a general consensus sequence in our previously characterized sensor, which works by recapitulating glycan masking (23). The new sensors were activated in the absence of glycosylation and, after modification of the inserted sequences, yielded specific activation in response to ablation of their corresponding enzyme. The T2 sensor used a target sequence in ANGPTL3 and revealed the potential of a glycine substitution flanking the glycan addition site to restrict isoform specificity of the sensor. The T3 sensor was based on a target sequence in FGF23 and its signal to noise ratio was dramatically improved by substitution of a valine next to its glycan acceptor site. Thus, we have developed T2- and T3-specific sensors that will be valuable in both the study of GalNAc-transferase regulation and in high-throughput screening for potential therapeutic regulators of specific GalNAc-transferases.
Optimizing the sensors focuses on the signal to noise ratio and their isoform specificity. In relationship to the insert sequences of the linker domain, the signal is largely a function of the efficiency of glycosylation, masking, and cleavage. GalNAc-transferase isoform specificity relates to differential substrate recognition. Ideally, one would modify the linker sequence to achieve efficient and isoform-specific GalNAc-transferase glycosylation with the site-specific glycosylation placed in or adjacent to the protease recognition site for maximal effect, and a sequence design that contains efficient protease cleavage in the absence of glycosylation. The degree to which these goals are mutually compatible for a particular GalNAc-transferase isoform is critical to successful sensor development. Presumably, these same criteria impacted the evolution of glycan masking sites. In that vein, we were fortunate to have the ANGPTL3 and FGF23 glycan masking sites as starting points for the T2- and T3-specific sensors, particularly as they were already well characterized. We envision extending this to other GalNAc-transferase isoforms and proprotein convertases using known substrate specificities and iterative modifications of the sensors.
Although we did not directly quantify the extent of glycosylation, masking, and cleavage for these sensors, we can conclude that all three aspects were efficient for the ANGPTL3-based T2 sensor given its >500-fold activation. Further improvement seems unnecessary. With one caveat this was also true for the isoform specificity of the sensor. The caveat is that the ANGPTL3 sequence needed to be modified so that it would be specifically activated in HEK ΔT2 cells. Thus, for use in isoform-specific screening, the T225G substitution is imperative. Interestingly, glycine substitution at this position detectably activated the sensor indicated by an increase in the GFP/MG fluorescence ratio from 0.002 to 0.02. This implies that it was now less efficiently glycosylated and that further optimization might be possible, perhaps using alternative substitutions. Nevertheless, even with its higher level of background activation, the modified sensor was activated greater than 50-fold in ΔT2 cells arguing that further optimization is not really needed.
In contrast, the wild type FGF23-based T3 sensor was marginal, yielding only 3- and 2-fold activation in control and ΔT3 cells, respectively. This was due to background activation stemming from any of a number of possibilities including inefficient O-glycosylation, highly efficient furin cleavage, or regulation by phosphorylation. Generally, peptide preferences of the GalNAc-transferases are sensitive to residues from −3 to +3 relative to the glycosylation site with a preference for proline at positions +1 and +3 (27). Furthermore, as mentioned above, the residue at +2 (Ser180) impedes glycosylation when phosphorylated (31). Consistent with these considerations, substitution of glycine at +2 and proline at +3 caused a significant drop in background activation of the T3 sensor (Fig. 4M). However, this version of the sensor activated poorly suggesting the changes caused a problem with furin recognition. We had better luck with valine substitution of a histidine at position −1, which also potently reduced background but did not interfere with activation (Fig. 4M). The presumed increase in glycosylation is consistent with the in vitro preference for a hydrophobic residue at this position (27). We were also fortunate that the FGF23-based T3 sensors showed strong isoform specificity and recapitulated the lectin domain-mediated mechanism by which T3 glycosylates Thr178 in FGF233 thereby raising the possibility of screening for drugs targeting either the catalytic unit or the lectin domain of T3.
In terms of applying the sensors to screening, it is important to mention that just as with sensors of N-glycosylation in the ER that are based on protein folding (32, 33), anything contributing to successful glycosylation of the sensor is a potential drug target in their activation. This includes the extending enzymes, nucleotide sugar transporters, nucleotide sugar biosynthesis, trafficking components (critical for proper delivery of the sensor and localization of both the GalNAc-transferases and furin), and even factors controlling Golgi structure as Golgi structure can influence glycosylation efficiency (34). Interestingly, glycosylation initiated by the addition of GlcNAc is strongly influenced by the metabolic flux of its precursor UDP-GlcNAc (35) but whether availability of UDP-GalNAc features as prominently in regulating mucin-type O-glycosylation is unknown.
In any case, relatively simple controls can be used to identify these types of “off target” hits. One relies on the fact that the sensors are ratiometric and traffic in the secretory pathway. By simply verifying a strong GFP signal at the cell surface, any compound that affects synthesis and trafficking of the sensor can be excluded. More importantly, one can now take advantage of the availability of both T2- and T3-specific sensors. Any compound that activates both sensors is either inhibiting both GalNAc-transferases directly (pan-specific) or it is acting indirectly, perhaps by affecting one of the processes just mentioned. Although such hits could be quite significant, the highest priority should be given to compounds that specifically block a single sensor because they are likely to be acting directly on the corresponding GalNAc-transferase isoform. As we expand the sensor development to other isoforms this aspect will be further enhanced.
An exciting future application of the sensors is in determining conditions that alter a particular glycosylation activity of the isoform. This might be a consequence of a change in signaling state in a differentiated cell type or a change occurring during differentiation. Once a change is identified, the responsible signaling pathway could be dissected using the sensor as an assay. Again, the availability of multiple isoform-specific sensors will allow general (and possibly indirect) effects to be separated from those that are likely direct. The turn on aspect of the sensors we describe here is most suited to signaling that inhibits an active enzyme. One simple way to generate a behavior capable of detecting both up- and down-regulation of a particular enzyme is to weaken the glycosylation site with respect to activity but not isoform specificity. We already saw this with the unmodified FGF23 insert. The background level of activation is significantly higher than an inactive sensor thus conditions that enhance T3 activity will detectably lower the signal (as was the case when we modified the sensor). Thus, once again the extensive in vitro work dissecting the roles of particular residues surrounding the glycan acceptor site in terms of both enzyme binding and specificity of binding will be enormously useful for de-tuning the sensors.
As a criterion for isoform specificity of the sensors, we used specific activation upon deletion of the corresponding isozyme in HEK cells. As mentioned above, all but 6 isoforms have been detected in HEK, so it remains possible that the missing isozymes could also target these sensors, possibly diminishing their utility outside HEK. Nevertheless, the T2- and T3-specific sensors were activated in ΔT2 and ΔT3 cells, respectively, even though these cells likely continued to express isozymes that are close paralogs with highly similar properties (4). Further mitigating this concern is our ability to engineer cell systems with and without a given GalNAc-transferase expressed, as well as, with a repertoire of GalNAc-transferases (25, 30). This should enable sensor activity to be isoform-specific even if one or more isoforms may glycosylate the same substrate.
In summary, this report describes cell-based, fluorescent sensors for two isoforms of GalNAc-transferase. These sensors are promising for high-throughput screening for novel inhibitors. They can also be used to monitor isoform-specific O-glycan masking in living cells under a variety of conditions. Finally, the approach described here can readily be extended to the development of sensors for many of the remaining GalNAc-transferase isoforms. It continues to be our hope that this technology will contribute to therapy development for disorders arising from GalNAc-transferase misregulation.
Acknowledgments
We thank HaiBing Teng and Tim Jarvela for help with the imaging and Tina Lee and Emily Simon for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GM095549 and GM056779 (to A. D. L.), the Novo Nordisk Foundation and Danish National Research Foundation Grant DNRF107 (to H. C.), and the Danish Medical Research Council (to K. T. S.).
K. T. Schjoldager and H. Clausen, unpublished data.
- ANGPTL3
- angiopoietin-like 3
- BAG
- benzyl-N-acetyl-α-galactosaminide
- GalNAc
- N-acetylgalactosamine
- MG
- malachite green
- T1–T20
- UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase 1–20.
REFERENCES
- 1. Schjoldager K. T., Clausen H. (2012) Site-specific protein O-glycosylation modulates proprotein processing: deciphering specific functions of the large polypeptide GalNAc-transferase gene family. Biochim. Biophys. Acta 1820, 2079–2094 [DOI] [PubMed] [Google Scholar]
- 2. van der Post S., Subramani D. B., Bäckström M., Johansson M. E., Vester-Christensen M. B., Mandel U., Bennett E. P., Clausen H., Dahlén G., Sroka A., Potempa J., Hansson G. C. (2013) Site-specific O-glycosylation on the MUC2 mucin protein inhibits cleavage by the Porphyromonas gingivalis secreted cysteine protease (RgpB). J. Biol. Chem. 288, 14636–14646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bennett E. P., Mandel U., Clausen H., Gerken T. A., Fritz T. A., Tabak L. A. (2012) Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paulson J. C., Colley K. J. (1989) Glycosyltransferases. Structure, localization, and control of cell type-specific glycosylation. J. Biol. Chem. 264, 17615–17618 [PubMed] [Google Scholar]
- 6. Gill D. J., Clausen H., Bard F. (2011) Location, location, location: new insights into O-GalNAc protein glycosylation. Trends Cell Biol. 21, 149–158 [DOI] [PubMed] [Google Scholar]
- 7. Fritz T. A., Raman J., Tabak L. A. (2006) Dynamic association between the catalytic and lectin domains of human UDP-GalNAc:polypeptide α-N-acetylgalactosaminyltransferase-2. J. Biol. Chem. 281, 8613–8619 [DOI] [PubMed] [Google Scholar]
- 8. Yoshimura Y., Nudelman A. S., Levery S. B., Wandall H. H., Bennett E. P., Hindsgaul O., Clausen H., Nishimura S. (2012) Elucidation of the sugar recognition ability of the lectin domain of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase 3 by using unnatural glycopeptide substrates. Glycobiology 22, 429–438 [DOI] [PubMed] [Google Scholar]
- 9. Gerken T. A., Revoredo L., Thome J. J., Tabak L. A., Vester-Christensen M. B., Clausen H., Gahlay G. K., Jarvis D. L., Johnson R. W., Moniz H. A., Moremen K. (2013) The lectin domain of the polypeptide GalNAc transferase family of glycosyltransferases (ppGalNAc Ts) acts as a switch directing glycopeptide substrate glycosylation in an N- or C-terminal direction, further controlling mucin type O-glycosylation. J. Biol. Chem. 288, 19900–19914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gill D. J., Tham K. M., Chia J., Wang S. C., Steentoft C., Clausen H., Bard-Chapeau E. A., Bard F. A. (2013) Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc. Natl. Acad. Sci. U.S.A. 110, E3152–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng R. Q., Wan H. Y., Li H. F., Liu M., Li X., Tang H. (2012) MicroRNA-214 suppresses growth and invasiveness of cervical cancer cells by targeting UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase 7. J. Biol. Chem. 287, 14301–14309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kohsaki T., Nishimori I., Nakayama H., Miyazaki E., Enzan H., Nomoto M., Hollingsworth M. A., Onishi S. (2000) Expression of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase isozymes T1 and T2 in human colorectal cancer. J. Gastroenterol. 35, 840–848 [DOI] [PubMed] [Google Scholar]
- 13. Kitada S., Yamada S., Kuma A., Ouchi S., Tasaki T., Nabeshima A., Noguchi H., Wang K. Y., Shimajiri S., Nakano R., Izumi H., Kohno K., Matsumoto T., Sasaguri Y. (2013) Polypeptide N-acetylgalactosaminyl transferase 3 independently predicts high-grade tumours and poor prognosis in patients with renal cell carcinomas. Br. J. Cancer 109, 472–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao Y., Liu Z., Feng J., Sun Q., Zhang B., Zheng W., Ma W. (2013) Expression pattern of polypeptide N-acetylgalactosaminyltransferase-10 in gastric carcinoma. Oncol. Lett. 5, 113–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brockhausen I. (2006) Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep. 7, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brooks S. A., Carter T. M., Bennett E. P., Clausen H., Mandel U. (2007) Immunolocalisation of members of the polypeptide N-acetylgalactosaminyl transferase (ppGalNAc-T) family is consistent with biologically relevant altered cell surface glycosylation in breast cancer. Acta Histochem. 109, 273–284 [DOI] [PubMed] [Google Scholar]
- 17. Chefetz I., Sprecher E. (2009) Familial tumoral calcinosis and the role of O-glycosylation in the maintenance of phosphate homeostasis. Biochim. Biophys. Acta 1792, 847–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schjoldager K. T., Vester-Christensen M. B., Goth C. K., Petersen T. N., Brunak S., Bennett E. P., Levery S. B., Clausen H. (2011) A systematic study of site-specific GalNAc-type O-glycosylation modulating proprotein convertase processing. J. Biol. Chem. 286, 40122–40132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato K., Jeanneau C., Tarp M. A., Benet-Pagès A., Lorenz-Depiereux B., Bennett E. P., Mandel U., Strom T. M., Clausen H. (2006) Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 281, 18370–18377 [DOI] [PubMed] [Google Scholar]
- 20. Kathiresan S., Melander O., Guiducci C., Surti A., Burtt N. P., Rieder M. J., Cooper G. M., Roos C., Voight B. F., Havulinna A. S., Wahlstrand B., Hedner T., Corella D., Tai E. S., Ordovas J. M., Berglund G., Vartiainen E., Jousilahti P., Hedblad B., Taskinen M. R., Newton-Cheh C., Salomaa V., Peltonen L., Groop L., Altshuler D. M., Orho-Melander M. (2008) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 40, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Willer C. J., Sanna S., Jackson A. U., Scuteri A., Bonnycastle L. L., Clarke R., Heath S. C., Timpson N. J., Najjar S. S., Stringham H. M., Strait J., Duren W. L., Maschio A., Busonero F., Mulas A., Albai G., Swift A. J., Morken M. A., Narisu N., Bennett D., Parish S., Shen H., Galan P., Meneton P., Hercberg S., Zelenika D., Chen W. M., Li Y., Scott L. J., Scheet P. A., Sundvall J., Watanabe R. M., Nagaraja R., Ebrahim S., Lawlor D. A., Ben-Shlomo Y., Davey-Smith G., Shuldiner A. R., Collins R., Bergman R. N., Uda M., Tuomilehto J., Cao A., Collins F. S., Lakatta E., Lathrop G. M., Boehnke M., Schlessinger D., Mohlke K. L., Abecasis G. R. (2008) Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 40, 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boskovski M. T., Yuan S., Pedersen N. B., Goth C. K., Makova S., Clausen H., Brueckner M., Khokha M. K. (2013) The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 504, 456–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bachert C., Linstedt A. D. (2013) A sensor of protein O-glycosylation based on sequential processing in the Golgi apparatus. Traffic 14, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santiago Y., Chan E., Liu P. Q., Orlando S., Zhang L., Urnov F. D., Holmes M. C., Guschin D., Waite A., Miller J. C., Rebar E. J., Gregory P. D., Klug A., Collingwood T. N. (2008) Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 105, 5809–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schjoldager K. T., Vakhrushev S. Y., Kong Y., Steentoft C., Nudelman A. S., Pedersen N. B., Wandall H. H., Mandel U., Bennett E. P., Levery S. B., Clausen H. (2012) Probing isoform-specific functions of polypeptide GalNAc-transferases using zinc finger nuclease glycoengineered SimpleCells. Proc. Natl. Acad. Sci. U.S.A. 109, 9893–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schjoldager K. T., Vester-Christensen M. B., Bennett E. P., Levery S. B., Schwientek T., Yin W., Blixt O., Clausen H. (2010) O-Glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3: possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 285, 36293–36303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gerken T. A., Jamison O., Perrine C. L., Collette J. C., Moinova H., Ravi L., Markowitz S. D., Shen W., Patel H., Tabak L. A. (2011) Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J. Biol. Chem. 286, 14493–14507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hassan H., Reis C. A., Bennett E. P., Mirgorodskaya E., Roepstorff P., Hollingsworth M. A., Burchell J., Taylor-Papadimitriou J., Clausen H. (2000) The lectin domain of UDP-N-acetyl-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-T4 directs its glycopeptide specificities. J. Biol. Chem. 275, 38197–38205 [DOI] [PubMed] [Google Scholar]
- 29. Wandall H. H., Irazoqui F., Tarp M. A., Bennett E. P., Mandel U., Takeuchi H., Kato K., Irimura T., Suryanarayanan G., Hollingsworth M. A., Clausen H. (2007) The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology 17, 374–387 [DOI] [PubMed] [Google Scholar]
- 30. Steentoft C., Vakhrushev S. Y., Vester-Christensen M. B., Schjoldager K. T., Kong Y., Bennett E. P., Mandel U., Wandall H., Levery S. B., Clausen H. (2011) Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 8, 977–982 [DOI] [PubMed] [Google Scholar]
- 31. Tagliabracci V. S., Engel J. L., Wiley S. E., Xiao J., Gonzalez D. J., Nidumanda Appaiah H., Koller A., Nizet V., White K. E., Dixon J. E. (2014) Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. U.S.A. 111, 5520–5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Losfeld M. E., Soncin F., Ng B. G., Singec I., Freeze H. H. (2012) A sensitive green fluorescent protein biomarker of N-glycosylation site occupancy. FASEB J. 26, 4210–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Contessa J. N., Bhojani M. S., Freeze H. H., Ross B. D., Rehemtulla A., Lawrence T. S. (2010) Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin. Cancer Res. 16, 3205–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Puthenveedu M. A., Bachert C., Puri S., Lanni F., Linstedt A. D. (2006) GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 8, 238–248 [DOI] [PubMed] [Google Scholar]
- 35. Hardivillé S., Hart G. W. (2014) Nutrient Regulation of Signaling, Transcription, and Cell Physiology by O-GlcNAcylation. Cell Metab. 20, 208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]