

Background: Neuromyelitis optica autoantibodies target the aquaporin-4 (AQP4) aggregate named orthogonal arrays of particles (OAP).
Results: Mutation of AQP4 aspartate 69 (Asp69) impairs NMO-IgG binding leaving the water channel function unaltered as well as its aggregation into OAPs.
Conclusion: Asp69 is the key determinant for the formation of NMO-IgG epitopes.
Significance: Such evidence provides additional clues on NMO pathogenesis.
Keywords: Aquaporin, Astrocyte, Autoimmune Disease, Epitope Mapping, Neurodegenerative Disease, Aquaporin-4, Epitope Determinant, NMO-IgG, Neuromyelitis Optica, Orthogonal Array of Particles
Abstract
Neuromyelitis optica (NMO) is characterized by the presence of pathogenic autoantibodies (NMO-IgGs) against supra-molecular assemblies of aquaporin-4 (AQP4), known as orthogonal array of particles (OAPs). NMO-IgGs have a polyclonal origin and recognize different conformational epitopes involving extracellular AQP4 loops A, C, and E. Here we hypothesize a pivotal role for AQP4 transmembrane regions (TMs) in epitope assembly. On the basis of multialignment analysis, mutagenesis, NMO-IgG binding, and cytotoxicity assay, we have disclosed the key role of aspartate 69 (Asp69) of TM2 for NMO-IgG epitope assembly. Mutation of Asp69 to histidine severely impairs NMO-IgG binding for 85.7% of the NMO patient sera analyzed here. Although Blue Native-PAGE, total internal reflection fluorescence microscopy, and water transport assays indicate that the OAP Asp69 mutant is similar in structure and function to the wild type, molecular dynamic simulations have revealed that the D69H mutation has the effect of altering the structural rearrangements of extracellular loop A. In conclusion, Asp69 is crucial for the spatial control of loop A, the particular molecular conformation of which enables the assembly of NMO-IgG epitopes. These findings provide additional clues for new strategies for NMO treatment and a wealth of information to better approach NMO pathogenesis.
Introduction
Neuromyelitis optica (NMO)2 is an autoimmune disorder of the central nervous system (CNS), causing inflammatory demyelinating lesions mainly in the spinal cord and optic nerve (1, 2). NMO is the first example of aquaporinopathy, aquaporin-4 (AQP4) being the molecular target of the typical autoantibodies (NMO-IgGs) detectable in NMO patients' sera (1, 2).
AQP4 is the water channel protein with the highest single channel water permeability, controlling CNS water and ion homeostasis and expressed at the barriers protecting the brain at the blood and CSF interfaces (3). In particular, it is highly concentrated in the astrocyte processes forming the blood-brain barrier and those forming the glial limitans externa and interna (3). The plasma membrane organization of the AQP4 water channel is unique among AQPs for two reasons. First, all AQPs are formed by homotetramers, whereas AQP4 forms heterotetramers made of two similar isoforms, called AQP4-M1 and AQP4-M23 (4). Second, AQP4 heterotetramers further aggregate to form very well ordered supramolecular structures known as orthogonal arrays of particles (OAPs) (3, 5). AQP4 aggregation of itself into OAPs serves to enhance water permeability in those cellular domains, such as the perivascular one, where this physiological need is correlated to the fluid shifts occurring and to confer a higher level of plasma membrane stability to AQP4 water channels, confined in microdomains not delimited by tight junctions such as the glial processes (6).
Before the discovery of anti-AQP4 autoantibodies, NMO spectrum disorders were mis-classified as multiple sclerosis (MS) variants. In some cases, such as patients with non-longitudinally extensive transverse myelitis not fulfilling MRI criteria for MS or with longitudinal spinal cord lesions and MS-like brain lesions, anti-AQP4 antibodies are the only factor allowing discrimination between MS and NMO with high specificity (7). The NMO-IgG test for the existence of antibodies against the AQP4 antigen has been therefore included in the supportive criteria for NMO diagnosis (8), with 97.5% sensitivity (9). However, their contribution to NMO pathogenesis is still not completely clear. Interestingly, the organization into OAPs is essential for NMO-IgG epitope formation, as AQP4 tetramers are not generally recognized by NMO-IgG (2, 9). A complex OAP-dependent conformational epitope and the NMO-IgG polyclonal origin (10) make it difficult to identify the molecular details of NMO-IgG-mediated pathogenesis. Moreover, AQP4 epitopes may differ between patients. In particular, there are two major conformational extracellular epitopes, generated by OAP-specific inter-tetrameric interaction of extracellular loops A, C, and E (10). Mutations in loop A, or loop C, combined with loop E, reduce NMO-IgG binding, suggesting a key role of OAP-specific interactions at the level of extracellular loops (10). It has been reported that the interaction between NMO-IgG and AQP4-OAPs induces pathogenic mechanisms in NMO by complement-dependent cytotoxicity (CDC), and/or by antibody-dependent cellular cytotoxicity (11–15).
Currently used therapies include classical approaches, such as immunomodulation, immunosuppression, and plasmapheresis (15), whereas a trial is in progress for a monoclonal antibody complement inhibitor (eculizumab) (16). All currently used approaches are therefore not specific for NMO and are associated to negative effects due to the generic suppression of key mechanisms of the immune system. Consequently, it is important to explore alternative strategies, based on the use of specific competitors of NMO-IgG-OAP binding or able to modify the epitope. The aim of this study was research into NMO-IgG epitope key molecular determinants and the identification of AQP4 point mutations able to impair such an epitope, whereas preserving the water channel function and the OAP structure.
EXPERIMENTAL PROCEDURES
Construction of AQP4-AQP0 Chimeras and Site-directed Mutagenesis
The cloning of human AQP4-M23 and AQP0 coding DNA sequences (CDSs) into pTarget Expression Vector (Promega) and human AQP4-M23 into pmCherry-N1 (Clontech) was performed as previously reported (10). AQP0 CDS cloned into pTarget was used for AQP4-AQP0 chimera construction. Extracellular loops A, C, and E, intracellular loops B and D, and the N-terminal region were changed in the homologues of AQP4 by site-directed mutagenesis, using the QuikChange site-directed II kit (Stratagene), according to the manufacturer's instructions, changing up to seven amino acids in a single step. The C-terminal region of AQP0 was changed with the relative AQP4, by PCR and restriction techniques. The KpnI restriction site was used to link two PCR products, then the restriction site was deleted by site-directed deletion. For the generation of AQP4-mutants, AQP4 CDS cloned into pTarget and pmCherry-N1 were used as a template for single point mutation, using the QuikChange site-directed II kit (Stratagene), according to the manufacturers instructions. All constructs were fully sequenced.
Cell Cultures and Transfections
HeLa, V79, and U87MG were grown in Dulbecco's high glucose medium with added 10% fetal bovine serum and penicillin-streptomycin (Invitrogen). To perform transfection experiments the cells were plated at 90–95% confluence and transfected using Lipofectamine reagent (Invitrogen). To obtain stable transfected cell lines, cells were transfected and stable clones were selected by G418 treatment.
Antibodies
Anti-AQP4 antibodies were from Santa Cruz (Sc-9888). Anti-AQP0 antibodies were from ADI. The secondary antibodies for immunoblotting analysis were peroxidase-conjugated IgGs from Santa Cruz, whereas Alexa Fluor 488-conjugated IgGs were used for immunofluorescence.
Patient Sera
NMO patients were diagnosed according to Wingercuck's 2006 criteria (8) and were all relapsing. The multiple sclerosis diagnosis was established according to the McDonald criteria (17). 40 NMO sera samples were used. 21 of these sera were categorized, as reported (10), and called NMO-1 (sera: 2, 3, 5, 8, 15, 17, and 19), NMO-2 (sera: 1, 4, 10, 13, 14, 18, and 21), and NMO-3 (sera: 6, 7, 9, 11, 12, 16, and 20), based on the epitope recognized (Table 3). NMO-1 were those sera showing a predominant role of loop A (residues 61–64), NMO-2 of the tip of the loop C (residues 146–150), and NMO-3 had an equal role as loop A and the tip of the loop.
TABLE 3.
21 NMO sera and relative epitope categories
NMO-1, predominant role of loop A (residues 61–64); NMO-2, predominant role of the tip of the loop C (residues 146–150); NMO-3, equal role of loop A and the tip of loop C.
| Epitope category | NMO sera |
|---|---|
| NMO-1 | 2, 3, 5, 8, 15, 17, 19 |
| NMO-2 | 1, 4, 10, 13, 14, 18, 21 |
| NMO-3 | 6, 7, 9, 11, 12, 16, 20 |
Immunofluorescence and Microscopy Analysis
Briefly, HeLa, V79, or U87MG cells were first fixed with 4% paraformaldehyde in PBS for 10 min, permeabilized with 0.3% Triton X-100 in PBS for 10 min, and finally saturated with PBS added with 0.1% gelatin for 10 min. The incubation with primary and secondary antibodies was performed as previously described (9). For the immunofluorescence analysis of NMO-IgG binding, unfixed cells were used as previously described (9). In reported cases, PBS, pH 5.5, was used during NMO-IgG binding. For immunofluorescence analysis of oocytes, 3 days after the injection, they were fixed in 4% paraformaldehyde, 0.1 m sucrose for 4 h at room temperature, cryo-protected in 30% sucrose in PBS at 4 °C overnight, mounted in OCT, and sectioned in 10-μm slices. Immunostained cells were observed with a photomicroscope equipped for epifluorescence (DMRXA; Leica) and digital images were obtained with a DMX 1200 camera (Nikon, Tokyo, Japan).
Total Internal Reflection Fluorescence (TIRF) Microscopy Analysis
Transfected HeLa or V79 cells were stained with commercial AQP4 antibodies, or NMO serum, as described above, and analyzed as follows. A Nikon laser TIRF setup was used, consisting of a 488-nm argon laser mounted on a Nikon laser TE2000U microscope, which also allows phase-contrast and epifluorescence techniques to be combined with TIRF technology. An incidence angle greater than the critical angle was achieved by the use of a ×100 CFI Plan Apo with a numerical aperture of 1.45. Fluorescence excited by TIR evanescent field ∼100 nm) was collected with the same objective, and images were collected using a cooled charge-coupled device camera (Hamamatsu Orca). The TIRF signal was measured in 10 independent areas of transfected cells.
Purification of Anti-M23 D69H NMO-IgG
V79 cells stably transfected with AQP4 D69H-mCherry, grown into two 15-cm dishes, were trypsinized, washed twice in PBS, and incubated with 500 μg of purified NMO-IgG (Melon-IgG purification kit) for 1 h at room temperature, with a rotary shaker. Cells were collected by centrifugation (1,500 × g, 5′) at room temperature, the supernatant (NMO-IgG D69H(−)) was recovered and cells were washed twice with PBS. Elution of D69H high affinity NMO-IgG (NMO-IgG D69H(+)), was performed with 1 ml of 0.1 m NaCl, 0.1 m Gly-HCl, pH 2.5, for 10 min at room temperature in a rotary shaker. Immediately after elution, 1/10 volume of 1 m Tris-HCl, pH 8.5, was added to the solution to neutralize the pH.
CDC Assay
Live stable transfected cells were incubated for 1 h in a cell incubator (37 °C, 5% CO2), in Hanks' balanced salt solution (Invitrogen), 5% human complement (Sigma), and human NMO serum (1:50) or purified 50–1600 μg/ml of IgG (by Melon-IgG purification kit) was added. Cells were washed in PBS/Ca2+-Mg2+, and dead cells were stained with 4 μm ethidium homodimer (Invitrogen) in PBS/Ca2+-Mg2+. The percent of dead cells was evaluated by fluorescence microscopy analysis, by counting red-stained nuclei with respect to total cells visualized by phase contrast; 10 independent fields were acquired.
Blue Native-PAGE
Blue Native-PAGE was performed as previously reported (10). Briefly, cells were lysed into 7–10 volumes of BN buffer (BN buffer: 1% Triton X-100, 12 mm NaCl, 500 mm 6-aminohexanoic acid, 20 mm BisTris, pH 7.0, 2 mm EDTA, 10% glycerol), with added protease inhibitor mixture (Roche Diagnostic Gmbh, Mannheim, Germany) on ice vortexing every 10 min for 1 h and centrifuged at 22,000 × g for 30 min at 4 °C. Twenty micrograms of protein sample were mixed with 5% Coomassie Blue G-250 and loaded in polyacrylamide native gradient gels (4–9 or 3–13%). At the end of the run, the gel was blotted onto a PVDF (Millipore, Bedford, MA) membrane for Western blot analysis.
AQP4 cRNA Synthesis and Xenopus laevis Water Transport Assay
X. laevis oocyte preparation, cRNA synthesis, and oocyte injections were performed as described (18). Briefly, oocytes were surgically removed from anesthetized (2 g/liter of Tricaine; Sigma) X. laevis and defolliculated as reported (18). The mMessage mMachine T7 in vitro transcription kit (Ambion, Austin, TX) was used to produce cRNA from each construct for oocyte injection. cRNAs were injected into single oocytes using an automated microinjector (Nanoject; Drummond Scientific, Broomall, PA), and Pf was measured (18).
Water Transport Measure by Fluorescence Quenching Assay
Cells were seeded on black, clear bottom 96-well plates (Corning) at a density of 12,000 cells/well and used 24 h after plating. 80–85% confluent cells were washed with PBS and incubated at 37 °C for 45 min with 10 μm membrane permeable calcein-AM (Molecular Probes, Eugene, OR) as previously described (19). Calcein fluorescence was recorded on a FlexStation3 plate reader equipped with an integrated liquid handling module (Molecular Devices, MDS Analytical Technologies) able to transfer compounds from a source plate to the assay plate during data acquisition. Cells were rinsed in 60 μl of isosmolar PBS and osmotic gradients were applied 15 s after the beginning of each reading by addition of an appropriate volume of mannitol to reach 450 mosmol final osmolarity. Time course fluorescence data following mixing of cells with hyperosmotic solution were recorded over a 90-s period. Data acquisition was performed by SoftMaxPro software, and the data were analyzed using Prism (Graph Pad) software. The time constant of cell shrinkage was obtained by fitting the data to an exponential function.
Molecular Dynamics
The initial structure of AQP4 was obtained from the Protein Data Bank (PDB entry 3GD8) (20). The obtained crystal was first pretreated using the MAESTRO protein preparation module (version 9.5) (43), which enables missing hydrogen atoms to be added and the optimal protonation states for histidine residues to be determined. The simulation system was built as follows. A 120 × 120 Å2 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer patch was first built using the membrane plug-in for visual molecular dynamics (VMD) (21), with the membrane normal along the z axis. A tetramer of AQP4 was embedded in this bilayer and lipid molecules within 0.8 Å of heavy atoms of the protein were removed. To neutralize the system, 23 Na+ and 19 Cl− ions were added using the visual molecular dynamics autoionized plug-in, generating 100 mm ionic concentration and a final system of 135,833 atoms (number computed for wild type). Both mutated and WT protein structures were incorporated into a periodic box of TIP3P water molecules (22) extended by 18 Å in each direction from all protein atoms using the Add Solvation Box plug-in of VMD. To consider both tautomeric states of the histidine residue, two different mutated forms were built, namely D69HSD and D69HSE, in which the asparate in position 69 was substituted for histidine with a hydrogen atom at positions δ and ϵ, respectively. All molecular dynamics (MD) simulations were performed using NAMD 2.9 (23) and the CHARMM27 force field (24). The full system was minimized to remove steric clashes in the initial geometry and gradually heated up to 310 K within 500 ps of MD. The SHAKE algorithm was employed to constrain all R-H bonds. Periodic boundary conditions were applied in all directions. A non-bonded cut-off of 12 Å was used, whereas the Particle-Mesh-Ewald (25) was employed to include the contributions of long-range interactions. All simulations were performed in an isothermal-isobaric ensemble (1 atm, 310 K) with a Nosè–Hoover Langevin barostat (26, 27) (oscillation period 200 fs, decay coefficient 100 fs) and a Langevin thermostat (28) (damping coefficient 1 ps−1). The time step was set to 2 fs, and coordinates were saved every 5000 steps (10 ps). A MD trajectory of 20 ns was obtained for the wild type and each mutated form. For each investigated system, the equilibration of the structure required less than 5 ns and thus the first 5 ns were removed from the analysis. All simulations were performed on the FERMI supercomputer at CINECA, Italy.
Immunoprecipitation
Rat brain (cerebral cortex) membrane vesicles and skeletal muscle light microsome vesicles were prepared as reported (39). Both AQP4-transfected cells and plasma membrane vesicles were extracted using IP buffer (500 mm 6-aminohexanoic acid, 50 mm imidazole pH 7, 2 mm EDTA, 150 mm NaCl, 3% n-dodecyl β-d-maltoside), and added with protease inhibitor mixture (Roche Diagnostic Gmbh, Mannheim, Germany). To immunoprecipitate a comparable amount of brain and skeletal muscle total AQP4, we first assessed by Western blot that 1.25 mg of skeletal muscle extract contains the same AQP4 amount as 18 μg of brain. To immunoprecipitate the same amount of total protein, brain extract was added with 1.25 mg of AQP4-null mice brain extract. Samples were then adjusted to 1 ml with IP buffer and processed as reported (10), using AQP4 commercial antibody (1:500) and NMO serum (patient 5, 1:1000). Densitometric analysis of immunoprecipitated signals was performed with ImageJ software.
Molecular Visualization System
PyMOL software was used to visualize the AQP4 crystal structure and position of mutated residues (PDB entry 3GD8).
Ethics Statement, Animals
All experiments conformed to international guidelines on the ethical use of animals and were designed to minimize the number and suffering of animals used (European Council Directive of 24 November 1986 (86/609/EEC)). Experiments in this study were approved by the Italian Health Department (Art. 9 del Decreto Legislativo 116/92).
Statistical Analysis
Mean ± S.E. of groups were evaluated using GraphPad Prism 5 (GraphPad) by Student's t test. A p value < 0.05 was considered statistically significant.
RESULTS
NMO-IgGs Do Not Recognize Chimeras Made by AQP0 Transmembrane Domains and AQP4 Loops and C and N Termini
To gain insights into AQP4 epitopes recognized by NMO-IgG at the molecular level, we tried to reproduce step by step the epitope in another OAP forming AQP, namely AQP0, not recognized by NMO-IgG as previously demonstrated (2, 10). To this aim, seven AQP4-AQP0 chimeras were produced (Table 1) and expressed in HeLa cells to evaluate NMO-IgG binding by immunofluorescence using four high titer representative NMO sera (sera numbers 1, 2, 5, and 6). Fig. 1 shows the absence of NMO-IgG binding to the chimera where AQP0 N and C terminus and all loops were substituted with those of AQP4. These data suggest that AQP4 transmembrane (TM) domains may have a crucial role in the generation of AQP4-OAP conformational NMO-IgG epitopes.
TABLE 1.
AQP4-AQP0 chimeras
Complete list of AQP4-AQP0 chimeras analyzed by immunofluorescence for the NMO-IgG binding.
| AQP4 sequences | AQP0 sequences |
|---|---|
| Loops A-C-E | Loops B-D, TM1–2–3–4, C-terminal, N-terminal |
| Loops A-C-E, N-terminal | Loops B-D, TM1–2–3–4, C-terminal |
| Loops A-C-E, C-terminal | Loops B-D, TM1–2–3–4, N-terminal |
| Loops A-C-E, N- and C-terminal | Loops B-D, TM1–2–3–4 |
| Loops A-B-C-E, N- and C-terminal | Loops D, TM1–2–3–4 |
| Loops A-C-d-E, N- and C-terminal | Loops B, TM1–2–3–4 |
| Loops A-B-C-d-E, N- and C-terminal | TM1–2–3–4 |
FIGURE 1.
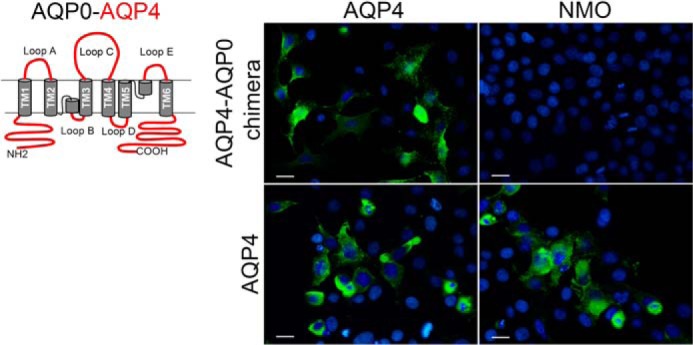
Immunofluorescence analysis of NMO-IgG binding using AQP4-AQP0 chimera. Left, AQP0-AQP4 chimera was made with AQP0 transmembrane regions (gray) and AQP4 loops, N and C terminus (red). Right, immunofluorescence was performed using anti-AQP4 antibodies (AQP4) or NMO sera (NMO), both in green. DAPI staining is in blue. NMO-IgG containing NMO sera did not recognize this chimera. Scale bars, 10 μm.
AQP4 Mutated at TM2 Assembles in OAPs but Is Not Recognized by NMO-IgG
The hypothesis that TMs could contribute to the spatial position of the extracellular loops, which are crucial for NMO-IgG epitope formation (10), was tested. A multialignment analysis of AQP4 from fish to mammals, and between mammals of different species, was performed. As shown in Fig. 2, the TMs are extremely conserved from fish to human, whereas extracellular loops are not. To test our hypothesis, we focused on TM amino acids at the anchor point with the extracellular loops. In particular, the highest conserved and AQP4 specific (data not shown) amino acids connected to loops A (D69MV) and C (H158GLL) were chosen because loops A and C play a crucial role in the formation of NMO-IgG epitopes (10) (Fig. 2). These residues were mutated in the homologues of human AQP0 and three mutants were produced, called AQP4-TM2, AQP4-TM4, and AQP4-TM2-TM4. In AQP4-TM2 the sequence D69MV of AQP4, was replaced with the H69VL of AQP0; in AQP4-TM4, the H158GLL of AQP4, was replaced with the Q158ATT of AQP0; both sequences were replaced in AQP4-TM2–TM4. Cells transfected with wild type and mutated AQP4 were analyzed by BN-PAGE and TIRF microscopy (Fig. 3), two techniques useful to study OAP characteristics (3). The biochemical BN-PAGE analysis (Fig. 3A) showed that the two mutants aggregate into supramolecular structures comparably to wild type AQP4. The TIRF microscopy analysis performed after staining the cells with AQP4 antibodies (Fig. 3B, top), confirmed correct plasma membrane targeting, and the typical OAP-like punctuated staining for both mutants, in line with BN-PAGE results and suggesting that the mutations do not grossly affect AQP4-OAP formation. The mutants were then tested for NMO-IgG binding using each serum (at both 1:100 and 1:1000 dilution) from a collection of 40 NMO sera. All these sera were able to recognize AQP4-TM4 but 37 of 40 were not able to bind AQP4-TM2 and AQP4-TM2-TM4.
FIGURE 2.

Multialignment analysis of AQP4 primary sequences from different species. The TM regions are conserved in different species. The residues indicated in TM2 and TM4 were identified as highly conserved in the close proximity of loops A and C.
FIGURE 3.

OAP analysis of AQP4-TM2 (D69MV > HVL) and AQP4-TM4 (H158GLL > QATT) by BN-PAGE and TIRF microscopy. A, BN-PAGE analysis shows that both mutants are able to form supramolecular structures similar to wild type AQP4. B, analysis by TIRF microscopy of cells transfected with AQP4, AQP4-TM2, and AQP4-TM4 and stained with AQP4 antibodies (AQP4) and NMO sera (NMO). Note that AQP4-TM2 is not recognized by NMO-IgG. Scale bars, 2.5 μm. C, the structure of the wild type and mutated tetramers as rendered by PyMOL. The mutated regions are indicated by white arrows.
A representative image of NMO-IgG binding of the majority of the sera to AQP4 mutants is shown in Fig. 3B, bottom. An analysis by PyMOL of the mutation position in the tetramer model is shown in Fig. 3C. Globally, these results indicate that TM2 plays an important role in NMO-IgG epitope formation without substantially affecting AQP4 tetramer aggregation into OAPs.
The Mutation of Aspartate 69 at TM2 Impairs NMO-IgG Epitope without Affecting AQP4-OAP Water Transport Properties
After the indication that three amino acids of the AQP4-TM2 are able to impair NMO-IgG binding, we tested the hypothesis whether a single amino acid residue may have a major role in disrupting the NMO-IgG conformational epitope. Three different mutated AQP4s were therefore produced accordingly (D69H, M70V, and V71L). Moreover, two other mutations were tested in parallel, V68I and Q86R, with the aim of testing the Asp69 upstream and downstream regions in NMO-IgG epitope assembly. All AQP4 mutants, expressed in cells and analyzed for NMO-IgG binding by immunofluorescence as described above, revealed that only D69H was able to strongly affect NMO-IgG binding of the 37 of 40 NMO patients, whereas no effect on NMO binding was observed for all other mutants. To obtain additional information on the role of Asp69 we generated another mutation, D69E, in which Asp69 was replaced with glutamate. A similar analysis of the D69E mutant showed a minor impact on the NMO epitope compared with D69H, NMO-IgG binding still being present although clearly reduced (Table 2, Fig. 4A).
TABLE 2.
Single amino acid mutants in AQP4 TM2 region tested for NMO-IgG binding by immunofluorescence
The sera were tested separately at 1:100 or 1:1000 dilutions.
| M23 mutants (AQP4 > AQP0) | Region | 40 NMO sera |
|---|---|---|
| Asp69 > His | TM2 (near loop A) | Strongly reduced binding, 37/40 |
| Asp69 > Glu | TM2 (near loop A) | Moderately reduced binding, 37/40 |
| Met70 > Val | TM2 (near loop A) | Yes binding, 40/40 |
| Val71 > Leu | TM2 (near loop A) | Yes binding, 40/40 |
| Val68 > Ile | Loop A | Yes binding, 40/40 |
| Gln86 > Arg | TM2 (central region of TM2) | Yes binding, 40/40 |
FIGURE 4.
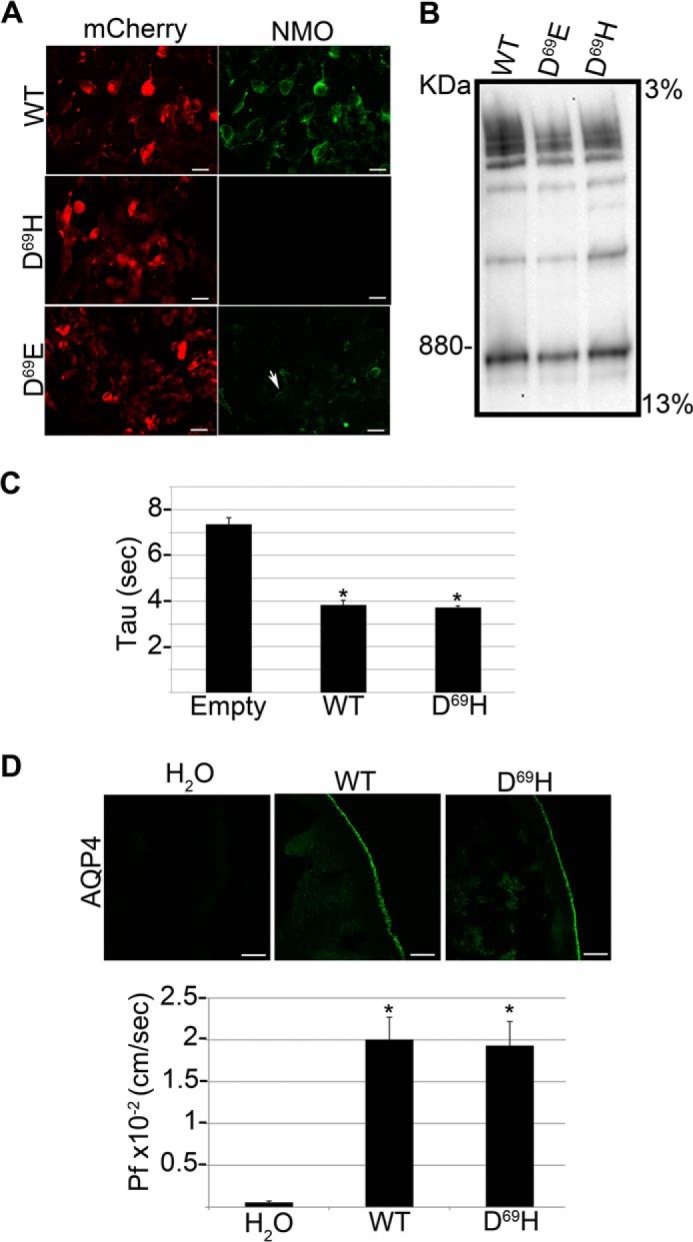
Analysis of NMO-IgG binding, supramolecular aggregates, and water transport properties of AQP4 D69H and D69E. A, immunofluorescence images of NMO-IgG binding (NMO, green) on mCherry tagged (mCherry, red) wild-type (WT), AQP4 D69E and D69H. Note that NMO-IgG binding is absent in D69H and strongly reduced in AQP4 D69E. Scale bars, 10 μm. B, BN-PAGE followed by AQP4 immunoblotting analysis of the same transfected cells shown in A. The supramolecular aggregates formed by the mutants are grossly comparable with those formed by wild type AQP4. C, water transport measured by a fluorescence quenching assay using empty vector (Empty), wild type AQP4 (WT), and AQP4 D69H (D69H)-transfected cells. The histogram shows the mean ± S.E. of the time constant of cell shrinkage (Tau) expressed in seconds. Tau is comparable between WT and D69H expressing cells (*, p < 0.005 versus Empty, n = 10). D, top panel: AQP4 immunofluorescence analysis of X. laevis oocytes injected with water (H2O), wild type AQP4 cRNA (WT), and AQP4 D69H cRNA (D69H). Bottom, histogram showing the mean ± S.E. of the corresponding Pf value. Both AQP4 D69H plasma membrane expression levels and Pf values are comparable with the wild type AQP4 (*, p < 0.005 versus H2O injected, n = 10).
The role of Asp69 was further evaluated on AQP4 suprastructures and water transport. BN-PAGE confirmed that the mutation did not induce any apparent alteration in OAPs (Fig. 4B). Furthermore, two different functional assays were used to measure the impact of the mutations on AQP4-mediated water transport. The TIRF microscopy assay, used to measure swelling kinetics of transfected cells, did not reveal any difference between the wild type and mutated AQP4 (Fig. 4C). This result was further confirmed by the X. laevis oocyte assay, used to measure the osmotic permeability coefficient (Pf) of oocytes injected with wild type and mutated AQP4 (Fig. 4D). These data demonstrate that Asp69 is a pivotal amino acid for NMO-IgG epitope formation, and that its mutation does not affect AQP4 capability to form supramolecular structures or AQP4 water channel properties.
AQP4-D69H Is Able to Reduce the AQP4 Binding Affinity for NMO-IgG in 85.7% of Patients, and Strongly Reduces NMO-IgG-mediated Cytotoxic Effect—
After the characterization of D69H as a mutation able to strongly affect NMO-IgG binding while preserving AQP4 function, the binding affinity of NMO-IgG to AQP4-D69H was analyzed in more detail using lower dilutions of the sera. To this end, the 21 sera characterized based on the epitope categories (10) (Table 3), were tested using an immunofluorescence limiting dilution assay, using dilutions ranging from 1:10 to 1:32,000, on AQP4 WT and D69H expressing cells. Fig. 5A shows the results obtained for each serum. A serum-specific limiting dilution binding for AQP4 and AQP4-D69H was identified. For 18 of 21 patients, D69H limiting dilution was very low, from 2 (serum 16), to 80 (serum 2) times lower, compared with those observed for WT, confirming a low affinity of NMO-IgG for the AQP4-D69H mutant. Only three sera (10, 20, and 21) showed different behavior; sera 20 and 21 revealed a ratio close to 1, and serum 10 showed an AQP4-D69H binding four times more efficient compared with WT. There was no correlation between those three sera and the epitope category (Fig. 5A, Table 3). These results indicate that the AQP4-D69H mutation is able to reduce the AQP4 binding affinity for NMO-IgG in the majority (85.7%) but not in all patients, further supporting a heterogeneous, as yet unclear, nature of the NMO-IgG epitope between patients.
FIGURE 5.
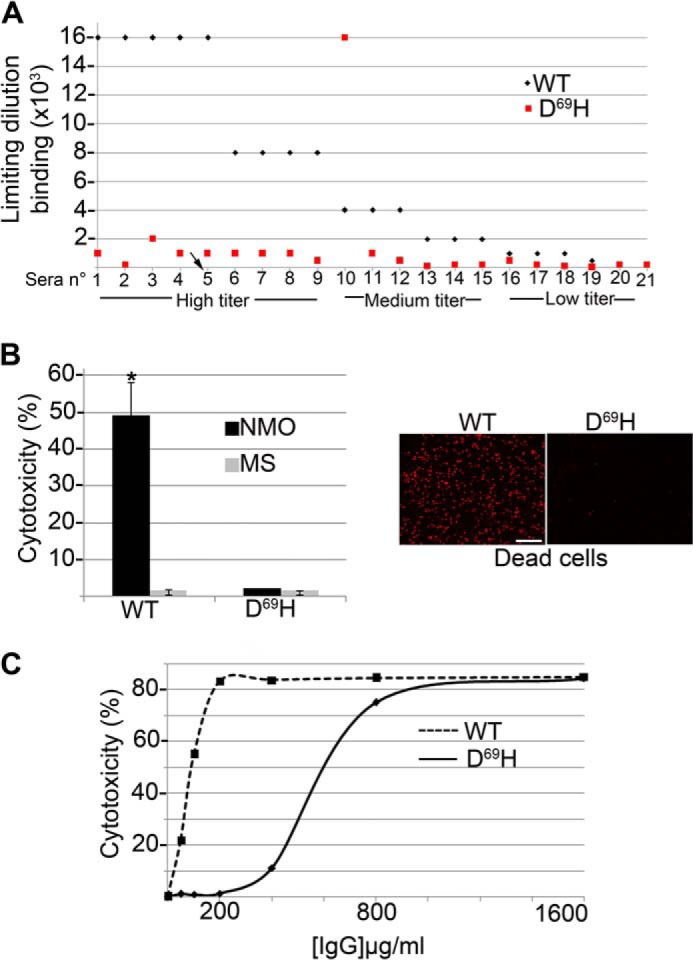
Quantitative analysis of NMO-IgG binding affinity and CDC to AQP4 D69H. A, limiting dilution measured by immunofluorescence using AQP4 (WT) and D69HAQP4 (D69H) expressing cells for 21 NMO patient sera with high (1–9), medium (10–15), and low (16–21) NMO-IgG titer. Sera number 5 (arrow) has been used for the assays shown in C. B, left: histogram showing the results of CDC assay performed on D69H and WT-transfected cells using NMO and control MS sera at 1:50 dilution (*, p < 0.005, n = 15). Right, representative images of dead cells (red stained nuclei). Scale bar, 40 μm. C, CDC assay using purified IgG from a representative NMO serum (patient 5, panel A). Note that the D69H mutation strongly reduces CDC that appears only at high NMO-IgG concentration.
Effect of the D69H mutation was finally tested in NMO-IgG-mediated CDC assays (Fig. 5, B and C). AQP4 and AQP4-D69H expressing cells were tested by CDC using 15 representative NMO, including sera 10, 20, and 21, and MS sera at the usual dilution for a CDC assay (1:50). None of the tested sera induced CDC on AQP4-D69H as for the WT AQP4 (Fig. 5B). Furthermore, when purified antibodies (from NMO patient 5, arrow in Fig. 5A) were used, the maximum cytotoxic effect of NMO-IgG on the D69H mutant was only obtained with very high concentrations of the NMO-IgG (Fig. 5C), with a shift of the IC50 from 50 to 700 μg/ml. These data confirm that the lower affinity of NMO-IgG for the D69H form compared with WT-AQP4 is associated to a strongly reduced cytotoxic effect on the mutant expressing cells.
The Effect of the Asp69 Mutation on NMO-IgG Binding Is pH Independent
In the physiological pH range, the aspartate lateral chain is always negatively charged, whereas the charge of the histidine side chain (imidazole ring) is pH-dependent. Consequently, the D69H mutation offers pH-mediated changes in the imidazole ring of histidine 69. To test whether the pH range is able to modify D69H NMO-IgG binding, immunofluorescence limiting dilution assay was performed using a representative NMO serum (patient 5), on AQP4 and D69H expressing cells. pH 5.5 and 7 were used because the imidazole ring is neutral at pH 7, whereas it is completely positively charged at pH 5.5. Fig. 6 shows that AQP4 versus D69H NMO-IgG binding is pH independent in the tested pH range. Despite a low pH value, pH 5.5 was tested, no alteration of NMO-IgG binding was identified.
FIGURE 6.
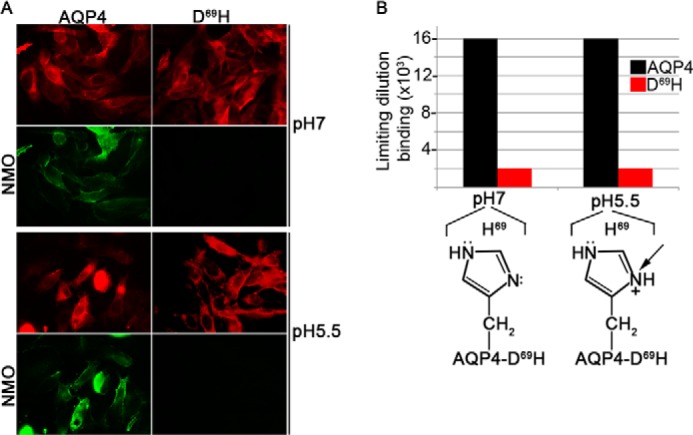
Analysis of NMO-IgG binding at pH 7 and 5.5 using AQP4 and D69H expressing cells. NMO-IgG binding, of serum from a representative NMO patient (patient 5), was tested by changing the histidine side chain from neutral (pH 7) to positive charge (pH 5.5), using AQP4 and D69H expressing cells. Panel A shows a representative result obtained at 1:4000. Panel B shows a dilution binding assay. Note that the NMO-IgG binding property is pH independent at tested pH.
Characterization of a Minor NMO-IgG Population Able to Bind AQP4-D69H
Although strongly reduced, we still found NMO-IgG binding to Asp69 expressing cells. The tested hypothesis was that NMO-IgGs are composed of a mixed population of IgG able to recognize different AQP4-OAP epitopes. In particular, populations of NMO-IgG not able to bind AQP4-D69H co-exist with different NMO-IgG populations whose binding capacity is not affected by the mutation under analysis. This second putative NMO-IgG population was purified (using total IgG from patient 5) by affinity using AQP4-D69H expressing cells. In particular, the affinity purification protocol allowed us to separate NMO-IgG D69H(+) (binding D69H expressing cells) and NMO-IgG D69H(−) (not binding D69H expressing cells). Fig. 7 shows the immunofluorescence experiments confirming the hypothesis of the existence of two NMO-IgG populations. We next tried to gain insight into the percent of the NMO-IgG D69H(+) population on the total of NMO-IgG by the limiting dilution experiments described in Table 4, reporting the frequency of NMO-IgG D69H(+) and relative affinity, measured by limiting dilution immunofluorescence. NMO-IgG D69H(+) represent up to around 2% of total NMO-IgG, and are able to bind AQP4 and AQP4-D69H with the same binding affinity. To test whether NMO-IgG D69H(+) were able to bind tetramer-specific AQP4 extracellular epitopes, analyses were carried out using immunofluorescence on cells selectively expressing AQP4-M1 tetramers (4). No binding was observed (data not shown), concluding that NMO-IgG D69H(+) recognizes a specific OAP-associated extracellular epitope.
FIGURE 7.
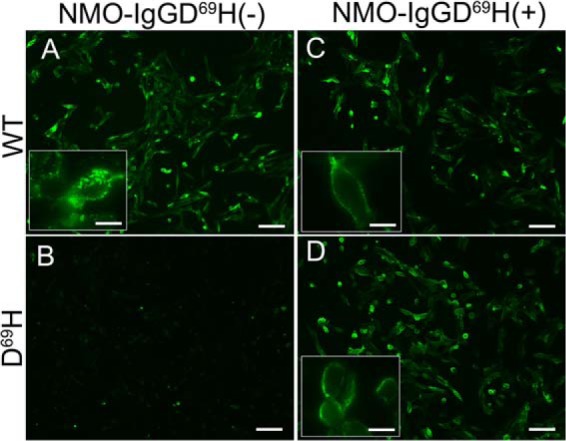
Immunofluorescence analysis of D69HAQP4 binding NMO-IgG population. Immunofluorescence experiments performed on AQP4 (WT) and D69HAQP4 (D69H) transfected cells using NMO-IgG D69H(+) and NMO-IgG D69H(−) were purified as described. Note that both NMO-IgG populations comparably bind AQP4 WT, whereas only NMO-IgG D69H(+) binds the mutant. Scale bars, 20 μm, 5 μm for insets.
TABLE 4.
Limiting dilution experiments for different NMO-IgG populations
Limiting dilution (in ng/μl) of total NMO-IgG (NMO-IgG), NMO-IgG not binding D69H expressing cells (NMO-IgG D69H(−)), and NMO-IgG binding D69H-AQP4 expressing cells (NMO-IgG D69H(+)), tested on WT-AQP4 (WT) and D69H-AQP4 (D69H) expressing cells. Note that NMO-IgG D69H (+) represent 2% of total NMO-IgG, and recognizes WT and D69H with the same affinity.
| Cell line | Limiting concentration (ng/μl) |
|
|---|---|---|
| WT | D69H | |
| % | ||
| NMO-IgG | 0.2 | 10 (2) |
| NMO-IgG D69H(−) | 0.2 | 83 (0.2) |
| NMO-IgG D69H(+) | 0.2 | 0.2 (100) |
Molecular Dynamics Simulations Suggest That Asp69 Is Crucial for Loop A Spatial Position
An in depth analysis of the obtained MD trajectories was carried out to obtain a molecular explanation of how the mutations in position Asp69 lead to the disruption of the epitope structures. Notably, beyond the wild type (WT), four different mutations were investigated: D69HSD, D69HSE, D69E, and M70V. M70V was analyzed as negative control of simulations, because it was found not to affect the NMO-IgG epitopes in the experiments (Table 2). As described under “Experimental Procedures,” the investigated tetramer is, by construction, symmetric with respect to the straight line passing through the center of the system and along the z axis (Fig. 8A). In other words, it is expected that the distance of a given residue with respect to the central z axis is almost equal in the four monomers. With this supposed symmetry in mind, the global effect of the considered mutations was investigated by focusing on the average distances between corresponding residues in the four monomers. Fig. 8B shows the values obtained when considering the distance between the Cα of a given residue of a given monomer and the Cα of the same residue belonging to the specular monomer (Fig. 8A). Notably, an average value along the considered trajectory was taken into account for both occurrences (monomer a versus monomer b and monomer c versus monomer d, see Fig. 8A) and the two obtained values were further averaged to obtain a single parameter for each residue, hereafter named CαAV. As itemized in Fig. 8B, data for the four mutated forms are almost equal with respect to the wild type except for few residues just upstream the mutation, ranging from 60 to 70. Fig. 7C shows the data concerning this segment of loop A. Although for all the considered forms, the mutation at position 69 (D69E, D69HSD, and D69HSE) or 70 (M70V) has a strong effect on the same residues, two different tendencies can be detected. Indeed, CαAV distances higher than wild type can be observed for all the mutated forms, except for M70V, the sole mutation, which has been experimentally proved not to affect AQP4 NMO-IgG binding. In this case, values slightly lower (from residue 62 to 65) and almost equal (from residue 66 to residue 70) with respect to WT (Fig. 7C) were detected. In this respect, the low S.D. values related to these average distances strongly support the non-randomness of the tendencies described herein, ranging from 0.018 Å (residue 70 in D69HSE) to 0.130 Å (residue 64 in D69E). This is also supported by the time-dependent evolution of the Cα distances within the time span of the simulations. An example is given by an extracted sample trajectory (from 15 to 20 ns) for residue 62 (monomer a versus monomer b) in WT, M70V, and D69HSD (Fig. 8D). It is then evident that D69HSD assumes a different state with respect to WT and M70V during the simulation. Importantly, the observed effect involves not only the backbone, but also the side chains of the considered residues, as shown in Fig. 8E, where the average distances between the centers of mass of each residue is considered. In summary, our MD simulations point out that histidine replacing Asp69 triggers a different conformational state for loop A, which is normally protruding inside the central part of the pore. In particular, the mutation in Asp69 increases the mobility of loop A thus affecting the conformational interactions involving loops C and E (Fig. 8F). This data suggests that the altered interaction between these extracellular loops might affect NMO-IgG conformational epitopes.
FIGURE 8.

Molecular dynamics simulations of WT, D69E, D69HSD, D69HSE, and M70V mutants. A, lateral and top view with monomer labeling of the simulated system. The water molecules, ions, and plasma membrane bilayer have been removed for simplicity. Each monomer of the tetramer is shown in a different color. B, CαAV values computed for all the residues of the investigated system: WT (black), D69E (red), D69HSD (blue), D69HSE (green), and M70V (magenta). All values are expressed in Å. C, close-up view of CαAV computed for residues 60–70. D, time-dependent evolution of Cα distance (monomer a versus monomer b) computed for residue 62 within a sample MD trajectory (from 15 to 20 ns) in WT, M70V, and D69HSD. E, close-up view of center of mass distance values computed for residues 60–70. F, lateral views of selected snapshots showing different conformations of loop A: WT (closed), D69E (open), D69HSD (open), D69HSE (open), and M70V (closed).
NMO-IgG Affinity for D69H and Skeletal Muscle AQP4
NMO-IgG lower affinity for D69H was also quantitatively analyzed by immunoprecipitation experiments performed as previously described (10). In parallel, to get insight into the tissue specificity of NMO pathogenesis, the same approach was also used to measure the difference in NMO-IgG affinity between brain and skeletal muscle AQP4. A representative NMO serum (patient 5) was used to immunoprecipitate AQP4 from cells transfected with WT and mutated AQP4 and from rat brain and skeletal muscle. Results obtained shown in Fig. 9 confirm lower NMO-IgG affinity for D69H and in parallel reveal a lower affinity of NMO-IgG for skeletal muscle compared with brain AQP4.
FIGURE 9.
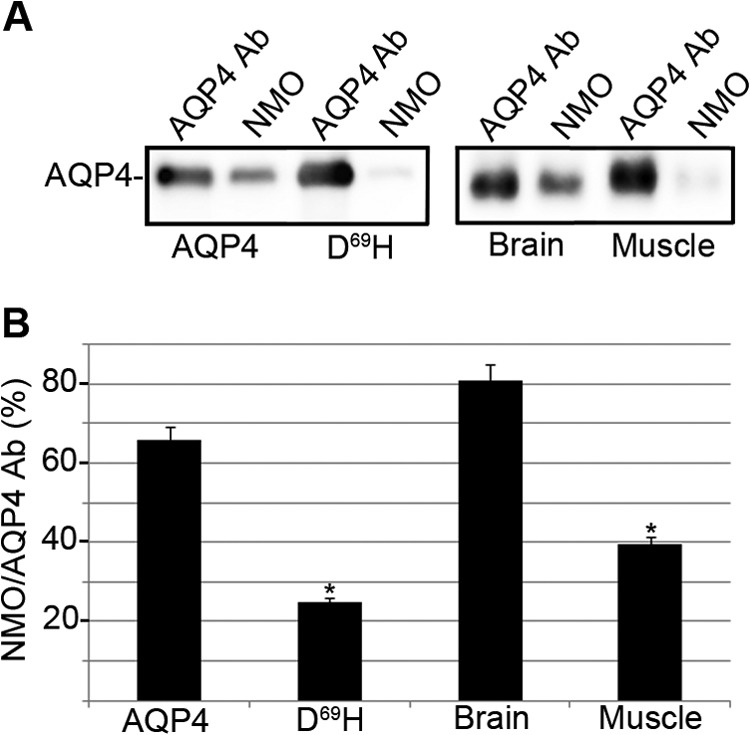
AQP4 immunoprecipitation from AQP4 and D69H expressing cells and from brain and muscle tissues using NMO serum. A, AQP4 IP experiments performed using NMO serum (NMO) and anti-AQP4 antibodies (AQP4 Ab) from AQP4 and D69H-transfected cells and from brain and skeletal muscle AQP4. B, densitometric analysis showing NMO/AQP4 Ab as the percentage of AQP4 immunoprecipitated with NMO-IgG (NMO) referred to the total AQP4 immunoprecipitated with anti-AQP4 antibodies (AQP4 Ab). Note: the lower affinity of NMO-IgG for D69H and skeletal muscle compared with WT AQP4 and brain (*, p < 0.01, n = 3).
DISCUSSION
The present work shows that mutating key amino acid residues of the AQP4 sequence affects NMO-IgG epitopes while preserving AQP4 aggregation into OAPs and its water channel function. Our investigation was based on previous evidence showing the key role of the AQP4 extracellular loops in generating NMO-IgG epitopes (8). We have identified two major AQP4-OAP-specific conformational NMO-IgG epitopes. In particular, we have distinguished sera able to recognize the G146VTTV150 sequence in loop C, sera characterized by a predominant role of the G61SEN64 sequence in loop A, and sera able to recognize both sequences (10). However, AQP0-AQP4 chimeras, made of AQP0 TM domains and AQP4 extracellular and intracellular loops, failed to generate the NMO-IgG epitope. We therefore focused on AQP4 TM sequences and finally identified Asp69 as being key for the generation of an NMO-IgG epitope common to the different serum categories here analyzed. Mutating Asp69 preserves the AQP4 water channel function and, more interestingly, the formation of OAPs as analyzed by BN-PAGE and TIRF microscopy.
MD simulations highlighted that the histidine replacing Asp69 leads to a different conformation of loop A, which normally protrudes inside the central pore lumen. In particular, the mutation under analysis increases the mobility of loop A therefore affecting conformational interactions involving loops C and E. This interpretation also finds a rationale in view of the absence of CDC shown for the unique serum (from patient 10) having higher binding affinity for D69H mutant than the WT. This unexpected behavior can be explained by the absence of complement activation starting with C1q binding to Fc regions of NMO-IgG (29). We can hypothesize that impact of the D69H mutation on the AQP4 extracellular loop spatial position affects the NMO-IgG Fc spatial position, therefore impairing correct C1q binding and complement activation (30–33).
The presence of a histidine offers pH-mediated changes in physiological relevant pH. Generally, histidine is considered uncharged at neutral pH, and positively charged at pH ≈ 6 and below. There are many examples of histidine protonation triggering structural changes at low pH (44), we therefore analyzed whether the autoantibody recognition was pH-sensitive, especially as adding a methylene group (D69E) did affect staining, but did not abolish it. This hypothesis was not confirmed by experiments performed at pH 5.5 and 7 demonstrating that histidine protonation does not affect the NMO-IgG binding capacity to D69H AQP4.
The main advance that this article provides to the field of NMO research is the molecular nature of the NMO-IgG epitope. On the discovery of AQP4 as the target for NMO-IgG (34), we have later added a key step forward by demonstrating that AQP4 tetramers per se do not contain the NMO-IgG epitope, which is in fact generated by AQP4 aggregation into OAPs (2). After several studies aimed at identifying the key determinant for such an epitope assembly (10, 35, 36), we demonstrate that AQP4 aggregation into OAPs is essential but alone not sufficient to generate such an epitope. We show that the epitope is generated within the OAPs by key inter-tetrameric interactions between extracellular loops A, C, and E.
Another important aspect that can be considered in view of the present data is the tissue specificity of NMO pathogenesis, which biases only spinal cord and optic nerve without compromising other AQP4-OAP expressing organs such as skeletal muscle. We have found that TIRF microscopy and BN-PAGE are unable to highlight subtle AQP4-OAP conformational changes, which are crucial for NMO-IgG binding. We therefore suggest that different tissues or different NMO patients, despite normal OAP expression, could have modifications at the Å-metric level, crucial to drive tissue- and patient-specific NMO pathogenesis. Single amino acid modifications able to induce key conformational changes at the molecular level, as shown for D69H, may impair NMO-IgG binding, without alteration of AQP4-OAPs in terms of dimension or water transport.
To, at least partially, explain the tissue specificity of NMO pathogenesis, the hypothesis was tested for a different NMO-IgG affinity for brain and skeletal muscle AQP4 by IP experiments. Our data for the first time show that NMO-IgG recognizes skeletal muscle AQP4 with a significantly lower affinity compared with brain AQP4, similarly to the different affinity between WT and D69H, indicating that fine structural differences between CNS and non-CNS AQP4 OAPs could be involved in tissue specificity of the NMO pathogenesis. If we consider that the AQP4 amino acid sequence is the same from CNS and non-CNS tissues, further studies will be necessary to address the nature of this difference. Our hypothesis is that different interacting proteins in the CNS versus skeletal muscle could play a key role in OAP quaternary structures therefore affecting OAP affinity for NMO-IgG. Alternatively, tissue-specific mechanisms may be involved such as mRNA editing (41) or expression of non-synonymous SNPs. No data exist for mRNA editing during AQP4 expression, in either physiological or pathological conditions, including NMO, whereas many non-synonymous SNPs have been reported for AQP4 CDS, potentially able to change the AQP4 amino acid (NCBI dbSNP). Interestingly, a non-synonymous SNP able to change aspartate 184 into glutamate (D184E) was identified as highly represented in NMO patients (42) and the same SNP has been associated to the reduced function of AQP4 associated with the altered mobility of the extracellular D loop despite displaying normal aggregation into OAPs (18), muscle, lung, and kidney (6, 15). In addition, we cannot exclude several other reasons independent of the OAP fine structural differences, playing a key role in this aspect. For example, Matiello and colleagues (40) have investigated the susceptibility of different AQP4 expressing tissues to NMO lesion and reasonably suggested that spinal cord and optic nerves are main targets of NMO-IgG due to more abundant AQP4 and AQP4-OAP expression (40). The tissue specificity could also be due to a lower accessibility of AQP4-OAPs from non-CNS tissues to NMO-IgG, or to the expression of the largest AQP4-OAPs in the CNS, which is also suggested by the presence of a different pool of AQP4 and only the dystrophin dependent one is the target of NMO-IgG (2, 37). However, by freeze-fracture electron miicroscopy (38) and BN-SDS/PAGE (39) it has been reported that kidney, skeletal muscle, and CNS express comparable AQP4-OAPs in terms of dimension.
In conclusion, this article represents the first attempt to explore the interaction between NMO-IgG and AQP4-OAPs at the Å-metric level. Data reported here indicates 1) a pivotal role of AQP4-TMs in NMO-IgG epitope assembly and 2) that changing Asp69 strongly affects the NMO-IgG epitope without consequences for OAP dimension or water transport. These findings add valuable knowledge on the molecular aspect of NMO-IgG pathogenesis and suggest the possibility of using the Asp69-dependent AQP4-OAP conformational properties to envisage new strategies for NMO treatment, based on selective epitope disassembly. Moreover, the data indicate the lower NMO-IgG affinity for skeletal muscle AQP4 compared with the brain as one cause at the base of tissue-specific NMO lesions. The results show improve the knowledge on NMO-IgG epitope structure and provide additional clues for more focused NMO therapy in view of a molecular control of AQP4 NMO-IgG binding leaving the water channel function unaltered.
Acknowledgments
We thank Richard Lusardi for English proofreading services. We acknowledge CINECA award HP10B4VZO7-epiNMO under the ISCRA initiative for the availability of high performance computing resources and support.
This work was supported by Italian Ministry of University and Research Project Firb “Futuro in Ricerca” RBFR12SJA8.
- NMO
- neuromyelitis optica
- CDC
- complement-dependent cytotoxicity
- OAP
- orthogonal arrays of particle
- AQP4
- aquaporin-4
- IP
- immunoprecipitation
- CDS
- coding DNA sequence
- TIRF
- total internal reflection fluorescence
- BN
- Blue Native
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TM
- transmembrane
- MD
- molecular dynamics
- VMD
- visual molecular dynamics.
REFERENCES
- 1. Lennon V. A., Wingerchuk D. M., Kryzer T. J., Pittock S. J., Lucchinetti C. F., Fujihara K., Nakashima I., Weinshenker B. G. (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364, 2106–2112 [DOI] [PubMed] [Google Scholar]
- 2. Nicchia G. P., Mastrototaro M., Rossi A., Pisani F., Tortorella C., Ruggieri M., Lia A., Trojano M., Frigeri A., Svelto M. (2009) Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 57, 1363–1373 [DOI] [PubMed] [Google Scholar]
- 3. Nicchia G. P., Rossi A., Mola M. G., Pisani F., Stigliano C., Basco D., Mastrototaro M., Svelto M., Frigeri A. (2010) Higher order structure of aquaporin-4. Neuroscience 168, 903–914 [DOI] [PubMed] [Google Scholar]
- 4. Rossi A., Pisani F., Nicchia G. P., Svelto M., Frigeri A. (2010) Evidences for a leaky scanning mechanism for the synthesis of the shorter M23 protein isoform of aquaporin-4: implication in orthogonal array formation and neuromyelitis optica antibody interaction. J. Biol. Chem. 285, 4562–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rossi A., Baumgart F., van Hoek A. N., Verkman A. S. (2012) Post-Golgi supramolecular assembly of aquaporin-4 in orthogonal arrays. Traffic 13, 43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nicchia G.P., Sparaneo A., Mola M.G., Basco D., Rossi A., Svelto M., Frigeri A. (2013) Aquaporin-4 orthogonal arrays of particles from a physiological and pathophysiological point of view. Wiley Interdisciplinary Reviews: Membrane Transport and Signaling [Google Scholar]
- 7. Jarius S., Paul F., Franciotta D., Aktas O., Hohlfeld R., Zipp F., Vincent A. (2007) Revised diagnostic criteria for neuromyelitis optica-incorporation of NMO-IgG status. Nat. Clin. Pract. Neurol. 3, E1. [DOI] [PubMed] [Google Scholar]
- 8. Wingerchuk D. M., Lennon V. A., Pittock S. J., Lucchinetti C. F., Weinshenker B. G. (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66, 1485–1489 [DOI] [PubMed] [Google Scholar]
- 9. Pisani F., Sparaneo A., Tortorella C., Ruggieri M., Trojano M., Mola M. G., Nicchia G. P., Frigeri A., Svelto M. (2013) Aquaporin-4 autoantibodies in neuromyelitis optica: AQP4 isoform-dependent sensitivity and specificity. PLoS One 8, e79185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pisani F., Mastrototaro M., Rossi A., Nicchia G. P., Tortorella C., Ruggieri M., Trojano M., Frigeri A., Svelto M. (2011) Identification of two major conformational aquaporin-4 epitopes for neuromyelitis optica autoantibody binding. J. Biol. Chem. 286, 9216–9224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalluri S. R., Illes Z., Srivastava R., Cree B., Menge T., Bennett J. L., Berthele A., Hemmer B. (2010) Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch. Neurol. 67, 1201–1208 [DOI] [PubMed] [Google Scholar]
- 12. Hinson S. R., Pittock S. J., Lucchinetti C. F., Roemer S. F., Fryer J. P., Kryzer T. J., Lennon V. A. (2007) Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 69, 2221–2231 [DOI] [PubMed] [Google Scholar]
- 13. Hinson S. R., Romero M. F., Popescu B. F., Lucchinetti C. F., Fryer J. P., Wolburg H., Fallier-Becker P., Noell S., Lennon V. A. (2012) Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc. Natl. Acad. Sci. U.S.A. 109, 1245–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ratelade J., Zhang H., Saadoun S., Bennett J. L., Papadopoulos M. C., Verkman A. S. (2012) Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 123, 861–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ratelade J., Verkman A. S. (2012) Neuromyelitis optica: aquaporin-4 based pathogenesis mechanisms and new therapies. Int. J. Biochem. Cell Biol. 44, 1519–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pittock S. J., Lennon V. A., McKeon A., Mandrekar J., Weinshenker B. G., Lucchinetti C. F., O'Toole O., Wingerchuk D. M. (2013) Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 12, 554–562 [DOI] [PubMed] [Google Scholar]
- 17. Polman C. H., Reingold S. C., Banwell B., Clanet M., Cohen J. A., Filippi M., Fujihara K., Havrdova E., Hutchinson M., Kappos L., Lublin F. D., Montalban X., O'Connor P., Sandberg-Wollheim M., Thompson A. J., Waubant E., Weinshenker B., Wolinsky J. S. (2011) Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69, 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nicchia G. P., Ficarella R., Rossi A., Giangreco I., Nicolotti O., Carotti A., Pisani F., Estivill X., Gasparini P., Svelto M., Frigeri A. (2011) D184E mutation in aquaporin-4 gene impairs water permeability and links to deafness. Neuroscience 197, 80–88 [DOI] [PubMed] [Google Scholar]
- 19. Mola M. G., Nicchia G. P., Svelto M., Spray D. C., Frigeri A. (2009) Automated cell-based assay for screening of aquaporin inhibitors. Anal. Chem. 81, 8219–8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho J. D., Yeh R., Sandstrom A., Chorny I., Harries W. E., Robbins R. A., Miercke L. J., Stroud R. M. (2009) Crystal structure of human aquaporin 4 at 1.8 Å and its mechanism of conductance. Proc. Natl. Acad. Sci. U.S.A. 106, 7437–7442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 22. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 23. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. MacKerell A. D., Jr., Banavali N., Foloppe N. (2000) Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56, 257–265 [DOI] [PubMed] [Google Scholar]
- 25. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 26. Martyna G. J., Tobias D. J., Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 [Google Scholar]
- 27. Feller S. E., Zhang Y., Pastor R. W., Brooks B. R. (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 [Google Scholar]
- 28. Adelman S. A., Doll J. D. (1976) Generalized Langevin equation approach for atom/solid-surface scattering: general formulation for classical scattering off harmonic solids. J. Chem. Phys. 64, 2375–2388 [Google Scholar]
- 29. Schumaker V. N., Hanson D. C., Kilchherr E., Phillips M. L., Poon P. H. (1986) A molecular mechanism for the activation of the first component of complement by immune complexes. Mol. Immunol. 23, 557–565 [DOI] [PubMed] [Google Scholar]
- 30. Hughes-Jones N. C., Gardner B., Rowlands J. (1977) Activation of human complement by glutaraldehyde-treated red cells. Nature 270, 613–614 [DOI] [PubMed] [Google Scholar]
- 31. Sledge C. R., Bing D. H. (1973) Binding properties of the human complement protein Clq. J. Biol. Chem. 248, 2818–2823 [PubMed] [Google Scholar]
- 32. Freedman J., Chaplin H., Johnson C. A., Hughes-Jones N. C. (1977) Comparison of low-molecular-weight products following reaction of C3-C3b with C3b inactivator and with trypsin. Vox Sang. 33, 212–220 [DOI] [PubMed] [Google Scholar]
- 33. Phuan P. W., Ratelade J., Rossi A., Tradtrantip L., Verkman A. S. (2012) Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J. Biol. Chem. 287, 13829–13839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lennon V. A., Kryzer T. J., Pittock S. J., Verkman A. S., Hinson S. R. (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 202, 473–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iorio R., Fryer J. P., Hinson S. R., Fallier-Becker P., Wolburg H., Pittock S. J., Lennon V. A. (2013) Astrocytic autoantibody of neuromyelitis optica (NMO-IgG) binds to aquaporin-4 extracellular loops, monomers, tetramers and high order arrays. J. Autoimmun. 40, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nelson P. A., Khodadoust M., Prodhomme T., Spencer C., Patarroyo J. C., Varrin-Doyer M., Ho J. D., Stroud R. M., Zamvil S. S. (2010) Immunodominant T cell determinants of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS One 5, e15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nicchia G. P., Rossi A., Nudel U., Svelto M., Frigeri A. (2008) Dystrophin-dependent and -independent AQP4 pools are expressed in the mouse brain. Glia 56, 869–876 [DOI] [PubMed] [Google Scholar]
- 38. Verbavatz J. M., Ma T., Gobin R., Verkman A. S. (1997) Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J. Cell Sci. 110, 2855–2860 [DOI] [PubMed] [Google Scholar]
- 39. Nicchia G. P., Cogotzi L., Rossi A., Basco D., Brancaccio A., Svelto M., Frigeri A. (2008) Expression of multiple AQP4 pools in the plasma membrane and their association with the dystrophin complex. J. Neurochem. 105, 2156–2165 [DOI] [PubMed] [Google Scholar]
- 40. Matiello M., Schaefer-Klein J., Sun D., Weinshenker B. G. (2013) Aquaporin 4 expression and tissue susceptibility to neuromyelitis optica. JAMA Neurol. 70, 1118–1125 [DOI] [PubMed] [Google Scholar]
- 41. Slotkin W., Nishikura K. (2013) Adenosine-to-inosine RNA editing and human disease. Genome Med. 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matiello M., Schaefer-Klein J. L., Hebrink D. D., Kingsbury D. J., Atkinson E. J., Weinshenker B. G. (2011) Genetic analysis of aquaporin-4 in neuromyelitis optica. Neurology 77, 1149–1155 [DOI] [PubMed] [Google Scholar]
- 43. Schrödinger (2013) Maestro, version 9.5, Release 2013-2, Schrödinger, LLC, New York [Google Scholar]
- 44. Nordlund H. R., Hytönen V. P., Laitinen O. H., Uotila S. T., Niskanen E. A., Savolainen J., Porkka E., Kulomaa M. S. (2003) Introduction of histidine residues into avidin subunit interfaces allows pH-dependent regulation of quaternary structure and biotin binding. FEBS Lett. 555, 449–454 [DOI] [PubMed] [Google Scholar]