

Background: SPARC is a matricellular protein that is increased in type 2 diabetes patients.
Results: SPARC is expressed by stromal cells within islets and inhibits growth factor responses and islet survival.
Conclusion: The matricellular protein SPARC is a novel regulator of islet survival.
Significance: The regulation of stromal-derived matricellular proteins represents a novel approach to promoting islet growth or survival.
Keywords: Cell Growth, Diabetes, Extracellular Matrix Protein, Growth Factor, Islet, Regeneration, Signaling, Stromal Cell
Abstract
Understanding the mechanisms regulating islet growth and survival is critical for developing novel approaches to increasing or sustaining β cell mass in both type 1 and type 2 diabetes patients. Secreted protein acidic and rich in cysteine (SPARC) is a matricellular protein that is important for the regulation of cell growth and adhesion. Increased SPARC can be detected in the serum of type 2 diabetes patients. The aim of this study was to investigate the role of SPARC in the regulation of β cell growth and survival. We show using immunohistochemistry that SPARC is expressed by stromal cells within islets and can be detected in primary mouse islets by Western blot. SPARC is secreted at high levels by pancreatic stellate cells and is regulated by metabolic parameters in these cells, but SPARC expression was not detectable in β cells. In islets, SPARC expression is highest in young mice, and is also elevated in the islets of non-obese diabetic (NOD) mice compared with controls. Purified SPARC inhibits growth factor-induced signaling in both INS-1 β cells and primary mouse islets, and inhibits IGF-1-induced proliferation of INS-1 β cells. Similarly, exogenous SPARC prevents IGF-1-induced survival of primary mouse islet cells. This study identifies the stromal-derived matricellular protein SPARC as a novel regulator of islet survival and β cell growth.
Introduction
Type 1 and type 2 diabetes are both characterized by the loss or insufficiency of insulin producing β cells (1). The mechanisms that determine whether β cells proliferate, survive or die, and thereby determine diabetes onset, are currently incompletely understood. Furthermore, understanding the regulation of β cell proliferation could allow the development of protocols to expand these cells to provide an unlimited supply of β cells and restore glycemic control.
Secreted Protein Acidic and Rich in Cysteine (SPARC)2 is a secreted protein that exists in the extracellular matrix (ECM) (2). Also known as osteonectin and BM40, SPARC is expressed during tissue remodeling and repair (3) and is also thought to be involved in the development of a number of different cancers (4). There is also evidence that SPARC expression is increased in newly diagnosed type 2 diabetes patients (5) and in gestational diabetes (6). Elevated SPARC is also associated with obesity and BMI (7, 8). Furthermore, SPARC protein expression in visceral adipose tissue explants is up-regulated by both leptin and insulin, suggesting that the metabolic conditions existing in type 2 diabetes patients may promote increased SPARC expression (7). Recently, SPARC expression was also found to be associated with insulin secretion (9). Additionally, there is evidence that SPARC expression may be involved in a number of diabetes complications. SPARC-null mice are protected against diabetic nephropathy (10), while elevated SPARC expression occurs in diabetes-associated vascular remodeling (11) and diabetic proliferative retinopathy (12).
SPARC has been shown to regulate growth factor signaling, cell proliferation and cell adhesion in various cell types in vitro (13) and is essential for matrix formation and remodeling in vitro and in vivo (13, 14). There is strong evidence that SPARC is important in the development of pancreatic cancer (15–22). However, the precise effects of SPARC are cell type dependent, and the effect of SPARC on the growth and survival of islet β cells has not previously been examined. We therefore investigated the expression of SPARC in islet tissue and determined the role of SPARC in regulating growth factor signaling in both β cells and in primary mouse islets, and in β cell proliferation and islet survival.
EXPERIMENTAL PROCEDURES
Animals
Adult female or male outbred ICR mice (21–25 g) were obtained from Harlan, Bicester, UK as were female C57BL/6 (B6) mice at 4 or 12 weeks of age. All animal procedures were undertaken in accordance with the UK Home Office Regulations. Pancreatic tissue for immunohistochemistry was kindly provided by Professor Nora Sarvetnick, The Scripps Research Institute. In this colony, over 70% of female NOD mice develop diabetes (23).
Islet Isolation
Islets were isolated from ICR mice using collagenase digestion followed by separation using density gradient. Mice were sacrificed by cervical dislocation and a laparotomy was performed. After clamping of the ampulla of Vater, ∼2 ml collagenase (1 mg/ml in minimal essential medium type XI, Sigma) was injected into the pancreas via the common bile duct and the pancreas was removed. Tubes containing up to three pancreases were incubated in a stationary water bath for 10 min at 37 °C. The islets were separated using Histopaque-1077 density gradient (Sigma) and centrifuged at 1170 × g for 25 min. After washing, islets were handpicked and cultured overnight at 37 °C and 5% CO2 in RPMI 1640 containing 11.1 mmol/liter glucose (Sigma) and supplemented with 10% FBS (Fisher Scientific), 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma).
Cell Culture
INS-1 β cells were cultured in RPMI 1640 containing 11.1 mmol/liter glucose and additionally supplemented with 10% FBS, 0.05 mm 2-mercaptoethanol, 10 mm HEPES, 1 mm sodium pyruvate, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Fisher Scientific). INS-1 cells were subcultured every 3–4 days, and used within 20 passages.
PS-1 cells are previously described human pancreatic stellate cells (24, 25). They were maintained in high glucose DMEM:Ham's F12 medium (1:1, both from PAA) supplemented with 10% FBS, 1 μg/ml puromycin (Sigma), 1 mm sodium pyruvate, 100 unit/ml penicillin and 100 μg/ml streptomycin, or in RPMI 1640 supplemented with 10% FBS, 0.1% l-glutamine, 100 unit/ml penicillin, and 100 μg/ml streptomycin. PS-1 cells were subcultured every 2–3 days and used within 10 passages.
For experiments involving incubation with specific concentrations of glucose, glucose-free RPMI 1640 medium was used. D(+)-glucose was obtained as a 0.56 m solution (Sigma). Human insulin (Santa Cruz Biotechnology) was obtained as 10 mg/ml solution in Hepes buffer and was diluted in PBS before use. Lyophilized rat leptin (R&D systems) was resuspended at 1 mg/ml in sterile 20 mm Tris-HCl at pH 8, and diluted in PBS before use.
Immunohistochemistry
Whole pancreas was removed from 4-week-old or 12-week-old female C57BL/6 and NOD mice, or ICR mice (21–25 g), then fixed in 10% NBF and embedded in paraffin. Sections (5 μm) for SPARC and FSP-1 staining were first subject to proteinase K treatment (50 μg/ml, 20 min at 37 °C; Sigma) before blocking with 10% normal horse serum in PBS containing 0.3% Triton-X-100.
For staining with single a single antibody, incubation with primary antibody was at ambient temperature overnight. Goat anti-SPARC antibody (R&D systems) was used at 1/25 dilution in blocking solution, and rabbit anti-FSP-1 antibody (Millipore) was used at 1/100 dilution. Positive areas were visualized with biotin-conjugated secondary antibody to goat or rabbit IgG using the ABC/DAB reagents according to the manufacturer's protocol (Vector Laboratories). Pancreas sections incubated with secondary antibody only served as controls. Sections were counterstained with hematoxylin. Digital images acquired using a Nikon Eclipse 80i microscope were analyzed with ImageJ software (26). SPARC-stained area, and total islet area were quantified in μm2 using the thresholding tool. All areas outside of the islet boundary were excluded from the analysis.
For co-staining experiments, sections were incubated with SPARC goat antibody (1/50, R&D Systems) overnight at 4 °C, followed by incubation with biotinylated anti-goat antibody (Vector labs) for 1 h at room temperature. SPARC-positive areas were detected using the ABC and DAB peroxidase substrate kits (Vector labs). Sections were then incubated with FSP-1 rabbit antibody (1/50, Millipore) for 1 h followed by incubation with alkaline phosphatase-conjugated anti-rabbit antibody (Vector Labs) for 30 min at room temperature. FSP-1-positive areas were detected with the enzyme substrate vector blue (Vector Labs). Digital images were acquired using the Nikon Eclipse 80i microscope.
Preparation of Samples for Western Blotting
For analysis of SPARC expression in conditioned medium from β cells (INS-1) and pancreatic stellate cells (PS-1), T25 flasks were seeded with 1 × 106 and 5 × 105 cells, respectively. Following overnight culture in low-serum (0.5% FBS) medium, the medium was removed and concentrated in a Vivapore concentrator to ∼100 times to determine the presence of SPARC secreted into the medium. To prepare cell lysates, after rinsing in PBS the cells were lysed in RIPA buffer for 30 min on ice. For similar analysis of SPARC expression in islets, 50 islets from ICR mice were cultured in reduced serum (1% FBS) overnight before medium was concentrated (to ∼40 times) in a Vivapore concentrator. After rinsing in PBS, islets were lysed in RIPA buffer (New England Biolabs) containing protease and phosphatase inhibitors (Thermo Scientific).
For experiments in which cells were cultured with glucose, insulin and leptin, protein quantification of cell lysates was performed using the BCA protein assay (Bio-Rad) and using BioTek Epoch plate reader to measure absorbance at 750 nm. Equal protein (10 μg/well) was loaded onto the gel.
For analysis of growth factor signaling, INS-1 cells were seeded in 48-well plates at a density of 1.2 × 105 cells per well and allowed to attach before serum-starving overnight (RPMI 1640 medium containing 0.5% FBS). Cells were treated with SPARC (0.5 - 2.5 μg/ml for 2 h) followed by either recombinant insulin-like growth factor (IGF-1) or recombinant hepatocyte growth factor (HGF), all obtained from R&D Systems. Similar protocols using SPARC have been published previously by other groups (27). Cells were harvested at the indicated time points by first washing in ice-cold PBS before lysis in cold RIPA buffer. SDS sample buffer was added to each sample before boiling. For islet signaling experiments, islets isolated from male ICR mice were cultured overnight then hand-picked into groups of equal amounts and sizes of islets (∼130 islets), and cultured for 1 h in RMPI-1640 with 1% FBS. Groups of islets were then treated with SPARC (2.5 or 20 μg/ml) for 1 h, before treatment with HGF or IGF-1 (200 ng/ml) for 7.5 min and lysis. For dispersed islet experiments, islets were centrifuged and incubated in Accutase at 37 °C with gentle pipetting for ∼10 min. After dispersal, equal volumes of the cell suspension were distributed into tubes to contain 150 islets/treatment. After starvation (1% FBS for 1 h), cells were pre-treated with SPARC (2.5 or 20 μg/ml) for 1 h before HGF treatment (20 ng/ml, 7.5 min) and lysis. Equal amounts of INS-1 or islet lysate proteins were loaded onto the gel.
Western Blotting
Lysates and concentrated medium were resolved by polyacrylamide gel electrophoresis (PAGE), and electrotransferred onto nitrocellulose membrane (GE Healthcare). Membranes were blocked in 5% nonfat milk and incubated with primary antibody at the following dilutions: anti-SPARC (1 in 1000; Santa Cruz/Insight Biotechnology), anti-phospho-Akt (1:500–1:1000), anti-phospho-p44/p42 MAPK (1:1000) and anti-β-tubulin (1:1000–1:10000). Incubations were performed overnight at ambient temperature (SPARC) or 4 °C (Akt and p44/p42 MAPK). Proteins were detected with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:2500). Where appropriate, after stripping the membrane, blots were probed for anti-pan-Akt (1:1000) and anti-total p44/p42 MAPK (1:1000). All Western blot antibodies were obtained from New England Biolabs unless otherwise indicated. Proteins were visualized using Supersignal West Pico chemiluminescent substrate (Pierce) using a Syngene GeneGnome imaging system with GeneSnap software. Quantitation of band intensity was carried out using GeneTools or ImageJ software.
Proliferation Assays
For measurement of cell density, the Sulforhodamine B (SRB) assay was used as described in Skehan et al. (28). INS-1 cells were plated at 5,000 cells per well in 96-well plates. After overnight incubation in reduced serum medium (0.5% FBS), cells were treated with SPARC followed by 200 ng/ml IGF-1. Separate plates of cells were fixed on day 0 and day 2 with 10% trichloroacetic acid for 1 h at 4 °C. After washing and sufficient drying, cells were incubated with SRB (0.4% in 1% acetic acid) for 30 min at room temperature. Cells were thoroughly washed with 1% acetic acid and left to dry. For analysis of protein content, plates were analyzed on the same day. SRB was solubilized with addition of 10 mm Tris base (pH 10) and the absorbance read at 565 nm on an Epoch Microplate Spectrophotometer (BioTek).
For measurement of DNA synthesis, the Roche colorimetric BrdU ELISA assay was used according to the manufacturer's instructions. INS-1 cells were plated at 10,000 cells/well in a 96-well plate and allowed to adhere overnight. Cells were then incubated in reduced serum medium (0.5% FBS) overnight before treatment with the indicated concentrations of IGF-1 and SPARC for 48 h. BrdU was added for the final 24 h of culture. Following antibody detection and addition of substrate, absorbance was measured at 370 nm.
Live/Dead Assay
Islets from male or female ICR mice were dispersed as above and seeded onto coverslips in medium containing 10% serum. After attachment, islet cells were then treated with medium containing 10% serum, 0% serum, or 0% serum plus combinations of SPARC (20 μg/ml) and IGF-1 (200 ng/ml).
After 2 days coverslips were removed from the plates and fluorescein diacetate (FDA) at 15 μg/ml and propidium iodide (PI) at 4 μg/ml (Sigma) were used to assess live/dead status of individual islet cells using fluorescence microscopy. Live cells (green) were calculated as a percentage of total cells visible as either green or red using ImageJ software (26).
Statistics
Results are expressed as means ± S.E. of the mean (S.E.). Statistical difference was assessed by analysis of variance using least significant difference post-hoc analysis. A p value ≤ 0.05 was considered to be significant.
RESULTS
Characterizing SPARC Expression in Mouse Islets
Immunostaining revealed that SPARC protein is present in the islets of both C57BL/6 (B6) and NOD mouse pancreas, and a similar overall staining pattern was observed with SPARC expressed in a minority of cells both within the islet and in the basement membrane surrounding the islet (Fig. 1A). All secondary antibody only controls were negative for staining (data not shown). SPARC is also expressed within ducts and vessels adjacent to islets (Fig. 1A, panels c and d, and Fig. 2A, panels c and d). Quantification of SPARC staining in islets shows that the area of SPARC expression is higher at 4 weeks than at 12 weeks in both strains (Fig. 1B). However, expression of SPARC in NOD islets at 4 weeks of age is almost double that of age-matched C57BL/6 mice (p = 0.008, n = 6), and a similar increase is also observed at 12 weeks of age (p = 0.001, n = 6). The expression of a fibroblast-lineage specific marker (FSP-1) (29) in adjacent islet sections indicates a similar, although not completely overlapping, distribution of SPARC and FSP-1 staining (Fig. 2A). Co-staining within the same section confirms that Fsp-1 positive cells also express SPARC in both islets and ducts (Fig. 2B). Again, the overlap between the two antibodies is not complete and SPARC expression is also observed in Fsp-1 negative cells. However, the SPARC staining pattern is distinct from that typically observed for β cells with insulin antibodies. This suggests that fibroblasts or other mesenchymal cells may be a primary source of SPARC within islets, rather than β cells. Consistent with this, SPARC expression was detected in both cellular lysates and in conditioned medium of stromal pancreatic stellate (PS-1) cells and islets, but not in the INS-1 β cell line (Fig. 2C). Although this data is not quantitative since appropriate loading controls for secreted proteins could not be identified, equal numbers of PS-1 and INS-1 cells were used. It is evident that despite the high protein content of the concentrated medium samples and very high SPARC expression in PS1-conditioned medium, no SPARC was detectable in INS-1 cells or medium. SPARC was also detectable in PS1 stromal cells but not INS-1 β cells by immunocytochemistry (Fig. 3).
FIGURE 1.
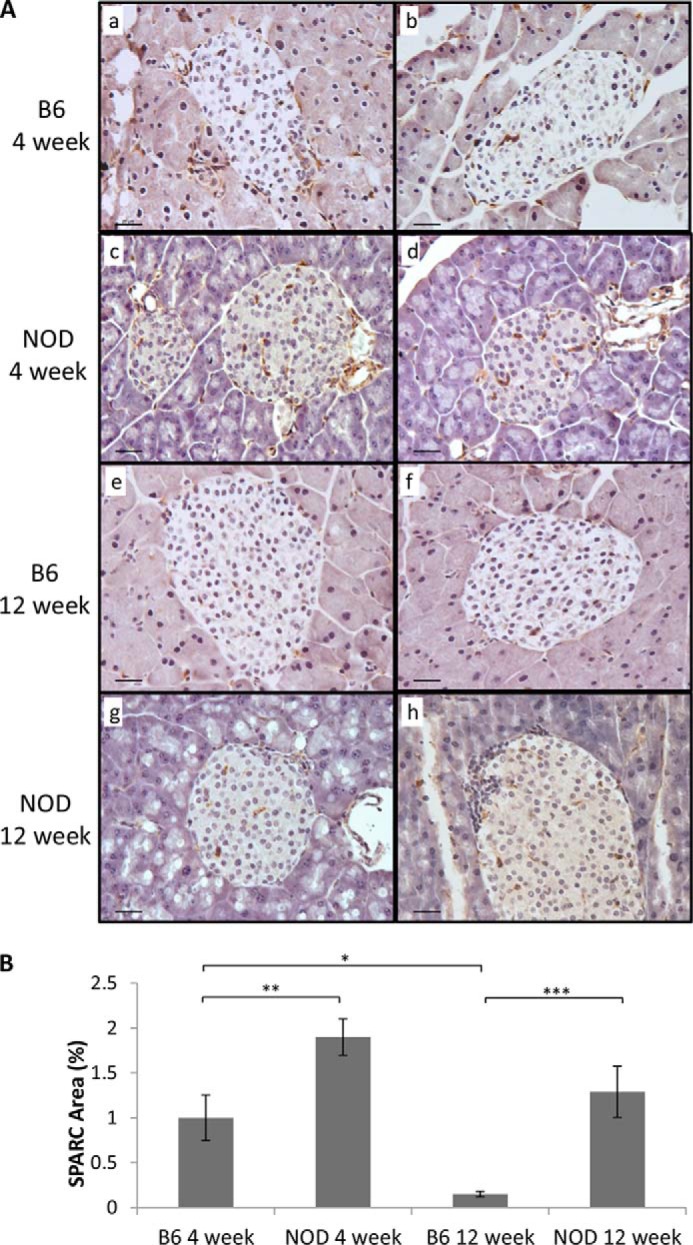
SPARC is expressed in the islets of mouse pancreas. Mouse pancreas sections (5 μm) from 4 and 12 week NOD or B6 mice were stained with anti-SPARC antibody followed by biotin-conjugated secondary antibody. Positive areas were visualized using the ABC/DAB method producing a brown color. A, two representative images of each age/strain are shown. Scale bar, 20 μm. B, area of SPARC staining as a percentage of total islet area within each section was quantified using ImageJ software. Areas of islet infiltration as well as peri-ductal and peri-vascular staining were excluded from the analysis. Bars represent mean ± S.E., n = 6, from 3 individual mice of each type. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
FIGURE 2.
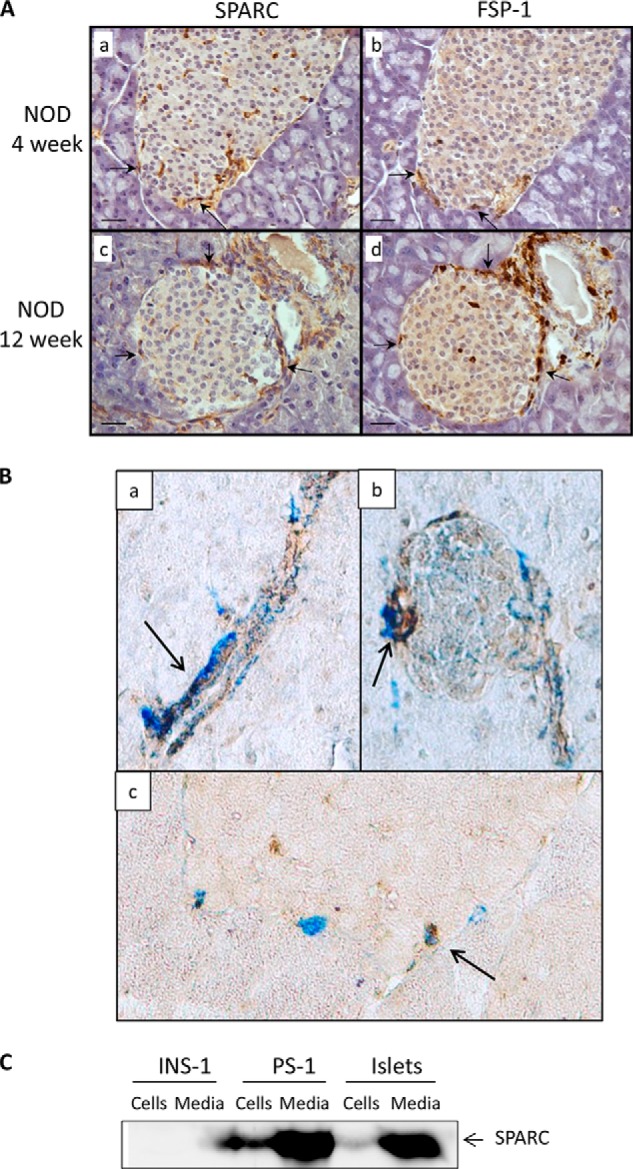
SPARC is expressed by stromal cells within islets. A, adjacent sections (5 μm) of 4- and 12-week NOD mouse pancreas were stained with antibodies to SPARC (left panels) and FSP-1 (right panels) followed by biotin-conjugated secondary antibody. Positive areas were visualized using the ABC/DAB method producing a brown color. Representative images are shown of multiple sections analyzed from at least three different mice. Arrowheads indicate examples of SPARC and FSP-1-positive cells. Scale bar, 20 μm. B, pancreatic sections from ICR mice were co-stained with antibodies to SPARC (brown DAB substrate) and Fsp-1 (Vector Blue substrate). Representative images of staining in ducts and islets are shown, taken with 20× objective and with additional 2× digital zoom in panels a and c. Arrows indicate areas of co-staining. In C, concentrated medium and cell lysates from equal numbers of INS-1 and PS-1 cells, or from intact islets (groups of 50 islets) were analyzed by Western blot and probed with antibodies to SPARC.
FIGURE 3.
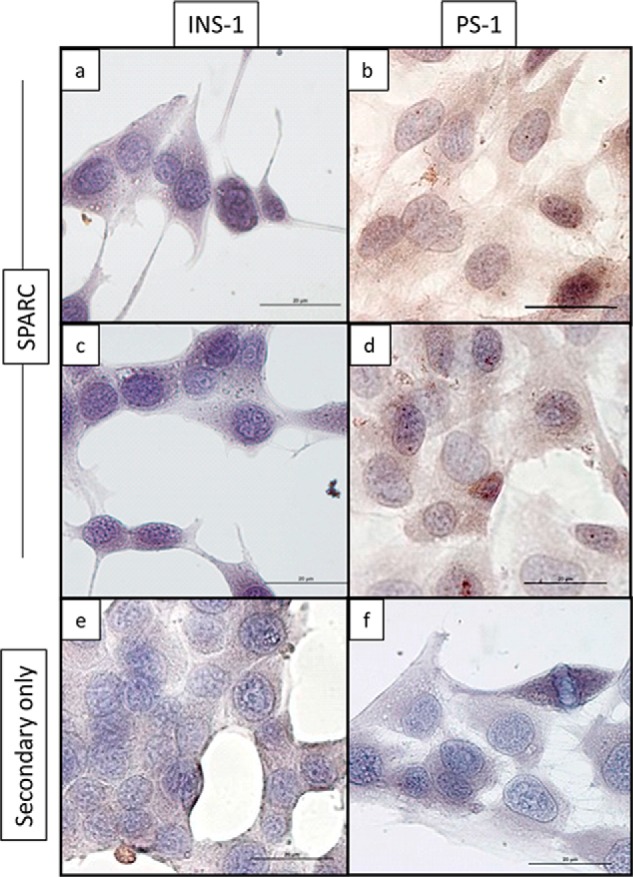
SPARC is detected in stromal PS1 cells but not INS-1 β cells by immunocytochemistry. INS-1 and PS1 cells were grown on coverslips, fixed using 10% NBF and stained using anti-SPARC antibody (panels a–d) or with biotinylated secondary antibody only (panels e, f). The ABC/DAB detection system was used, with brown color indicating the presence of bound antibody. Bar, 20 μm.
SPARC Expression in Pancreatic Stromal Cells Is Regulated by Insulin and Glucose
An interesting study by Kos et al. showed that SPARC expression in adipose tissue is regulated by metabolic parameters (7). Within both islets and the exocrine pancreas SPARC is expressed primarily by stromal cells (Figs. 1 and 2 and Ref. 30). We therefore wished to test whether SPARC expression in pancreatic stromal cells is similarly regulated by metabolic parameters. PS1 stellate cells were incubated with various concentrations of glucose, insulin, and leptin, and SPARC protein expression was determined by Western blot. Quantitation of SPARC band intensity relative to β-tubulin loading control was performed using ImageJ software. As shown in Fig. 4, increasing the glucose concentration inhibits SPARC expression in PS1 cells (p = 0.01, n = 5). In contrast, increasing insulin stimulates an increase in SPARC expression (p = 0.03, n = 3–4). Treatment with leptin also elevated SPARC expression, although this difference was not statistically significant. Metabolic parameters can therefore regulate SPARC expression in pancreatic stromal cells, similar to that observed in adipose tissue by Kos et al. (7).
FIGURE 4.
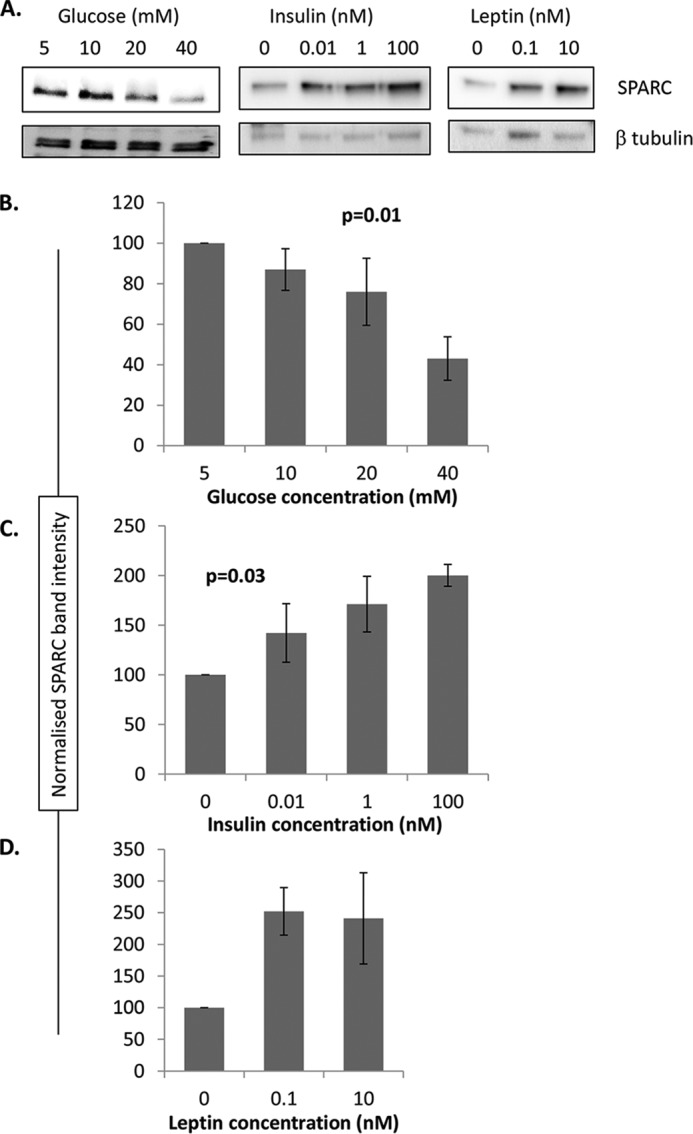
SPARC expression in pancreatic stromal cells is regulated by metabolic parameters. PS-1 cells were seeded at 5 × 104 cells/well in 48-well plates and cultured for 24 h at the indicated concentrations of glucose and insulin, or for 48 h at the indicated concentrations of leptin. SPARC expression was then quantitated by Western blot and standardized to β-tubulin to control for loading variation. Equal protein was loaded (10 μg) in each well. Representative blots are shown in A, and mean relative SPARC expression (standardized to β-tubulin) ± S.E. is shown for glucose (B), insulin (C), and leptin (D). Quantitated data are pooled from 3–5 independent experiments, each with single data point for each treatment. p values indicate statistical analysis using one-way ANOVA.
SPARC Inhibits IGF-1-induced Signaling
The kinases Akt and extracellular signal-regulated kinases (ERK) 1/2 are key components of growth factor-induced pathways in the β cell that result in cellular changes such as proliferation, gene transcription, survival, and insulin secretion (31–35). Initial validation experiments were carried out to determine the optimal conditions for activation of these signaling pathways by growth factors (Fig. 5). We then determined the effects of SPARC on IGF-1 induced signaling. Phosphorylation of Akt and ERK 1/2 following addition of SPARC alone is comparable to that in unstimulated cells (Fig. 6). However, treatment of INS-1 cells with IGF-1 (20 ng/ml) increased phosphorylation of both Akt and ERK 1/2 at 7.5 min (Fig. 6, A and B). The 2.5-fold increase in Akt phosphorylation observed following IGF-1 exposure is inhibited by pre-incubation with SPARC for 2 h in a concentration-dependent manner, from a level comparable to IGF-1 stimulated levels using 0.1 μg/ml SPARC down to basal levels using 2.5 μg/ml SPARC. Treatment with both SPARC and IGF-1 also results in an inhibition of ERK 1/2 activation compared with IGF-1 treatment alone, with the 2.5-fold stimulation of ERK 1/2 phosphorylation by IGF-1 alone again inhibited to background levels by pre-incubation with 2.5 μg/ml SPARC (Fig. 6, A and B). The inhibition of IGF-1 induced Akt and ERK-1 signaling by SPARC was statistically significant when analyzed across multiple experiments using one-way ANOVA (p = 0.01, n = 3–6 and p = 0.002, n = 3–5, respectively, Fig. 6E).
FIGURE 5.
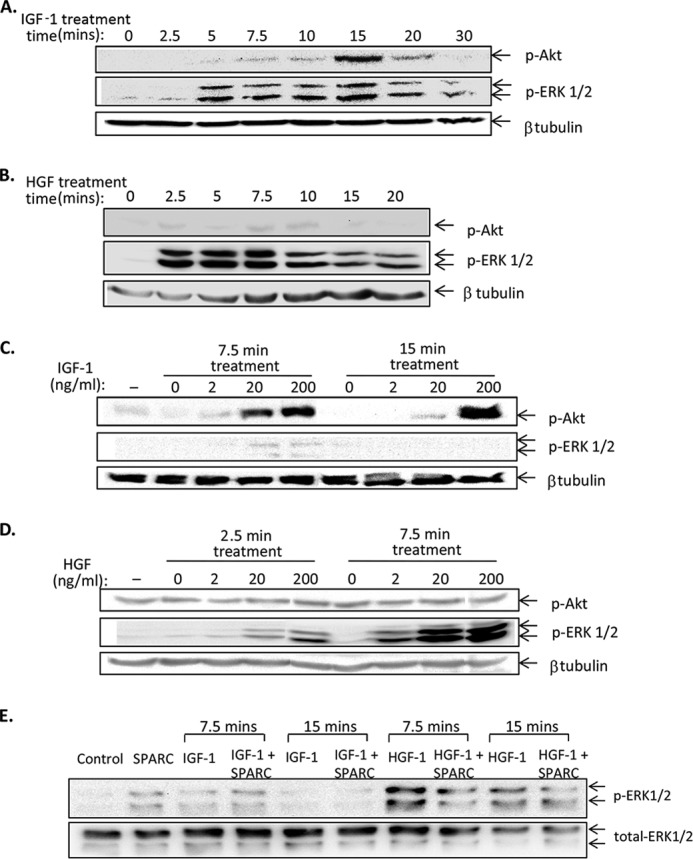
Validation of conditions for growth factor-induced signaling in β cells. INS-1 cells were plated at 1.2 × 105 cells/well in 48-well plates and cultured in reduced-serum conditions (0.5% FBS) overnight before treatment for the indicated times with 20 ng/ml IGF-1 (A) or 20 ng/ml HGF (B). Protein lysates were analyzed by Western blot using antibodies to phospho-Akt, phospho-ERK1/2, and β-tubulin as a loading control. Similarly, the concentration of IGF-1 (C) and HGF (D) was titrated at the indicated timepoints in INS-1 cells. In E, equal numbers of intact primary mouse islets were treated with 200 ng/ml IGF-1 or HGF in the presence and absence of 2.5 μg/ml SPARC for the timepoints indicated. Lysates were analyzed by Western blot with antibodies to phospho-ERK1/2 and total-ERK1/2 as a loading control. Each blot is representative of two independent experiments.
FIGURE 6.
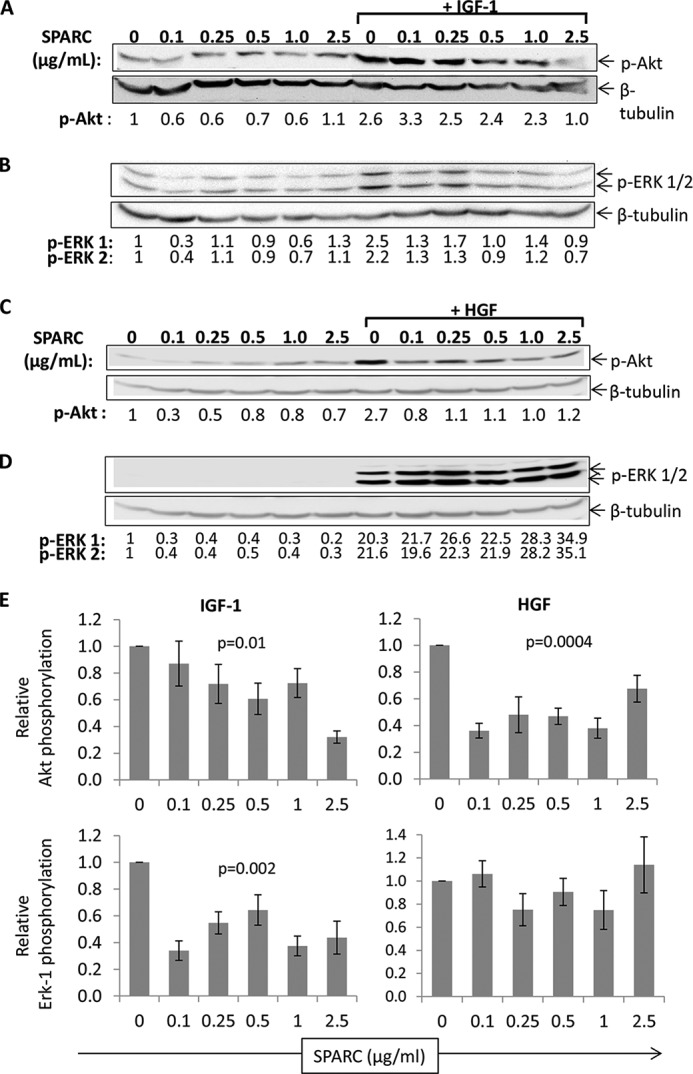
SPARC inhibits growth factor signaling in INS-1 β cells. INS-1 cells treated for 7.5 min with 20 ng/ml IGF-1 (A, B) or 20 ng/ml HGF (C, D) and the indicated concentration of SPARC were analyzed by Western blot to detect phosphorylated Akt (A, C), phosphorylated ERK 1/2 (B, D), and β-tubulin as a loading control. The numbers below the blot indicate band intensities standardized to β-tubulin and the untreated control in lane 1. Representative blots are shown in A–D. The graphs in E show pooled data from multiple experiments for samples treated with growth factor in addition to the indicated concentration of SPARC. Plots show mean ERK-1/Akt phosphorylation of samples treated with growth factor plus SPARC relative to growth factor alone ± S.E. (n = 3–6 for IGF-1 data, n = 5 for HGF data) p values indicate statistical analysis using one-way ANOVA.
SPARC Inhibits HGF-induced Akt Phosphorylation
INS-1 cells were also treated with HGF in order to determine whether inhibition of growth factor signaling by SPARC was specific to IGF-1 or more general. HGF stimulates a 2.7-fold increase in Akt phosphorylation over basal levels at 7.5 min (Fig. 6C). However, pre-incubation with SPARC for 2 h inhibits this HGF-dependent activation of Akt, an effect seen at all concentrations of SPARC tested (0.1–2.5 μg/ml). Again, this inhibition was statistically significant when data were analyzed across multiple experiments (p = 0.0004, n = 5, Fig. 6E). As before, SPARC treatment alone does not consistently affect phosphorylation of Akt or ERK 1/2, though some inhibition of background signaling is occasionally observed. Treatment with HGF causes a strong, 20-fold, stimulation of ERK 1/2 phosphorylation in INS-1 cells (Fig. 6D). However, in this case no inhibition of signaling was observed by SPARC.
SPARC Inhibits HGF-induced ERK Signaling in Islets
To test whether similar inhibition of growth factor signaling occurs in primary islet tissue, islets were isolated from ICR mice to test the effect of SPARC on IGF-1 and HGF signaling. Phosphorylated and total ERK 1/2 were detected by Western blot as above. In islets, the total ERK 1 bands detected are stronger than the total ERK 2 bands, however, this antibody is known to have reduced affinity for ERK 2 (as indicated by manufacturer). The stimulation of ERK 1/2 phosphorylation by HGF (20 ng/ml) in islets is decreased to basal levels by SPARC (20 μg/ml) in dispersed islets (Fig. 7A). Across multiple experiments, SPARC inhibited HGF signaling on average by over 50%, and this result was highly statistically significant (p = 6 × 10−5, n = 4, Fig. 7B). IGF-1 did not induce phosphorylation of ERK 1/2 in primary islets (Fig. 5E). In all experiments, treatment with SPARC alone had no effect on ERK 1/2 phosphorylation (Fig. 7). SPARC treatment therefore inhibits growth factor signaling in both INS-1 β cells and primary mouse islets.
FIGURE 7.
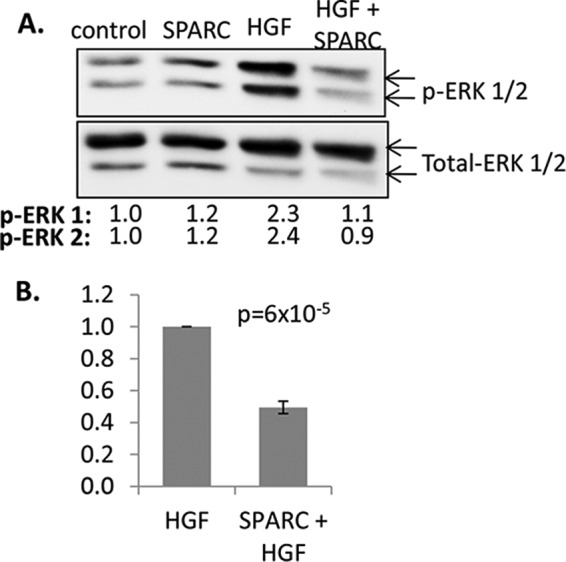
SPARC inhibits growth factor signaling in primary mouse islets. Dispersed islets (groups of 150) were pre-incubated with SPARC (20 μg/ml for 1 h) before treatment with 20 ng/ml HGF. Cell lysates were analyzed by Western blotting using antibodies against phosphorylated and total ERK 1/2. A representative blot is shown in A. The numbers below the blot indicate band intensities standardized to total ERK 1 and relative to the control in lane 1. In B, pooled data shows mean standardized ERK-1 phosphorylation of samples treated with HGF plus SPARC relative to HGF alone ± S.E. (n = 4). p values indicate statistical analysis using one-way ANOVA.
SPARC Inhibits IGF-1-induced INS-1 Cell Growth and Proliferation
IGF-1 signaling can induce expansion in the INS-1 cell line (37). The effect of SPARC on IGF-1 induced β cell expansion was therefore examined using the SRB assay to determine protein content as an indirect measure of cell density (28). INS-1 cells were seeded at equal cell density and treated with IGF-1 (200 ng/ml) and SPARC (2.5 μg/ml and 20 μg/ml), or with IGF-1 alone. Cell density was measured after 2 days and growth relative to the plating density was determined. As shown in Fig. 8A, treatment with 2.5 μg/ml SPARC inhibits IGF-1 induced cell growth by 25%, and 20 μg/ml SPARC inhibits cell growth 60% (p < 0.001, n = 24) when compared with IGF-1 alone, demonstrating a dose-dependent effect. No increase in cell growth by HGF was observed under the conditions tested (data not shown). SPARC had a small but significant inhibition of cell growth in the absence of IGF-1 i.e. in 0.5% FBS alone, presumably due to inhibition of residual growth due to the presence of low serum (p < 0.001, n = 24; Fig. 8B).
FIGURE 8.
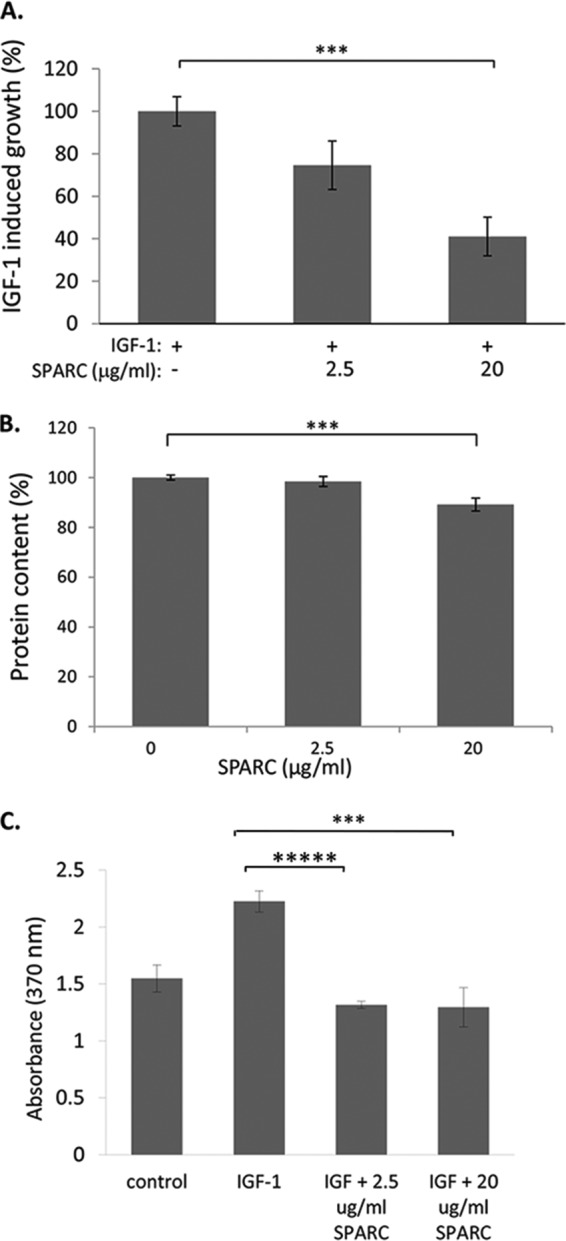
SPARC inhibits IGF-1 induced INS-1 cell growth and proliferation. INS-1 cells were treated with IGF-1 (200 ng/ml) and the indicated concentration of SPARC. A, SRB assay was used at day 2 as an indirect measurement of cell number. Results are expressed as mean percentage of IGF-1 induced growth (relative to no IGF-1) ± S.E. (n = 24, pooled from four independent experiments). B, analysis of mean protein content ± S.E., plotted relative to untreated cells indicates that SPARC reduces INS-1 cell growth in the presence of reduced serum alone. C, DNA synthesis was determined using colorimetric BrdU ELISA. The graph shows mean absorbance ± S.E. (n = 6, representative of three independent experiments). For all graphs, ***, p = ≤ 0.001; *****, p = ≤ 0.00001.
To test whether the inhibition of IGF-1 induced cell growth by SPARC was due to inhibition of cell proliferation, the effect of SPARC on BrdU incorporation was determined. IGF-1 treatment induced ∼50% increase in BrdU incorporation in INS-1 cells, yet this was completely abolished by pre-treatment with either 2.5 or 20 μg/ml SPARC (Fig. 8C). SPARC therefore inhibits growth factor induced proliferation in INS-1 β cells.
The Effect of SPARC on Islet Cell Survival
To test whether SPARC similarly inhibits downstream growth factor responses in primary islet tissue, a fluorescent live/dead assay was used to measure islet cell survival in response to growth factor treatment. Dispersed islets were used to overcome issues associated with diffusion into the islet mass. As a baseline control against which to measure growth factor responsiveness, dispersed mouse islets were cultured in serum-free medium to induce cell death. Initial validation experiments confirmed that a significant reduction in dispersed islet survival was observed after 2 days in serum-free medium (p < 0.001, n = 4; Fig. 9), and that this reduction in survival could be rescued by the addition of 200 ng/ml IGF-1, but not HGF (Fig. 10A). Therefore, this assay could be used to measure the effect of SPARC on islet growth factor responses, specifically IGF-1 induced islet survival. On average, 38 ± 3.6% live cells were observed after 2 days in serum-free medium (Fig. 10B). In contrast, addition of 10% FBS to the medium as a positive control increased this to 52 ± 3.0% live cells, and similarly treatment with IGF-1 (200 ng/ml) resulted in 54 ± 1.0% live cells. However, treatment with SPARC in addition to IGF-1 reduced this to 46 ± 3.4% live cells, i.e. SPARC inhibits the IGF-1 induced increase in islet survival by ∼50%. The effect of exogenous SPARC in reducing IGF-1-induced islet survival is therefore consistent with the reduction in IGF-1-induced β cell growth observed using INS-1 cells (Fig. 8).
FIGURE 9.

Validation of assay to detect islet cell death. Islets were dispersed and equal numbers of cells adhered to coverslips. Islet cells were then treated with medium containing 10% FBS, 0% FBS (untreated) or 10% FBS plus high glucose (33.3 mm) for either one or 2 days. FDA/PI staining was used to determine the number of live/dead cells using fluorescence microscopy and quantitation was performed using ImageJ. Representative images of FDA (green/live), PI (red/dead), and merged images are shown in A, and the mean percentage of live cells ± S.E. shown at day 1 and day 2 is shown in B. Islets were pooled from 4 ICR mice, and n = 4 per treatment. **, p < 0.01; ***, p < 0.001.
FIGURE 10.
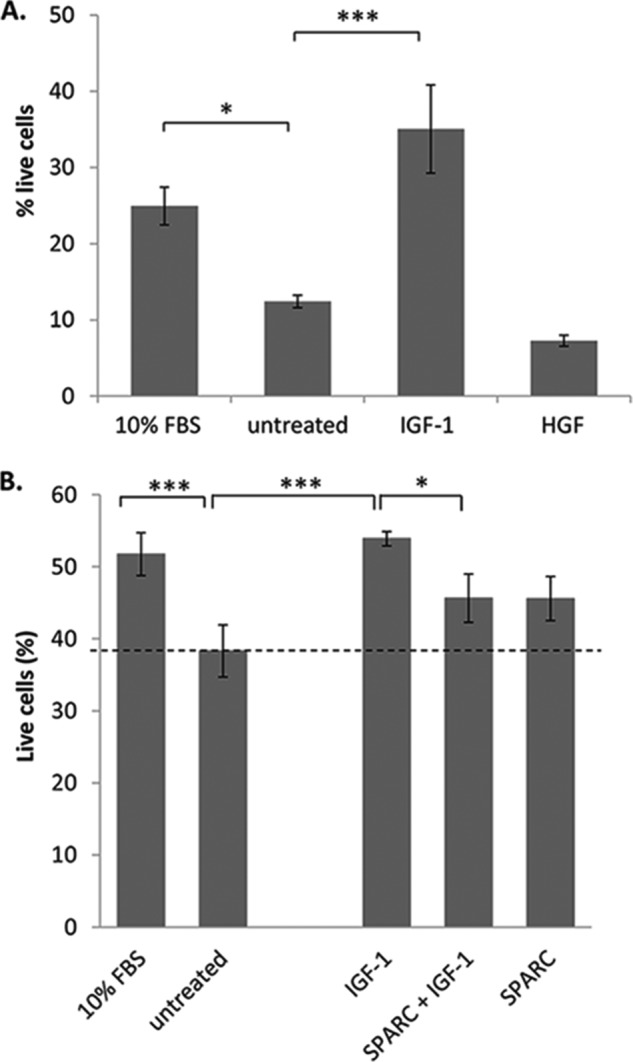
IGF-1 rescue of islet cell death is inhibited by SPARC. Dispersed islets were adhered to coverslips in serum-free medium then treated for 2 days (A) with either 10% FBS, untreated (0% FBS), 200 ng/ml IGF-1 or 200 ng/ml HGF. Islet cells were stained with FDA (green/live) and PI (red/dead) and the %live cells determined using ImageJ. The mean percentage of live islet cells ± S.E. is shown. Islets were pooled from 4 ICR mice, and n = 4 per treatment. B, islets were cultured with either 10% FBS, untreated (0% FBS), IGF-1 (200 ng/ml), SPARC (20 μg/ml), or both IGF-1 and SPARC and stained as in A. Results are pooled from three separate experiments. In both graphs the mean percentage of live islet cells ± S.E. is shown. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
DISCUSSION
Adult β cells are well differentiated and hence have a low mitotic rate and are difficult to expand in vitro (38). However, under certain in vivo conditions, such as pregnancy, obesity, and islet inflammation, β cells are able to proliferate at an increased rate (39–41). This proliferative capacity suggests that it may be possible to stimulate β cell growth to treat diabetes patients. SPARC is known to regulate the proliferation, adhesion, and migration of cells (42). However, the effect of SPARC on islet cells has not previously been examined. We have shown that SPARC is expressed by stromal cells within islets and can regulate β cell growth and survival.
Our results show that SPARC is expressed in islets with a staining pattern suggestive of a non-β cell population and consistent with fibroblast-type cell staining. This is consistent with previous studies showing SPARC expression in the pancreas by stromal fibroblasts (30). However, SPARC staining did not completely overlap with that of the Fsp-1 fibroblast cell marker we used, and some intra-islet SPARC staining may be explained by SPARC expression in endothelial cells (43). Furthermore, we have shown that SPARC is strongly expressed and secreted by pancreatic stellate cells, while we were unable to detect SPARC expression in INS-1 β cells. As in other tissues, SPARC within islets is therefore likely to be expressed by stromal cells including fibroblasts and stellate cells, rather than in β cells. A study by Harries et al. showed SPARC mRNA expression in islets, but SPARC protein expression in normal β cells was not described (9). However, while we were unable to detect SPARC protein in β cells, we have not examined SPARC mRNA expression and cannot exclude the possibility that SPARC gene transcription in β cells has regulatory effects at the non-protein level.
We also observed an increased area of SPARC expression in the islets of NOD mice compared with wild type controls, suggesting that changes in SPARC expression may be associated with the development of type 1 diabetes. This could be explained by the expansion or recruitment of a population of SPARC positive cells, or by induction of SPARC expression in a cell type not normally expressing SPARC. SPARC was detected in tissues of both ages examined but expression was strongest in young (4-week) old mice. In mice, developmental changes associated with islet formation can be detected up to 3 weeks of age (44) and waves of β cell apoptosis occur from 2 weeks of age (45, 46). In NOD mice these changes correlate with early infiltration of various innate immune cells (47–49). The early expression of high levels of SPARC suggests the importance of SPARC in islets at this critical stage when β cell neogenesis, replication, and apoptosis occur, consistent with SPARC's role in tissue remodeling in other contexts (3, 50).
Recombinant SPARC had an inhibitory effect on growth factor responses in β cells, reducing IGF-1 induced proliferation by up to 60%, and inhibiting IGF-1 induced phosphorylation of Akt and ERK in a dose-dependent manner. A similar effect was seen with HGF-induced stimulation of Akt in β cells. We have also shown that SPARC prevents HGF-induced phosphorylation of ERK 1/2 in primary mouse islets. Others have shown that SPARC inhibits signaling in response to a range of growth factors in various cell types (27, 51, 52). In some cases, such as with VEGF and PDGF, SPARC binds directly to the growth factor to prevent association with the receptor (52, 53). However, in the case of IGF-1 it has been previously shown that SPARC can regulate signaling even in the absence of evidence for direct interaction (27). It is not currently known whether SPARC interacts with HGF, and this is the first study in which the regulation of HGF signaling by SPARC has been examined.
The effects of SPARC on cell proliferation and survival are complex, and there is evidence that SPARC can both promote and inhibit cell survival in different cell types (13, 54). Our data show that exogenous SPARC inhibits both the growth factor-dependent proliferation of β cells and the survival of primary islets. The recent study by Harries et al. showed that ectopic expression of SPARC in β cells increases insulin release in response to high glucose levels (9). However, our data would suggest that SPARC is not normally expressed at high levels in β cells, and it may be that SPARC present intracellularly in β cells has a distinct function from SPARC acting extracellularly, as used in our study. The possibility of distinct activity of intracellular SPARC has been previously suggested, particularly since, as a calcium binding protein, SPARC is regulated by the high Ca2+ concentrations present extracellularly (55–57). We propose a model in which SPARC secreted by stromal cells is present in the islet matrix environment and regulates β cell responsiveness to growth factors, as well as potentially other extracellular signals.
Following on from the study by Kos et al. (7) showing that SPARC expression in adipose tissue is increased by high levels of insulin and leptin, our data suggests that stromal SPARC expression in the pancreas may be similarly regulated by metabolic parameters. This raises the possibility that pancreatic SPARC expression could be induced in type 2 diabetes patients and may contribute to the development of β cell loss or insufficiency, and potentially also the risk of pancreatic cancer, although these hypotheses remain to be tested. Furthermore, the identification of SPARC as a novel regulator of β cell growth and survival underlines the importance of understanding the role of stromal-derived matricellular proteins such as SPARC in β cell expansion and the pathogenesis of diabetes.
Acknowledgments
We thank Dr. James Bowe and Dr. Chloe Rackham at the Diabetes Research Group, King's College London, for assistance with islet procedures.
This work was supported by the Diabetes UK and the Biomedical and Pharmaceutical Sciences Research Group at Kingston University.
- SPARC
- secreted protein acidic and rich in cysteine
- NOD
- non-obese diabetic
- ECM
- extracellular matrix
- PI
- propidium iodide
- FDA
- fluorescein diacetate
- HGF
- hepatocyte growth factor.
REFERENCES
- 1. Matveyenko A. V., Butler P. C. (2008) Relationship between beta-cell mass and diabetes onset. Diabetes Obes. Metab. 10, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brekken R. A., Sage E. H. (2001) SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 19, 816–827 [DOI] [PubMed] [Google Scholar]
- 3. Sage H., Vernon R. B., Decker J., Funk S., Iruela-Arispe M. L. (1989) Distribution of the calcium-binding protein SPARC in tissues of embryonic and adult mice. J Histochem. Cytochem. 37, 819–829 [DOI] [PubMed] [Google Scholar]
- 4. Arnold S. A., Brekken R. A. (2009) SPARC: a matricellular regulator of tumorigenesis. J. Cell Commun. Signal 3, 255–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu D., Li L., Yang M., Liu H., Yang G. (2011) Elevated plasma levels of SPARC in patients with newly diagnosed type 2 diabetes mellitus. Eur. J. Endocrinol. 165, 597–601 [DOI] [PubMed] [Google Scholar]
- 6. Xu L., Ping F., Yin J., Xiao X., Xiang H., Ballantyne C. M., Wu H., Li M. (2013) Elevated plasma SPARC levels are associated with insulin resistance, dyslipidemia, and inflammation in gestational diabetes mellitus. PLoS One 8, e81615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kos K., Wong S., Tan B., Gummesson A., Jernas M., Franck N., Kerrigan D., Nystrom F. H., Carlsson L. M., Randeva H. S., Pinkney J. H., Wilding J. P. (2009) Regulation of the fibrosis and angiogenesis promoter SPARC/osteonectin in human adipose tissue by weight change, leptin, insulin, and glucose. Diabetes 58, 1780–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee S. H., Lee J. A., Park H. S., Song Y. S., Jang Y. J., Kim J. H., Lee Y. J., Heo Y. (2013) Associations among SPARC mRNA expression in adipose tissue, serum SPARC concentration and metabolic parameters in Korean women. Obesity 21, 2296–2302 [DOI] [PubMed] [Google Scholar]
- 9. Harries L. W., McCulloch L. J., Holley J. E., Rawling T. J., Welters H. J., Kos K. (2013) A role for SPARC in the moderation of human insulin secretion. PLoS One 8, e68253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taneda S., Pippin J. W., Sage E. H., Hudkins K. L., Takeuchi Y., Couser W. G., Alpers C. E. (2003) Amelioration of diabetic nephropathy in SPARC-null mice. J. Am. Soc. Nephrol. 14, 968–980 [DOI] [PubMed] [Google Scholar]
- 11. Jandeleit-Dahm K., Rumble J., Cox A. J., Kelly D. J., Dziadek M., Cooper M. E., Gilbert R. E. (2000) SPARC gene expression is increased in diabetes-related mesenteric vascular hypertrophy. Microvasc. Res. 59, 61–71 [DOI] [PubMed] [Google Scholar]
- 12. Watanabe K., Okamoto F., Yokoo T., Iida K. T., Suzuki H., Shimano H., Oshika T., Yamada N., Toyoshima H. (2009) SPARC is a major secretory gene expressed and involved in the development of proliferative diabetic retinopathy. J. Atheroscler. Thromb. 16, 69–76 [DOI] [PubMed] [Google Scholar]
- 13. Chlenski A., Cohn S. L. (2010) Modulation of matrix remodeling by SPARC in neoplastic progression. Semin. Cell Dev. Biol. 21, 55–65 [DOI] [PubMed] [Google Scholar]
- 14. Barker T. H., Baneyx G., Cardó-Vila M., Workman G. A., Weaver M., Menon P. M., Dedhar S., Rempel S. A., Arap W., Pasqualini R., Vogel V., Sage E. H. (2005) SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase activity. J. Biol. Chem. 280, 36483–36493 [DOI] [PubMed] [Google Scholar]
- 15. Neuzillet C., Tijeras-Raballand A., Cros J., Faivre S., Hammel P., Raymond E. (2013) Stromal expression of SPARC in pancreatic adenocarcinoma. Cancer Metastasis Reviews 32, 585–602 [DOI] [PubMed] [Google Scholar]
- 16. Miyoshi K., Sato N., Ohuchida K., Mizumoto K., Tanaka M. (2010) SPARC mRNA expression as a prognostic marker for pancreatic adenocarcinoma patients. Anticancer Res. 30, 867–871 [PubMed] [Google Scholar]
- 17. Chen G., Tian X., Liu Z., Zhou S., Schmidt B., Henne-Bruns D., Bachem M., Kornmann M. (2010) Inhibition of endogenous SPARC enhances pancreatic cancer cell growth: modulation by FGFR1-III isoform expression. Br. J. Cancer 102, 188–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edmonds C., Cengel K. A. (2008) Tumor-Stroma interactions in pancreatic cancer: Will this SPARC prove a raging fire? Cancer Biol. Ther. 7, 1816–1817 [DOI] [PubMed] [Google Scholar]
- 19. Prenzel K. L., Warnecke-Eberz U., Xi H., Brabender J., Baldus S. E., Bollschweiler E., Gutschow C. A., Hölscher A. H., Schneider P. M. (2006) Significant overexpression of SPARC/osteonectin mRNA in pancreatic cancer compared to cancer of the papilla of Vater. Oncology Reports 15, 1397–1401 [PubMed] [Google Scholar]
- 20. Puolakkainen P. A., Brekken R. A., Muneer S., Sage E. H. (2004) Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell apoptosis. Mol. Cancer Res. 2, 215–224 [PubMed] [Google Scholar]
- 21. Infante J. R., Matsubayashi H., Sato N., Tonascia J., Klein A. P., Riall T. A., Yeo C., Iacobuzio-Donahue C., Goggins M. (2007) Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 25, 319–325 [DOI] [PubMed] [Google Scholar]
- 22. Sato N., Fukushima N., Maehara N., Matsubayashi H., Koopmann J., Su G. H., Hruban R. H., Goggins M. (2003) SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene 22, 5021–5030 [DOI] [PubMed] [Google Scholar]
- 23. Hill N. J., Stotland A., Solomon M., Secrest P., Getzoff E., Sarvetnick N. (2007) Resistance of the target islet tissue to autoimmune destruction contributes to genetic susceptibility in Type 1 diabetes. Biology Direct 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Froeling F. E., Mirza T. A., Feakins R. M., Seedhar A., Elia G., Hart I. R., Kocher H. M. (2009) Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, beta-catenin, and Ezrin expression in tumor cells. Am. J. Pathol. 175, 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li N. F., Kocher H. M., Salako M. A., Obermueller E., Sandle J., Balkwill F. (2009) A novel function of colony-stimulating factor 1 receptor in hTERT immortalization of human epithelial cells. Oncogene 28, 773–780 [DOI] [PubMed] [Google Scholar]
- 26. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Francki A., Motamed K., McClure T. D., Kaya M., Murri C., Blake D. J., Carbon J. G., Sage E. H. (2003) SPARC regulates cell cycle progression in mesangial cells via its inhibition of IGF-dependent signaling. J. Cell. Biochem. 88, 802–811 [DOI] [PubMed] [Google Scholar]
- 28. Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D., Warren J. T., Bokesch H., Kenney S., Boyd M. R. (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107–1112 [DOI] [PubMed] [Google Scholar]
- 29. Strutz F., Okada H., Lo C. W., Danoff T., Carone R. L., Tomaszewski J. E., Neilson E. G. (1995) Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 130, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guweidhi A., Kleeff J., Adwan H., Giese N. A., Wente M. N., Giese T., Büchler M. W., Berger M. R., Friess H. (2005) Osteonectin influences growth and invasion of pancreatic cancer cells. Annals Surgery 242, 224–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tuttle R. L., Gill N. S., Pugh W., Lee J. P., Koeberlein B., Furth E. E., Polonsky K. S., Naji A., Birnbaum M. J. (2001) Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nature Med. 7, 1133–1137 [DOI] [PubMed] [Google Scholar]
- 32. Wrede C. E., Dickson L. M., Lingohr M. K., Briaud I., Rhodes C. J. (2002) Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 277, 49676–49684 [DOI] [PubMed] [Google Scholar]
- 33. Frödin M., Sekine N., Roche E., Filloux C., Prentki M., Wollheim C. B., Van Obberghen E. (1995) Glucose, other secretagogues, and nerve growth factor stimulate mitogen-activated protein kinase in the insulin-secreting beta-cell line, INS-1. J. Biol. Chem. 270, 7882–7889 [DOI] [PubMed] [Google Scholar]
- 34. Dickson L. M., Lingohr M. K., McCuaig J., Hugl S. R., Snow L., Kahn B. B., Myers M. G., Jr., Rhodes C. J. (2001) Differential activation of protein kinase B and p70(S6)K by glucose and insulin-like growth factor 1 in pancreatic beta-cells (INS-1). J. Biol. Chem. 276, 21110–21120 [DOI] [PubMed] [Google Scholar]
- 35. Briaud I., Lingohr M. K., Dickson L. M., Wrede C. E., Rhodes C. J. (2003) Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes 52, 974–983 [DOI] [PubMed] [Google Scholar]
- 36. Chlenski A., Guerrero L. J., Salwen H. R., Yang Q., Tian Y., Morales La Madrid A., Mirzoeva S., Bouyer P. G., Xu D., Walker M., Cohn S. L. (2011) Secreted protein acidic and rich in cysteine is a matrix scavenger chaperone. PLoS One 6, e23880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hügl S. R., White M. F., Rhodes C. J. (1998) Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J. Biol. Chem. 273, 17771–17779 [DOI] [PubMed] [Google Scholar]
- 38. Kayali A. G., Flores L. E., Lopez A. D., Kutlu B., Baetge E., Kitamura R., Hao E., Beattie G. M., Hayek A. (2007) Limited capacity of human adult islets expanded in vitro to redifferentiate into insulin-producing beta-cells. Diabetes 56, 703–708 [DOI] [PubMed] [Google Scholar]
- 39. Sherry N. A., Kushner J. A., Glandt M., Kitamura T., Brillantes A. M., Herold K. C. (2006) Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes 55, 3238–3245 [DOI] [PubMed] [Google Scholar]
- 40. Marynissen G., Aerts L., Van Assche F. A. (1983) The endocrine pancreas during pregnancy and lactation in the rat. J. Dev. Physiol. 5, 373–381 [PubMed] [Google Scholar]
- 41. Hull R. L., Kodama K., Utzschneider K. M., Carr D. B., Prigeon R. L., Kahn S. E. (2005) Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48, 1350–1358 [DOI] [PubMed] [Google Scholar]
- 42. Bradshaw A. D., Sage E. H. (2001) SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J. Clin. Invest. 107, 1049–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sage H., Johnson C., Bornstein P. (1984) Characterization of a novel serum albumin-binding glycoprotein secreted by endothelial cells in culture. J. Biol. Chem. 259, 3993–4007 [PubMed] [Google Scholar]
- 44. Miller K., Kim A., Kilimnik G., Jo J., Moka U., Periwal V., Hara M. (2009) Islet formation during the neonatal development in mice. PLoS One 4, e7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mathis D., Vence L., Benoist C. (2001) beta-Cell death during progression to diabetes. Nature 414, 792–798 [DOI] [PubMed] [Google Scholar]
- 46. Turley S., Poirot L., Hattori M., Benoist C., Mathis D. (2003) Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med. 198, 1527–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diana J., Simoni Y., Furio L., Beaudoin L., Agerberth B., Barrat F., Lehuen A. (2013) Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nature Med. 19, 65–73 [DOI] [PubMed] [Google Scholar]
- 48. Jansen A., Homo-Delarche F., Hooijkaas H., Leenen P. J., Dardenne M., Drexhage H. A. (1994) Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes 43, 667–675 [DOI] [PubMed] [Google Scholar]
- 49. Rosmalen J. G., Homo-Delarche F., Durant S., Kap M., Leenen P. J., Drexhage H. A. (2000) Islet abnormalities associated with an early influx of dendritic cells and macrophages in NOD and NODscid mice. Laboratory Investigation 80, 769–777 [DOI] [PubMed] [Google Scholar]
- 50. Sage H., Vernon R. B., Funk S. E., Everitt E. A., Angello J. (1989) SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2-dependent binding to the extracellular matrix. J. Cell Biol. 109, 341–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Francki A., McClure T. D., Brekken R. A., Motamed K., Murri C., Wang T., Sage E. H. (2004) SPARC regulates TGF-beta1-dependent signaling in primary glomerular mesangial cells. J. Cell. Biochem. 91, 915–925 [DOI] [PubMed] [Google Scholar]
- 52. Kupprion C., Motamed K., Sage E. H. (1998) SPARC (BM-40, osteonectin) inhibits the mitogenic effect of vascular endothelial growth factor on microvascular endothelial cells. J. Biol. Chem. 273, 29635–29640 [DOI] [PubMed] [Google Scholar]
- 53. Raines E. W., Lane T. F., Iruela-Arispe M. L., Ross R., Sage E. H. (1992) The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF)-AB and -BB and inhibits the binding of PDGF to its receptors. Proc. Natl. Acad. Sci. U. S. A. 89, 1281–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi Q., Bao S., Maxwell J. A., Reese E. D., Friedman H. S., Bigner D. D., Wang X. F., Rich J. N. (2004) Secreted protein acidic, rich in cysteine (SPARC), mediates cellular survival of gliomas through AKT activation. J. Biol. Chem. 279, 52200–52209 [DOI] [PubMed] [Google Scholar]
- 55. Delostrinos C. F., Hudson A. E., Feng W. C., Kosman J., Bassuk J. A. (2006) The C-terminal Ca2+-binding domain of SPARC confers anti-spreading activity to human urothelial cells. J. Cell. Physiol. 206, 211–220 [DOI] [PubMed] [Google Scholar]
- 56. Yan Q., Weaver M., Perdue N., Sage E. H. (2005) Matricellular protein SPARC is translocated to the nuclei of immortalized murine lens epithelial cells. J. Cell. Physiol. 203, 286–294 [DOI] [PubMed] [Google Scholar]