

Background: Due to its hydrophobic character, the adenylate cyclase (CyaA) toxin from Bordetella pertussis is prone to aggregate into multimeric forms.
Results: We define the experimental conditions required to fold CyaA into a monomeric state.
Conclusion: Molecular confinement, post-translational acylation, and calcium binding are critical for CyaA folding into a monomeric and cytotoxic form.
Significance: Monomeric CyaA opens the way for structural and functional studies.
Keywords: Adenylate Cyclase (Adenylyl Cyclase), Bacterial Toxin, Bordetella pertussis, Calcium-binding Protein, Chromatography, Macromolecular Crowding, Protein Acylation, Protein Aggregation, Protein Folding, Molecular Confinement
Abstract
The adenylate cyclase (CyaA) toxin, a multidomain protein of 1706 amino acids, is one of the major virulence factors produced by Bordetella pertussis, the causative agent of whooping cough. CyaA is able to invade eukaryotic target cells in which it produces high levels of cAMP, thus altering the cellular physiology. Although CyaA has been extensively studied by various cellular and molecular approaches, the structural and functional states of the toxin remain poorly characterized. Indeed, CyaA is a large protein and exhibits a pronounced hydrophobic character, making it prone to aggregation into multimeric forms. As a result, CyaA has usually been extracted and stored in denaturing conditions. Here, we define the experimental conditions allowing CyaA folding into a monomeric and functional species. We found that CyaA forms mainly multimers when refolded by dialysis, dilution, or buffer exchange. However, a significant fraction of monomeric, folded protein could be obtained by exploiting molecular confinement on size exclusion chromatography. Folding of CyaA into a monomeric form was found to be critically dependent upon the presence of calcium and post-translational acylation of the protein. We further show that the monomeric preparation displayed hemolytic and cytotoxic activities suggesting that the monomer is the genuine, physiologically active form of the toxin. We hypothesize that the structural role of the post-translational acylation in CyaA folding may apply to other RTX toxins.
Introduction
The adenylate cyclase (CyaA) toxin produced by Bordetella pertussis, the causative agent of whooping cough, is one of the major virulence factors of this organism (1–3). CyaA plays an important role in the early stages of respiratory tract colonization by B. pertussis. CyaA is able to invade eukaryotic target cells, where it is activated by an endogenous protein, calmodulin (CaM),4 and produces high levels of cAMP that, in turn, alters the cellular physiology.
CyaA is a 1706-residue long protein organized in a modular fashion (Fig. 1A). The ATP-cyclizing, CaM-activated, catalytic domain (ACD) is located in the 400 N-terminal residues (4), whereas the C-terminal 1306 residues are responsible for the ACD translocation and the hemolytic phenotype of B. pertussis (5–7). Both activities can function independently as adenylate cyclase and hemolysin, respectively. Several domains can be identified in the C-terminal region. The so-called translocation region, spanning residues 400–500, is crucial for the translocation of ACD across the plasma membrane (8) and exhibits properties related to membrane-active peptides (9). The hydrophobic region, spanning residues 500–750, contains several hydrophobic segments predicted to adopt α-helical structures. The acylation region, spanning residues 800–1000, contains two post-translational modification sites that are essential for the cytotoxic activities of CyaA (10–12). The toxin is indeed synthesized as an inactive precursor, pro-CyaA that is converted into the active CyaA toxin upon specific acylation of Lys-860 and Lys-983 by a dedicated acyltransferase, CyaC (10, 11, 13). The C-terminal part of CyaA is the cell receptor-binding domain (RD; residues 1000–1706). This domain consists of ∼40 copies of a calcium-binding, glycine- and aspartate-rich nonapeptide repeat that is characteristic of a large family of bacterial cytolysins known as RTX (repeat-in-toxin) toxins (14, 15). These motifs constitute the main Ca2+ binding sites of the protein (16). The RTX motifs are intrinsically disordered in the absence of calcium (17–20). The intrinsic disorder predictors (21–24) (Fig. 1, B–D) show that RD is characterized by structural disorder and a significant negative mean net charge, which is partially neutralized upon calcium binding, as experimentally reported elsewhere (25). In the presence of calcium, RTX proteins fold into a structure called the β-roll, as revealed in the three-dimensional structures of several RTX proteins (26–28). CyaA is secreted across the bacterial envelope by a dedicated type I secretion machinery (29) made of CyaB, CyaD, and CyaE proteins (5, 30). Once secreted, CyaA binds in a calcium-dependent manner to the CD11b/CD18 integrin expressed on myeloid cells, such as macrophages, neutrophils, dendritic cells, and natural killer cells, that are the primary targets of this toxin in vivo (31). However, CyaA can also efficiently intoxicate a variety of cell types lacking this receptor (16, 32–35).
FIGURE 1.

Domain organization and structural disorder propensity of CyaA. A, schematic representation of CyaA localizing the ACD, the translocation region (T), the hydrophobic region (HR), the acylation region (AR), and RD. The structural disorder propensity of CyaA was analyzed by FoldIndex (B), IUPred (C), and GlobPlot (D).
One of the main originalities of CyaA, with respect to other bacterial or plant toxins, stems from its unique mechanism of penetration into eukaryotic cells; CyaA is the only known toxin able to translocate its catalytic domain directly across the plasma membrane of the target cells, from the extracellular side into the cytosol (36–41). The molecular mechanism by which CyaA enters the target cells remains, however, largely unknown. It is believed that after binding of RD to the CD11b/CD18 receptor, the hydrophobic regions of CyaA may insert into the plasma membrane of the target cells. The catalytic domain is then delivered through the plasma membrane, possibly through a transient and local destabilization of the membrane integrity (8, 9, 42, 43), to reach the cytosol, where, upon binding to the endogenous CaM, its enzymatic activity is stimulated to generate supraphysiologic levels of cAMP (37, 44). Moreover, CyaA, after insertion into the membrane, can also form cation-selective pores, which impair membrane impermeability and ultimately cause cell lysis (6, 40, 45). This membrane-damaging activity is thought to synergize with the cAMP intoxication ability, thus increasing the overall cytotoxicity of the toxin (33, 40, 46, 47).
Although CyaA has been extensively studied by various cellular and molecular approaches and used in several biotechnological applications, the structural and functional states of the toxin remain poorly characterized (1, 2). Indeed, CyaA is a large protein made of several distinct domains and exhibits a pronounced hydrophobic character, making it prone to aggregation into multimeric complexes (37, 48–50). Up to now, CyaA has been mainly extracted from B. pertussis with a chaotropic reagent (urea) or overproduced in Escherichia coli, where it accumulates as inclusion bodies that also need to be solubilized with urea. After purification, CyaA is usually stored in denaturing conditions, typically in the presence of 6 m urea (37, 48, 51–53).
Here, we defined the experimental conditions to refold CyaA into a monomeric and functional species. We found that CyaA, when refolded by dialysis, dilution, or buffer exchange, mainly produced multimers, as reported earlier (37, 48). However, by exploiting molecular confinement on size exclusion chromatography (SEC), we were able to obtain a monomeric form of CyaA that was stable in buffer without urea. Noticeably, this monomeric form exhibited much higher hemolytic and cell-invasive activities than the multimeric ones. CyaA refolding into the monomeric state was critically dependent upon the presence of calcium and the post-translational acylation. The structural role of both calcium and acyl chains in CyaA folding demonstrated here may possibly apply to other RTX cytolysins that are activated by selective acylation and are dependent upon calcium for their cytolytic activities.
EXPERIMENTAL PROCEDURES
CyaA Production and Purification
CyaA was produced and purified from inclusion bodies as described previously (53, 54). Briefly, the inclusion bodies were solubilized in about 50 ml of 20 mm Hepes, 8 m urea, pH 7.4, by overnight solubilization under constant stirring with a magnet at 4 °C. After centrifugation at 12,000 rpm for 20 min, the supernatant was supplemented with 0.14 m NaCl and incubated for 1 h at room temperature with 75 ml of Q-Sepharose resin equilibrated with 20 mm Hepes, 140 mm NaCl, 8 m urea, pH 7.4. The resin, retaining the CyaA protein, was then loaded onto a column, and contaminants were washed out. After an extensive wash with the same buffer, the CyaA protein was eluted in 20 mm Hepes, 500 mm NaCl, 8 m urea, pH 7.4. The eluate was then diluted in 20 mm Hepes, 8 m urea to decrease salt down to 140 mm NaCl and loaded onto a second Q-Sepharose column (50 ml). Washing and elution were performed in the same conditions as described above with the first Q-Sepharose column. This last step further removed contaminants and concentrated the CyaA protein. Proteins eluted from the second Q-Sepharose column were diluted 5 times with 20 mm Hepes, 1 m NaCl, pH 7.4, and applied onto a 70-ml phenyl-Sepharose column equilibrated with the same buffer. Resin was washed with 20 mm Hepes, 1 m NaCl, with 20 mm Hepes, and then with 50% isopropyl alcohol. The isopropyl alcohol washing step allowed the removal of many contaminants and LPS. After an extensive wash with 20 mm Hepes, the toxin was eluted with Hepes 20 mm, urea 8 m. The eluate was finally loaded onto a Sephacryl S-500 column (GE Healthcare, HIPREP 26/60) equilibrated in 20 mm Hepes, 8 m urea. CyaA was then concentrated by ultrafiltration to 1–2 mg/ml and stored at −20 °C in 20 mm Hepes, 8 m urea. All toxins purified by this method were more than 90% pure as judged by SDS-PAGE analysis and contained less than 1 endotoxin unit of LPS/μg of protein as determined by a standard Limulus amebocyte lysate assay (Lonza). CyaA toxin concentrations were determined spectrophotometrically using a molecular extinction coefficient of 144,000 m−1 cm−1 at 280 nm. Altogether, the overall recovery from a 1.6-liter fermenter varies from 20 to 40 mg of pure CyaA proteins.
Refolding of Urea-denatured CyaA
The renaturation of CyaA from its denatured state in 8 m urea was followed by tryptophan intrinsic fluorescence, ANS fluorescence, and circular dichroism in the far-UV ranges at 25 °C. Tryptophan intrinsic fluorescence was used to follow the changes of tryptophan environment, and ANS fluorescence was used to follow the changes of the hydrophobic environment (55), whereas ellipticity changes at 220 nm were used to follow secondary structural changes (19).
Intrinsic Disorder Predictions
The primary sequence of CyaA was used to analysis the intrinsic structural disorder. The disorder predictions were first checked with MeDor (24), and the results presented herein were obtained with GlobPlot, IUPred, and FoldIndex (21–24).
Fluorescence Spectroscopy
Measurements were performed with an FP-6200 spectrofluorimeter (Jasco, Tokyo, Japan) in a Peltier-thermostated cell holder, using a Quartz SUPRASIL 105.251-QS cuvette (Hellma) as described elsewhere (56). A bandwidth of 5 nm was used for the excitation and emission beams. For tryptophan intrinsic fluorescence and tryptophan fluorescence quenching by KI, the excitation wavelength was fixed at 290 nm. The emission spectra were recorded at 25 °C, from 300 to 400 nm at a scan rate of 125 nm·min−1. For ANS fluorescence (5 μm ANS; 0.5 μm CyaA), the excitation wavelength was fixed at 360 nm. The emission spectra were recorded from 450 to 550 nm.
The renaturation of CyaA was initiated by directly diluting the CyaA protein (10 μm stored in 8 m urea, 20 mm Hepes, pH 7.4) to a final concentration of 0.5 μm into either buffer A (20 mm Hepes, 150 mm NaCl, pH 7.4) or buffer B (buffer A plus 2 mm CaCl2) supplemented with the appropriate quantity of the chaotropic agent to obtain the final urea concentration. Samples were equilibrated for 2 h at 25 °C before fluorescence measurements. The buffer A (or buffer B) supplemented with the targeted urea concentration was used as a blank, and its spectrum was subtracted to each protein fluorescence spectrum. The maximum emission wavelength (λmax) values represent the average of three values obtained from emission spectra that were corrected for blank measurements.
Synchrotron Radiation Circular Dichroism Spectroscopy
Synchrotron radiation CD spectra were recorded on the Disco beamline at the synchrotron facility SOLEIL, (Gif-sur-Yvette, France). The synchrotron radiation CD experiments were carried out at 25 °C, an integration time of 1200 ms, and a bandwidth of 1 nm with a 1 nm resolution step. Each far-UV spectrum represents the average of three scans of CyaA at 5 μm. Optical cells with a 26-μm path length and CaF2 windows (Hellma) were used for recording CD signals in the far-UV region (from 180 to 260 nm). The CD units used were the mean residue ellipticity, expressed in kilodegrees·cm2/dmol·amino acid and calculated as described previously (57). The far-UV CD spectrum was deconvoluted using K2D3 (58), and secondary structure predictions were performed with SOPMA (59).
Circular Dichroism Spectroscopy
CD spectra were recorded on an Aviv circular dichroism spectrometer model 215, equipped with a water-cooled Peltier unit as described elsewhere (17). CD measurements were carried out at a scan rate of 0.5 nm/s (step, 0.5 nm; integration time, 1 s) with a time constant of 100 ms and a bandwidth of 1 nm. Each far-UV and near-UV CD spectrum represents the average of at least 5 scans. Far-UV and near-UV CD spectra (CyaA, 1 μm) were recorded in rectangular quartz Suprasil cells of 0.1 mm and 1-cm path lengths (106.QS and 114B.QS, Hellma). To follow the renaturation of CyaA, the stock solution (10 μm in 8 m urea, 20 mm Hepes, pH 7.4) was directly diluted to a final concentration of 1 μm either in buffer A or in buffer B adjusted with the appropriate concentration of urea. CD spectra were recorded after 1 h of equilibration at 25 °C. The buffer A or buffer B supplemented with the final urea concentrations was used as a blank, and its spectrum was subtracted from each protein CD spectrum.
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed on a Beckman XL-A analytical ultracentrifuge (Beckman Coulter) in an AN60-Ti rotor at 25 °C. The samples were filtrated on 0.2-μm filters before experiments. Detection of the protein concentration as a function of radial position and time was performed by optical density measurements at a wavelength of 280 nm. The buffer was buffer A or buffer B. The computed viscosity and density of this buffer were (SEDNTERP 1.09) 0.908 centiPoise and 1.004 g·ml−1 at 25 °C, respectively. The CyaA stock solution (10 μm in 8 m urea, 20 mm Hepes, pH 7.4) was loaded onto a G25 column equilibrated in buffer A to remove urea, and the collected CyaA samples (diluted to 400 μl at 1.4 μm) were supplemented or not with 2 mm CaCl2, loaded into a 1.2-mm-thick two-channel epoxy centerpiece, and spun at 20,000 rpm. Data were analyzed with the Sedfit software using a continuous size distribution c(s) model (17). We used the Svedberg equation to estimate the molecular mass of the species identified by sedimentation velocity, assuming a frictional ratio ranging from 1.2 to 3 as acceptable values (60).
Size Exclusion Chromatography Coupled On-line to a Tetra Detector Array
SEC was carried out on TSK 4000SWxl (TOSOH; rigid spherical silica; particle size, 8 μm), Superdex 200 10/300 and 5/150 (GE Healthcare; cross-linked agarose and dextran; particle size, 9–13 μm), Superose 6HR (GE Healthcare; agarose; particle size, 11–15 μm), and Sephacryl S-200 (GE Healthcare; copolymer of allyl dextran and N,N-methylenebisacrylamide; particle size, 25–75 μm) media. SEC was controlled by a GPCmax module connected on-line to a tetra detector array (TDA) model 302 (Malvern Instruments Ltd). The oven of the TDA contained (i) a static light scattering cell with two photodiode detectors, at 7º for low angle and at 90° for right angle laser light scattering; (ii) a deflection refractometer; (iii) a photometer; and (iv) a differential viscometer. Protein concentration was determined using both the photometer and the deflection refractometer. The right angle laser light scattering data coupled to the concentration provided the molecular mass. The SEC is also coupled on-line to a QELS detector (μV, Malvern Instruments Ltd.), which provided the hydrodynamic radius. CyaA samples in 8 m urea or after dialysis, dilution, or desalting were analyzed by SEC-TDA, following the procedures described elsewhere (25, 60, 61). Briefly, all solutions were filtered on 0.2-μm filters and allowed to equilibrate at 20 °C before SEC experiments. Sample analyses were performed at 25 °C in the oven of the TDA. All experimental sequences comprised calibration injections of BSA and polyethylene oxide used for TDA calibration (200 μl at 2 mg/ml). All data were acquired and processed using the Omnisec software (Malvern Instruments Ltd.).
The protein batches were prepared as follows. The dialyzed CyaA in buffer A or B was obtained by incubating an aliquot of CyaA in 8 m urea, 20 mm Hepes, pH 7.4, in a Float-A-Lyzer G2 cassette. The desalted CyaA in buffer A or B was prepared using a 5-ml bed volume G25SF column.
Hemolysis and Intoxication Assays
The hemolytic and cytotoxic (i.e. ability to raise cAMP inside target cells) activities of the toxin were determined on sheep erythrocytes as described previously (8). Sheep erythrocytes were washed several times with buffer B and resuspended in this buffer at 5 × 108 cells/ml. The different CyaA samples (i.e. CyaA renatured by G25 buffer exchange and oligomeric and monomeric CyaA species collected after refolding on the TSK column) were directly diluted into the erythrocyte suspension to reach the final concentration (ranging from 0.03 to 30 μg/ml for hemolytic activity and from 1 to 1000 ng/ml for the invasive activity). Control experiments were performed in the presence of excess EDTA (4 mm). The hemolytic activity was measured after an overnight incubation at 37 °C by quantifying the amount of hemoglobin released at 540 nm (and of intracellular content released at 405 nm). Complete lysis was obtained by the addition of 0.1% Triton X-100.
The invasive activity was determined by measuring the intracellular cAMP accumulation. The erythrocyte suspensions were incubated with CyaA at 37 °C for 20 min, and then cells were chilled on ice, centrifuged at 4 °C at 2 500 rpm for 5 min, and resuspended in buffer A supplemented with 4 mm EDTA. The cells were centrifuged similarly, and the pellets were resuspended in 200 μl of buffer A plus 4 mm EDTA. After transfer into clean tubes, the samples were lysed with 400 μl of 0.1 n HCl and boiled for 5 min at 100 °C (to inactivate any remaining adenylate cyclase). The solutions were then neutralized by the addition of 400 μl of 0.1 n NaOH, and the insoluble material was removed by centrifugation at 14,000 rpm for 10 min. The intracellular cAMP content was determined by a competitive immunoassay using the HitHunter® cAMP XS+ assay kit (DISCOVERX) following the manufacturer's instructions.
RESULTS
Folding of CyaA by Dilution, Dialysis, or Desalting
We first characterized the folding process of the CyaA toxin starting from the unfolded state in 8 m urea at neutral pH. The recombinant CyaA toxin used hereafter was produced in E. coli and purified essentially as described previously (see “Experimental Procedures”); an additional step of SEC in the presence of 8 m urea was added to improve the protein purity (see “Experimental Procedures”). The CyaA toxin was stored at −20 °C in 20 mm Hepes, 8 m urea, pH 7–8. We first investigated the refolding process of CyaA as a function of urea concentration in the absence (apo-state) and in the presence (holo-state) of calcium by intrinsic fluorescence of tryptophan, by ANS fluorescence and by far-UV circular dichroism (Fig. 2). CyaA contains 15 tryptophan residues that can be used as a macroscopic probe of protein folding, whereas ANS fluorescence is sensitive to the presence of solvent-exposed apolar surfaces made of organized hydrophobic residues on the protein. The folding process was studied in 20 mm Hepes, 150 mm NaCl, pH 7.4, in the absence (buffer A) or in the presence of 2 mm CaCl2 (buffer B).
FIGURE 2.

CyaA refolding followed by fluorescence and circular dichroism. Refolding of urea-denaturated apo-CyaA (open circles) or holo-CyaA (black circles) was followed by tryptophan intrinsic fluorescence (A), ANS fluorescence (B), and CD in the far-UV region (C and D). Maximum emission wavelength values are reported for both tryptophan and ANS fluorescence. C, far-UV CD spectra of CyaA (1 μm) in the absence (dashed line) and in the presence (solid line) of 2 mm calcium are shown in the presence of 6 m urea and after extensive dialysis against buffer A or buffer B. Changes of mean residual ellipticity of the n-π* band followed at 220 nm are shown in D. For all spectra and mean residue ellipticity changes, the contributions from the buffer were subtracted.
Both tryptophan and ANS fluorescence data showed that CyaA was unfolded in the presence of urea at concentrations higher than 4 m (Fig. 2, A and B) with an intrinsic fluorescence maximum emission wavelength at 355 nm typical of tryptophan side chains fully exposed to the solvent and a maximum emission wavelength of ANS at 520 nm, indicating the absence of solvent-exposed hydrophobic surface. Below 4 m urea, both tryptophan and ANS maximum emission wavelength of fluorescence changed, indicating that tryptophan residues were less exposed to the solvent, reaching a more apolar environment (Fig. 2A) and that solvent-exposed hydrophobic patches were formed (Fig. 2B). The main refolding steps of CyaA in the presence of calcium were initiated at 4 m and essentially completed at 2 m urea, whereas in the absence of calcium, the refolding appeared less cooperative, occurring between 4 and 0 m urea.
The refolding of CyaA was then explored by circular dichroism in the far-UV region. The CD spectra of CyaA in 6 m urea both with and without calcium were typical of unfolded proteins (Fig. 2C). The far-UV CD spectra of CyaA after extensive dialysis against buffer in the absence or in the presence of 2 mm calcium showed that CyaA had acquired significant secondary structure elements. The presence of a split π-π* band (above and below 200 nm) and the higher intensity of the n-π* band around 220 nm of the CD spectrum of the protein in the presence of calcium suggest a higher content of helical structure as compared with CyaA in the absence of calcium. The secondary structure content of CyaA upon refolding by dilution of urea was then followed at 220 nm as a function of urea concentration (Fig. 2D). The secondary structure changes as a function of urea concentrations were similar to the tryptophan or ANS fluorescence changes, suggesting that the folding of secondary and tertiary structures occurred in a concerted manner.
We further analyzed the various CyaA batches by SEC followed by a TDA. The right angle static laser light scattering, combined with the UV detector signals, allows molecular mass determination, whereas the quasielastic light scattering provides the hydrodynamic radius (RH) of the eluting species. CyaA (in 8 m urea) when loaded on a TSK column equilibrated in 20 mm Hepes, 150 mm NaCl, 4 m urea, pH 7.4, eluted as a broad peak with a molecular mass of 180 ± 10 kDa (Fig. 3A) and an averaged RH of 12 ± 2 nm (Fig. 3B and Table 1), indicating that CyaA in these conditions was monomeric and unfolded. It is noteworthy that this RH value of urea-unfolded CyaA corresponds to the expected value for a urea-unfolded protein of that size (62) (see Table 1). CyaA was then dialyzed against buffer A or B (i.e. in the absence or in the presence of 2 mm calcium) and analyzed similarly by SEC-TDA. The CyaA dialyzed without calcium eluted mainly as multimeric species with molecular masses ranging between 500 and 2000 kDa (Fig. 3C). A similar profile was observed with the CyaA dialyzed in the presence of calcium, albeit a weak peak around 14 ml probably corresponding to a monomeric form could be detected (Fig. 3D).
FIGURE 3.

Size exclusion chromatography of CyaA in the presence and in the absence of urea after dialysis or desalting. A and B, SEC of CyaA on a TSK 4000SWxl column equilibrated in 4 m urea in buffer A (without calcium) with the molecular mass (A) and hydrodynamic radius (B) distributions measured by TDA. C–F, SEC analysis on the TSK 4000SWxl column of CyaA samples after dialysis on a Float-A-Lyzer G2 cassette against buffer A (C) or buffer B (D) or after CyaA desalting on G25SF in buffer A (E) or buffer B (F).
TABLE 1.
Comparison of expected and measured hydrodynamic radii
Hydrodynamic radius values of CyaA were measured in 6 m urea, 20 mm Hepes, pH 7.4, and in the monomeric state in 20 mm Hepes, 150 mm NaCl, 2 mm CaCl2, pH 7.4. The data were compared with the expected values for a protein of 177 kDa in various folding states. The relations from molecular masses and folding states and hydrodynamic radii were taken from Ref. 62. The RH of the monomeric state of CyaA fits to a molten globule state. Several hypotheses may explain this observation: (i) some regions might be partially folded in the folded monomer; (ii) CyaA is a multidomain protein and could hence increase its friction with the solvent; and/or (iii) the molecular shape of the monomeric CyaA is far from a spherical shape. All hypotheses are not mutually exclusive.
| State of the protein | RH of a 177-kDa protein | RH of CyaA |
|---|---|---|
| nm | nm | |
| Native | 4.7 | |
| Molten globule | 5 | |
| Monomeric CyaA | 5.2 ± 0.3 | |
| Premolten globule | 7 | |
| Natively unfolded | 10.9 | |
| CyaA unfolded in 6 m urea | 12 ± 2 | |
| Unfolded in urea | 12.2 | |
| Unfolded in guanidinium | 13.4 |
In another set of experiments, the urea was removed from the CyaA samples by using a rapid buffer exchange by chromatography on a Sephadex G-25 Superfine desalting column instead of the dialysis procedure. The desalted CyaA samples were then analyzed similarly by SEC-TDA on the TSK column. As shown in Fig. 3, E and F, CyaA refolded by buffer exchange in the absence of calcium eluted mainly as multimers, whereas a significant quantity of monomeric forms could be detected in the sample desalted in the presence of calcium. Collectively, these data indicate that although the urea-unfolded state was monomeric, upon removal of urea by dialysis or by a desalting procedure, CyaA formed mainly multimeric species both in calcium-free and in calcium-containing buffers, in agreement with prior studies (37, 48).
Analytical ultracentrifugation was performed to further analyze the CyaA samples obtained by refolding through the gel filtration buffer exchange procedure (Fig. 3, E and F). Analytical ultracentrifugation was done either in the absence (Fig. 4, A and B) or in the presence of 2 mm calcium (Fig. 4, C and D). The distribution of sedimentation coefficients (Fig. 4E) shows that in the absence of calcium, the main population is centered on 12 S, corresponding to multimers, mainly dimers to tetramers, whereas a weak population around 6 S may correspond to a monomeric species in the apo-state. In the presence of calcium, a broad distribution of multimers was observed from 10 to 45 S with a minor peak at 7–8 S possibly corresponding to the monomeric population of holo-CyaA (see “Experimental Procedures”).
FIGURE 4.
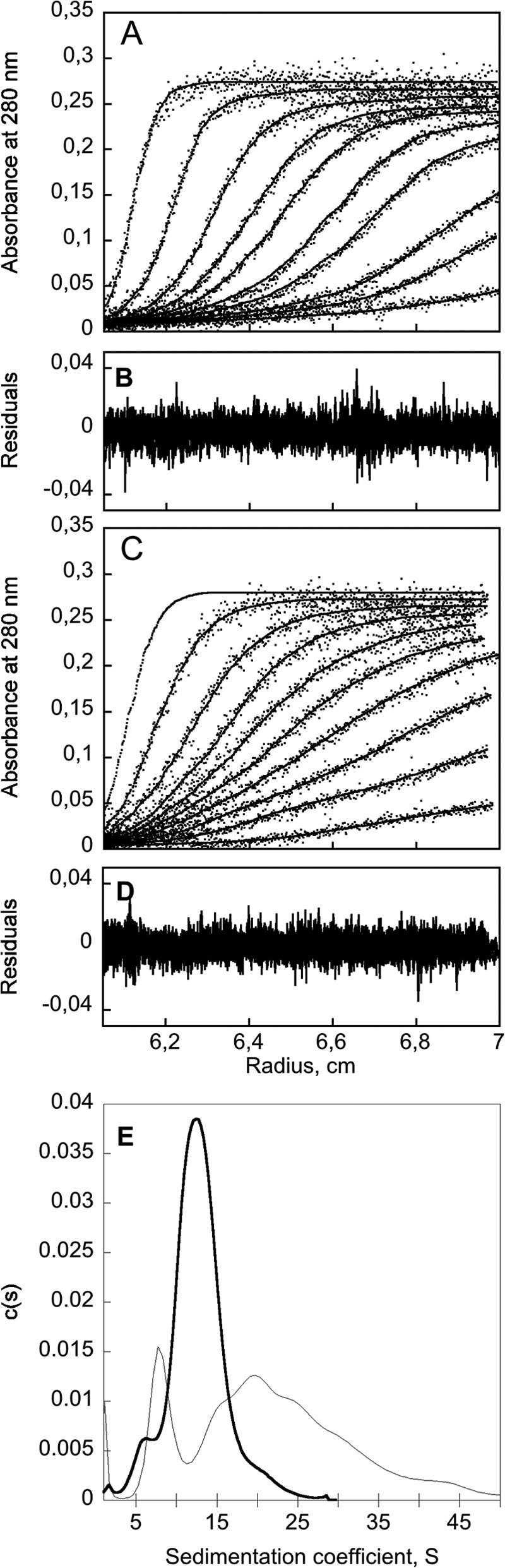
Analytical ultracentrifugation of CyaA after urea removal by desalting on G25. CyaA (in 8 m urea) was chromatographed on G25SF equilibrated in buffer A or buffer B. The sedimentation distribution profile of CyaA (1.4 μm) was analyzed with a Beckman-Coulter XL-I analytical ultracentrifuge using an AnTi rotor. Experimental data of sedimentation velocity (dots) in the absence (A) and in the presence (C) of 2 mm calcium were fitted with the Lamm equation (lines), and the distributions of the residual values are shown in B and D, respectively. E, sedimentation coefficient distribution of apo-CyaA (dashed line), and holo-CyaA (thick line) deduced from the fitted curves.
Molecular Confinement Is Required to Produce Monomeric CyaA
The results described above showed that CyaA folding is initiated below 4 m urea with a concomitant formation of secondary and tertiary structures and the appearance of solvent-exposed hydrophobic surfaces that are probably involved in the aggregation process leading to the multimeric states of CyaA. We hypothesized that molecular confinement of CyaA during the refolding process could decrease the intermolecular interactions between the polypeptides and could thus reduce potential aggregation. An experimental approach to confine proteins is to use gel filtration on matrix characterized by small particle and pore sizes. To test this hypothesis, we directly loaded the unfolded CyaA (in 8 m urea) onto a TSK 4000SWxl column made of a matrix with a particle size of 8 μm and pore size dimension of ∼45 nm (see “Experimental Procedures”) and equilibrated in buffer B. Fig. 5A shows the UV and molecular mass profiles of CyaA chromatographed on the TSK column. In these conditions, we observed multimeric forms eluting from 9 to 13 ml and a significant fraction of a monomeric species eluting between 13 and 15 ml (molecular mass, 180 ± 20 kDa). The hydrodynamic radius of the monomeric CyaA species measured by dynamic light scattering was 5.2 ± 0.3 nm (Table 1). This RH value is slightly higher than expected for a folded and globular native protein of 1706 residues (62) (see Table 1). This excess of friction might be related to CyaA shape and/or hydration (see Ref. 61 for details; see the Table 1 legend). We further showed that the monomeric holo-CyaA toxin was stable for long term storage at −20 °C because its hydrodynamic radius and its retention volume by SEC were identical before and after thawing.
FIGURE 5.

Refolding of CyaA into monomeric species by SEC-TDA. A, SEC-TDA analysis of CyaA (5 μm in 8 m urea) directly injected on a TSK 4000SWxl column equilibrated in buffer B (with 2 mm calcium). Black line, UV profile; red line, molecular mass. B, SEC-TDA in the same conditions of CyaA (in 8 m urea) loaded at the following concentrations: 1 μm (blue), 2.5 μm (green), 5 μm (red), and 12 μm (black). C, the UV profiles shown in B are normalized to the monomer peak (∼14 ml) intensity to highlight the concentration-dependent distribution of monomers and multimers.
We further analyzed the impact of the initial concentration of CyaA on the multimer/monomer ratio. Samples of CyaA at a concentration of 12, 5, 2.5, or 1 μm in 8 m urea were loaded on the TSK column equilibrated in buffer B. Fig. 5C shows that the proportion of monomers versus multimers was dependent upon the initial concentration of the urea-unfolded CyaA loaded onto the column. The four SEC profiles of CyaA show that the fraction of multimers increased with the initial CyaA concentration, suggesting that the multimer formation is an aggregative process.
Fig. 6A compares the UV profiles on SEC-TDA obtained by the three methods used to refold CyaA (i.e. by dialysis, by buffer exchange onto G-25, or by direct loading onto the TSK column). It is noteworthy that the proportion of monomers appears to be related to the molecular confinement during refolding because a higher fraction of monomeric CyaA was obtained with the TSK matrix with a smaller particle size (8 μm) as compared with the G-25 matrix (52 μm) or with the absence of confinement in the case of the dialysis procedure. The TSK column is made of rigid spherical silica beads bonded with hydrophilic groups, whereas the Sephadex G-25 is a bead-formed gel made of epichlorohydrin-cross-linked dextran. To determine whether the chemical nature of the matrix may also contribute to the formation of monomeric CyaA in addition to the confinement effect, we compared results obtained on the TSK column with those obtained on a Superdex 200 resin, made of cross-linked dextran and agarose with a particle size of 11 μm. As shown in Fig. 6B, the urea-denatured CyaA loaded and refolded onto the Superdex 200 10/300 column eluted as both multimers (8–11 ml) and monomer (12 ml), as observed with the TSK column. To further test the confinement-dependent refolding, CyaA was analyzed by SEC on Superdex 200 after protein dialysis in the presence of calcium. The Superdex 200 chromatogram clearly shows that dialysis of CyaA produced only multimers (Fig. 6B). These results indicate that the confinement properties of the matrix (TSK or Superdex 200) rather than its chemical nature are important for efficient folding of CyaA into a monomeric form.
FIGURE 6.

Molecular confinement favors CyaA folding into monomeric species. A, SEC of CyaA on TSK 4000SWxl (particle size, 8 μm) after urea removal by dialysis (dashed trace), desalting on G25SF (thin gray trace) and direct refolding on TSK 4000SWxl (heavy trace). B, SEC of CyaA on Superdex 200 10/300 after urea removal by dialysis (dashed trace) and by direct refolding (heavy trace) on the column. C, SEC of CyaA directly loaded on Superdex 200 10/300 (heavy trace; particle size, 11 ± 2 μm) or on Sephacryl S-200 (dashed trace; particle size, 50 ± 25 μm). Both columns had a bed volume of 24 ml. The O and M arrows indicate the oligomeric and monomeric fractions collected on the Superdex 200 10/300 chromatography. D, SEC of CyaA loaded on Superose 6HR (particle size, 13 ± 2 μm; total volume of 24 ml). Samples of 200 μl of 5 μm CyaA in 8 m urea, 20 mm Hepes, pH 7.4, were loaded in all experiments of direct refolding on the column. All SECs were carried out in buffer B.
The confinement effect was further evaluated by comparing the CyaA folding process on Superdex 200 and on a Sephacryl S-200 column, the latter having a similar chemical nature (agarose and dextran) and similar optimum protein separation range as the Superdex resin but with larger particle size (50 ± 25 μm for Sephacryl versus 11 μm for Superdex). The UV profiles obtained with the Sephacryl S-200 and the Superdex 200 (packed in the same XK16/60 column to strictly compare the experiments) are superimposed in Fig. 6C and clearly demonstrate that the confinement provided by the Superdex matrix by reducing intermolecular interactions strongly favored monomer formation as compared with the Sephacryl medium, which only produced multimers. Refolding of CyaA on a Superose 6HR column made of agarose beads with a particle size of 13 ± 2 μm (similar to Superdex 200) also favored the folding of the protein into a monomeric state (Fig. 6D). Taken together, these data indicate that molecular confinement during CyaA refolding on SEC may reduce intermolecular interactions between proteins and thus favor folding of the toxin into a monomeric state. The confinement properties appear rather independent of the chemical composition of the matrix (silica, agarose, and dextran).
Calcium and Acylation Are Required for CyaA Folding into a Monomeric Form
Besides molecular confinement, we investigated the parameters that could affect the efficiency of formation of CyaA monomers. CyaA is known to require calcium and acylation for its toxic activities (i.e. cell lysis (hemolysis) and delivery of its catalytic domain across the plasma membrane into the cytosol to produce cAMP (intoxication)). The effect of calcium on CyaA folding is illustrated by the SEC profiles of CyaA refolded on a Superdex 200 5/150 column equilibrated in the absence or in the presence of 2 mm calcium (Fig. 7A). The comparison of the SEC profiles shows that the presence of calcium was crucial for CyaA refolding. The refolding properties of pro-CyaA (i.e. the toxin without acyl chains) were then investigated (Fig. 7B). The post-translational acylation of pro-CyaA, leading to the mature CyaA protein, is well known to be essential for toxin activity. However, its impact on the folding of CyaA has never been investigated. We first measured the molecular mass of pro-CyaA in 4 m urea. The SEC-TDA data showed that pro-CyaA was monomeric in 4 m urea (not shown), as observed for the acylated CyaA (Fig. 3A). The SEC profile of pro-CyaA upon refolding onto Superdex 200 in the presence of 2 mm calcium shows two species (Fig. 7B, thick trace), the main peak corresponding to large multimers and the second peak, appearing as a shoulder on the main peak of multimers, corresponding to species of smaller sizes. However, in marked contrast to the acylated CyaA (Fig. 7A, thick trace), no distinct peak corresponding to a monomeric pro-CyaA species could be evidenced. Finally, refolding of CyaA and pro-CyaA in the absence of calcium was also examined and showed that both proteins refolded in EDTA led to the formation of multimers (Fig. 7, A and B, dashed traces). Collectively, these data indicate that besides molecular confinement, both acylation and calcium are required to produce high yields of CyaA monomers.
FIGURE 7.
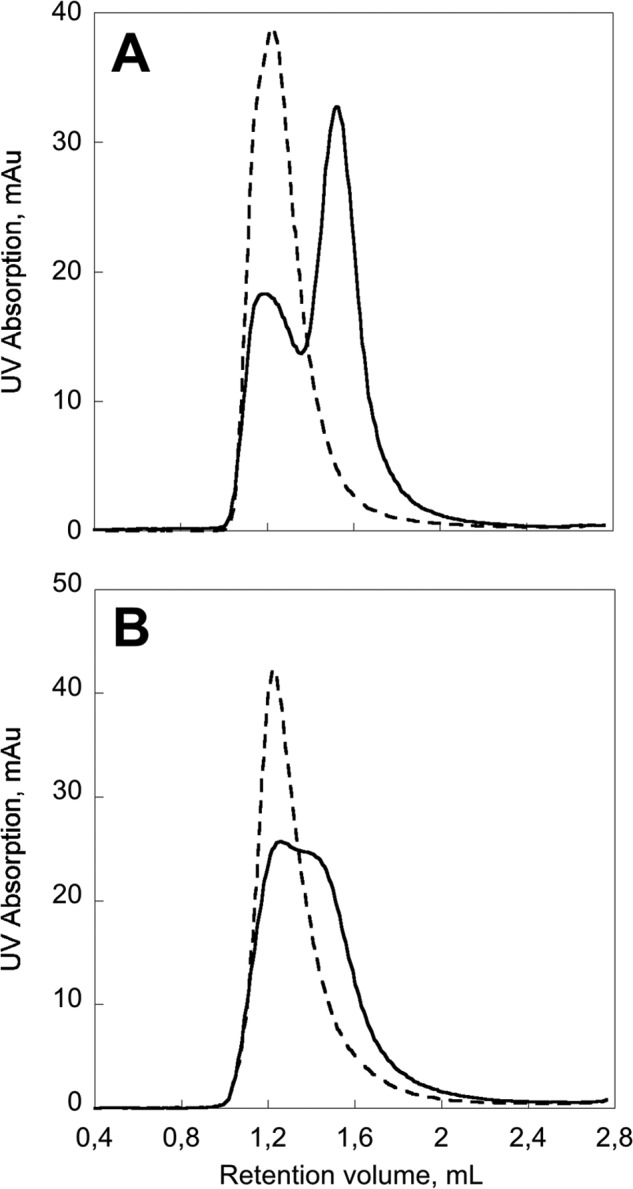
Post-translational acylation of CyaA and calcium binding are required to produce CyaA monomers. Shown is SEC of CyaA (A) and pro-CyaA (B) refolded on Superdex 200 5/150 in the absence (dashed traces; buffer A) and in the presence (heavy traces; buffer B) of 2 mm calcium. Samples of 50 μl of 5 μm CyaA or pro-CyaA (in 8 m urea, 20 mm Hepes, pH 7.4) were loaded in each experiment.
Structural and Functional Characterization of the Monomeric State of CyaA
We then analyzed by synchrotron radiation circular dichroism the secondary structure content of the monomeric CyaA species isolated from the SEC-assisted folding procedure (monomeric fraction M, eluting at 12–13 ml on the Superdex 200 chromatography shown in Fig. 6C). The far-UV CD spectrum of CyaA is typical of an α/β protein (Fig. 8), as suggested by the CD spectrum deconvolution performed by K2D3. The secondary structure content estimation obtained from the deconvolution is close to the secondary structure content of CyaA predicted by SOPMA (see legend to Fig. 8). We also analyzed the far-UV CD spectrum of the multimeric CyaA species (oligomeric fraction O, eluting at 7–9 ml in Fig. 6C). Both monomers and multimers exhibit similar far-UV CD spectra (Fig. 8, inset), indicating that the secondary structure content of CyaA is not significantly affected by the oligomerization state (quaternary structure) of the molecule.
FIGURE 8.

Circular dichroism and tryptophan quenching of CyaA. A, far-UV synchrotron radiation CD spectrum of holo-CyaA monomers in buffer B. Deconvolution by K2D3 provides the following secondary structure content estimation: 24% of helix and 27% of β-sheets. Inset, comparison between far-UV CD spectra of the monomeric (solid line) and multimeric (dashed line) forms of CyaA acquired on an Aviv spectropolarimeter, showing that n-π* bands of both species exhibit similar intensities. B, near-UV CD of the urea-unfolded (dashed lines), multimeric (dotted lines), and monomeric (heavy lines) CyaA. Shown are row data (C) and Stern-Volmer analysis (D) of KI quenching of intrinsic tryptophan fluorescence of l-Trp (crosses), monomeric (black circles), and multimeric (open circles) CyaA species.
The tertiary structure contents of CyaA in the urea-unfolded, monomeric, or multimeric states were then analyzed by near-UV CD (Fig. 8B). The near-UV CD spectrum of the urea-unfolded CyaA shows weak dichroic phenylalanine 1Lb bands at 262 and 268 nm and tryptophan 1Lb bands around 285 and 292 nm. Such weak intensities are expected for a urea-unfolded protein, indicating a low content of tertiary structure. The near-UV CD spectra of the folded monomeric and multimeric CyaA toxins exhibit similar strong dichroic bands, although the monomeric state provides more intense dichroic bands. The near-UV CD spectra show broad tyrosine 1Lb bands centered at 280 nm and tryptophan 1Lb bands around 285 and 292 nm (shifted to 295 nm due to the slope of the tyrosine 1Lb band). These near-UV CD data indicate that once CyaA is refolded, several aromatic side chains are involved in tertiary structure constraints. These results also indicate that the monomeric and oligomeric states of CyaA cannot be distinguished from their CD spectra either in the far-UV or in the near-UV region.
We also probed the accessibility of tryptophans by fluorescence quenching using KI as quencher. As shown in Fig. 8, C and D, the tryptophan side chains are more accessible to KI quenching in the monomeric CyaA than in the oligomeric ones. This suggests that, as a result of multimerization, some tryptophans of CyaA became buried in oligomers and thus less accessible to collisional quenching than in the monomeric state.
Finally, the biological activities of the different CyaA species were compared using sheep erythrocytes as model target cells. The oligomeric (O) and monomeric (M) CyaA fractions, eluted from the Superdex 200 column (Fig. 6C), were independently pooled, concentrated, and assayed for activity. For each species, both the hemolytic activity (i.e. ability to lyse cells) and the capacity of the toxin to increase intracellular cAMP (invasive activity) were monitored as a function of protein concentrations. CyaA refolded by rapid buffer exchange on G25 in the absence or in the presence of calcium was also tested in parallel. Fig. 9 shows that the refolded monomeric CyaA (fraction M) displayed the highest hemolytic and cytotoxic activities; its specific hemolytic and cytotoxic activities were about 20–25 times higher than that of the multimeric fractions (fraction O). CyaA refolded by rapid buffer exchange on G25 in the presence of calcium exhibited similar hemolytic activity as the oligomers from SEC and was rather inefficient at inducing cAMP production (Fig. 9, triangles). CyaA refolded by rapid buffer exchange on G25 in the absence of calcium exhibited neither hemolytic nor cytotoxic activities, in agreement with previous data showing that calcium is required for both activities.
FIGURE 9.
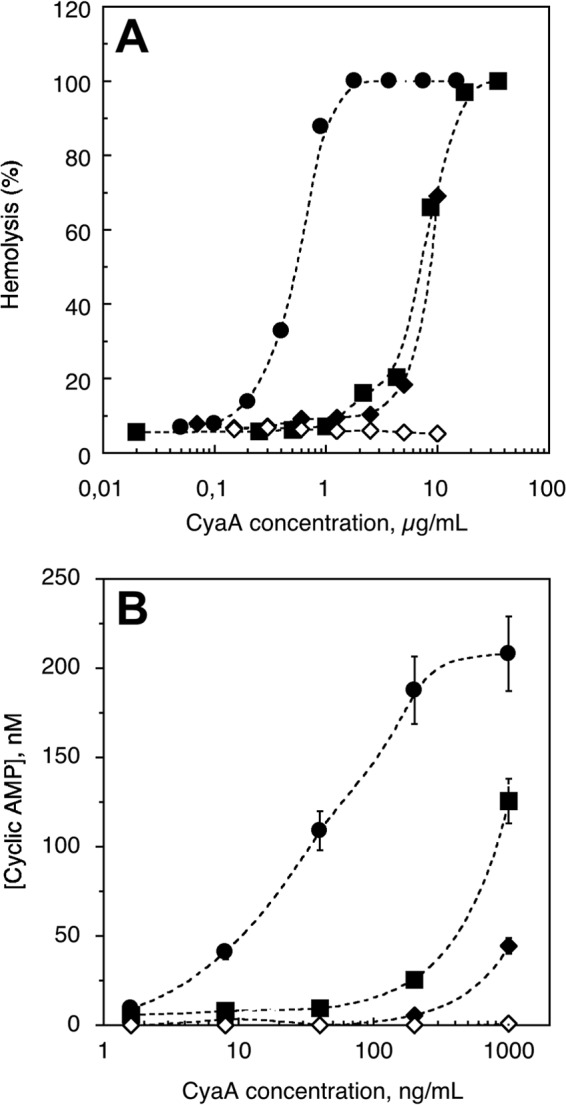
Hemolytic and cAMP-inducing activities of the various CyaA preparations. The different CyaA samples (i.e. CyaA renatured by G25 buffer exchange in the presence of calcium (black diamonds), CyaA renatured by G25 buffer exchange in the absence of calcium (open diamonds), and the oligomeric (black squares) and monomeric (black circles) CyaA species collected after refolding on TSK column in the presence of calcium (see the O and M fractions in Fig. 5C)) were directly diluted into an erythrocyte suspension (5 × 108 cells/ml in buffer B) to reach the indicated final concentrations. All hemolysis (A) and cAMP production experiments (B) were performed in the presence of 2 mm calcium except for the G25 buffer-exchanged CyaA in the absence of calcium, which was tested in the presence of 4 mm EDTA. The hemolytic activity and intracellular cAMP accumulation were determined as described under “Experimental Procedures.” Error bars, S.D.
It should be noted that the hemolytic activity shows a cooperativity (n ∼3) in agreement with prior reports suggesting that pore formation requires oligomerization of the protein in the membrane. In contrast, the accumulation of intracellular cAMP did not display any cooperativity, in line with the idea that the competent form able to translocate its catalytic domain across the plasma membrane is the monomeric state of CyaA. All together, these data indicate that the refolded, monomeric state of CyaA exhibits the highest hemolytic and cell-invasive activities and might therefore be considered as the genuine, functionally active form of the toxin.
DISCUSSION
We report here a procedure to produce a monomeric form of B. pertussis CyaA toxin that exhibits high hemolytic and cytotoxic activities and that can be stably maintained in this functional monodisperse state in the absence of any chaotropic agent. Since the original description of the adenylate cyclase in B. pertussis in the early 1980s, numerous reports have highlighted the heterogeneity of the molecular forms of this toxin that appeared to be prone to aggregation into multimeric non-functional complexes (37, 48, 52). Indeed, CyaA is a large protein of 1706 amino acid residues that contains several markedly hydrophobic regions as well as two post-translationally added acyl groups. Due to these characteristics, CyaA has been mostly extracted, purified, and stored in the presence of high concentrations of chaotropic agent (usually urea) to prevent its aggregation. In initial works, CyaA was purified from urea extracts from wild-type B. pertussis bacteria (6, 48, 51, 52). Later, after cloning of the cyaA gene in the late 1980s, CyaA was largely produced as a recombinant protein in E. coli, also co-expressing CyaC to permit its post-translational acylation (53, 63). In these recombinant cells, CyaA mainly accumulates as inclusion bodies requiring denaturing conditions for its solubilization. In most of these studies, the protein batches were stored after purification in buffers containing urea (higher than 6 m) to maintain it in a soluble form. The cytotoxic activities of CyaA have usually been tested by directly diluting the stock solution of toxin into cell suspensions without taking special care about its refolding process and/or its final oligomeric status. Even biological assays of CyaA toxin or CyaA-based vaccines (64–66) in animals have been carried out with recombinant CyaA preparations that were extemporaneously diluted in physiological buffers just before administration. How the toxin refolds and what are the precise molecular forms achieved by CyaA in these conditions remained largely unexplored.
Here we have characterized the refolding process of CyaA with the hope of identifying conditions that might favor its folding as a monomeric form suitable for further structural, biochemical, and cellular characterization. Our results showed that CyaA is unfolded and monomeric in urea concentrations higher than 4 m (Fig. 3 and Table 1), with a hydrodynamic radius of 12 ± 2 nm, in good agreement with the expected hydrodynamic radius for a protein of 177 kDa unfolded in urea (25, 62, 67). The folding of CyaA in the presence of calcium, as revealed by the acquisition of secondary structure elements and the formation of solvent-exposed hydrophobic patches, occurs at urea concentrations lower than 4 m and is essentially completed at 2 m (Fig. 2). Characterization of the hydrodynamic properties of the protein refolded upon urea removal either through dialysis, dilution, or rapid buffer exchange on a desalting column, indicated that in all of these conditions, CyaA mainly formed multimers, from tetramers to higher order oligomers, as reported earlier (37, 48).
With the aim of decreasing CyaA intermolecular interactions during urea removal and thus reducing the aggregative processes, we explored an alternative approach that involved molecular confinement of the protein during its refolding process. We found that refolding of urea-unfolded CyaA by size exclusion chromatography on resins with small particle and pore sizes (beads of different chemical nature with diameters from 8 to 15 μm on average) resulted in a significant fraction of the molecules eluting as a monomeric species with an apparent hydrodynamic radius of 5.2 ± 0.3 nm, as determined by dynamic light scattering. Interestingly, the fraction of monomeric species thus obtained appeared to be inversely correlated with the protein concentration of the sample loaded on the SEC column, suggesting that the formation of CyaA multimers is probably an aggregative process. This aggregation propensity is probably due to the appearance of solvent-exposed hydrophobic surfaces during the refolding process, as indicated by the ANS studies. More importantly, the folding of CyaA into a monomeric form was found to be critically dependent upon the presence of calcium as well as on the post-translational acylation of the protein. We further showed that, although the secondary structure content of the monomeric and multimeric species is rather similar, the monomeric form displayed about 20–25 times higher hemolytic and cytotoxic activities than the multimeric ones. This suggests that the refolded CyaA monomer is the genuine, physiologically active form of the toxin.
Interestingly, the hemolytic activity shows a marked cooperativity (nH ∼3) as a function of CyaA monomer concentration, indicating that, in accordance with prior studies, the membrane permeabilizing capacity requires oligomerization of the toxin. The fact that the monomeric state, in solution, of CyaA exhibits a much higher lytic potency than the multimeric ones suggests that the oligomerization of the protein might take place within the lipid bilayer, once monomers have partitioned into membranes. The conformations of membrane-inserted oligomers of CyaA are probably distinct from CyaA multimers formed in solution because the latter are less hemolytic. Previously, Sebo and colleagues (40) proposed that the lytic activity of CyaA may be carried out by a dedicated oligomeric conformation of CyaA, preexisting in solution, whereas the invasive activity would be mainly displayed by a distinct monomeric conformation. At variance, our present observations suggest that the monomeric state in solution, rather than the oligomeric state, is endowed with both the lytic and cytotoxic activities of CyaA. CyaA oligomerization within the membrane might then lead to the pore-forming state, whereas the monomeric CyaA is probably the competent form able to translocate its catalytic domain across the plasma membrane of the target cells, as suggested by the lack of cooperativity of intracellular cAMP accumulation.
We thus propose the following model. The CyaA monomer in solution is the functional state that partitions into the membrane mainly via hydrophobic forces, through its hydrophobic regions. Once CyaA is membrane-inserted, the translocation region may locally destabilize the lipid bilayer, favoring the passage of the catalytic domain across the plasma membrane (8, 9). Meanwhile, CyaA monomers can interact and reorganize into oligomers within the membrane. These CyaA oligomers may lead to pore formation that ultimately induces cell lysis (1). This sequential model fits nicely with the observed delay between the kinetics of cAMP accumulation and of hemolysis (6). However, further works will be needed to characterize the oligomerization process of CyaA within the membrane and its relation to functions.
Our present data provide a rational explanation for many prior observations on CyaA structure-function relationships and may also have important implications for the understanding of the role of calcium and acylation in the biological functions of other RTX cytolysins (14). Indeed, it has been known for many years that RTX cytolysins, such as CyaA, HlyA, LktA, etc., are synthetized as inactive precursors that are converted to their cytotoxic forms upon acylation by dedicated acyltransferase (CyaC, HlyC, LktC, etc …) and that their cytotoxic/cytolytic activities are critically dependent upon the presence of calcium (14, 15). We and others previously showed that calcium is essential for folding of the CyaA RTX domain, RD, which is important for protein interaction with the CyaA receptor and/or with target cell membrane. Our present work indicates that calcium binding to the RD directly contributes to the overall folding of the full-length toxin into a monomeric, active species. It is likely that in the presence of calcium, RD can acquire its calcium-bound three-dimensional structure that can then serve as a nucleation site for further folding of the upstream CyaA regions (i.e. the central hydrophobic and N-terminal catalytic domains). Incidentally, this proposed sequential refolding scheme fits with the idea that CyaA, like other RTX proteins, is secreted by the type I secretion machinery (T1SS) in a vectorial C- to N-terminal way, with its C terminus reaching first the external, calcium-rich environment. Calcium-dependent folding of the secreted RD probably begins as it emerges form the secretion channel and while the remaining CyaA polypeptide is still in transit in the T1SS machinery. The folded RD may thus drive the progressive folding of the upstream CyaA regions as they exit the secretory channel. This co-secretional folding of CyaA at the mouth of the T1SS machinery could contribute to the confinement of the toxin molecules and thus prevent their potential aggregation at the bacterial surface. Interestingly, Gray et al. showed that only the toxin that is actively secreted by B. pertussis by the T1SS pathway is able to invade eukaryotic cells (68). It is tempting to speculate that this invasive, actively secreted species described by these authors indeed corresponds to the monomeric CyaA toxin.
Besides calcium and molecular confinement, a major finding reported here is that CyaA acylation appears to be critical for toxin folding into the monomeric form. Indeed, we were not able to produce pro-CyaA monomers or isolate them from multimers, as we did with the acylated CyaA. These results thus establish for the first time a direct structural role for the acyl chains in the acquisition of a biologically functional conformation by CyaA, independently of, or in addition to, their putative role(s) in membrane interaction. This result is in line with the seminal observation of Rogel et al. (37), who first reported the purification of the toxic form of adenylate cyclase from B. pertussis extracts. By gel filtration, they resolved the enzymatic activity into two peaks, one major peak of high molecular mass (>700 kDa) and a minor peak, with an apparent size of 200 kDa. They showed that, although both fractions contained the same CyaA polypeptide, only the small sized fraction could induce cAMP accumulation in target cells (37). Because the CyaA polypeptide expressed in E. coli also lacks the ability to induce cAMP accumulation in target cells, they postulated that a post-translational modification should occur in B. pertussis but not in E. coli, to confer cytotoxic capabilities to CyaA. The CyaC-modifying acyltransferase and the specific acylation of CyaA on lysine residues 860 and 983 were later described by Hewlett and colleagues (10). How this modification could convert the non-cytotoxic pro-CyaA into an invasive and hemolytic protein has remained elusive until now (14, 15). Similar observations have been reported for other RTX cytolysins, in particular for HlyA, but the putative contribution of the acyl chains in the cytolytic activities of these toxins is also still unknown. It is generally assumed that the acyl chains could insert into the lipid bilayer to favor toxin partitioning to the plasma membrane of target cells (69). Alternatively, the acyl groups might play a structural role in maintaining the toxins in a partially folded conformation competent for membrane insertion (14, 15). No strong experimental evidence for this hypothesis has been obtained thus far. Herlax and Bakas (70) tentatively proposed that the acylation may maintain HlyA in a molten globule conformation favorable for membrane insertion, but their experimental data barely support such a conclusion.
Our present work suggests possible molecular mechanisms by which the acyl chains and calcium binding may accelerate the kinetics of CyaA refolding and thus favor the overall formation of functional monomeric species at the expense of the less active multimeric ones. The presence of hydrophobic acyl chains on CyaA may significantly modify the local free energy landscape of the polypeptide chain. Shielding of these acyl chains into a nascent hydrophobic core may have a major thermodynamic contribution in restricting the temporal and conformational spaces accessible to CyaA upon refolding. Similarly, we provide direct evidence that calcium binding is mandatory for the formation of monomers, probably by kinetically favoring native folding and burying hydrophobic regions, whereas in the absence of calcium, a slower kinetics of folding into the apo-state may favor intermolecular interactions between folding intermediates and would thus increase the population of multimer species. Further studies will be needed to unravel the respective contributions of calcium and acylation to the CyaA folding pathways.
Finally, the ability to produce CyaA as a functional and monodisperse species that appears to be sufficiently stable for in depth biophysical, structural, and cellular characterizations will be invaluable to clarify the fundamental basis of its mode of action. In addition, our current results will also be instrumental for further biotechnological improvements of CyaA as a vaccine vehicle for antigen delivery to dendritic cells, a novel vaccine strategy that is currently being evaluated in clinical trials.
Acknowledgments
We acknowledge SOLEIL for provision of synchrotron radiation facilities (Proposal ID 20110586 and 20120444), and we thank Frank Wien for assistance in using the Disco beamline. We thank Agnes Ullmann for critical reading of the manuscript.
This work was supported by the Agence Nationale de la Recherche, program Jeunes Chercheurs (Grant ANR-09-JCJC-0012), the Institut Pasteur Projet Transversal de Recherche PTR#374, the Jacob class of Pasteur-Paris University International Ph.D. Program from Institut Pasteur, and CNRS (UMR 3528, Biologie Structurale et Agents Infectieux).
- CaM
- calmodulin
- ACD
- ATP-cyclizing, CaM-activated, catalytic domain
- RD
- cell receptor-binding domain
- ANS
- 1-anilino-8-naphthalenesulfonate
- SEC
- size exclusion chromatography
- TDA
- tetra detector array
- RH
- hydrodynamic radius
- T1SS
- type I secretion machinery.
REFERENCES
- 1. Ladant D., Ullmann A. (1999) Bordetella pertussis adenylate cyclase: a toxin with multiple talents. Trends Microbiol. 7, 172–176 [DOI] [PubMed] [Google Scholar]
- 2. Vojtova J., Kamanova J., Sebo P. (2006) Bordetella adenylate cyclase toxin: a swift saboteur of host defense. Curr. Opin. Microbiol. 9, 69–75 [DOI] [PubMed] [Google Scholar]
- 3. Carbonetti N. H. (2010) Pertussis toxin and adenylate cyclase toxin: key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 5, 455–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glaser P., Ladant D., Sezer O., Pichot F., Ullmann A., Danchin A. (1988) The calmodulin-sensitive adenylate cyclase of Bordetella pertussis: cloning and expression in Escherichia coli. Mol. Microbiol. 2, 19–30 [PubMed] [Google Scholar]
- 5. Glaser P., Sakamoto H., Bellalou J., Ullmann A., Danchin A. (1988) Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J. 7, 3997–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bellalou J., Sakamoto H., Ladant D., Geoffroy C., Ullmann A. (1990) Deletions affecting hemolytic and toxin activities of Bordetella pertussis adenylate cyclase. Infect. Immun. 58, 3242–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sakamoto H., Bellalou J., Sebo P., Ladant D. (1992) Bordetella pertussis adenylate cyclase toxin. Structural and functional independence of the catalytic and hemolytic activities. J. Biol. Chem. 267, 13598–13602 [PubMed] [Google Scholar]
- 8. Karst J. C., Barker R., Devi U., Swann M. J., Davi M., Roser S. J., Ladant D., Chenal A. (2012) Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 287, 9200–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subrini O., Sotomayor-Pérez A. C., Hessel A., Spiaczka-Karst J., Selwa E., Sapay N., Veneziano R., Pansieri J., Chopineau J., Ladant D., Chenal A. (2013) Characterization of a membrane-active peptide from the Bordetella pertussis CyaA toxin. J. Biol. Chem. 288, 32585–32598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barry E. M., Weiss A. A., Ehrmann I. E., Gray M. C., Hewlett E. L., Goodwin M. S. (1991) Bordetella pertussis adenylate cyclase toxin and hemolytic activities require a second gene, cyaC, for activation. J. Bacteriol. 173, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hackett M., Guo L., Shabanowitz J., Hunt D. F., Hewlett E. L. (1994) Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 266, 433–435 [DOI] [PubMed] [Google Scholar]
- 12. Masin J., Basler M., Knapp O., El-Azami-El-Idrissi M., Maier E., Konopasek I., Benz R., Leclerc C., Sebo P. (2005) Acylation of lysine 860 allows tight binding and cytotoxicity of Bordetella adenylate cyclase on CD11b-expressing cells. Biochemistry 44, 12759–12766 [DOI] [PubMed] [Google Scholar]
- 13. Westrop G. D., Hormozi E. K., Da Costa N. A., Parton R., Coote J. G. (1996) Bordetella pertussis adenylate cyclase toxin: proCyaA and CyaC proteins synthesised separately in Escherichia coli produce active toxin in vitro. Gene 180, 91–99 [DOI] [PubMed] [Google Scholar]
- 14. Welch R. A. (2001) RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Curr. Topics Microbiol. Immunol. 257, 85–111 [DOI] [PubMed] [Google Scholar]
- 15. Linhartová I., Bumba L., Mašín J., Basler M., Osička R., Kamanová J., Procházková K., Adkins I., Hejnová-Holubová J., Sadílková L., Morová J., Sebo P. (2010) RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 34, 1076–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rose T., Sebo P., Bellalou J., Ladant D. (1995) Interaction of calcium with Bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 270, 26370–26376 [DOI] [PubMed] [Google Scholar]
- 17. Chenal A., Guijarro J. I., Raynal B., Delepierre M., Ladant D. (2009) RTX calcium binding motifs are intrinsically disordered in the absence of calcium: implication for protein secretion. J. Biol. Chem. 284, 1781–1789 [DOI] [PubMed] [Google Scholar]
- 18. Szilvay G. R., Blenner M. A., Shur O., Cropek D. M., Banta S. (2009) A FRET-based method for probing the conformational behavior of an intrinsically disordered repeat domain from Bordetella pertussis adenylate cyclase. Biochemistry 48, 11273–11282 [DOI] [PubMed] [Google Scholar]
- 19. Chenal A., Karst J. C., Sotomayor Pérez A. C., Wozniak A. K., Baron B., England P., Ladant D. (2010) Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys. J. 99, 3744–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sotomayor-Pérez A. C., Subrini O., Hessel A., Ladant D., Chenal A. (2013) Molecular crowding stabilizes both the intrinsically disordered calcium-free state and the folded calcium-bound state of a repeat in toxin (RTX) protein. J. Am. Chem. Soc. 135, 11929–11934 [DOI] [PubMed] [Google Scholar]
- 21. Linding R., Russell R. B., Neduva V., Gibson T. J. (2003) GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 31, 3701–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prilusky J., Felder C. E., Zeev-Ben-Mordehai T., Rydberg E. H., Man O., Beckmann J. S., Silman I., Sussman J. L. (2005) FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21, 3435–3438 [DOI] [PubMed] [Google Scholar]
- 23. Dosztányi Z., Csizmók V., Tompa P., Simon I. (2005) The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 347, 827–839 [DOI] [PubMed] [Google Scholar]
- 24. Lieutaud P., Canard B., Longhi S. (2008) MeDor: a metaserver for predicting protein disorder. BMC Genomics 9, Suppl. 2, S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sotomayor-Pérez A. C., Ladant D., Chenal A. (2011) Calcium-induced folding of intrinsically disordered repeat-in-toxin (RTX) motifs via changes of protein charges and oligomerization states. J. Biol. Chem. 286, 16997–17004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baumann U., Wu S., Flaherty K. M., McKay D. B. (1993) Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two-domain protein with a calcium binding parallel β roll motif. EMBO J. 12, 3357–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meier R., Drepper T., Svensson V., Jaeger K. E., Baumann U. (2007) A calcium-gated lid and a large β-roll sandwich are revealed by the crystal structure of extracellular lipase from Serratia marcescens. J. Biol. Chem. 282, 31477–31483 [DOI] [PubMed] [Google Scholar]
- 28. Satchell K. J. (2011) Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 65, 71–90 [DOI] [PubMed] [Google Scholar]
- 29. Holland I. B., Schmitt L., Young J. (2005) Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review). Mol. Membr. Biol. 22, 29–39 [DOI] [PubMed] [Google Scholar]
- 30. Masure H. R., Au D. C., Gross M. K., Donovan M. G., Storm D. R. (1990) Secretion of the Bordetella pertussis adenylate cyclase from Escherichia coli containing the hemolysin operon. Biochemistry 29, 140–145 [DOI] [PubMed] [Google Scholar]
- 31. Guermonprez P., Khelef N., Blouin E., Rieu P., Ricciardi-Castagnoli P., Guiso N., Ladant D., Leclerc C. (2001) The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the α(M)β(2) integrin (CD11b/CD18). J. Exp. Med. 193, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogel A., Hanski E. (1992) Distinct steps in the penetration of adenylate cyclase toxin of Bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J. Biol. Chem. 267, 22599–22605 [PubMed] [Google Scholar]
- 33. Basler M., Knapp O., Masin J., Fiser R., Maier E., Benz R., Sebo P., Osicka R. (2007) Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 282, 12419–12429 [DOI] [PubMed] [Google Scholar]
- 34. Eby J. C., Ciesla W. P., Hamman W., Donato G. M., Pickles R. J., Hewlett E. L., Lencer W. I. (2010) Selective translocation of the Bordetella pertussis adenylate cyclase toxin across the basolateral membranes of polarized epithelial cells. J. Biol. Chem. 285, 10662–10670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paccani S. R., Finetti F., Davi M., Patrussi L., D'Elios M. M., Ladant D., Baldari C. T. (2011) The Bordetella pertussis adenylate cyclase toxin binds to T cells via LFA-1 and induces its disengagement from the immune synapse. J. Exp. Med. 208, 1317–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gordon V. M., Young W. W., Jr., Lechler S. M., Gray M. C., Leppla S. H., Hewlett E. L. (1989) Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J. Biol. Chem. 264, 14792–14796 [PubMed] [Google Scholar]
- 37. Rogel A., Schultz J. E., Brownlie R. M., Coote J. G., Parton R., Hanski E. (1989) Bordetella pertussis adenylate cyclase: purification and characterization of the toxic form of the enzyme. EMBO J. 8, 2755–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guermonprez P., Ladant D., Karimova G., Ullmann A., Leclerc C. (1999) Direct delivery of the Bordetella pertussis adenylate cyclase toxin to the MHC class I antigen presentation pathway. J. Immunol. 162, 1910–1916 [PubMed] [Google Scholar]
- 39. Bauche C., Chenal A., Knapp O., Bodenreider C., Benz R., Chaffotte A., Ladant D. (2006) Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 281, 16914–16926 [DOI] [PubMed] [Google Scholar]
- 40. Vojtova-Vodolanova J., Basler M., Osicka R., Knapp O., Maier E., Cerny J., Benada O., Benz R., Sebo P. (2009) Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J. 23, 2831–2843 [DOI] [PubMed] [Google Scholar]
- 41. Bumba L., Masin J., Fiser R., Sebo P. (2010) Bordetella adenylate cyclase toxin mobilizes its β2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 6, e1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Veneziano R., Rossi C., Chenal A., Devoisselle J. M., Ladant D., Chopineau J. (2013) Bordetella pertussis adenylate cyclase toxin translocation across a tethered lipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 110, 20473–20478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uribe K. B., Etxebarria A., Martín C., Ostolaza H. (2013) Calpain-mediated processing of adenylate cyclase toxin generates a cytosolic soluble catalytically active N-terminal domain. PLoS One 8, e67648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heveker N., Ladant D. (1997) Characterization of mutant Bordetella pertussis adenylate cyclase toxins with reduced affinity for calmodulin: implications for the mechanism of toxin entry into target cells. Eur. J. Biochem. 243, 643–649 [DOI] [PubMed] [Google Scholar]
- 45. Benz R., Maier E., Ladant D., Ullmann A., Sebo P. (1994) Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 269, 27231–27239 [PubMed] [Google Scholar]
- 46. Hewlett E. L., Donato G. M., Gray M. C. (2006) Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: more than just making cyclic AMP!. Mol. Microbiol. 59, 447–459 [DOI] [PubMed] [Google Scholar]
- 47. Fiser R., Masín J., Basler M., Krusek J., Spuláková V., Konopásek I., Sebo P. (2007) Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin. Membrane translocation of AC domain polypeptide promotes calcium influx into CD11b+ monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 282, 2808–2820 [DOI] [PubMed] [Google Scholar]
- 48. Gentile F., Knipling L. G., Sackett D. L., Wolff J. (1990) Invasive adenylyl cyclase of Bordetella pertussis: physical, catalytic, and toxic properties. J. Biol. Chem. 265, 10686–10692 [PubMed] [Google Scholar]
- 49. Hewlett E. L., Gray L., Allietta M., Ehrmann I., Gordon V. M., Gray M. C. (1991) Adenylate cyclase toxin from Bordetella pertussis. Conformational change associated with toxin activity. J. Biol. Chem. 266, 17503–17508 [PubMed] [Google Scholar]
- 50. Cheung G. Y., Kelly S. M., Jess T. J., Prior S., Price N. C., Parton R., Coote J. G. (2009) Functional and structural studies on different forms of the adenylate cyclase toxin of Bordetella pertussis. Microb. Pathog. 46, 36–42 [DOI] [PubMed] [Google Scholar]
- 51. Hewlett E. L., Urban M. A., Manclark C. R., Wolff J. (1976) Extracytoplasmic adenylate cyclase of Bordetella pertussis. Proc. Natl. Acad. Sci. U.S.A. 73, 1926–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hewlett E. L., Gordon V. M., McCaffery J. D., Sutherland W. M., Gray M. C. (1989) Adenylate cyclase toxin from Bordetella pertussis. Identification and purification of the holotoxin molecule. J. Biol. Chem. 264, 19379–19384 [PubMed] [Google Scholar]
- 53. Karimova G., Fayolle C., Gmira S., Ullmann A., Leclerc C., Ladant D. (1998) Charge-dependent translocation of Bordetella pertussis adenylate cyclase toxin into eukaryotic cells: implication for the in vivo delivery of CD8+ T cell epitopes into antigen-presenting cells. Proc. Natl. Acad. Sci. U.S.A. 95, 12532–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ladant D., Brezin C., Alonso J. M., Crenon I., Guiso N. (1986) Bordetella pertussis adenylate cyclase: purification, characterization, and radioimmunoassay. J. Biol. Chem. 261, 16264–16269 [PubMed] [Google Scholar]
- 55. Semisotnov G. V., Rodionova N. A., Razgulyaev O. I., Uversky V. N., Gripas' A. F., Gilmanshin R. I. (1991) Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31, 119–128 [DOI] [PubMed] [Google Scholar]
- 56. Sotomayor Pérez A. C., Karst J. C., Davi M., Guijarro J. I., Ladant D., Chenal A. (2010) Characterization of the regions involved in the calcium-induced folding of the intrinsically disordered RTX motifs from the Bordetella pertussis adenylate cyclase toxin. J. Mol. Biol. 397, 534–549 [DOI] [PubMed] [Google Scholar]
- 57. Karst J. C., Sotomayor Pérez A. C., Guijarro J. I., Raynal B., Chenal A., Ladant D. (2010) Calmodulin-induced conformational and hydrodynamic changes in the catalytic domain of Bordetella pertussis adenylate cyclase toxin. Biochemistry 49, 318–328 [DOI] [PubMed] [Google Scholar]
- 58. Louis-Jeune C., Andrade-Navarro M. A., Perez-Iratxeta C. (2011) Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 10.1002/prot.23188 [DOI] [PubMed] [Google Scholar]
- 59. Geourjon C., Deléage G. (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684 [DOI] [PubMed] [Google Scholar]
- 60. Sotomayor-Pérez A. C., Karst J. C., Ladant D., Chenal A. (2012) Mean net charge of intrinsically disordered proteins: experimental determination of protein valence by electrophoretic mobility measurements. Methods Mol. Biol. 896, 331–349 [DOI] [PubMed] [Google Scholar]
- 61. Karst J. C., Sotomayor-Pérez A. C., Ladant D., Chenal A. (2012) Estimation of intrinsically disordered protein shape and time-averaged apparent hydration in native conditions by a combination of hydrodynamic methods. Methods Mol. Biol. 896, 163–177 [DOI] [PubMed] [Google Scholar]
- 62. Uversky V. N. (2002) What does it mean to be natively unfolded? Eur. J. Biochem. 269, 2–12 [DOI] [PubMed] [Google Scholar]
- 63. Sebo P., Glaser P., Sakamoto H., Ullmann A. (1991) High-level synthesis of active adenylate cyclase toxin of Bordetella pertussis in a reconstructed Escherichia coli system. Gene 104, 19–24 [DOI] [PubMed] [Google Scholar]
- 64. Saron M. F., Fayolle C., Sebo P., Ladant D., Ullmann A., Leclerc C. (1997) Anti-viral protection conferred by recombinant adenylate cyclase toxins from Bordetella pertussis carrying a CD8+ T cell epitope from lymphocytic choriomeningitis virus. Proc. Natl. Acad. Sci. U.S.A. 94, 3314–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dadaglio G., Morel S., Bauche C., Moukrim Z., Lemonnier F. A., Van Den Eynde B. J., Ladant D., Leclerc C. (2003) Recombinant adenylate cyclase toxin of Bordetella pertussis induces cytotoxic T lymphocyte responses against HLA*0201-restricted melanoma epitopes. Int. Immunol. 15, 1423–1430 [DOI] [PubMed] [Google Scholar]
- 66. Préville X., Ladant D., Timmerman B., Leclerc C. (2005) Eradication of established tumors by vaccination with recombinant Bordetella pertussis adenylate cyclase carrying the human papillomavirus 16 E7 oncoprotein. Cancer Res. 65, 641–649 [PubMed] [Google Scholar]
- 67. Uversky V. N., Gillespie J. R., Fink A. L. (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41, 415–427 [DOI] [PubMed] [Google Scholar]
- 68. Gray M. C., Donato G. M., Jones F. R., Kim T., Hewlett E. L. (2004) Newly secreted adenylate cyclase toxin is responsible for intoxication of target cells by Bordetella pertussis. Mol. Microbiol. 53, 1709–1719 [DOI] [PubMed] [Google Scholar]
- 69. Welch R. A. (1991) Pore-forming cytolysins of Gram-negative bacteria. Mol. Microbiol. 5, 521–528 [DOI] [PubMed] [Google Scholar]
- 70. Herlax V., Bakas L. (2007) Fatty acids covalently bound to α-hemolysin of Escherichia coli are involved in the molten globule conformation: implication of disordered regions in binding promiscuity. Biochemistry 46, 5177–5184 [DOI] [PubMed] [Google Scholar]