

Background: Mechanistic and structural conservation between evolutionarily distant Srv2/CAP homologs has remained unclear.
Results: Mouse CAP1 forms hexameric structures that autonomously bind F-actin, enhance cofilin-mediated severing, and catalyze nucleotide exchange on actin.
Conclusion: Yeast and mouse CAP share similar structures and functions.
Significance: The role of CAP in actin regulation is remarkably well conserved.
Keywords: Actin, Cofilin, Electron Microscopy (EM), Fluorescence, Microscopic Imaging, Srv2, Cyclase-associated Protein, Severing
Abstract
Srv2/CAP is a conserved actin-binding protein with important roles in driving cellular actin dynamics in diverse animal, fungal, and plant species. However, there have been conflicting reports about whether the activities of Srv2/CAP are conserved, particularly between yeast and mammalian homologs. Yeast Srv2 has two distinct functions in actin turnover: its hexameric N-terminal-half enhances cofilin-mediated severing of filaments, while its C-terminal-half catalyzes dissociation of cofilin from ADP-actin monomers and stimulates nucleotide exchange. Here, we dissected the structure and function of mouse CAP1 to better understand its mechanistic relationship to yeast Srv2. Although CAP1 has a shorter N-terminal oligomerization sequence compared with Srv2, we find that the N-terminal-half of CAP1 (N-CAP1) forms hexameric structures with six protrusions, similar to N-Srv2. Further, N-CAP1 autonomously binds to F-actin and decorates the sides and ends of filaments, altering F-actin structure and enhancing cofilin-mediated severing. These activities depend on conserved surface residues on the helical-folded domain. Moreover, N-CAP1 enhances yeast cofilin-mediated severing, and conversely, yeast N-Srv2 enhances human cofilin-mediated severing, highlighting the mechanistic conservation between yeast and mammals. Further, we demonstrate that the C-terminal actin-binding β-sheet domain of CAP1 is sufficient to catalyze nucleotide-exchange of ADP-actin monomers, while in the presence of cofilin this activity additionally requires the WH2 domain. Thus, the structures, activities, and mechanisms of mouse and yeast Srv2/CAP homologs are remarkably well conserved, suggesting that the same activities and mechanisms underlie many of the diverse actin-based functions ascribed to Srv2/CAP homologs in different organisms.
Introduction
While mechanisms for cellular actin assembly have been extensively studied, the counter-balancing mechanisms underlying actin filament disassembly and turnover are only now beginning to be understood. At the heart of the disassembly process is the actin-binding protein cofilin, which severs filaments, thereby amplifying the number of filament ends for depolymerization (1–4). Cofilin is essential in vivo for rapid actin disassembly (5, 6), and three decades of biochemical research have produced a rich understanding of its mechanism (7–9). Cofilin binds cooperatively to F-actin, alters the conformation of actin subunits, and induces filament twisting by about 5 degrees per subunit, reducing the helical crossover distance (10, 11). In addition, cofilin decoration changes the mechanical properties of filaments, reducing filament persistence length by about 4-fold, and thus creates phase boundaries between decorated and undecorated regions, which induces severing events (12, 13).
Although cofilin is sufficient to sever filaments in vitro, as visualized in real time by TIRF microscopy (14), its activities in vivo are further regulated and enhanced by additional factors, including Aip1, coronin, and Srv2/CAP (cyclase-associated protein)2 (15, 16). The physiological importance of these disassembly co-factors has been clearly demonstrated through numerous genetic studies; however, their underlying mechanisms remain only partially understood. Further, different protein activities have been reported in some cases depending on which species is used, which has raised questions about how well the mechanisms are conserved across distant species. Here, we address the mechanism and conservation of Srv2/CAP in regulating cofilin-mediated actin disassembly.
Yeast and mammalian Srv2/CAP proteins have two distinct functions in stimulating actin turnover. The C-terminal-half of the protein displaces cofilin from ADP-G-actin and catalyzes monomer nucleotide exchange (17–19), whereas the N-terminal-half enhances cofilin-mediated severing of filaments, even in the absence of actin monomers (20, 21). Recently, it was shown that the two halves of yeast Srv2 can even be physically separated and function in a largely autonomous manner in vitro and in vivo (22). The N-terminal-half of yeast Srv2 (N-Srv2) has also been shown to self-associate into hexameric structures resembling shurikens (20), although it has been unclear whether other species of Srv2/CAP adopt similar structures. There is also some disagreement about the mechanism by which the C-terminal-half of Srv2/CAP catalyzes actin monomer recycling. Studies on yeast Srv2 show that the ability to catalyze nucleotide exchange on cofilin-bound ADP-actin monomers requires both the WH2 and β-sheet domains (23). In contrast, recent studies on mouse CAP1 reported that the β-sheet domain alone was sufficient to catalyze nucleotide exchange on ADP-actin monomers, and have called into question the role of the WH2 domain in C-CAP1 functions (24). Importantly, these assays were performed in the absence of cofilin, precluding direct comparison of the two studies. This has left the role of the WH2 domain in C-CAP1 function elusive.
To address the conservation of Srv2/CAP mechanism between yeast and mammals, here we dissected the structure and function of mouse CAP1 using a combination of mutagenesis, bulk fluorescence assays, TIRF microscopy, and electron microscopy. Our results reveal that N-CAP1 forms hexameric structures that bind autonomously to F-actin using evolutionarily conserved surfaces, alter the twist of F-actin, and enhance the severing effects of cofilin. Moreover, the ability of C-CAP1 to catalyze nucleotide exchange on cofilin-bound ADP-actin monomers requires both its WH2 and β-sheet domains. These findings indicate that the activities and mechanisms of distantly related Srv2/CAP homologs are highly conserved, and suggest that the diverse members of this protein family may have similar cellular functions in regulating the actin cytoskeleton.
EXPERIMENTAL PROCEDURES
Hydrodynamic Analysis of Endogenous Human CAP1-Actin Complex
HEK293T cells were maintained at 37 °C under a humidified atmosphere containing 5% CO2 in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% (v/v) heat-inactivated fetal bovine serum, glucose (4.5 g/liter), penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells were harvested in PBS, collected by centrifugation at 1000 × g for 5 min, and lysed by douncing in 50 mm Tris/HCl, pH 7.5, 150 mm NaCl, and 1% (v/v) Triton X-100. The lysate was cleared by centrifugation at 14,000 rpm at 4 °C, then loaded on top of a 12-ml sucrose gradient (3–30%) in PBS. Size standards were fractionated in parallel. After centrifugation in a SW40 Ti rotor (Beckman) at 30,000 rpm for 18 h at 4 °C, 500-μl fractions were collected and analyzed by immunoblotting with CAP1 antibodies (MaxPab D01, Abnova, Taiwan) or Coomassie staining for protein standards. Peak CAP1-positive fractions from sucrose gradients were concentrated and fractionated on a Superose 6 gel filtration column, with protein standards fractionated in parallel. Using the sedimentation coefficient and Stokes radius for CAP1, obtained from the analyses above, the molecular weight of native CAP1 complex was calculated using the formula: M = (6πη0Nas)/(1 − νρ), with M = molecular weight, η0 (viscosity of water) = 1.002 × 10−2g/(cm*s), n = Avogadro's number, a = Stokes radius, s = sedimentation coefficient, ν (partial specific volume of an average particle) = 0.725 cm3/g, ρ (density of water) = 0.998 g/cm2.
Plasmids
pHAT2-N-CAP1 and pGAT2-C-CAP1 were kindly provided by Pekka Lappalainen (Univ. Helsinki) and used to purify N-CAP1 (residues 1–217) and C-CAP1 (residues 216–475) from Escherichia coli (24). Inserts from these plasmids were combined to reconstitute a full-length CAP1 plasmid for expression and purification from yeast. The β-sheet of CAP1 was PCR amplified from a C-CAP1 plasmid and cloned into the pET28a vector. Mutant N-CAP1 and C-CAP1 constructs were generated by site-directed mutagenesis. All constructs were verified by DNA sequencing. The plasmid for expressing human cofilin 1 (HsCof1) in E. coli was generously provided by David Kovar (Univ. Chicago). Plasmids for expressing yeast cofilin (yCof1) and N-Srv2 have been described elsewhere (20).
Protein Purification
Rabbit skeletal muscle actin was purified as previously described in detail (25). His6-tagged polypeptides (N-Srv2, N-CAP1, C-CAP1, B-CAP1, and mutants) were expressed in BL21 (pRARE) E. coli cells. Cultures were grown to log phase at 37 °C and induced for 16 h with 0.4 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) at 18 °C. Cells were harvested by centrifugation and lysed by sonication in 20 mm phosphate buffer pH 7.4, 300 mm NaCl, 1 mm DTT (lysis buffer) supplemented with 10 mm imidazole and a standard mixture of protease inhibitors. Clarified lysates were incubated with Ni2+-NTA beads (Qiagen, Valencia, CA) for 90 min at 4 °C and then transferred to a poly-prep chromatography column (Bio-Rad). The resin was washed with 10 column volumes of lysis buffer supplemented with 50 mm imidazole. Proteins were eluted with 5 column volumes of lysis buffer supplemented with 250 mm imidazole, concentrated, and purified further on a Superose 6 gel filtration column (GE Healthcare) equilibrated in 20 mm Tris pH 8.0, 100 mm NaCl, and 1 mm DTT. Full-length CAP1 was expressed under control of the GAL promotor in a protease-deficient yeast strain (BGY502). Two liters of cells were grown at 30 °C in synthetic medium without uracil and with 2% raffinose to an OD600 of 0.8–0.9, then expression was induced for 16 h at 30 °C by addition of 2% galactose. Cells were harvested by centrifugation, washed twice with 100 ml water, and resuspended in 10 ml of water per 2.5 grams cells. The cell suspension was drop frozen in liquid N2, then lysed by mechanical sheering in a coffee blender under liquid N2, and stored as a lysed powder at −80 °C. For purifications, 20 g of yeast powder was thawed in 20 ml of 2× lysis buffer supplemented with 20 mm imidazole and protease inhibitors. The cleared lysate was added to Ni2+-NTA beads and purified as described above for the other Srv2/CAP constructs. HsCof1 was expressed in E. coli as above and purified as follows. Cells were lysed by sonication in 20 mm Tris pH 8.0, 50 mm NaCl, 1 mm DTT, and protease inhibitors. Lysates were cleared and applied to a 5-ml HiTrap HP Q column (GE Healthcare). The flow-through fraction contained HsCof1, and was collected and dialyzed into 20 mm Hepes pH 6.8, 25 mm NaCl, and 1 mm DTT. Then the protein was applied to a 5 ml of HiTrap SP FF column (GE Healthcare) and eluted with a linear gradient of 25 to 500 mm NaCl. The fractions containing HsCof1 were concentrated and dialyzed to 20 mm Tris, pH 8.0, 50 mm NaCl, and 1 mm DTT, aliquoted, snap-frozen in liquid N2, and stored at −80 °C until use.
Electron Microscopy and Single Particle Analysis
To image actin filaments by electron microscopy (EM), Ca2+-ATP-G-actin (24 μm) was polymerized by addition of 2 mm MgCl2 and 50 mm KCl and incubation for 1 h at 25 °C. F-actin was diluted to 2 μm in F-buffer (50 mm KCl, 2 mm MgCl2, 0.2 mm EGTA, 1 mm DTT, 5 mm Tris, pH.8) and incubated for 15 min at 25 °C with control buffer (for undecorated filaments), or with 5 μm HsCof1, N-CAP1, and/or N-CAP1–91. Samples were diluted 2-fold in F-buffer and adsorbed to glow discharged formvar-carbon coated 200 mesh copper grids for 15–20 s, blotted to remove excess solution, negatively stained with 1% (w/v) uranyl acetate for 1 min, blotted again, and allowed to air-dry. For single particle analysis, 3 μm N-CAP1 or N-Srv2 was applied to grids and negative stained as above. For both analyses, images were captured using an FEI Morgani 268 transmission electron microscope at an acceleration voltage of 80 kV and magnifications of 14,000, 18,000, or 22,000. For single particle analysis, 4068 N-CAP1 particles were selected from the EM images using Boxer (26) and windowed into 80 × 80 pixel images. These were then filtered and normalized to a standard deviation of 1, and processed for reference-free classification in IMAGIC (27). Iterative classification yielded 40 classes, which were used for the three-dimensional reconstruction by the angular reconstitution method (28) and the back-projection algorithm to obtain a first rough three-dimensional model. This model was reprojected onto two-dimensional space for refining using iterative procedures. Further improvements in three-dimensional reconstructions and correction for CTF were performed using Frealign program (29). 6-fold symmetry was applied to the final reconstruction, as the majority of class-sum averages possessed 6-fold symmetry. Resolution of the final structure, defined by 0.5 FSC, was 25 Å. For scoring N-CAP1 particles bound to actin filaments, particles were identified by their unique structure and size and categorized as bound if in direct contact with a filament. Densities of particles and filaments under these conditions were low, such that particles in contact with filaments are likely to represent specifically bound molecules, in agreement with the specificity of binding observed in co-sedimentation assays (below).
Bulk F-actin Disassembly Assays
At time 0 in the assays, preassembled F-actin (2 μm final, 10% pyrene labeled) was mixed with the indicated proteins or control buffers, 100 nm CapZ, and 3 μm vitamin D-binding protein (VDBP)/human plasma Gc-globulin (Sigma-Aldrich) in F-buffer (20 mm Tris, pH 7.5, 50 mm KCl, 0.2 mm ATP, 1 mm MgCl2, and 1 mm DTT). Decrease in fluorescence was monitored for 900 s at 25 °C at 365-nm excitation and 407-nm emission in a fluorescence spectrophotometer (Photon Technology International, Lawrenceville, NJ).
Total Internal Reflection Fluorescence (TIRF) Microscopy
In all experiments, 24 × 60 mm coverslips (Fisher Scientific) were first cleaned by sonication in detergent for 60 min, followed by successive sonications in 1 m KOH and 1 m HCl for 20 min each, then sonication in ethanol for at least 60 min. Coverslips were then washed extensively with ddH2O, dried in an N2-stream, layered with 200 μl of 80% ethanol pH 2.0, 2 mg/ml methoxy-poly (ethylene glycol)-silane and 2 μg/ml biotin-poly (ethylene glycol)-silane (Laysan Bio Inc., Arab, AL), and incubated for 16 h at 70 °C. Flow cells were assembled by rinsing PEG-coated coverslips extensively with ddH2O, then attaching it to a flow chamber (Ibidi, Martinsried, Germany) with double-sided tape (2.5 cm × 2 mm × 120 μm) and 5 min epoxy resin. Oregon Green (OG)-labeled actin was prepared as described (30). For filament severing assays, flow cells were incubated for 5 min with HBSA (HEK buffer with 1% BSA), followed by 30 s incubation with 0.1 mg/ml streptavidin in PBS. Flow cells were washed with 5 chamber volumes (∼50 μl) HBSA, then equilibrated with 1× TIRF buffer (10 mm imidazole, 50 mm KCl, 1 mm MgCl2, 1 mm EGTA, 0.2 mm ATP, 10 mm DTT, 15 mm glucose, 20 μg/ml catalase, 100 μg/ml glucose oxidase, and 0.5% methylcellulose (4000 cP), pH 7.5). Reactions were initiated by rapidly diluting actin monomers (1 μm final, 10% OG-labeled, 0.5% biotinylated) into 1× TIRF buffer and transferring the mixture to a flow chamber. The filaments were left polymerizing till 10–15 μm, after which the reaction mixture was replaced with TIRF buffer containing Cof1 and/or CAP1 polypeptides, and lacking actin monomers. Time-lapse TIRFM of OG-actin filaments was performed using a Nikon-Ti200 inverted microscope equipped with a 150 milliwatt Ar-Laser (Mellot Griot, Carlsbad, CA), a TIRF-objective with a N.A. of 1.49 (Nikon Instruments Inc., New York, NY), and an EMCCD camera (Andor Ixon, Belfast, Northern Ireland). During measurements, optimal focus was maintained using the perfect focus system (Nikon Instruments Inc.). Images were captured every 5 s. The pixel size corresponded to 0.27 μm. Filament severing efficiency, expressed as severing events μm−1 s−1, was determined by measuring the lengths of individual filaments prior to Cof1 addition in ImageJ, and scoring severing events over time after flowing in cofilin and/or N-CAP1.
F-actin Cosedimentation Assays
Preformed actin filaments were incubated with variable concentrations of N-CAP1 or N-CAP1–91 for 30 min at room temperature in F-buffer (20 mm Tris, pH 7.5, 50 mm KCl, 0.2 mm ATP, 1 mm MgCl2, and 1 mm DTT). Reactions were centrifuged at 350,000 × g for 30 min at 20 °C. Supernatant and pellet fractions were analyzed on gels by Coomassie staining and quantified by scanning densitometry. Each concentration of N-CAP1 and N-CAP1–91 was processed in parallel reactions lacking F-actin to control against nonspecific pelleting. The binding affinity of N-CAP1 for F-actin was determined in assays as above using F-buffer at two different pH values (7.5 and 8.0). For each, a binding curve was fit, and the Kd was determined by non-linear regression analysis using Prism 5.0.
Nucleotide Exchange Assays
Nucleotide exchange rates on ADP-G-actin were determined by measuring the increase in fluorescence upon incorporation of ϵ-ATP (Sigma-Aldrich). ADP-G-actin was generated by removing bound ATP by dowex treatment and overnight incubation at 4 °C in the presence of hexokinase and an excess of ADP. Next, 2 μm of ADP-G-actin was mixed with proteins in CDT buffer (0.2 mm CaCl2, 0.2 mm DTT, 10 mm Tris pH 8.0) or buffer alone and added to 50 μm ϵ-ATP. The reaction was monitored for 200 s at 350-nm excitation and 410-nm emission at 25 °C in a fluorescence spectrophotometer (Photon Technology International). Exchange rates were calculated from linear fitting of the first 50 s of each reaction curve.
RESULTS
N-CAP1 Forms a Conserved Hexameric Structure
There has been some controversy about whether CAP homologs from evolutionarily distant species of yeast, plants, and animals have similar or distinct structures, activities, and mechanisms (16). To begin addressing this question, we investigated the oligomeric state of endogenous human CAP1 by fractionating HEK293 cell lysates both on sucrose gradients (Fig. 1A) and gel filtration columns (Fig. 1B), and detecting CAP1 by immunoblotting. Endogenous CAP1 had a sedimentation coefficient of 17.3 S and a Stokes radius of 7.7 nm, similar to the values obtained for endogenous yeast Srv2-actin complex in cell lysates (17). Using these values, we calculated the MW of the CAP1-actin complex to be 549 kDa. Since CAP binds to actin monomers with a 1:1 stoichiometry, this suggested formation of a hexameric complex consisting of 6 molecules of CAP1 (51.9 kDa) and 6 molecules of actin (41.7 kDa), similar to the yeast Srv2-actin complex (31).
FIGURE 1.

Mammalian CAP1 forms a hexameric complex with 6-fold symmetry. A, sedimentation velocity analysis of endogenously expressed human CAP1. HEK293 cell lysates were fractionated on sucrose gradients, and fractions were immunoblotted with CAP1 antibodies. The sedimentation coefficient (S) for CAP1 (red triangle) was determined by comparison to size standards (blue diamonds): 1, thyroglobulin, MW, 670,000, 19.4S, 8.5 nm; 2, gamma globulin, MW, 158,00, 7.4S, 5.22 nm; 3, ovalbumin, MW, 44,000, 3.6S, 3.05 nm; 4, myoglobin, MW, 17,000, 2S, 2.08 nm; 5, vitamin B12, MW, 1,350, 0.75 nm). B, gel filtration analysis of endogenously expressed human CAP1 from HEK293 cell lysates. The Stokes radius (in nm) for CAP1 (red triangle) was determined by comparison to the same size standards as in A (blue diamonds). C, representative electron micrographs of negatively stained, purified mouse N-CAP1, and yeast N-Srv2. Bar, 20 nm. D, single particle analysis of mouse N-CAP1: raw images (top row), two-dimensional projections of class averages (middle two rows), and three-dimensional reconstructions of each class (bottom row). These data were used to generate a final three-dimensional reconstruction of N-CAP1, which is compared with the three-dimensional reconstruction of N-Srv2 previously determined (20). Bar, 10 nm.
We next purified the N-terminal-half of mouse CAP1 (N-CAP1) and examined its structure by negative stain electron microscopy and single particle analysis. The raw images showed that N-CAP1 forms discrete structures with six protruding arms, similar to yeast N-Srv2 (Fig. 1C). Particles were categorized into class averages with different orientations and used to determine a three-dimensional reconstruction of N-CAP1 (Fig. 1D), revealing in more detail the 6-fold symmetry. N-CAP1 is slightly more compact (diameter = 12.3 nm ± 0.87) compared with N-Srv2 (diameter = 14.3 nm ± 1.2), consistent with N-CAP1 having a slightly lower MW (by ∼5 kDa). Interestingly, these molecules differ by an acidic N-terminal sequence that mediates Ras-signaling in N-Srv2, and is lacking in N-CAP1 (32, 33). Thus, the overall architecture of the N-Srv2/N-CAP1 hexamer is remarkably well conserved between yeast and mice, yet in yeast Srv2 also allows the insertion of some residues that introduce new functional capabilities.
N-CAP1 Strongly Enhances Cofilin-mediated Severing of Actin Filaments
We next purified full-length CAP1, N-CAP1, and C-CAP1 (Fig. 2A) to compare their effects on cofilin-mediated F-actin disassembly in bulk assays (Fig. 2B). Both FL-CAP1 and N-CAP1, but not C-CAP1, enhanced cofilin-mediated disassembly; however, they had no effect on actin disassembly in the absence of cofilin (Fig. 2B). Further, the ability of N-CAP1 to enhance cofilin-mediated disassembly was concentration-dependent (Fig. 2C). Mutant N-CAP1–91, which targets a conserved surface on the HFD domain required for yeast N-Srv2 activity (20, 31), abolished N-CAP1 stimulatory effects on actin disassembly (Fig. 2B). These results indicate that the enhanced disassembly activity of the N-terminal-half of CAP depends on surfaces in the HFD domain that are well conserved between yeast and mammals, and agrees with our results showing that N-Srv2 and N-CAP1 form highly similar structures.
FIGURE 2.

Effects of CAP1 on cofilin-mediated F-actin disassembly. A, schematic of domains in CAP and constructs used in these experiments. OD, oligomerization domain; HFD, helical folded domain; P, polyproline region; WH2, WASP-homology 2 domain. Arrow indicates position of CAP1–91 mutation in the HFD. B, effects of 250 nm human cofilin (HsCof1) and/or 750 nm mouse N-CAP1 constructs on disassembly of 2 μm F-actin (10% pyrene-labeled) in the presence of 100 nm CapZ and 3 μm vitamin d-binding protein. C, bulk F-actin disassembly assays, as in B, testing a range of concentrations of mouse N-CAP1 both in the presence and absence of 250 nm HsCof1. D, TIRF microscopy analysis of actin filament severing by cofilin with and without N-CAP1. Filaments were polymerized from 1 μm G-actin (10% Oregon Green, 0.5% biotinylated) and tethered through biotin-streptavidin conjugation. After filaments were polymerized to lengths of ∼15 μm, then 250 nm HsCof1 and/or 750 nm N-CAP1 was flowed in without actin monomers (indicated by black arrow), and filaments were monitored for 200 s. In the montage shown, severing events are indicated by yellow arrows. Bar, 5 μm. E, average number of severing events per μm filament at 50 s, quantified 50 s after flow in. Averages for each condition are from at least 40 individual filaments obtained from 3 independent trials. Error bars represent S.D. (n = 3).
To better understand the mechanism by which CAP1 enhances cofilin-mediated actin disassembly, we monitored the effects of cofilin and/or CAP1 on filaments in real time using TIRF microscopy (Fig. 2, D and E). Fluorescently labeled actin filaments (10% Oregon Green-labeled; 0.5% biotin-labeled) were polymerized and tethered through biotin-streptavidin-biotin-PEG interactions to the coverslip surface. Human cofilin (HsCof1) and/or N-CAP1 were flowed in, and severing events were monitored for 200 s (Fig. 2D). Quantification revealed that 50 s after flow-in, ∼8-fold more severing events per μm of filament had occurred in the presence of cofilin and N-CAP1 than in the presence of cofilin alone (Fig. 2E). N-CAP1 alone failed to induce severing (see “Experimental Procedures”). Thus, N-CAP1 strongly increases the efficiency of cofilin-mediated severing. Importantly, there are no actin monomers present in these reactions when severing was being monitored, which excludes the possibility that enhanced disassembly stems from N-CAP1 recycling cofilin from actin monomers. These activities are similar to the effects reported for both yeast N-Srv2 (20) and full-length bovine CAP1 (21), and again suggest a conserved mechanism.
N-CAP1 Hexamers Autonomously Bind F-actin
To probe the mechanism by which N-CAP1 enhances cofilin-mediated severing, we asked whether N-CAP1 and/or N-CAP1–91 bind directly to F-actin. In the absence of cofilin, N-CAP1 co-sedimented with F-actin, in a concentration-dependent manner, demonstrating a direct interaction (Fig. 3, A and B). In contrast, N-CAP1–91 showed minimal association, indicating that binding requires the conserved functional surface on the HFD domain targeted by the CAP1–91 mutation. At a fixed concentration of N-CAP1 (1 μm), the fraction of bound N-CAP1 appears to saturate at 0.4 μm (Fig. 3, C and D), and the F-actin binding affinity of N-CAP1 was measured (Kd = 7.6 μm at pH 7.5; Kd = 1.6 μm at pH 8.0). Further, by performing these assays at a higher concentration of F-actin (10 μm) and variable concentrations of N-CAP1, we found that binding saturates at ∼0.3 moles of N-CAP1 per mol of F-actin (Fig. 3D). It is not yet clear why binding of N-CAP1 saturates at 40%, but this may reflect heterogeneity in the population of N-CAP1 molecules, including the possibility of N-CAP1 existing in equilibrium between different conformations that do and do not bind F-actin.
FIGURE 3.

N-CAP1 binding to F-actin. A, co-sedimentation of N-CAP1 constructs with F-actin. Reactions contained 2.5 μm F-actin and/or 10 μm N-CAP1 or N-CAP1-91. Pellets and supernatants were analyzed on Coomassie-stained gels. B, concentration-dependent binding of N-CAP1 to F-actin (2.5 μm) at pH 7.5. Co-sedimentation assays, as in A, were performed over a range of concentrations of N-CAP1 and N-CAP1-91. The concentration of N-CAP1 protein bound to filaments was determined by scanning densitometry of bands on Coomassie-stained gels. C, binding affinity of N-CAP1 for F-actin. Co-sedimentation assays were performed using 1 μm N-CAP1 and a range of F-actin concentrations at both pH 7.5 and pH 8.0. The concentration of N-CAP1 bound to filaments was determined by densitometry of bands on Coomassie-stained gels. Each data point shown is an average from three independent trials. For each pH, the binding curve was fit, and the Kd was measured by non-linear regression analysis using Prism 5.0 (Rsquare at pH 7.5 = 0.97, Rsquare at pH 8.0 = 0.95). Error bars, S.D. D, molar ratio of binding of N-CAP1 to F-actin. Co-sedimentation assays were performed using 10 μm F-actin and variable concentrations of N-CAP1 at pH 8.0. The molar concentration of N-CAP1 bound to F-actin was determined by densitometry of bands on Coomassie-stained gels. Using a linear curve fit (shown), the molar ratio of binding was determined from the slope (0.312 ± 0.018) E, representative electron micrograph of negatively stained actin filament decorated by N-CAP1 (top) or N-Srv2 (bottom). Shown for each is the raw micrograph and a replica micrograph with the N-CAP1 and N-Srv2 particles shaded (purple and blue, respectively). F, distribution of the distances measured between N-CAP1 particles on the actin filaments. G, distribution of N-CAP1 particle association with F-actin (n = 76). Representative images of each binding mode are shown, along with percentage of particles that fell into each class.
Consistent with the binding analysis above, we were able to visualize by electron microscopy N-CAP1 (and N-Srv2) hexamers decorating actin filaments in the absence of cofilin (Fig. 3E). The majority of N-CAP1 particles (81%) was associated with the sides of filaments and were spaced 35–54 nm apart (Fig. 3, F and G). Further examination of the filaments decorated by cofilin, N-CAP1, or both (Fig. 4, A and B) provided structural insights into the enhanced severing mechanism. Undecorated filaments were long and smooth with an average crossover length of 36.65 ± 0.61 nm. Cofilin-decorated filaments were fragmented, and had regions of cofilin-decoration marked by an increase in filament diameter (10.07 ± 0.44 nm versus 8.19 ± 0.22 nm for undecorated filaments) and filament 'twisting', or a reduction in helical crossover distance (26.9 ± 1.09 nm), as reported previously (11). In contrast, N-CAP1 decoration led to an increase, rather than decrease in filament crossover distance (41.01 ± 1.02 nm), opposite to the effects of cofilin. In the presence of both CAP1 and cofilin, filaments were reduced to very short and heavily decorated fragments, making it difficult to measure crossover length (Fig. 4B). These results show not only that N-CAP1 binds autonomously to F-actin, but that its binding interactions may alter the structure of the filament.
FIGURE 4.

Structural effects of N-CAP1 and cofilin on actin filaments. A, representative electron micrographs of negatively stained actin filaments decorated with HsCof1 and/or N-CAP1. Higher magnification insets show regions of cofilin decoration (yellow arrows) or bound N-CAP1 particles (purple). B, comparison of mean crossover length for undecorated actin filaments versus filaments decorated by HsCof1 or N-CAP1. C, model showing how CAP1 binding to F-actin may enhance cofilin-mediated severing. The two proteins bind independently to F-actin and induce distinct structural effects that may lead to local discontinuities in filament topology and accelerate fragmentation.
The WH2 and β-Sheet Domains of CAP1 Are Required for Recycling Cofilin-bound ADP-actin Monomers
The yeast counterpart of CAP1, Srv2, has been shown to bind with high affinity to ADP-G-actin and to catalyze the conversion of cofilin-bound ADP-G-actin to ATP-G-actin (17, 18). This activity requires conserved surfaces on both the WH2 and β-sheet domains (18, 20, 23, 31). Thus, neither domain is sufficient for this function, and instead the two actin-binding domains work in concert to displace cofilin and stimulate nucleotide exchange on G-actin. Recently, it was reported that the β-sheet domain of CAP1 is sufficient to promote nucleotide exchange on ADP-G-actin (24), however, these assays were performed in the absence of cofilin. This prompted us to directly test the importance of the WH2 and β-sheet domains in C-CAP1-catalyzed actin monomer recycling in the presence of cofilin.
We first compared the effects of full-length CAP1 and each separate half of the protein on the nucleotide exchange rate of ADP-actin monomers in the presence and absence of cofilin. Under both conditions, catalysis of nucleotide exchange was observed for full-length CAP1 and C-CAP1, but not for N-CAP1 (Fig. 5, B and C). We then tested the separate contributions of the WH2 and β-sheet domains of C-CAP1 to this activity by mutating conserved surface residues on these domains, which disrupt the nucleotide exchange activity of yeast C-Srv2 (18, 24, 31). Following the strategy of Makkonen et al. (24), we introduced mutations at two actin-binding surfaces on the β-sheet domain, producing C-CAP1–108/109. Following the strategy of Chaudhry et al. (24), we mutated all four residues of the LKHV motif in the WH2 domain, producing C-CAP1–98. At 100 nm, wild type C-CAP1 stimulated nucleotide exchange on ADP-actin monomers, but C-CAP1–108/109 and C-CAP1–98 showed no activity, either in the presence or absence of cofilin (Fig. 1, E and F). At much higher concentrations (4 μm), the β-sheet alone showed nucleotide exchange activity in the absence of cofilin (Fig. 5E), but its effects were minimal in the presence of cofilin (Fig. 5D). These results show conclusively that both the WH2 and β-sheet domains of C-CAP1 are required to stimulate recycling of cofilin-bound ADP-actin monomers.
FIGURE 5.

C-CAP1 catalyzes nucleotide exchange on ADP-actin monomers in the presence and absence of cofilin. A, schematic of constructs and mutants used in this analysis. Arrows designate locations of point mutants. B and C, nucleotide exchange rate of 2 μm ADP-actin monomers in the presence of 100 nm full-length CAP1, N-CAP1, or C-CAP1, with (B) or without (C) 5 μm HsCof1. D and E, nucleotide exchange rate of 2 μm ADP-actin monomers in the presence of 100 nm C-CAP1, B-CAP1, and C-CAP1 mutants, with (D) or without (E) 5 μm HsCof1. In all bar graphs, data were averaged from three independent experiments. Error bars represent S.D. (n = 3).
Cross-species Compatibility of Srv2/CAP and Cofilin Activities
Finally, based on the results above, we asked whether mouse CAP1 could function with yeast cofilin, and conversely whether yeast Srv2 could function with mammalian cofilin in promoting actin turnover. In F-actin disassembly assays, mouse CAP1 enhanced the effects of yeast cofilin (Fig. 6A, yCof1), and conversely, yeast Srv2 strongly enhanced the effects of human cofilin (Fig. 6B, HsCof1). Further, the magnitude of the enhancement was similar for cross-species reactions compared with same-species reactions. Similarly, in nucleotide exchange assays, mouse CAP1 strongly catalyzed recycling of actin monomers bound by yCof1, and yeast Srv2 strongly catalyzed recycling of actin monomers bound by HsCof1 (Fig. 6C). These results strengthen the view that the Srv2/CAP mechanisms in promoting actin turnover are conserved, and further suggest that the underlying molecular interactions among Srv2/CAP, actin, and cofilin are also conserved.
FIGURE 6.
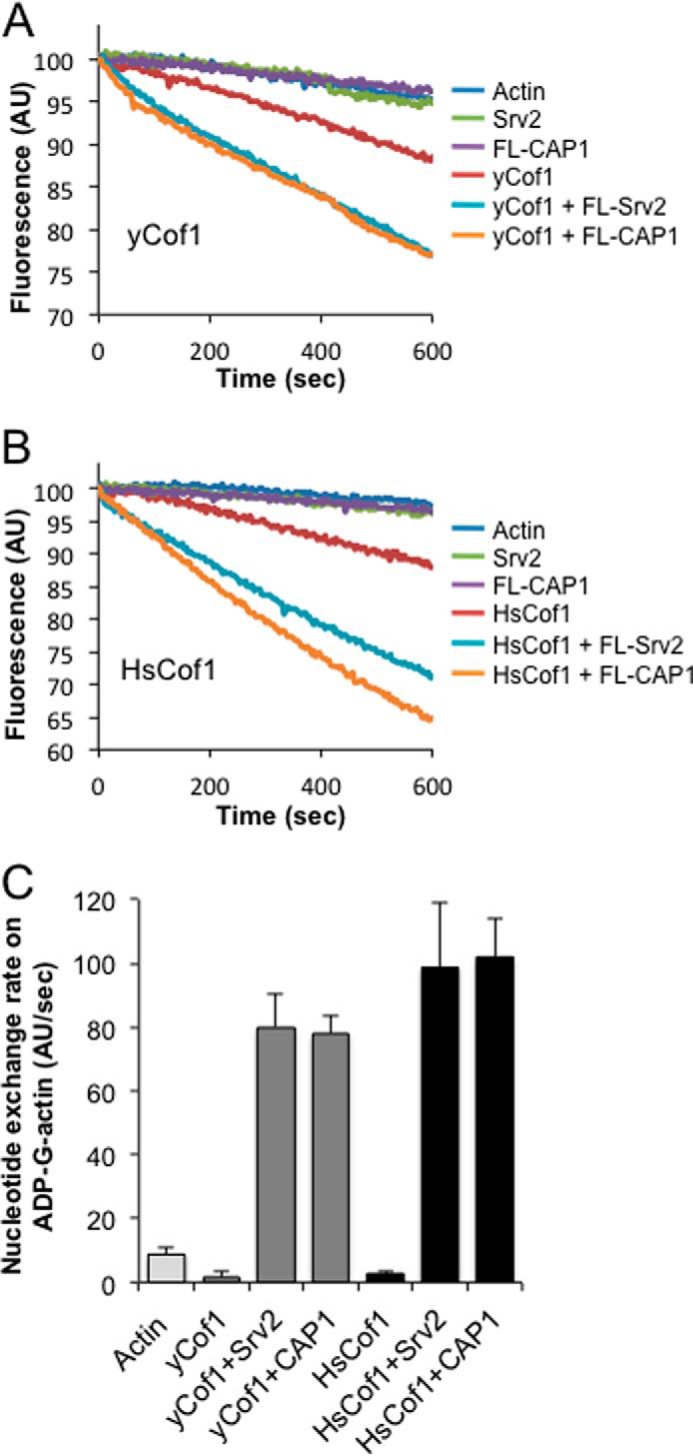
Testing cross-species compatibility of Srv2/CAP and cofilin activities. A and B, comparative effects of full-length CAP1, full-length Srv2, N-CAP1, and N-Srv2 on the disassembly of 2 μm F-actin (10% pyrene-labeled) in the presence of 100 nm yeast cofilin (yCof1) (A) or 250 nm HsCof1 (B). Reactions also contain 100 nm CapZ and 3 μm vitamin d-binding protein. C, comparative effects of full-length CAP1 or Srv2 (200 nm each) on the nucleotide exchange rate of ADP-actin monomers (2 μm) with 5 μm HsCof1 or yeast cofilin. Error bars represent S.D. (n = 3).
DISCUSSION
Srv2/CAP and cofilin are two of the most highly conserved components of the eukaryotic actin cytoskeleton, and have clear homologs in a diverse range of plants, animals, and fungi. A wealth of genetic studies in different systems has established the physiological importance of Srv2/CAP in regulating actin dynamics and organization (reviewed in Ref. 16). However, the mechanisms underlying these cellular functions remain only partially understood. Biochemical studies have shown that Srv2/CAP homologs have two separate activities, one in enhancing cofilin-mediated severing of filaments, and one in displacing cofilin from ADP-actin monomers and catalyzing nucleotide exchange on actin (17–21). For yeast Srv2, these two functions were assigned to separate halves of the protein that function in a largely autonomous manner (22). The N-terminal-half mediates enhanced severing, which depends on its ability to form large hexameric structures (20), while the C-terminal-half mediates actin monomer recycling, which depends on actin-binding surfaces located in both its WH2 and β-sheet domains (18, 23). One question that has remained open is how the hexameric Srv2/CAP structures enhance cofilin-mediated severing of filaments. A second important question is whether CAP homologs from other species adopt related structures and/or promote F-actin severing and G-actin recycling by similar mechanisms.
Our data reveal that the N-terminal-half of mouse CAP1 (N-CAP1) forms a hexameric structure with six symmetrical protrusions, highly similar to yeast N-Srv2. N-CAP1 was sufficient to enhance cofilin-mediated severing by 8-fold, and this activity was abolished by mutations at conserved surfaces on the HFD domain. These results suggest that both the structure and activity of the N-terminal-half of Srv2/CAP are well conserved across distant species. To better understand the mechanism by which N-CAP1 enhances cofilin-mediated severing, we investigated its interactions with F-actin, and found that N-CAP1 hexamers bind autonomously to F-actin, independent of cofilin (Kd = 7.6 μm at pH 7.5; Kd = 1.6 μm at pH 8.0). Under these conditions, we also observed that N-CAP1 binding alters actin filament structure, leading to an increase in filament crossover distance and opposite to cofilin's twisting effects. These observations raise the possibility that N-CAP1 enhances severing by introducing local discontinuities in filament topology to accelerate cofilin-mediated fragmentation (34). Our ultrastructural analysis also showed that most N-CAP1 particles contact actin filaments with only one or two of their HFD protrusions. Thus, hexamerization may serve primarily to create a multivalent structure where avidity effects increase the likelihood of binding between HFD domains and actin filaments.
Our data also shed important light on the mechanism by which mammalian CAP1 stimulates recycling of cofilin and actin monomers. Previously it was shown that the ability of yeast Srv2 to catalyze recycling of ADP-actin monomers in the presence of cofilin depends on both its WH2 and β-sheet domains. However, a more recent study has suggested that mammalian CAP1 may use a different mechanism (24). Specifically, it was shown that CAP1 β-sheet domain alone is sufficient to promote nucleotide exchange on ADP-actin monomers, and that this activity is abolished by mutations at conserved actin-binding residues on this domain (referred to herein as CAP1–108/109). It is critical to note however, that these assays were performed in the absence of cofilin, which is abundant in cells and has a strong inhibitory effect on nucleotide exchange (35, 36). For this reason, we directly compared the activities of C-CAP1 and its β-sheet domain alone (B-CAP1) on nucleotide exchange both in the presence and absence of cofilin. In the absence of cofilin, C-CAP1 and B-CAP1 each stimulated nucleotide exchange, although high concentrations of B-CAP1 were required for this activity. In the presence of cofilin, C-CAP1 effectively catalyzed nucleotide exchange, whereas B-CAP1 did not, demonstrating that the WH2 domain is crucial for this function when cofilin is bound to ADP-actin monomers. Further, C-CAP1–98 (which mutates a key actin-binding surface on the WH2 domain) almost completely abolished the activity. These results demonstrate that the WH2 domain of CAP1 is critical for actin monomer recycling specifically in the presence of cofilin, similar to what has been observed for yeast Srv2 (23). Thus, the mechanisms used by yeast and mammalian CAP homologs for catalyzing actin monomer recycling appear to be highly conserved.
We also note that in the above-mentioned study on C-CAP1 (24) a point mutant in the WH2 domain targeting the conserved Lys in the LKHV motif disrupted binding to ATP-actin monomers, but failed to disrupt C-CAP1 stimulation of nucleotide exchange on ADP-actin monomers. However, earlier studies on yeast Srv2 showed that mutating the central pair of Lys residues in its LKKV motif had little effect on function either in vitro or in vivo (18, 23). Instead, mutating both of the flanking hydrophobic residues (Leu and Val) was required to disrupt WH2 function in vitro and in vivo (23). Similarly here, we were able to abolish WH2 function in CAP1 by targeting all four residues in its LKHV motif. Thus, future studies addressing WH2 function in other CAP homologs should use a similar mutational strategy. Additionally, these results show that while the central charged residues are important for binding ATP-actin monomers, the flanking hydrophobic residues are critical for the nucleotide exchange function (ATP for ADP), and therefore may be important for binding ADP-G-actin.
Overall, our results demonstrate that the structure, activities, and mechanisms of Srv2/CAP in stimulating actin turnover are remarkably well conserved between species as evolutionarily distant as yeast and mice. This is supported further by our observation of cross-species compatibility in the activities of yeast and mammalian Srv2/CAP and cofilin. A broad implication of our findings is that many of the diverse cellular and physiological functions reported for CAP in different systems may stem from the same underlying activities and mechanisms in stimulating actin dynamics. It is our hope that the tools generated here by dissection of CAP1 will inspire and facilitate additional genetic testing of CAP function and mechanism in different organisms.
Acknowledgments
We thank Dr. Chen Xu for assistance with the Morgani electron microscope. The three-dimensional reconstruction of N-CAP1 was accomplished with grant support from the Russian Scientific Foundation (Project 14-14-00234) (to O. S.).
This work was supported, in whole or in part, by National Institutes of Health Grant GM063691 (to B. G.).
- CAP
- cyclase-associated protein
- TIRF
- total internal reflection fluorescence
- EM
- electron microscopy
- HsCof
- human cofilin.
REFERENCES
- 1. Blanchoin L., Pollard T. D. (1999) Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J. Biol. Chem. 274, 15538–15546 [DOI] [PubMed] [Google Scholar]
- 2. Mabuchi I. (1983) An actin-depolymerizing protein (depactin) from starfish oocytes: properties and interaction with actin. J. Cell Biol. 97, 1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maciver S. K., Zot H. G., Pollard T. D. (1991) Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 115, 1611–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nishida E., Maekawa S., Sakai H. (1984) Characterization of the action of porcine brain profilin on actin polymerization. J. Biochem. 95, 399–404 [DOI] [PubMed] [Google Scholar]
- 5. Hotulainen P., Paunola E., Vartiainen M. K., Lappalainen P. (2005) Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell 16, 649–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lappalainen P., Drubin D. G. (1997) Cofilin promotes rapid actin filament turnover in vivo. Nature 388, 78–82 [DOI] [PubMed] [Google Scholar]
- 7. Bamburg J. R., McGough A., Ono S. (1999) Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 9, 364–370 [DOI] [PubMed] [Google Scholar]
- 8. Elam W. A., Kang H., De la Cruz E. M. (2013) Biophysics of actin filament severing by cofilin. FEBS Lett. 587, 1215–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ono S. (2007) Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int. Rev. Cytol. 258, 1–82 [DOI] [PubMed] [Google Scholar]
- 10. Galkin V. E., Orlova A., Kudryashov D. S., Solodukhin A., Reisler E., Schröder G. F., Egelman E. H. (2011) Remodeling of actin filaments by ADF/cofilin proteins. Proc. Natl. Acad. Sci. U. S.A. 108, 20568–20572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McGough A., Pope B., Chiu W., Weeds A. (1997) Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J. Cell Biol. 138, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCullough B. R., Blanchoin L., Martiel J. L., De la Cruz E. M. (2008) Cofilin increases the bending flexibility of actin filaments: implications for severing and cell mechanics. J. Mol. Biol. 381, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suarez C., Roland J., Boujemaa-Paterski R., Kang H., McCullough B. R., Reymann A. C., Guérin C., Martiel J. L., De la Cruz E. M., Blanchoin L. (2011) Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr. Biol. 21, 862–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andrianantoandro E., Pollard T. D. (2006) Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 24, 13–23 [DOI] [PubMed] [Google Scholar]
- 15. Brieher W. (2013) Mechanisms of actin disassembly. Mol. Biol. Cell 24, 2299–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ono S. (2013) The role of cyclase-associated protein in regulating actin filament dynamics - more than a monomer-sequestration factor. J. Cell Sci. 126, 3249–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Balcer H. I., Goodman A. L., Rodal A. A., Smith E., Kugler J., Heuser J. E., Goode B. L. (2003) Coordinated regulation of actin filament turnover by a high-molecular-weight Srv2/CAP complex, cofilin, profilin, and Aip1. Curr. Biol. 13, 2159–2169 [DOI] [PubMed] [Google Scholar]
- 18. Mattila P. K., Quintero-Monzon O., Kugler J., Moseley J. B., Almo S. C., Lappalainen P., Goode B. L. (2004) A high-affinity interaction with ADP-actin monomers underlies the mechanism and in vivo function of Srv2/cyclase-associated protein. Mol. Biol. Cell 15, 5158–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moriyama K., Yahara I. (2002) The actin-severing activity of cofilin is exerted by the interplay of three distinct sites on cofilin and essential for cell viability. Biochem. J. 365, 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaudhry F., Breitsprecher D., Little K., Sharov G., Sokolova O., Goode B. L. (2013) Srv2/cyclase-associated protein forms hexameric shurikens that directly catalyze actin filament severing by cofilin. Mol. Biol. Cell 24, 31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Normoyle K. P., Brieher W. M. (2012) Cyclase-associated protein (CAP) acts directly on F-actin to accelerate cofilin-mediated actin severing across the range of physiological pH. J. Biol. Chem. 287, 35722–35732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chaudhry F., Jansen S., Little K., Suarez C., Boujemaa-Paterski R., Blanchoin L., Goode B. L. (2014) Autonomous and in trans functions for the two halves of Srv2/CAP in promoting actin turnover. Cytoskeleton 71, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chaudhry F., Little K., Talarico L., Quintero-Monzon O., Goode B. L. (2010) A central role for the WH2 domain of Srv2/CAP in recharging actin monomers to drive actin turnover in vitro and in vivo. Cytoskeleton 67, 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makkonen M., Bertling E., Chebotareva N. A., Baum J., Lappalainen P. (2013) Mammalian and malaria parasite cyclase-associated proteins catalyze nucleotide exchange on G-actin through a conserved mechanism. J. Biol. Chem. 288, 984–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graziano B. R., Jonasson E. M., Pullen J. G., Gould C. J., Goode B. L. (2013) Ligand-induced activation of a formin-NPF pair leads to collaborative actin nucleation. J. Cell Biol. 201, 595–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 27. van Heel M., Harauz G., Orlova E. V., Schmidt R., Schatz M. (1996) A new generation of the IMAGIC image processing system. J. Struct. Biol. 116, 17–24 [DOI] [PubMed] [Google Scholar]
- 28. Van Heel M. (1987) Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy 21, 111–123 [DOI] [PubMed] [Google Scholar]
- 29. Grigorieff N. (2007) FREALIGN: high-resolution refinement of single particle structures. J. Struct. Biol. 157, 117–125 [DOI] [PubMed] [Google Scholar]
- 30. Kuhn J. R., Pollard T. D. (2005) Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophys. J. 88, 1387–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quintero-Monzon O., Jonasson E. M., Bertling E., Talarico L., Chaudhry F., Sihvo M., Lappalainen P., Goode B. L. (2009) Reconstitution and dissection of the 600-kDa Srv2/CAP complex: roles for oligomerization and cofilin-actin binding in driving actin turnover. J. Biol. Chem. 284, 10923–10934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mintzer K. A., Field J. (1994) Interactions between adenylyl cyclase, CAP and RAS from Saccharomyces cerevisiae. Cell. Signal. 6, 681–694 [DOI] [PubMed] [Google Scholar]
- 33. Shima F., Okada T., Kido M., Sen H., Tanaka Y., Tamada M., Hu C. D., Yamawaki-Kataoka Y., Kariya K., Kataoka T. (2000) Association of yeast adenylyl cyclase with cyclase-associated protein CAP forms a second Ras-binding site which mediates its Ras-dependent activation. Mol. Cell. Biol. 20, 26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCullough B. R., Grintsevich E. E., Chen C. K., Kang H., Hutchison A. L., Henn A., Cao W., Suarez C., Martiel J. L., Blanchoin L., Reisler E., De La Cruz E. M. (2011) Cofilin-linked changes in actin filament flexibility promote severing. Biophys. J. 101, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blanchoin L., Pollard T. D. (1998) Interaction of actin monomers with Acanthamoeba actophorin (ADF/cofilin) and profilin. J. Biol. Chem. 273, 25106–25111 [DOI] [PubMed] [Google Scholar]
- 36. Nishida E. (1985) Opposite effects of cofilin and profilin from porcine brain on rate of exchange of actin-bound adenosine 5′-triphosphate. Biochemistry 24, 1160–1164 [DOI] [PubMed] [Google Scholar]