

Background: Lysine acetylation is an important regulatory modification of STAT3.
Results: Acetylation of Lys-685 is critical for gene expression driven by unphosphorylated STAT3 (U-STAT3) but not by STAT3 phosphorylated on Tyr-705 (Y-P-STAT3).
Conclusion: Distinct modifications regulate U-STAT3 and Y-P-STAT3 differentially.
Significance: Lys-685 acetylation could be targeted to block U-STAT3-specific functions, such as activation of oncogene expression.
Keywords: Angiotensin II, Gene Expression, Microarray, Post-translational Modification (PTM), STAT3, U-STAT3
Abstract
STAT3 is a pleiotropic transcription factor that is activated by the phosphorylation of tyrosine 705 in response to many cytokines and growth factors. STAT3 without Tyr-705 phosphorylation (U-STAT3) is also a potent transcription factor, and its concentration in cells increases greatly in response to STAT3 activation because the STAT3 gene can be driven by phosphorylated STAT3 dimers. We have now searched for post-translational modifications of U-STAT3 that might have a critical role in its function. An analysis by mass spectroscopy indicated that U-STAT3 is acetylated on Lys-685, and the integrity of Lys-685 is required for the expression of most U-STAT3-dependent genes. In contrast, we found only a very minor role for Lys-685 in gene expression induced in response to tyrosine-phosphorylated STAT3. U-STAT3 plays an important role in angiotensin II-induced gene expression and in the consequent development of cardiac hypertrophy and dysfunction. Mutation of Lys-685 inhibits this function of STAT3, providing new information on the role of U-STAT3 in augmenting the development of heart failure.
Introduction
STAT3 plays a crucial role in many cellular processes, including growth, regulation of apoptosis, and motility, and knock-out of the STAT3 gene in mice is embryonically lethal. STAT3 is activated by phosphorylation of Tyr-705 in response to members of the IL-6 family of cytokines, leptin, interferons, growth factors, and non-receptor tyrosine kinases. The binding of phosphorylated Tyr-705 to the SH2 domain of a second STAT3 monomer promotes dimerization and nuclear translocation, activating target gene transcription (1). The diverse biological functions of tyrosine-phosphorylated STAT3 dimers are regulated by several different post-translational modifications. Ser-727, which lies in the transactivation domain, is phosphorylated in different situations to facilitate gene expression (2). Several different post-translational modifications of lysine residues of STAT3 have been reported to affect function. Yuan et al. (3) reported that the acetylation of Lys-685 is necessary for STAT3 dimerization and activation in response to oncostatin M, a conclusion that was disputed by others (4) and is further called into question by our current data. Additionally, IL-6-induced gene expression was reported to require p300-mediated acetylation of Lys-49 and Lys-87 of STAT3 (5). Lysine polyubiquitination facilitates STAT3 degradation, with important cellular consequences (6, 7). Recently, Ray et al. (8) reported that STAT3 mono-ubiquitination at Lys-97 is a key mediator of BRD4-dependent antiapoptotic and pro-proliferative gene expression. Several transcription factors, including STAT3, NF-κB,2 and p53, are reversibly methylated on lysine residues by histone-modifying enzymes, with profound consequences for their functions (9). STAT3 is methylated on Lys-140 and Lys-180 by the histone methyl transferases SET9 and EZH2, respectively (10, 11).
Previously, we reported that STAT3 lacking tyrosine phosphorylation (U-STAT3) also activates gene expression (12). Phosphorylated STAT3 dimers drive the expression of the STAT3 gene itself, leading to a large ligand-dependent increase in the intracellular concentration of U-STAT3, which drives expression of a set of genes distinct from the set activated by phosphorylated STAT3 dimers. For some genes, a heterologous transcription factor comprising U-STAT3 and unphosphorylated NF-κB binds to the κB elements of a small subset of NF-κB-target genes and activates them (13), but how additional U-STAT3 targets are regulated remains unknown. U-STAT3 also participates in gene expression even when its concentration is not increased in response to STAT3 activation, and both serine phosphorylation and lysine acetylation have been reported to be involved. When topoisomerase I is inhibited, STAT3 is phosphorylated by CDK5 on Ser-727 (and not on Tyr-705), leading to increased expression of EME1, an endonuclease involved in the processing of damaged replication forks (14). Constitutive Ser-727 phosphorylation without Tyr-705 phosphorylation was reported to be a hallmark of chronic lymphocytic leukemia (15). Moreover, STAT3 activation by means of Ser-727 phosphorylation without Tyr-705 phosphorylation was observed in non-tumorigenic cells as well (16, 17). Recently, Waitkus et al. (18) demonstrated that STAT3 can be activated independently of Tyr-705 phosphorylation by simultaneous phosphorylation of Thr-714 and Ser-727. Lee et al. (19) demonstrated that, in the absence of cytokines and growth factors, U-STAT3 is activated upon engagement of the cell surface receptor CD44 through the acetylation of Lys-685, leading to increased target gene expression.
Thus, it is evident that post-translational modifications of U-STAT3 have substantial effects on its ability to induce target gene expression. We now show that the acetylation of Lys-685 is critical for gene expression that is driven by increased intracellular concentrations of U-STAT3. We also demonstrate that the ability of U-STAT3 to support the expression of many genes in response to angiotensin II depends upon the integrity of Lys-685, consistent with a previous study of Yue et al. (20) that activation of the angiotensin II type 1 receptor (AT1R) leads to nuclear accumulation of U-STAT3 and promotes its interaction with the acetyl transferase p300, inducing the expression of proteins involved in cardiac hypertrophy and dysfunction.
EXPERIMENTAL PROCEDURES
Cell Lines
Immortalized human mammary epithelial cells (hTERT-HME1) were grown in mammary epithelial growth medium (catalog number CC3151) supplemented with bovine pituitary extract, gentamycin, hydrocortisone, insulin, and human epidermal growth factor (BulletKit; catalog number CC4136), all from Lonza. The human DLD1 and its STAT3-null derivative A4 cells were grown in McCoy's 5A medium with l-glutamine (HyClone; catalog number SH300200.01), supplemented with 5% FBS, penicillin (100 units/ml) and streptomycin (100 μg/ml) (Invitrogen). PC3 cells were grown in RPMI 1640 medium supplemented with 5% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml) (Invitrogen).
Reagents
Human IL-6 (catalog number 200-06 and soluble IL-6Rα (catalog number 200-06R) were from PeproTech. Antibodies against total STAT3 (catalog number D1A5 XP), Tyr-705-phosphorylated STAT3 (phospho-Tyr-705-STAT3)(catalog number 9131), and HA-Tag (catalog number 2367) were from Cell Signaling Technology. Anti-GAPDH (catalog number sc-25578) was from Santa Cruz Biotechnology.
Constructs and Viral Transduction
Using the pLPCX retroviral plasmid (Clontech), the K49R, K140R, K685R, and S727A STAT3 mutant constructs were generated from WT STAT3 using the QuikChange II XL site-directed mutagenesis kit (Stratagene catalog number 200521). To obtain infectious retroviral stocks, each construct was transfected into 293T cells along with the packaging plasmid using Lipofectamine and Plus reagents (Invitrogen). Stable cell pools of each mutant cell line were obtained by infecting hTERT-HME1 cells with the virus collected after 48 h followed by selection in puromycin at 4 μg/ml.
pLEGFP-N1 retroviral plasmid (Clontech) was used to generate the K685R, Y705F, and K685R/Y705F from WT STAT3 using the QuikChange II XL site-directed mutagenesis kit (Stratagene catalog number 200521). Infectious retroviral stocks were obtained by transfecting each construct into 293T cells along with the packaging plasmid using the Lipofectamine and Plus reagents (Invitrogen). A stable cell pool of each mutant cell line was obtained by infecting either PC3 or A4 cells with the 48-h virus followed by selection in neomycin G418 at 500 μg/ml.
Western Analyses
Cells were cultured to ∼85% confluence before treatment. Cell pellets were collected and processed as described previously (12) to obtain whole cell lysates. Protein (20–30 μg) was loaded onto 10% SDS-PAGE gels. The separated proteins were then transferred to PVDF membranes (Millipore). Following transfer, the membranes were incubated overnight at 4 °C with primary antibody (in 5% milk or BSA) and with secondary antibody in milk for 1 h at room temperature and then developed with Pierce ECL reagent.
PCR and Quantitative Real-time PCR
cDNA samples were prepared by reverse PCR from total RNA extracted by using the SuperScript III first-strand synthesis system (Invitrogen) as described in the random hexamer protocol. Samples were either amplified according to standard PCR procedures or analyzed using FastStart Universal SYBR Green Master (ROX) (Roche Diagnostics) quantitative PCR (qPCR) reactions. Primers were either obtained from PrimerBank – MGH-PGA or designed using AlleleID 6 software. The primer list can be found in the supplemental Method.
hAT1R Cloning and Viral Transduction
Human angiotensin I receptor (hAT1R) ORF from Open Biosystems (catalog number MHS1010-98075368) was cloned in the pLCMV-PL4-puro lentiviral vector. Virus production and cell infection established stably transduced hAT1R-expressing cell pools.
To obtain infectious lentivirus stocks, the lentiviral expression vector pLCMV-PL4-Neo, harboring the hAT1R cDNA, was packaged into 293T cells along with the packaging constructs pCMV-dR8.74 and pMD2G. Supernatant medium containing virus collected after 36–48 h was filtered and supplemented with 4 μg/ml Polybrene before being added on to A4 cells expressing either WT or mutant (K685R, Y705F, and K685R/Y705F) STAT3 (at 50% confluence). The medium was replaced 24 h after infection, and the cells were subjected to puromycin selection at 3 μg/ml after another 24 h.
Calcium Measurement Assay
Assays were performed using the FLIPR Calcium 5 assay kit (Molecular Devices, Sunnyvale, CA) according to the manufacturer's instructions. Cells were plated onto 96-well clear bottom black cell culture plates that were precoated with poly-l-lysine at a density of 105 cells/well in 100 μl of medium. The plate was incubated in a cell culture incubator for 24 h at 37 °C. The cells were then serum-starved for 4 h. Following serum starvation, 50 μl of calcium assay reagent was added. The plates were incubated for 30 min at 37 °C and allowed to return to room temperature 30 min before reading. Fluorescence was measured every 2 s for a total time of 2 min on a fluorometric imaging plate reader (FlexStation 3, Molecular Devices). The cells were stimulated by adding 1 μm angiotensin II at the 18th second. The data generated during the calcium flux assay in the presence of 1 μm angiotensin II were analyzed by averaging maximum-minimum relative fluorescence units in GraphPad PrismR.
Gene Expression Analysis
Total RNA was isolated using the Qiagen RNeasy mini kit according to the manufacturer's protocol. 1 μg of this RNA was used for microarray analysis on an Illumina Human Ht-12 V4 Expression BeadChip kit. The data were analyzed by using the Illumina GenomeStudio software and normalized by the quantile method. Genes were selected according to the criteria mentioned in each figure legend.
Protein Digestion and LC Tandem Mass Spectrometry
FLAG-STAT3 was immunoprecipitated from hTERT-HME1 cells expressing high levels of WT STAT3 by using EZ-FLAG beads (EZviewTM Red ANTI-FLAG® M2 affinity gel, Sigma catalog number F2426) according to the manufacturer's protocol. The immunoprecipitated FLAG-STAT3 was run in an SDS-PAGE gel and stained with Coomassie Blue.
For protein digestion, bands were cut from the gel as closely as possible with a punch and washed/destained in 50% ethanol, 5% acetic acid. The gel pieces were then dehydrated in acetonitrile, dried in a speed vacuum, and digested with trypsin by adding 5 μl of 10 ng/μl trypsin in 50 mm ammonium bicarbonate and incubating overnight at room temperature. The resulting peptides were extracted from the polyacrylamide in two aliquots of 30 μl each of 50% acetonitrile, 5% formic acid. These extracts were combined and evaporated to <10 μl in the speed vacuum and then resuspended in 1% acetic acid to a final volume of ∼30 μl, for LC-MS analysis.
The LC-MS system was a Finnigan LTQ-Orbitrap Elite hybrid system. The HPLC column was a Dionex 15 cm × 75-μm inner diameter Acclaim PepMap C18, 2-μm, 100 Å reversed phase capillary chromatography column. 5-μl volumes of extract were injected, and the peptides were eluted from the column by using an acetonitrile, 0.1% formic acid gradient at a flow rate of 0.25 μl/min, and were introduced into the source of the mass spectrometer on-line. The microelectrospray ion source is operated at 2.5 kV. The digest was analyzed by using the data-dependent multitask capability of the instrument, acquiring full scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in successive instrument scans. This mode of analysis produces ∼15,000 collision-induced dissociation spectra of ions, ranging in abundance over several orders of magnitude. Note that not all the collision-induced dissociation spectra are derived from peptides.
The data were analyzed by using all collision-induced dissociation spectra collected in the experiment to search the National Center for Biotechnology Information (NCBI) non-redundant database with the search program Mascot, using a human taxonomy filter. All matching spectra were verified by manual interpretation. The interpretation process was aided by additional searches using the programs Sequest and Blast, as needed.
RESULTS
Mass Spectroscopy Indicates That U-STAT3 Is Acetylated on Lys-685
Many recent studies reveal crucial roles for signal-dependent post-translational modifications of transcription factors. Because STAT3 can drive gene expression even without tyrosine phosphorylation, we investigated post-translational modifications of U-STAT3 that might be critical for its functions by mass spectroscopy, using FLAG-tagged STAT3 purified from immortalized human mammary epithelial cells (hTERT-HME1 cells) expressing a level of this protein (Fig. 1A) that corresponds to the increased level seen 36 h after IL-6 treatment (Fig. 1B). The LYPDIPKEEAFGKY peptide (residues 673–686) was identified in a chymotryptic digest of STAT3. The MS/MS spectrum contains several sequence-specific ions, including the C-terminal y2 fragment ion at 352 Da, consistent with acetylation of the KY sequence (Fig. 1C).
FIGURE 1.

U-STAT3 is acetylated on Lys-685. A, hTERT-HME1 cells were infected with retroviral constructs expressing WT STAT3, and stable pools of cells were selected with G418. The level of STAT3 expression corresponds to the previously reported increase of STAT3 expression 36 h after treatment with IL-6 (12). B, hTERT-HME1 cells were treated with IL-6 (200 ng/ml) and s-IL-6R (250 ng/ml) for the indicated times, and levels of total STAT3 and Tyr-705 phospho-STAT3 were measured. This figure is reproduced from Yang et al. (12). C, WT STAT3 was immunoprecipitated from hTERT-HME1 cells with high expression of the FLAG-tagged protein. Mass spectrometric analysis of chymotryptic peptides of STAT3 was performed. MS/MS spectra were obtained for the acetylated chymotryptic peptide LYPDIPKEEAFGKAceY. This peptide, representing residues 673–686 of STAT3, was of low abundance and was only identified in a targeted LC-MS/MS experiment. The doubly charged peptide has an observed m/z of 856.4378 Da and is within 6 ppm of the expected mass. This spectrum is dominated by fragmentation N-terminal to the Pro residues; however, the spectrum includes the C-terminal y2 ion at 352 Da, which is consistent with acetylation on the C-terminal portion of the peptide. The most likely site of modification is Lys-685. Several unidentified ions present in this spectrum are probably derived from the isobaric ion, which elutes at the same time and has a mass of 856.3903 (see inset).
Microarray Reveals Differential Regulation of Gene Expression by K685R for U-STAT3
To examine the differential effect of acetylation of Lys-685 on gene expression, we made stable pools of hTERT-HME1 cells expressing high levels of FLAG-tagged wild-type STAT3 or several different mutant proteins, including K685R (Fig. 2A). For comparison, we also included the K49R and K140R mutations because modifications of these lysine residues have previously been shown to affect STAT3 function, as well as the S727A mutation, because phosphorylation of this serine residue is a positive regulator of STAT3-dependent transcription. As before, the STAT3 levels correspond to those induced naturally 36 h after IL-6 treatment of these cells. We then performed an Illumina array experiment to analyze the roles of each of the mutated residues in U-STAT3-mediated gene expression, scoring genes whose expression was increased by 2-fold or more (details under “Experimental Procedures”) in hTERT-HME1 cells upon increased expression of wild-type STAT3. The expression of 124 of the ∼35,000 genes on the array increased by 2-fold or more in response to increased expression of wild-type STAT3. The K685R mutation inhibited the expression of 84 of these genes by 2-fold or more. In contrast, the expression of only 21, 2, or 12 of the 125 genes induced by wild-type STAT3 was reduced by the S727A, K140R, or K49R mutations, respectively (Fig. 2B; complete list in supplemental Table S1). The induction of three representative genes from the array (IL7R, CSF2, and CDC45L) by wild-type STAT3 in response to IL-6 and the effects of the four mutations were confirmed by real-time quantitative PCR (Fig. 2C). We propose that the K685R mutation impairs U-STAT3-regulated gene expression.
FIGURE 2.

The K685R mutation impairs expression of U-STAT3-induced genes. A, hTERT-HME1 cells were infected with retroviral constructs expressing WT STAT3 or the STAT3 mutants K49R, K140R, K685R, or S727A. Stable pools of cells were selected with G418. Western analysis of total cell lysates was done to analyze STAT3 expression in these cells. B, the heat map of microarray performed to compare genes induced in hTERT-HME1 cells expressing wild-type STAT3 or STAT3 mutants shows the increased expression of 124 genes in response to increased levels of WT STAT3 and the changes in expression of these genes in cells with increased levels of each of the STAT3 mutants. In the heat map, red corresponds to high expression, green corresponds to low expression, and black corresponds to intermediate expression. We include only genes whose expression changed by >2-fold, with differential p ≤ 0.05, in WT cells when compared with vector control cells, and with average signals greater than 100 in WT cells. C, qPCR analysis of relative expression of CDC45L, IL7R, and CSF2 genes was done. Values are the means, with standard deviations, from triplicate experiments.
The K685R Mutation Has Only a Marginal Effect on the Function of Phosphorylated STAT3
Yuan et al. (3) reported that Lys-685 is acetylated by p300 in response to oncostatin M, a member of the IL-6 family of cytokines. They studied STAT3-deficient PC3 cells in which wild-type or K685R mutant STAT3 was expressed, stating that Lys-685 acetylation is crucial for the ability of STAT3 to form stable dimers, which is required for DNA binding and cytokine-stimulated transcriptional regulation, and to enhance the expression of cell growth-related genes. STAT3 was also shown to be acetylated on Lys-685 by p300 in response to IL-6 in HEK293 cells (21). To re-examine the importance of Lys-685 for the function of tyrosine-phosphorylated STAT3, we determined the effect of the K685R mutation on IL-6-induced gene expression in PC3 cells. Cells expressing either wild-type or K685R mutant STAT3 were treated with IL-6 for 4 h (Fig. 3A), and the expression of the SOCS3 and CCND1 mRNAs was measured. The K685R mutation did not inhibit the induced expression of these two mRNAs at all (Fig. 3B). Because the IL-6-mediated induction of SOCS3 mRNA in PC3 cells expressing wild-type STAT3 was much weaker than in immortalized HME cells (Fig. 3C), we thought it was best to use a STAT3-null cell line other than PC3, in which the response was more robust, for additional experiments. To this end, we stably expressed wild-type or K685R mutant STAT3 in A4 cells (Fig. 4A), which were derived from the human colon cancer cell line DLD1 by deletion of the STAT3 gene by homologous recombination (10). A4 cells have a strong response to IL-6 when wild-type STAT3 is re-expressed, comparable with the response of the parental DLD1 cells (10). We confirmed the strong induction of SOCS3 mRNA in response to IL-6 in A4 cells with restored expression of wild-type STAT3 (Fig. 4B) and also confirmed the finding of Yuan et al. (3) that the K685R mutation does not impair the phosphorylation of STAT3 on Tyr-705 (Fig. 4C). We then performed a microarray analysis to identify genes whose expression was increased in response to treatment with IL-6. Of the ∼35,000 genes on the array, the expression of 59 increased by 2-fold or more in this experiment. The expression of 41 of these 59 genes was remarkably similar when the response of A4 cells expressing the K685R mutant was compared with that in cells expressing wild-type STAT3 (Fig. 4D, complete list in supplemental Table S2). Moreover, for 11 of the 18 genes whose expression was reduced by the K685R mutation, the degree of inhibition was small. The lack of effect of the K685R mutation on the IL-6-induced expression of several genes from the array, including BCL3, SERPINA1, GADD45G, and SOCS3, was confirmed by real-time quantitative PCR (Fig. 4E). We conclude that the K685R mutation has little or no effect on IL-6-induced gene expression in the cells we have studied.
FIGURE 3.
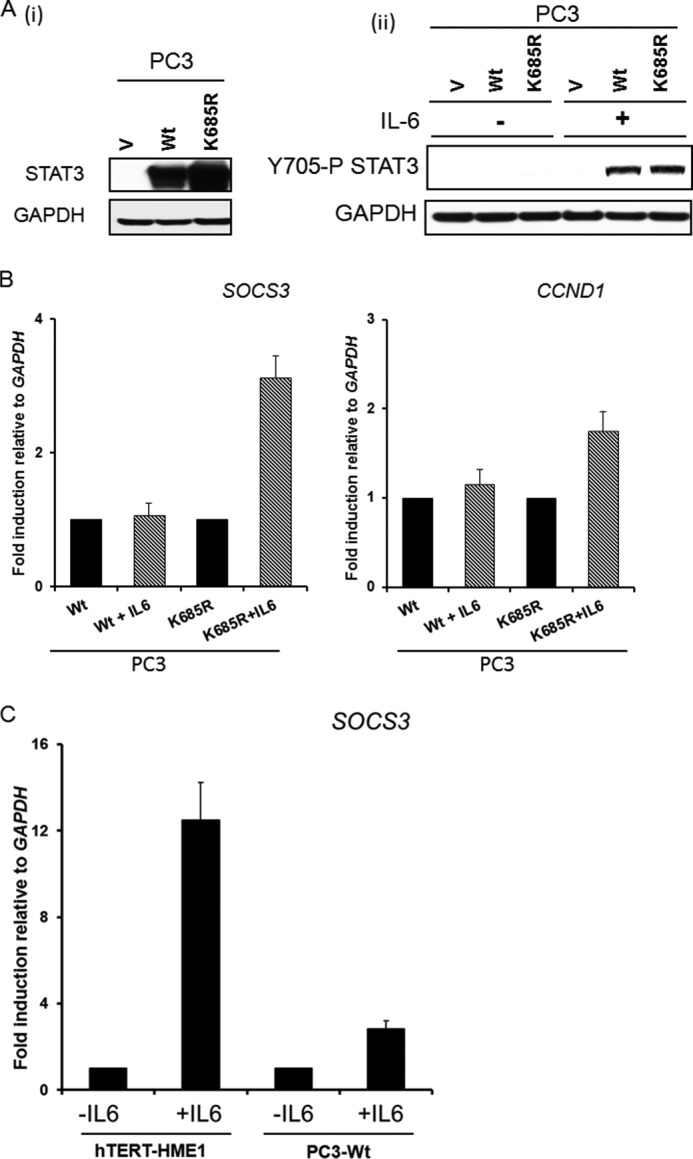
The K685R mutation does not affect the IL-6 dependent induction of SOCS3 and CCND1 genes in PC3 cells. A, panel i, STAT3-null PC3 cells were infected with retroviral constructs expressing WT STAT3 or K685R STAT3, and stable pools of cells were selected with G418. Panel ii, PC3 vector control (V), WT, and K685R cells were treated with IL-6 (200 ng/ml) and sIL-6R (250 ng/ml) for 4 h, and total cell lysates were analyzed for Tyr-705 phosphorylation. B, PC3-WT and -K685R cells were treated with IL-6 (200 ng/ml) for 4 h, and the relative expression of the SOCS3 and CCND1 genes was determined by qPCR. Values are the means, with standard deviations, of triplicate experiments. C, PC3 and hTERT-HME1 cells were treated with IL-6 (200 ng/ml) and sIL-6R (250 ng/ml) for 4 h, and relative expression of SOCS3 was determined by qPCR to compare. Values are the means, with standard deviations, of triplicate experiments.
FIGURE 4.

The K685R mutation has only a marginal effect on IL-6-dependent STAT3 phosphorylated on Tyr-705 (Y705-P STAT3)-mediated gene expression in A4 cells. A, STAT3-null A4 cells were infected with retroviral constructs expressing either WT STAT3 or the K685R mutant and stable pools were selected with G418. A4-WT and A4-K685R cells express similar levels of wild-type or mutant STAT3 proteins when compared with parental DLD1 cells. B, the cells were treated with IL-6 (200 ng/ml) and sIL-6R (250 ng/ml) for 4 h. PCR (left) and qPCR (right) analyses of relative expression of SOCS3 are shown. Values are the means, with standard deviations, from triplicate experiments. C, A4-WT and A4-K685R cells were treated with IL-6 (200 ng/ml) and sIL-6R (250 ng/ml) for 4 h, and total cell lysates were analyzed for Tyr-705 phosphorylation. D, array was performed to compare genes induced in A4-WT STAT3 and A4-K685R STAT3 cells upon IL-6 treatment. The heat map shows the induced expression of 59 genes. We include only signals that changed by >2-fold, with differential p ≤ 0.05, in WT cells when compared with vector control cells, and with average signals greater than 30 in WT cells. E, A4-WT and A4-K685R cells were treated with IL-6 (200 ng/ml) and sIL-6R (250 ng/ml) for 4 h, and the relative expression of the BCL3, SERPINA1, GADD45G, and SOCS3 genes was determined by qPCR. Values are the means, with standard deviations, from triplicate experiments.
Angiotensin II-dependent Gene Expression Requires Lys-685 of STAT3
Yue et al. (20) reported that the level of U-STAT3, unphosphorylated at residues Tyr-705 and Ser-727, correlates with the development of cardiac hypertrophy and dysfunction in AT1R transgenic mice. They also found that the STAT3 Y705F mutant protein binds to p300 in HEK cells expressing AT1R upon treatment with angiotensin II. To evaluate the role of Lys-685 in angiotensin II-dependent gene expression, we expressed FLAG-tagged versions of Y705F STAT3 and the K685R/Y705F double mutant in A4 cells (Fig. 5A), at levels similar to those of wild-type and K685R STAT3 in earlier experiments. The HA-tagged human angiotensin II receptor was also expressed stably in these cells, and the normal hAT1R-dependent response to angiotensin II was confirmed in a calcium flux assay (Fig. 5B). A microarray analysis revealed that the expression of 73 of the ∼35,000 genes on the array was enhanced by 2-fold or more in response to angiotensin II in cells expressing Y705F-STAT3 when compared with angiotensin II-treated vector control cells (Fig. 5C; complete list in supplemental Table S3), confirming the dependence on U-STAT3. The expression of 36 of these 73 genes was diminished by 2-fold or more by the K685R mutation, but the expression of most of the remaining genes was diminished only modestly (2-fold or less). The expression of three randomly selected genes that pass the threshold criteria (SOS2, LTA, and PLA2G10) was confirmed by real-time quantitative PCR, which showed induction levels similar to those determined in the microarray experiment (Fig. 5D). These results show that the integrity of Lys-685 is important for the ability of U-STAT3 to participate in the angiotensin II-induced expression of many but not all genes. It is interesting that no gene induced by increased expression of U-STAT3 in hTERT-HME1 cells is also induced in response to angiotensin II in A4 cells (see supplemental Tables S1 and S3), indicating that U-STAT3 participates in these two pathways by distinctly different mechanisms.
FIGURE 5.

The K685R mutation impairs the induction of some angiotensin II-dependent genes in A4 cells. A, A4-V, -WT, -K685R, -Y705F, and -K685R/Y705F cells were infected with lentiviral constructs expressing hAT1R, and stable pools of cells were selected with puromycin. Western analysis of total cell lysates confirms human AT1R expression in each pool. IB, immunoblot. B, the hAT1R-expressing A4 cells were stimulated with 1 μm angiotensin II, and their response to angiotensin II-induced calcium mobilization was determined by measuring their respective calcium flux. C, the heat map of microarray done to compare genes induced in A4-Y705F STAT3 and A4-K685R/Y705F STAT3 cells expressing hAT1R upon angiotensin II (Ang II) treatment shows the increased expression of 73 genes. We include only genes whose expression changed by 2-fold, with differential p < 0.05 in Y705F STAT3/hAT1R cells when compared with vector control cells, and with average signals greater than 30 in WT cells, following treatment with 1 μm angiotensin II for 4 h. D, A4-Y705F and A4-K685R/Y705F cells were treated with angiotensin II (1 μm for 4 h), and the relative expression of the SOS2, LTA, and PLA2G10 genes was determined by qPCR. Values are the means, with standard deviations, from triplicate experiments. *, p ≤ 0.05; **, p ≤ 0.005.
DISCUSSION
Role of Lysine Acetylation in the Function of Tyrosine-phosphorylated STAT3
STAT3 phosphorylation and functions in liver were shown to be tightly regulated by the acetylation of four key C-terminal lysine residues (Lys-679, Lys-685, Lys-707, and Lys-709) but not by acetylation of Lys-685 alone (22). In contrast, cytokine-stimulated Lys-685 acetylation was reported to be critical for the ability of STAT3 to form stable dimers, to bind to DNA, and to regulate transcription (3). However, O'Shea et al. (4) criticized this conclusion at the time, arguing that it was unlikely that Lys-685 acetylation would block the cytokine-dependent formation of STAT3 dimers because this residue was not close enough to the dimer interface in the crystal structure of STAT3. In a separate study, p300 was shown to acetylate STAT3 at Lys-685 in HepG2 and HEK293 cells in response to IL-6, and it was claimed that this modification was required for sequence-specific DNA binding and transactivation (21). However, the K685R mutant of STAT3 was expressed together with wild-type endogenous STAT3, complicating interpretation of the results. Lysine acetylation at Lys-49 and Lys-87 has been associated with positive regulation of gene expression and DNA binding (5). Furthermore, activation of STAT3-dependent transcription by IL-6 and leukemia inhibitory factor (LIF) was enhanced in HepG2, MCF7, and HEK cells following inhibition of the histone deacetylase SIN3A, which interacted more strongly with the K49Q/K87Q double mutant of STAT3 (where Gln mimics acetyl lysine) than with the K685Q mutant. Further analysis of single STAT3 lysine mutations revealed Lys-87 acetylation as the main regulator of the STAT3-SIN3A interaction (23). To clarify the role of Lys-685 in gene expression driven by tyrosine-phosphorylated STAT3 dimers, we investigated the function of the K685R mutant in response to IL-6. Gene expression data from IL-6-stimulated PC3 and A4 cells reveal that Lys-685 does not play an essential role in the expression of the great majority of phospho-STAT3-dependent genes (Figs. 3 and 4).
Role of Lysine Acetylation in the Signal-dependent Activity of U-STAT3
Yang et al. (12) demonstrated that the high level of STAT3 that accompanies its constitutive activation in many tumors is responsible for the increased expression of several genes (MET, MRAS, CDC2, CCND1, and RANTES) whose protein products are important in oncogenesis, cell cycle control, and the immune response. A major stimulus for increased STAT3 expression is the ability of phosphorylated STAT3 dimers to activate expression of the STAT3 gene. We have now begun to analyze the contribution of post-translational modification of U-STAT3 on its ability to induce target gene transcription, and our mass spectrometry analysis reveals that U-STAT3 is acetylated on Lys-685 in hTERT-HME1 cells (Fig. 1). A global analysis revealed that more than 70% of the gene expression induced by a high level of U-STAT3 was impaired by 2-fold or more in cells expressing the K685R mutant (Fig. 2). Therefore, we conclude that the majority of U-STAT3-dependent gene expression depends upon the ability of Lys-685 to become acetylated.
Prior studies have shown that p300 is responsible for acetylating STAT3. Yuan et al. (3) revealed the acetylation of Lys-685 by MS analysis of STAT3 purified from 293T cells. Because the cells were not treated, we believe that the STAT3 was not phosphorylated. In a separate experiment, the same authors showed that a truncated STAT3 mutant protein, lacking Lys-685, failed to bind to p300, and that Lys-685 was the only lysine residue in the full-length protein that was acetylated. In another study, also carried out in untreated cells in which STAT3 was not phosphorylated, Lee et al. (19) demonstrated that wild-type STAT3 in complex with CD44 and p300 is acetylated and that WT STAT3, but not the K685R mutant, binds to p300. Finally, Yue et al. (20) showed that the phosphorylation-defective Y705F mutant of STAT3 is bound to p300 in HEK-AT1R cells. In summary, we conclude that p300 binds to U-STAT3 to acetylate Lys-685 but does not bind to the K685R mutant protein
U-STAT3 is required for the angiotensin II-dependent expression of genes implicated in the pathogenesis of heart failure, and it associates with p300 in response to angiotensin II (20). Furthermore, Samraj et al. (24) have found that the accumulation of U-STAT3 in cerebral arteries plays a pivotal role in orchestrating the expression of genes that are involved in late cerebral ischemia and related pathogenesis following a subarachnoid hemorrhage. These two studies provide evidence that U-STAT3 is involved in important pathological situations outside of cancer. To elucidate the relevance of Lys-685 modification for U-STAT3-dependent gene expression in response to angiotensin II, we evaluated induced gene expression in human A4 cells expressing AT1R, together with either the Y705F or the K685R/Y705F mutant of STAT3. We conclude that the integrity of Lys-685 is important for the expression of about half of the genes activated in response to angiotensin II (Fig. 5), thus providing new information on how U-STAT3 may contribute to the development of cardiac hypertrophy following angiotensin II stimulation.
Additional Therapeutic Implications
Lee et al. (25) reported that, by reducing STAT3 acetylation, ERα gene expression can be reactivated in tumors in which the ERα promoter had previously been methylated. Moreover, because inactivation of the ERα gene by DNA methylation strongly correlates with poor prognosis as well as an aggressive phenotype in cancers such as melanoma and the basal subtype of breast cancer, these data provide a rationale for targeting acetylated STAT3 for cancer therapy (26, 27). In non-small cell lung cancer, hyaluronan promotes the CD44-dependent association of the stem cell marker NANOG with both the phosphorylated and the unphosphorylated forms of STAT3, resulting in chemo-resistance that depends upon induced expression of the microRNA miR-21 (28). Lee et al. (19) have demonstrated that the transmembrane glycoprotein CD44, recently recognized to be a signature of cancer stem cells, is internalized following its binding to osteopontin or Hermes 3, an anti-CD44 antibody. In the nucleus, CD44, in complex with STAT3 and p300, activates the CCND1 promoter, thus stimulating cell proliferation. The acetylation of Lys-685 of STAT3, mediated by p300, is crucial for this function of STAT3. Our laboratory has previously shown that overexpression of the oncoproteins MET and MRAS, found in many cancers, is driven by overexpression of U-STAT3 (12). Therefore, inhibitors that selectively target the acetylation of Lys-685 have the potential to inhibit tumorigenesis. Pena et al. (29) reported that STAT3-conditional knock-out mice have an overwhelming susceptibility to sepsis due to loss of the anti-inflammatory activity of U-STAT3, providing new insight into the role of U-STAT3 in infectious diseases. Further understanding of the diverse signaling pathways that lead to U-STAT3-dependent gene expression will lead to additional opportunities to evaluate the importance of the acetylation of Lys-685 of U-STAT3 in different physiological and pathological settings.
Supplementary Material
Acknowledgments
We thank Yuxin Wang for critical reading of the manuscript and Josephine Kam Tai Dermawan for help with figure preparation.
This work was supported, in whole or in part, by National Institutes of Health Grant CA062220 (to G. R. S.) and National Institutes of Health Shared Instrument Grant 1S10RR031537-01 (to B. W.) for purchasing the Orbitrap Elite instrument.

This article contains supplemental Tables S1–S3 and a supplemental Method.
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cells
- U-STAT3
- STAT3 unphosphorylated on Tyr-705
- hAT1R
- human angiotensin type I receptor
- qPCR
- quantitative PCR
- sIL-6R
- soluble IL-6 receptor
- ERα
- estrogen receptor α
- HME
- human mammary epithelial.
REFERENCES
- 1. Akira S., Nishio Y., Inoue M., Wang X. J., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., Kishimoto T. (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71 [DOI] [PubMed] [Google Scholar]
- 2. Wen Z., Zhong Z., Darnell J. E., Jr. (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250 [DOI] [PubMed] [Google Scholar]
- 3. Yuan Z. L., Guan Y. J., Chatterjee D., Chin Y. E. (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273 [DOI] [PubMed] [Google Scholar]
- 4. O'Shea J. J., Kanno Y., Chen X., Levy D. E. (2005) Cell signaling: Stat acetylation—a key facet of cytokine signaling? Science 307, 217–218 [DOI] [PubMed] [Google Scholar]
- 5. Ray S., Boldogh I., Brasier A. R. (2005) STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 129, 1616–1632 [DOI] [PubMed] [Google Scholar]
- 6. Perry E., Tsruya R., Levitsky P., Pomp O., Taller M., Weisberg S., Parris W., Kulkarni S., Malovani H., Pawson T., Shpungin S., Nir U. (2004) TMF/ARA160 is a BC-box-containing protein that mediates the degradation of Stat3. Oncogene 23, 8908–8919 [DOI] [PubMed] [Google Scholar]
- 7. Tanaka T., Yamamoto Y., Muromoto R., Ikeda O., Sekine Y., Grusby M. J., Kaisho T., Matsuda T. (2011) PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci. Signal. 4, ra85. [DOI] [PubMed] [Google Scholar]
- 8. Ray S., Zhao Y., Jamaluddin M., Edeh C. B., Lee C., Brasier A. R. (2014) Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell. Signal. 26, 1445–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stark G. R., Wang Y., Lu T. (2011) Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 21, 375–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J., Huang J., Dasgupta M., Sears N., Miyagi M., Wang B., Chance M. R., Chen X., Du Y., Wang Y., An L., Wang Q., Lu T., Zhang X., Wang Z., Stark G. R. (2010) Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 21499–21504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim E., Kim M., Woo D. H., Shin Y., Shin J., Chang N., Oh Y. T., Kim H., Rheey J., Nakano I., Lee C., Joo K. M., Rich J. N., Nam D. H., Lee J. (2013) Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang J., Chatterjee-Kishore M., Staugaitis S. M., Nguyen H., Schlessinger K., Levy D. E., Stark G. R. (2005) Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 65, 939–947 [PubMed] [Google Scholar]
- 13. Yang J., Liao X., Agarwal M. K., Barnes L., Auron P. E., Stark G. R. (2007) Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 21, 1396–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Courapied S., Sellier H., de Carné Trécesson S., Vigneron A., Bernard A. C., Gamelin E., Barré B., Coqueret O. (2010) The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J. Biol. Chem. 285, 26765–26778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hazan-Halevy I., Harris D., Liu Z., Liu J., Li P., Chen X., Shanker S., Ferrajoli A., Keating M. J., Estrov Z. (2010) STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 115, 2852–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Androutsellis-Theotokis A., Leker R. R., Soldner F., Hoeppner D. J., Ravin R., Poser S. W., Rueger M. A., Bae S. K., Kittappa R., McKay R. D. (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442, 823–826 [DOI] [PubMed] [Google Scholar]
- 17. Liu H., Ma Y., Cole S. M., Zander C., Chen K. H., Karras J., Pope R. M. (2003) Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood 102, 344–352 [DOI] [PubMed] [Google Scholar]
- 18. Waitkus M. S., Chandrasekharan U. M., Willard B., Tee T. L., Hsieh J. K., Przybycin C. G., Rini B. I., Dicorleto P. E. (2014) Signal integration and gene induction by a functionally distinct STAT3 phosphoform. Mol. Cell. Biol. 34, 1800–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J. L., Wang M. J., Chen J. Y. (2009) Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J. Cell Biol. 185, 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yue H., Li W., Desnoyer R., Karnik S. S. (2010) Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc. Res. 85, 90–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang R., Cherukuri P., Luo J. (2005) Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 280, 11528–11534 [DOI] [PubMed] [Google Scholar]
- 22. Nie Y., Erion D. M., Yuan Z., Dietrich M., Shulman G. I., Horvath T. L., Gao Q. (2009) STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Icardi L., Mori R., Gesellchen V., Eyckerman S., De Cauwer L., Verhelst J., Vercauteren K., Saelens X., Meuleman P., Leroux-Roels G., De Bosscher K., Boutros M., Tavernier J. (2012) The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl. Acad. Sci. U.S.A. 109, 12058–12063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Samraj A. K., Müller A. H., Grell A. S., Edvinsson L. (2014) Role of unphosphorylated transcription factor STAT3 in late cerebral ischemia after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 34, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee H., Zhang P., Herrmann A., Yang C., Xin H., Wang Z., Hoon D. S., Forman S. J., Jove R., Riggs A. D., Yu H. (2012) Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. U.S.A. 109, 7765–7769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mori T., Martinez S. R., O'Day S. J., Morton D. L., Umetani N., Kitago M., Tanemura A., Nguyen S. L., Tran A. N., Wang H. J., Hoon D. S. (2006) Estrogen receptor-α methylation predicts melanoma progression. Cancer Res. 66, 6692–6698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brinkman J. A., El-Ashry D. (2009) ER re-expression and re-sensitization to endocrine therapies in ER-negative breast cancers. J. Mammary Gland Biol. Neoplasia 14, 67–78 [DOI] [PubMed] [Google Scholar]
- 28. Bourguignon L. Y., Earle C., Wong G., Spevak C. C., Krueger K. (2012) Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene 31, 149–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peña G., Cai B., Liu J., van der Zanden E. P., Deitch E. A., de Jonge W. J., Ulloa L. (2010) Unphosphorylated STAT3 modulates α7 nicotinic receptor signaling and cytokine production in sepsis. Eur. J. Immunol. 40, 2580–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.