

Background: The roles of SMS isoforms (SMS1 and SMS2) in membrane fusion have not yet been reported.
Results: Overexpression of Sms2 in Sms-deficient cells increased membrane fusion susceptibility and F-actin polymerization in the region of membrane fusion.
Conclusion: SMS2 could promote membrane fusion by regulating HIV-1 Env-triggered F-actin polymerization.
Significance: Our findings provide new insights into the role of SMS2 in membrane fusion.
Keywords: Human Immunodeficiency Virus (HIV), Membrane Fusion, Membrane Lipid, Phospholipid, Sphingolipid, Cell-Cell Fusion Assay, Sphingomyelin Synthase
Abstract
Membrane fusion between the viral envelope and plasma membranes of target cells has previously been correlated with HIV-1 infection. Lipids in the plasma membrane, including sphingomyelin, may be crucially involved in HIV-1 infection; however, the role of lipid-metabolic enzymes in membrane fusion remains unclear. In this study, we examined the roles of sphingomyelin synthase (SMS) in HIV-1 Env-mediated membrane fusion using a cell-cell fusion assay with HIV-1 mimetics and their target cells. We employed reconstituted cells as target cells that stably express Sms1 or Sms2 in Sms-deficient cells. Fusion susceptibility was ∼5-fold higher in Sms2-expressing cells (not in Sms1-expressing cells) than in Sms-deficient cells. The enhancement of fusion susceptibility observed in Sms2-expressing cells was reversed and reduced by Sms2 knockdown. We also found that catalytically nonactive Sms2 promoted membrane fusion susceptibility. Moreover, SMS2 co-localized and was constitutively associated with the HIV receptor·co-receptor complex in the plasma membrane. In addition, HIV-1 Env treatment resulted in a transient increase in nonreceptor tyrosine kinase (Pyk2) phosphorylation in Sms2-expressing and catalytically nonactive Sms2-expressing cells. We observed that F-actin polymerization in the region of membrane fusion was more prominent in Sms2-expressing cells than Sms-deficient cells. Taken together, our research provides insight into a novel function of SMS2 which is the regulation of HIV-1 Env-mediated membrane fusion via actin rearrangement.
Introduction
Membrane dynamics play crucial roles in fundamental processes in living cells, including exocytosis, endocytosis, membrane recycling, protein trafficking, and organelle biogenesis (1). A number of protein factors, such as clathrin, COP, and Rho GTPase, have been shown to regulate membrane dynamics (2–4). These protein factors as well as other specific phospholipids, such as phosphatidic acid and lysophospholipids, can regulate the fusion, fission, or tubulation of membranes. Furthermore, various enzymes, including phospholipases and acyltransferases, have been suggested to play a role in membrane dynamics by producing and removing fusogenic lipids (5, 6).
Membrane dynamics, including fusion, are involved in physiological as well as pathological processes. Many pathogens are known to “hijack” the membrane dynamic machinery of host cells for processes, such as infection, survival, symbiosis, and replication. HIV-1 is one of the best known pathogens that utilizes a membrane fusion mechanism (7). Because HIV-1 is surrounded by a viral envelope composed of phospholipid bilayers, membrane fusion between the viral envelope and the plasma membranes of target cells is essential for its entry.
Several proteins in virus and target cells are involved in HIV-1-mediated membrane fusion. HIV-1 envelope proteins (Env)2 are composed of gp120 and gp41 subunits that specifically recognize and bind to CD4, the HIV-1 receptor, and subsequently to one of two chemokine receptors, either CCR5 or CXCR4, which acts as the HIV-1 co-receptor on the surface of target cells. CCR5 can be used by the macrophage tropic HIV-1 strains (HIVJRFL) during early infection, whereas CXCR4 can be used by the T cell tropic HIV-1 strains (HIVNL4-3) during late stage infection (8). This tropism change has previously been associated with mutations in the third variable loop domain of the gp120 subunit (9). Binding of the Env subunit gp120 to the HIV-1 receptor and co-receptor induces a conformational change in the Env and unmasks the second subunit gp41. The exposed hydrophobic domain of gp41 is subsequently inserted into the target cell membrane, resulting in the fusion of HIV-1 Env with target cells. Finally, the HIV-1 genome and proteins enter the target cells.
Several lines of evidence have reported that membrane lipids, particularly sphingolipids, are involved in the fusion of HIV-1 to target cells. Dihydrosphingomyelin, which is identical to SM except that it lacks a 4,5-trans double bond, was previously shown to regulate formation of rigid domains (10). The genetic and pharmacological blockade of dihydroceramide desaturase replaced SM with dihydrosphingomyelin in cultured cells. This increase in dihydrosphingomyelin levels led to more rigid membrane domains that were resistant to the insertion of the gp41 peptide, which consequently inhibited virus-cell membrane fusion. These findings demonstrated that the physicochemical properties of SM (or its analog) affect HIV-1 infection.
Cellular levels of ceramide are also suggested to be important for the entry of HIV-1 into cells (11, 12). The treatment of target cells with a pharmacological activating agent of the ceramide synthesis pathway markedly inhibited the fusion of HIV-1 to target cell membranes (11). The bacterial sphingomyelinase from Bacillus cereus also inhibited the entry of HIV-1, which indicated that ceramide derived from the degradation of SM may reduce the susceptibility of cells to membrane fusion. Ceramide was previously shown to translocate cholesterol from lipid rafts to the liquid-disordered phase in the supported lipid bilayer, which decreases the diffusion coefficient in this phase (13). Additionally, treatment of target cells with sphingomyelinase was shown to restrict the lateral diffusion of CD4 and subsequently inhibited HIV-1 fusion (12).
Another sphingolipid, glycosphingolipid, was also reported to be a potential lipid involved in HIV-1 infection; HIV-1-mediated membrane fusion was reduced by treating target cells with a ceramide glucosyltransferase inhibitor, and the reconstitution of globotriaosylceramide restored the susceptibility of cells to membrane fusion (14). Furthermore, a glycerolipid from Acholeplasma laidlawii was able to bind to HIV-1 and accelerate the infection of target cells (15). Although the importance of membrane lipids for the entry of HIV-1 into target cells has been confirmed, the roles of lipid-metabolic enzymes in membrane fusion and their regulation have not yet been elucidated in detail.
SM is synthesized de novo from serine and palmitoyl coenzyme A by the sequential reactions of various enzymes. The final step of its synthesis is catalyzed by SM synthase (SMS), which transfers the phosphorylcholine moiety from PC to the primary hydroxy of ceramide, resulting in the production of SM and diacylglycerol. This enzyme has two isoforms, SMS1 and SMS2 (16). SMS1 is mainly localized in the Golgi apparatus, although SMS2 is localized in both the Golgi apparatus and plasma membrane (16). Previous studies revealed that SM produced by SMS1 and/or SMS2 played important roles in various metabolic diseases, including atherosclerosis, insulin secretion, and obesity (17–19). However, the roles of SMS isoforms in pathogen infection have not yet been reported.
In this study, we attempted to determine the involvement of SM and SMS isoforms in HIV-1 Env-mediated membrane fusion using a cell-cell fusion assay. This fusion assay is a reproducible method that can be used to analyze the membrane fusion process of HIV-1 infection (20–22) and does not need to be carried out in a P3 class facility. By using this assay, we showed that SMS2, but not SMS1, augmented membrane fusion susceptibility. More importantly, we found that the SMS2 protein itself, but not SM generated by SMS activity, was involved in this process. The results of this study demonstrate for the first time that lipid-metabolizing enzymes are involved in HIV-1 Env-mediated membrane fusion, regardless of their enzyme activities.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The mouse anti-His6 (clone 9F2) antibody was obtained from Wako Pure Chemicals (Japan). The mouse anti-FLAG (clone M2) and rabbit anti-V5 antibodies as well as anti-FLAG M2 affinity gel were obtained from Sigma. The rat anti-HA antibody (clone 3F10) was from Roche Applied Science, and the goat anti-rat IgG-HRP antibody was from Santa Cruz Biotechnology. The anti-HA affinity gel was obtained from Thermo Scientific, and the anti-Pyk2 and anti-phospho-Pyk2 (Tyr-402) antibodies were obtained from Cell Signaling Technology. The goat anti-mouse IgG-HRP, anti-rat IgG-AlexaFluor 546, anti-mouse IgG-AlexaFluor 488, and anti-rabbit IgG-AlexaFluor 405 antibodies as well as phalloidin-AlexaFluor 546 and CellTrackerTM Blue CMAC were obtained from Invitrogen. The rabbit anti-GAPDH antibody was from GeneTex, and the goat anti-rabbit IgG-HRP antibody was from MBL. Anti-CD4 IgG-APC (clone RPA-T4) for FACS analysis was obtained from eBioscience, and anti-CCR5 IgG-PE (clone 3A9), anti-CXCR4 IgG-PE (clone 12G5), and goat anti-mouse Ig-PE (multiple adsorption) were obtained from Pharmingen.
Plasmids
The expression vector for the nontoxic SM probe, the EGFP fusion protein of the lysenin deletion mutant (pQE30-EGFP-lysenin(161–297)), was kindly provided by Dr. T. Kobayashi (RIKEN, Japan). pCXN/EnvJRFL, pCXN/EnvNL4-3, pCDNA6.2/HIV-tat, pcDNA3.1/CD4, pCDNA6.2/CCR5, pcDNA3.1/CXCR4, and pLTR-LucE were kindly provided by Prof. H. Mitsuya (National Institutes of Health, Bethesda). YUgp140 (−/GCN4)/pcDNA3.1(−) was provided by the AIDS Research and Reference Reagent Program (National Institutes of Health). pRL-SV40 was purchased from Promega.
Plasmids for the retroviral expression of mouse Sms isoforms (pQCXIP/Sms1-V5 and pQCXIP/Sms2-V5) were constructed as described previously (23). The mutated open reading frame (ORF) of Sms2 (H229A) that lacked SMS activity was prepared by QuikChange site-directed mutagenesis (Stratagene). The expression vectors of SMS isoforms (pCMV-3Tag-8/SMS1-V5 and pCMV-3Tag-8/SMS2-V5) or human CD4 (pCMV-3Tag-8/CD4-HA) were constructed as follows. The ORFs of human SMS1 and SMS2 were amplified from the cDNA library derived from HEK293 cells using specific primers containing sequences corresponding to the V5 epitope in front of the stop codon. CD4 cDNA was amplified from pcDNA3.1/CD4 as a template using specific primers containing sequences corresponding to the HA epitope in front of the stop codon. PCR products were subcloned into pCMV-3Tag-8 (Stratagene). The expression vectors of human CCR5 and CXCR4 (pCMV-3Tag-8/CCR5–3×FLAG and pCMV-3Tag-8/CXCR4–3×FLAG) were created as described below. The ORFs of human CCR5 and CXCR4 without a stop codon were amplified by PCR using pCDNA6.2/CCR5 and pcDNA3.1/CXCR4 as templates and then subcloned into pCMV-3Tag-8.
RNA Expression Analysis
Jurkat and Molt4 cells, which are human T cell lines, were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 μg/ml streptomycin, and 100 units/ml penicillin. Total RNA was isolated from cells with the RNeasy mini kit (Qiagen, Canada). cDNAs were synthesized from 0.5 μg of RNA with the SuperScript III First-strand Synthesis System for RT-PCR (Invitrogen). In the semi-quantitative analysis, cDNA products were amplified with PrimeSTAR HS DNA polymerase (Takara, Japan) with the primers for SMS1 (sense, 5′-ATGAAGGAAGTGGTTTATTGGTC-3′; antisense, 5′-AGGGTTTCTATCATGTCC-3′) and SMS2 (sense, 5′-ATGGATATCATAGAGACAGCAAAAC-3′; antisense, 5′- CCTCTCATGTACAACTGT-3′), which amplified 200- and 300-bp products, respectively. The PCR program consisted of 45 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 20 s. Relative quantification was estimated by GAPDH amplification. Amplified PCR fragments were separated on a 4% agarose gel.
Assay of SMS Activity in Vitro
The assay of SMS activity was performed as described elsewhere (23). C6-NBD-ceramide (400 pmol) and lecithin (6.5 nmol) were mixed in 100 μl of ethanol, and the solvent was then evaporated. Aqueous solution (20 μl) was added, and the mixture was sonicated to form liposomes. The reaction mixture (40 μl) containing 20 μl of an appropriate amount of cell lysate and 20 μl of C6-NBD-ceramide liposomes were incubated at 37 °C for 1 h. The reaction was stopped by the addition of 200 μl of chloroform/methanol (2:1, v/v) and vigorous mixing. Lipids containing the reaction product were extracted in the lower layer using the Bligh and Dyer method after a 5-min centrifugation at 18,000 × g. After the solvents were evaporated, the lipids were dissolved in 10 μl of chloroform/methanol (2:1, v/v). The lipids were separated by TLC in chloroform/methanol/water (65:25:4, v/v/v) as a solvent. Each band of C6-NBD-ceramide and C6-NBD-SM was quantified using the fluorescence imaging analyzer, Typhoon 9000 (GE Healthcare).
Expression of Sms1, Sms2, or the Sms2 Mutant in Mouse Embryonic Fibroblasts from Sms1/Sms2 Double Knock-out Mice
Sms-deficient immortalized embryonic fibroblasts (ZS cells) were isolated from Sms1 and Sms2 double-deficient mice as described previously (23). Sms1, Sms2, or the catalytically inactive Sms2 mutant (Sms2-H229A) was stably expressed in ZS cells as parental cells by the retroviral vector system. The enzymes were all designed as V5 tag fusion proteins at the C terminus. Each isolated cell line was named ZS/Sms1, ZS/Sms2, or ZS/Sms2-H229A, respectively. All cell types were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C in a humidified incubator containing 5% CO2.
Lysenin Treatment and Cell Mortality Measurement
Cultured cells (1 × 104 cells) were washed twice with phosphate-buffered saline (PBS) and incubated for 30 min at 37 °C with various concentrations of lysenin (Peptide Institute, Japan), a cytotoxic protein derived from the earthworm that specifically binds to SM-rich domains. Cell viability was measured using the WST-1 cell proliferation reagent (Roche Applied Science) according to the manufacturer's instructions.
Cell Staining with Nontoxic EGFP-Lysenin
The procedure for the expression and purification of nontoxic EGFP-lysenin was performed as described previously (24). Cultured cells (3 × 105 cells) were washed twice with PBS and incubated with 3 μg/ml EGFP-lysenin for 30 min at 4 °C. After washing three times with PBS, the binding of EGFP-lysenin to the cells was quantified by FACS Aria (Pharmingen).
HIV-1 Env-mediated Cell-Cell Fusion Assay
The HIV-1 Env-mediated cell-cell fusion assay was performed as described previously (25, 26) with minor modifications. To prepare the effector cells, the envelope expression vector (1 μg of plasmid of pCXN/EnvJRFL, pCXN/EnvNL4-3, or pVSV-G) and HIV-1 “Trans-activator of Transcription” (Tat) expression vector (1 μg of plasmid of pCDNA6.2/HIV-tat) were transfected into HEK293 cells (5 × 105 cells) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. To generate target cells, the expression vectors of the HIV-1 receptor·co-receptor complex (2.5 μg of pcDNA3.1/CD4 and 3.5 μg of pCDNA6.2/CCR5 or pcDNA3.1/CXCR4) and the reporter genes (3 μg of pLTR-LucE and 2 μg of pRL-SV40) were introduced into the cultured cells (1 × 106 cells), i.e. ZS, ZS/Sms1, ZS/Sms2, or ZS/Sms2-H229A cells using the NEPA21 electroporator (NEPA Gene, Japan). pLTR-LucE and pRL-SV40 were used as the reporter gene for Tat and an internal control, respectively. In some experiments, target cells were treated with maraviroc (Sigma), AMD3100 (Sigma), cytochalasin D (Wako, Japan), or tyrphostin A9 (Sigma) immediately before the cell-cell fusion assay. The effector and target cells were harvested 24 h post-transfection, and 2 × 104 cells from each were mixed in the wells of a 96-well plate. After incubating at 37 °C for 4 h, the luciferase activity in each well was detected using the Dual-Glo Luciferase Assay System (Promega), and its luminescence level was measured using PowerscanHT (DS Pharma Biomedical, Japan). Nonspecific luciferase activity was determined according to the luminescence level using effector cells expressing only Tat and not HIV-1 Env. The value of the nonspecific luminescence level was subtracted from each experimental luminescence level. Relative light units were calculated as the quotient of the firefly and Renilla activity values and were indicative of cell-cell fusion.
Analysis of Cell-surface Expression of CD4, CCR5, and CXCR4
The target cells (1 × 105 cells) were incubated with monoclonal antibodies against CD4, CCR5, or CXCR4 (1:10 dilution). After incubating for 30 min at 4 °C, cells were washed twice in PBS with 0.1% BSA and analyzed by FACS Aria to determine the level of cell-surface expression.
Sms2 mRNA Silencing
Sms2 gene silencing was performed using Stealth RNAiTM siRNA (Invitrogen) against the following target sequences: siSms2-1, 5′-CCACACUGUCGUGCUCACACUUACU-3′; siSms2-2, 5′-CCCAGUGGCUCUUUCUGCGUUACAA-3′. Control experiments used StealthTM control siRNA. ZS/Sms2 cells (1 × 106 cells) were transiently introduced with 1 μm siRNA using the NEPA21 electroporator. After 2 days, cells were dissociated with Cellstripper (Cellgro), and the cells collected (1 × 106 cells) were transfected with the plasmids (pcDNA3.1/CD4, pCDNA6.2/CCR5, or pcDNA3.1/CXCR4, pLTR-LucE, and pRL-SV40) as described above. These cells were counted 24 h post-transfection and assayed for Sms2 levels by Western blotting, cell-surface SM levels, and the cell-cell fusion assay.
gp140 Binding Assay
Three gp120-gp41 heterodimers combine in a trimer to form the mature HIV-1 Env, which mediates binding to the HIV-1 receptor/co-receptors. The soluble gp140 trimer is produced by modifications in the gp120-gp41 cleavage site and introduction of the GCN4 trimeric motif (27). The plasmid (3 μg) of YUgp140 (−/GCN4)/pcDNA3.1(−), which encoded soluble gp140 with a His6 tag at the C terminus, was transfected into HEK293 cells using Lipofectamine 2000 as described previously (27).
In the negative control study, the empty vector was transfected using similar procedures. The cell culture supernatants were collected after 2 days, passed through a 0.45-μm membrane filter, and used as the gp140 solution. The target cells (1.5 × 105, CD4−/CCR5-expressing ZS, ZS/Sms1, and ZS/Sms2 cells) were incubated with gp140 solution for 1 h at 4 °C. After washing three times with 1% FBS in PBS, the cells were incubated with the anti-His antibody (1:500) for 1 h at 4 °C, followed by incubation with the anti-mouse Ig-PE (multiple adsorption) secondary antibody (1:1000) for 1 h at 4 °C. After washing three times with 1% FBS in PBS, the amount of gp140 that bound to the cell surface was quantified by FACS Aria. Nonspecific binding was observed using the culture supernatants of empty vector-transfected cells.
Co-immunoprecipitation Analysis of HIV-1 Receptor, Its Co-receptor, and SMS Isoforms
Co-immunoprecipitation was performed as described previously (28) with minor modifications. COS7 cells were co-transfected with plasmids (pCMV-3Tag-8/CD4-HA and pCMV-3Tag-8/CCR5–3×FLAG or CXCR4–3×FLAG and pCMV-3Tag-8/SMS1-V5 or SMS2-V5), or HEK cells stably expressing SMS2-V5 were transfected with plasmids (pCMV-3Tag-8/CD4-HA and/or pCMV-3Tag-8/CCR5–3×FLAG) using Lipofectamine 2000. Cells were incubated 24 h post-transfection with HIV-1 EnvJRFL- or EnvNL4-3-expressing HEK293 cells at 37 °C for 10 min. After washing with PBS three times, the cells were incubated on ice with lysis buffer containing 50 mm Tris-HCl, pH 8.0, CompleteTM protease inhibitor (Roche Applied Science), 150 mm NaCl, and 1% CHAPS. The lysate was incubated with anti-FLAG M2 or anti-HA beads at 4 °C for 4 h. The beads were then washed four times with lysis buffer and eluted with sample buffer. Co-immunoprecipitated proteins were immunoblotted with specific antibodies, such as anti-FLAG, anti-HA, or anti-V5 antibodies. Signals were detected with the Western BLoT Quant HRP substrate (Takara, Japan) and analyzed using the Light-capture II System and CS Analyzer 3.0 software (ATTO, Japan).
Immunocytochemistry and Fluorescent Microscopy
COS7 cells were co-transfected with plasmids (pCMV-3Tag-8/CD4-HA, pCMV-3Tag-8/CCR5–3×FLAG, or CXCR4–3×FLAG and pCMV-3Tag-8/SMS1-V5 or SMS2-V5) using Lipofectamine 2000. Cells were cultured on glass-bottom dishes 24 h post-transfection (Matsunami Glass, Japan) and then fixed with 3% paraformaldehyde in PBS for 15 min. After being rinsed with 50 mm NH4Cl in PBS, cells were permeabilized with 0.1% Triton X-100 in PBS. After being treated with blocking buffer (2% FBS in PBS) for 15 min, the samples were incubated with anti-HA, anti-FLAG, and anti-V5 antibodies diluted 1:2000 with blocking buffer at 4 °C overnight followed by incubation with AlexaFluor-conjugated secondary antibodies diluted 1:1000 with blocking buffer at room temperature for 2 h. Cells were observed with the confocal laser-scanning fluorescent microscope, FV10i (Olympus, Japan).
Analysis of F-actin Formation in Cell-Cell Contacts
F-actin analysis of conjugates between target cells and effector cells was performed as described previously (29) with minor modifications. ZS, ZS/Sms1, and ZS/Sms2 cells expressing CD4/CCR5 were stained with CellTrackerTM Blue CMAC according to the manufacturer's instructions. Target cells were incubated with COS7 cells that expressed HIV-1 EnvJRFL and EGFP at 37 °C for 10 min on poly-l-lysine-coated glass-bottom dishes. Cells were fixed with paraformaldehyde, permeabilized, and stained with phalloidin-AlexaFluor 546. Cells were observed with the confocal laser-scanning fluorescent microscope, FV10i, and the fluorescent signal of phalloidin was measured in plasma membrane. The region of interest in target cells was defined as the contacting and noncontacting regions between effector cells in the plasma membrane. F-actin polymerization was quantified as the ratio between the mean fluorescence intensity per area in the region of interest from noncontacting to contacting regions.
Statistical Analysis
All analyses were performed with GraphPad PRISM 6 (GraphPad Software). Statistical comparisons were performed using a one-way ANOVA and Tukey-Kramer multiple comparison test; p < 0.05 was considered significant.
RESULTS
Evaluation of HIV-1 Env-mediated Membrane Fusion by Cell-Cell Fusion Assay with HIV-1 Mimetics
The cell-cell fusion assay was conducted using effector cells (HIV-1 mimetics) and target cells to evaluate the effects of membrane lipids and their metabolizing enzymes in HIV-1 Env-mediated membrane fusion. The effector cells were prepared by the expression of the viral envelope protein Env (gp120 and gp41 subunits) in HEK293 cells as follows: EnvJRFL for CCR5-tropic HIV-1 mimetics and EnvNL4-3 for CXCR4-tropic HIV-1 mimetics. These HIV-1 mimetics also possessed the Tat gene, and the target cells had trans-activating response element-dependent luciferase. The Tat protein was introduced by the fusion of HIV-1 mimetics with the target cells, and luciferase was subsequently induced. This membrane fusion was entirely dependent on the correct combination of Env tropism as well as type of CD4 and co-receptor (see below). Using this assay system, we first examined the effects of various acyltransferases and phospholipases in HIV-1 Env-mediated membrane fusion. However, the overexpression or knockdown of these lipid-metabolism enzymes (membrane-bound O-acyltransferases, 1-acylglycerol-3-phosphate O-acyltransferases, and phospholipase A1) did not significantly affect the membrane fusion susceptibility (data not shown).
SMS Expression and Activity in T Cell Lines
We then attempted to examine the effects of SM and its synthesizing enzymes SMS1 and SMS2 on HIV-1 Env-mediated membrane fusion. Before the experiments were conducted, we confirmed the expression of SMS isoforms in Jurkat and Molt4 cells as human CD4+ T cells that are the target cells for HIV-1. As shown in Fig. 1A, the expression of SMS1 and SMS2 mRNA was observed in these T cell lines by semi-quantitative RT-PCR analysis. Real time RT-PCR showed that the major isoform of SMS was SMS1 in these cells (data not shown). In addition, SMS activities were detected in the cell lysates using C6-NBD-ceramide as the substrate (Fig. 1B). By contrast, ZS cells isolated from Sms1 and Sms2 double-deficient mice did not have SMS activity.
FIGURE 1.
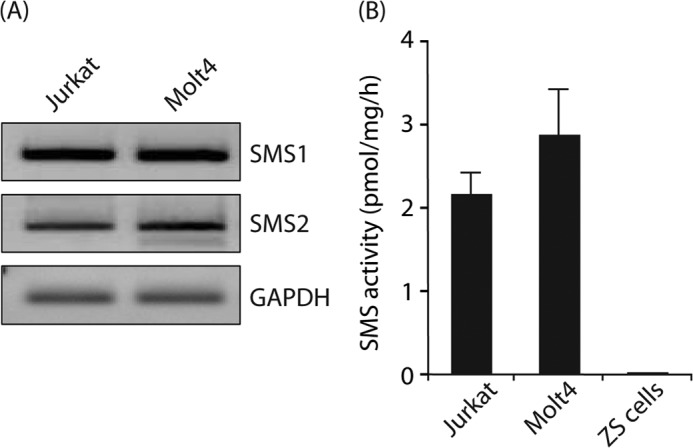
SMS expression and activity in the CD4+ T cell line. A, mRNA levels of SMS1, SMS2, and GAPDH in Jurkat and Molt4 cells were detected by semi-quantitative RT-PCR, as described under “Experimental Procedures.” One representative experiment is shown, and similar results were obtained in three independent experiments. B, SMS activities in Jurkat cells, Molt4 cells, and ZS cells were determined using C6-NBD-ceramide as a substrate. Reaction mixtures containing cell lysates (50 μg of protein/lane) were incubated at 37 °C for 1 h. Values represent the mean ± S.D. from three independent experiments.
Ectopic Expression of Sms1 or Sms2 in Mouse Embryonic Fibroblasts from Sms1/Sms2 Double Knock-out Mice
To examine the roles of SMS1 and SMS2 in HIV-1 Env-mediated membrane fusion, we employed reconstituted cells as the target cells that express V5-tagged Sms1 or Sms2 stably in Sms-deficient cells, thereby excluding the effects of endogenous Sms1 and Sms2. The isolated Sms1- and Sms2-expressing cells were termed ZS/Sms1 and ZS/Sms2 cells, respectively.
First, the expression of Sms was confirmed by Western blot analysis with the anti-V5 antibody. As shown in Fig. 2A, no significant differences were observed in Sms protein levels between ZS/Sms1 and ZS/Sms2 cells based on the intensity of the Sms bands. SM levels on the cell surface were then determined by cell mortality analysis with toxic lysenin and FACS analysis with nontoxic EGFP-lysenin staining. Lysenin is a toxic protein that is derived from earthworms and binds specifically to SM-rich domains. As shown in Fig. 2B, ZS/Sms1 and ZS/Sms2 cells were highly sensitive to the lysenin treatment; however, the same treatment did not affect the parent ZS cells. The half-maximal cytotoxic concentrations (CC50) were 0.14 ± 0.05 μg/ml for ZS/Sms1 cells and 0.05 ± 0.01 μg/ml for ZS/Sms2 cells. Similar results were observed in the FACS analysis using nontoxic EGFP-lysenin (Fig. 2C). The filled histograms show that EGFP-lysenin-staining cells shifted more to the right in ZS/Sms1 and ZS/Sms2 cells than in parent ZS cells. Collectively, these results demonstrate that Sms1 or Sms2 were functionally active in ZS cells and the enhancement in SMS activity increased the amounts of cell surface SM in ZS/Sms1 and ZS/Sms2 cells. Despite the deficiency in SM synthesis in ZS cells, significant amounts of SM were present on the cell surface in ZS cells (Fig. 2C). Cell surface SM in ZS cells may have been derived from serum in the culture medium.
FIGURE 2.

Levels of Sms expression and cell-surface SM in Sms-reconstituted cells. A, cell lysates (22 μg of protein/lane) from SM-deficient mouse embryonic fibroblasts (ZS cells) and Sms1- or Sms2-expressing ZS cells (ZS/Sms1 and ZS/Sms2 cells, respectively) were subjected to Western blotting with the anti-V5 antibody to confirm their expression levels. Western blotting was also performed with the anti-GAPDH antibody for the loading control. IB, immunoblot. B, amount of SM on the cell surface was determined by the sensitivity of lysenin. Cell numbers were determined by the WST-1 cell proliferation reagent after ZS (filled square), ZS/Sms1 (open triangle), and ZS/Sms2 cells (filled circle) were cultivated with various amounts of lysenin (0–10 μg/ml). Values represent the mean ± S.D. from three independent experiments. C, amount of SM on the cell surface was also determined using a probe for SM (EGFP-lysenin). Each cell was stained with 3 μg/ml EGFP-lysenin and analyzed by flow cytometry. Ten thousand cells were measured in each sample. The filled histogram represented cells that bound EGFP-lysenin. In contrast, the open histogram indicates cells without staining for EGFP-lysenin. One representative experiment is shown, and similar results were obtained in three independent experiments.
Sms2 Increases HIV-1 Env-mediated Membrane Fusion Susceptibility
We examined the effects of SMS1 and SMS2 on HIV-1 Env-mediated membrane fusion. The target cells were prepared by the expression of CD4/CCR5 or CD4/CXCR4 in the three types of cells described above: ZS, ZS/Sms1, and ZS/Sms2 cells. After each HIV-1 mimetic was incubated with the corresponding target cells, susceptibility to membrane fusion was determined by measuring luciferase activity (Fig. 3A).
FIGURE 3.

Sms2 augmented HIV-1 Env-mediated cell-cell fusion susceptibility. A, HIV-1 Env-mediated cell-cell fusion assay was performed using Sms1- or Sms2-reconstituted cells as the target cells. The target cells (ZS, ZS/Sms1, and ZS/Sms2 cells) that expressed CD4/CCR5 and the LTR-luciferase gene were cultured for 4 h with the effector HEK293 cells that expressed EnvJRFL (CCR5-tropic) and the HIV-1 Tat gene (left panel). The target cells that expressed CD4/CXCR4 and the reporter gene were also cultured for 4 h with other effector HEK293 cells that expressed EnvNL4-3 (CXCR4-tropic) and the HIV-1 Tat gene (right panel). Fusion susceptibility was determined from luciferase activity using the reporter gene activation assay. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01; NS, not significant. B, VSV-G Env-mediated cell-cell fusion assay was performed. The target cells (ZS, ZS/Sms1, and ZS/Sms2 cells) that expressed the LTR-luciferase gene were cultured for 4 h with the effector HEK293 cells that expressed VSV-G Env and the HIV-1 Tat gene. Fusion susceptibility was determined from luciferase activity using the reporter gene activation assay. Values are represented as the mean ± S.D. from three independent experiments. C, cell-surface expression levels of CD4/CCR5 or CD4/CXCR4 in each target cell (ZS, ZS/Sms1, and ZS/Sms2 cells) were analyzed by flow cytometry using each specific antibody (filled histogram). Open histograms indicate the isotype control. Ten thousand cells were measured in each sample. D, values represent the mean ± S.D. from three independent experiments in C.
When the CCR5-tropic HIV-1 mimetic (EnvJRFL-expressing HEK293 cells) was incubated with CD4/CXCR5-expressing ZS cells, a small amount of luciferase activity was induced (Fig. 3A, left panel). The same experiment employing Sms2-expressing ZS cells revealed that the expression of Sms2 augmented the induction of luciferase activity (∼5-fold higher than that in Sms-deficient ZS cells). These results clearly indicate that SMS2 was involved in HIV-1 Env-mediated membrane fusion. In contrast, luciferase activity was not augmented by Sms1 expression, indicating that the expression of Sms1 did not increase the susceptibility of cells to membrane fusion. Similar results were also observed in the combination of the CXCR4-tropic HIV-1 mimetic (EnvNL4-3-expressing HEK293 cells) and CD4/CXCR4-expressing target cells; hence, Sms2, but not Sms1, augmented membrane fusion susceptibility (Fig. 3A, right panel). We further examined the membrane fusion susceptibility using VSV-G-expressing HEK293 cells. There were no differences in membrane fusion susceptibility between ZS, ZS/Sms1, and ZS/Sms2 cells incubated with effector cells expressing VSV-G (Fig. 3B). Although the VSV-G protein is known to be a low pH-activated viral fusion protein (30), we employed a cell-cell fusion assay using VSV-G in the normal medium, not acidic condition, to prevent acidic damage to the cells. The increase of luciferase activity in effector cells expressing Tat and VSV-G was observed compared with effector cells expressing only Tat and not VSV-G, due presumably to either gradual acidification of the medium (31) or the high sensitivity of this cell-cell fusion assay. These results indicate that the augmentation of membrane fusion susceptibility by SMS2 was specific to HIV-1 Env-mediated membrane fusion.
Because membrane fusion was strictly dependent on the cell-surface levels of the HIV-1 receptor and its co-receptors (32), the effects of SMS isoforms on the expression of CD4, CCR5, and CXCR4 in the target cells were determined. No significant differences were observed in the expression levels of CD4, CCR5, and CXCR4 on the cell surface between ZS, ZS/Sms1, and ZS/Sms2 cells (Fig. 3, C and D). These results clearly indicate that the increase of membrane fusion susceptibility in ZS/Sms2 was not attributed to the expression levels of the HIV receptor or its co-receptors on the cell surface.
Sms2 Silencing Inhibited HIV-1 Env-mediated Membrane Fusion
To confirm the roles of SMS2 in HIV-1 Env-mediated membrane fusion, we further examined the effects of Sms2 knockdown. Two stealth siRNAs, siSms2-1 and siSms2-2, for different target sequences were shown to be potent and specific for Sms2 silencing; the transfection of siSms2-1 and siSms2-2 in ZS/Sms2 cells each reduced Sms2 protein levels but did not affect the housekeeping gene product GAPDH (Fig. 4A). The stealth siRNAs for Sms2 also reduced SM levels on the cell surface of ZS/Sms2 cells (Fig. 4B). Furthermore, HIV-1 Env-mediated membrane fusion susceptibility was ∼60% lower with the stealth siRNAs for Sms2 than with negative control siRNA (Fig. 4E) without affecting the cell-surface expression levels of CD4, CCR5, and CXCR4 (Fig. 4, C and D). These results again demonstrated that SMS2 was involved in HIV-1 Env-mediated membrane fusion.
FIGURE 4.

Interference of Sms2 in ZS/Sms2 cells decreased HIV-1 Env-mediated cell-cell fusion susceptibility. StealthTM control siRNA, siSms2-1, or siSms2-2 were transfected into ZS/Sms2 cells by electroporation. After 2 days, the expression vectors of CD4/CCR5 or CD4/CXCR4 were transfected into these cells. These cells were harvested 24 h post-transfection and examined for Sms2 levels by Western blotting, cell-surface SM levels, and cell-cell fusion assay. A, Sms2 knockdown was assessed by Western blot analysis with the anti-V5 antibody in total cell lysates (5 μg of protein/lane). The same blots were also probed with an anti-GAPDH antibody for the loading control. One representative experiment is shown, and similar results were obtained in three independent experiments. IB, immunoblot. B, cell-surface SM levels of Sms2 knockdown cells were determined by flow cytometry after EGFP-lysenin staining. Ten thousand cells were measured in each sample. C and D, cell-surface expression levels of CD4/CCR5 or CD4/CXCR4 in Sms2 knockdown cells were analyzed by flow cytometry using each specific antibody (filled histograms). Open histograms indicate the isotype control. Ten thousand cells were measured in each sample. E, Sms2 knockdown cells that expressed the CD4/CCR5/reporter gene or CD4/CXCR4/reporter gene were cultured for 4 h with HEK293 cells that expressed HIV-1 Tat and EnvJRFL- or EnvNL4-3. Fusion susceptibility was determined by the amount of luciferase activity using the reporter gene activation assay. The values were expressed as the percentage of Env-induced fusion using control siRNA-transfected cells as the reference value. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01.
Catalytically Nonactive Sms2 Also Increases HIV-1 Env-mediated Membrane Fusion Susceptibility
SMS1 could not augment membrane fusion, even though its expression increased SM on the cell surface (Figs. 2 and 3). Furthermore, no correlation was observed between fusion susceptibility and cell surface SM (Figs. 2–4). These results prompted us to question whether the SMS2 protein itself, but not SM produced by SMS2, played an important role in HIV-1 Env-mediated membrane fusion. Therefore, we examined the effects of the SMS2 mutant on HIV-1 Env-mediated membrane fusion.
Because the His-229 amino residue in SMS2 is known to be responsible for SMS activity (33), we employed catalytically inactive Sms2-expressing ZS cells. Western blot analysis revealed no significant differences in Sms protein levels between Sms2 and Sms2-H229A cells (Fig. 5A). However, the assay for SMS activity using a fluorescent-labeled substrate (C6-NBD-ceramide) indicated that the mutant Sms2 completely abolished SMS activity (Fig. 5B). The loss of SMS activity was also demonstrated by the lower SM levels on the cell surface of ZS/Sms2-H229A cells than on ZS/Sms2 cells (Fig. 5C). More importantly, the HIV-1 Env-mediated membrane fusion susceptibility of ZS/Sms2-H229A cells was similar to that of ZS/Sms2 cells (Fig. 5E), and no significant difference was noted in cell-surface CD4, CCR5, and CXCR4 expression levels (Fig. 5D). These results clearly indicate that the SMS2 protein itself, but not SM generated by SMS2, is involved in the augmentation of HIV-1 Env-mediated membrane fusion.
FIGURE 5.

Catalytic nonactive Sms2 also augmented HIV-1 Env-mediated cell-cell fusion susceptibility. Sms2 and its mutant (Sms2-H229A) were expressed in ZS cells by a retroviral expression system. To be used as target cells, CD4 and its corresponding co-receptor were also expressed in these cells. A, cell lysates (11 μg of protein/lane) from each cell were subjected to Western blotting with the anti-V5 antibody to evaluate the expression of Sms2 and Sms2-H229A. The same blots were also probed with an anti-GAPDH antibody for the loading control. IB, immunoblot. B, SMS activities in the lysates from each cell were determined using C6-NBD-ceramide as a substrate. Reaction mixtures containing cell lysates (75 μg of protein) were incubated at 37 °C for 1 h. The reaction products were applied to the TLC plate, which was developed with chloroform/methanol/water (65:25:4, v/v/v), followed by visualization with a fluorescence imaging analyzer. One representative experiment is shown, and similar results were observed in three independent experiments. C, cell-surface SM levels of each cell were assessed by flow cytometry with EGFP-lysenin staining. Ten thousand cells were analyzed in each sample. NS, not significant. D, CD4/CCR5 or CD4/CXCR4 cell-surface expression levels in each cell were analyzed by flow cytometry using each specific antibody (filled histogram). Open histograms indicate the isotype control. Ten thousand cells were measured in each sample. MIF, mean intensity of fluorescence. E, HIV-1 Env-mediated cell-cell fusion susceptibility was determined employing Sms2 and Sms2-H229A-expressing ZS cells as target cells. Fusion susceptibility was determined with luciferase activity using the reporter gene activation assay. Data are expressed as the percentage of Env-induced fusion of ZS cells as the reference value. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01.
Sms2 Was Not an Entry Receptor and Could Not Facilitate HIV-1 Env Attachment
We then investigated the mechanism by which SMS2 promoted HIV-1 Env-mediated membrane fusion. Based on the results that membrane fusion could occur even in Sms-null ZS cells (Fig. 3A), SMS2 appeared to be a promoting, but not essential, factor for membrane fusion susceptibility. Although the entry of HIV-1 occurs via the binding of HIV-1 Env to CD4 and its co-receptors, HIV-1 is also known to enter target cells by other pathways using the mannose receptor for CD4-independent infection in astrocytes (34). To investigate whether HIV-1 Env-mediated membrane fusion in ZS/Sms2 cells occurred via the CD4/co-receptor-dependent pathway, we examined the requirement of CD4 and its co-receptors in this cell-cell fusion assay. Target cells were prepared by the expression of various combinations of CD4/CCR5/CXCR4 in ZS/Sms2 cells. As shown in Fig. 6A, HIV-1 Env-mediated fusion only occurred in CD4- and co-receptor-expressing ZS/Sms2 cells. However, membrane fusion hardly occurred in the absence of any CD4 or co-receptors and in the wrong combination of co-receptor and HIV-1 Env-tropism.
FIGURE 6.
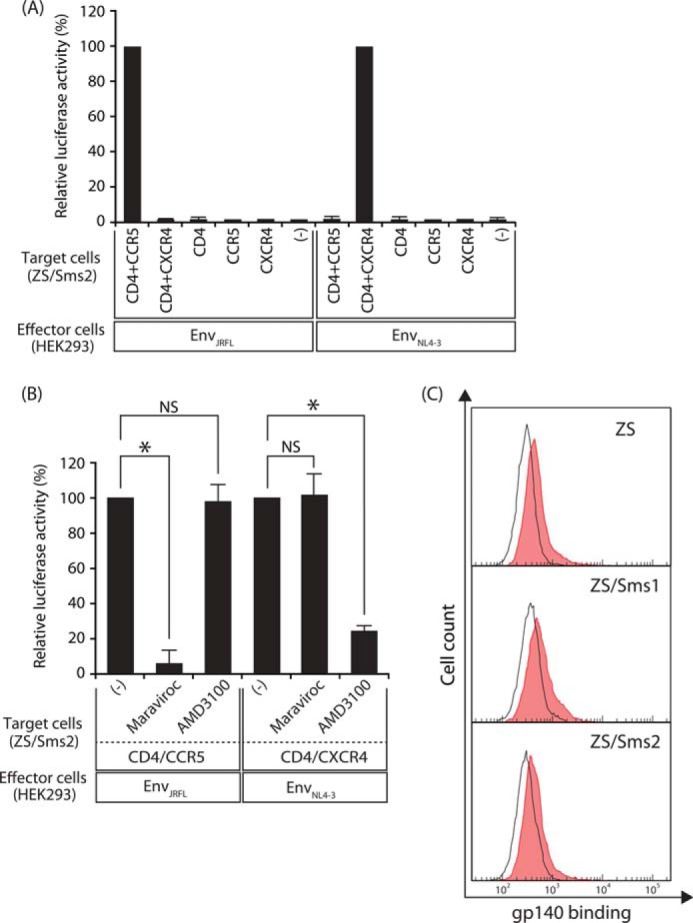
Augmentation of membrane fusion by Sms2 was dependent on CD4/co-receptors, and Sms2 itself was not an entry receptor. A, cell-cell fusion assay was conducted by employing ZS/Sms2 cells that expressed in various combinations of CD4/CCR5/CXCR4 as indicated. HIV-1 EnvJRFL (CCR5-tropic)-expressing or EnvNL4-3 (CXCR4-tropic)-expressing HEK293 cells were cultured with the indicated target cells. Fusion susceptibility was determined with luciferase activity using the reporter gene activation assay. B, effects of co-receptor antagonists on HIV-1 Env-mediated cell-cell fusion in ZS/Sms2 cells. ZS/Sms2 cells that expressed CD4 and co-receptor (CCR5 or CXCR4) were incubated with medium containing 50 nm maraviroc (CCR5 antagonist) or 100 nm AMD3100 (CXCR4 antagonist) for 30 min, and cells were then harvested. The cell-cell fusion assay was conducted by incubating antagonist-treated target cells with the corresponding HIV-1 mimetics (HEK293 cells that expressed EnvJRFL or EnvNL4-3). Fusion susceptibility was determined with luciferase activity using the reporter gene activation assay. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01; NS, not significant. C, binding of HIV-1 Env to target cells was determined using the soluble Env subunit gp140 with the C-terminal His6 tag. The target cells (ZS, ZS/Sms1, and ZS/Sms2) that expressed CD4/CCR5 were incubated with gp140 solution for 1 h at 4 °C. The binding of gp140 to the target cells was determined by flow cytometry after staining with the mouse anti-His6 antibody and goat anti-mouse Ig-PE (filled histogram). Open histograms indicate cells that were incubated with medium from the mock transfectant (absence of soluble gp140). Ten thousand cells were measured in each sample. One representative experiment is shown, and similar results were obtained in three independent experiments.
The effects of antagonists for each co-receptor were further examined. Maraviroc, an antagonist for CCR5, specifically inhibited the fusion of CCR5-tropic HIV-1 mimetics to CCR5-expressing target cells, whereas maraviroc could not inhibit the fusion of CXCR4-tropic HIV-1 mimetics to CXCR4-expressing target cells (Fig. 6B). Alternatively, AMD3100, an antagonist for CXCR4, specifically inhibited the fusion of CXCR4-tropic HIV-1 mimetics to CXCR4-expressing target cells, although AMD3100 could not inhibit the fusion of CCR5-tropic HIV-1 mimetics to CCR5-expressing target cells. These results indicate that HIV-1 Env-mediated membrane fusion in ZS/Sms2 cells occurred in a manner that was dependent on CD4 and its co-receptors.
We also examined the possibility that SMS2 could facilitate the attachment of HIV-1 to target cells, because other proteins, such as the gut-homing receptor integrin α4β7, were reported to be promoting factors for virion attachment to target cells but not as entry receptors for HIV-1 (35). Binding of the soluble gp140 protein, truncated at the Env transmembrane domain, was investigated to determine whether SMS2 acted as an entry receptor or promoting factor for HIV-1 binding. As shown in Fig. 6C, no significant difference was observed between the binding of gp140 to ZS, ZS/Sms1, and ZS/Sms2 cells. These results collectively indicate that SMS2 was not an entry receptor or promoting factor for HIV-1 Env to attach to target cells.
SMS2 Co-localized and Is Constitutively Associated with the HIV-1 Receptor·Co-receptor Complex in the Plasma Membrane
We next determined whether SMS2 is involved in the HIV-1 gp120-induced association of the HIV-1 receptor·co-receptor in the plasma membrane, which is a process directly related to efficient viral fusion (28). Therefore, the cellular localization of SMS2 and the HIV-1 receptor·co-receptor complex was examined by confocal microscopy. Immunofluorescence staining using confocal microscopy demonstrated that SMS2 was localized in the cell surface and concentrated structures in the perinuclear region, whereas SMS1 was only localized in the perinuclear region (Fig. 7). These locations were consistent with the findings of a previous study, in which SMS1 was localized in the Golgi complex only, and SMS2 was localized in both the plasma membrane and Golgi complex (16). Similar staining patterns revealed that the HIV-1 receptor CD4 and its co-receptors CCR5/CXCR4 were mainly localized to the plasma membrane as well as intracellular dot and reticular structures. The perinuclear regions of CD4/CCR5/CXCR4 were due to the intracellular pool during trafficking to the cell surface. Confocal images revealed that SMS2 was co-localized with CD4 and CCR5 or CXCR4 in the plasma membrane and Golgi complex, whereas SMS1 was co-localized with CD4 and CCR5 or CXCR4 in only the Golgi complex.
FIGURE 7.
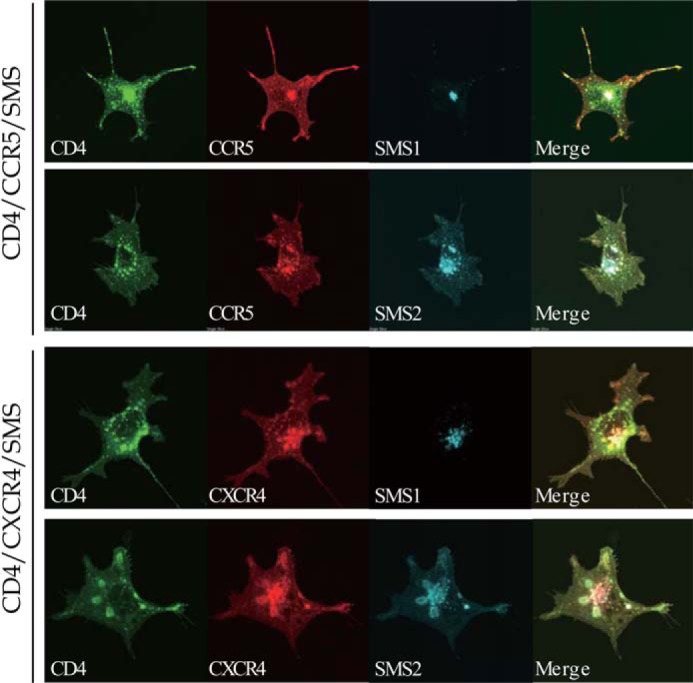
SMS2 co-localized with HIV-1 receptor and co-receptor in the plasma membrane. HA-tagged CD4, 3×FLAG-tagged CCR5 or CXCR4 and V5-tagged SMS1 or SMS2 were co-expressed in COS7 cells in several combinations. Subcellular localization of CD4, CCR5, or CXCR4 and SMS1 or SMS2 was examined by confocal laser microscopy after each protein was stained with the corresponding antibody. CD4, green; CCR5 or CXCR4, red; SMS1 or SMS2, blue. Confocal images suggested the partial co-localization of the SMS2 with the CD4·CCR5 or CD4·CXCR4 complex in the plasma membrane (white signal in Merge).
To further assess the association between the SMS2 and HIV-1 receptor·co-receptor complex, immunoprecipitation assays were performed. The target cells were co-incubated with HIV-1 Env-expressing cells prior to lysis and immunoprecipitation. The immunoprecipitation by the anti-FLAG beads precipitated the HIV-1 co-receptors FLAG-tagged CCR5/CXCR4 as well as the HIV-1 receptor HA-tagged CD4 (Fig. 8A). SMS2 was also precipitated when CCR5 or CXCR4 were immunoprecipitated, which indicated that SMS2 bound to the CD4·CCR5 or CD4·CXCR4 complex. Although another isoform, SMS1, also co-immunoprecipitated with CCR5 or CXCR4, the ratio of precipitated SMS2 to CCR5 or CXCR4 was markedly higher than those of SMS1 (Fig. 8, A and B). These results indicate that the binding affinity between SMS2 and the HIV-1 receptor·co-receptor complex was higher than SMS1. Immunoprecipitation revealed another important result. Fig. 8, A and B, shows that the association between HIV-1 receptor and co-receptor was unchanged in the presence or absence of SMS2, which indicates that SMS2 bound to the HIV-1 receptor·co-receptor complex, whereas the binding of SMS2 could not facilitate the association between HIV-1 receptor and co-receptor. More importantly, the association levels of SMS2·CD4·CCR5 were not significantly different, even in the absence of HIV-1 Env stimulation (Fig. 8C). Additionally, SMS2 could associate with CCR5 even in the absence of CD4, and SMS2 was also associated with CD4 in the absence of CCR5 expression (Fig. 8D). Collectively, these results demonstrate that SMS2 could be constitutively associated with the HIV-1 receptor and co-receptor, respectively.
FIGURE 8.

SMS2 constitutive association with HIV-1 receptor and co-receptor. A, HA-CD4-, 3×FLAG-CCR5-, or CXCR4- and V5-SMS1-, or SMS2-expressing COS7 cells were incubated for 10 min with HIV-1 EnvJRFL- or EnvNL4-3-expressing HEK293 cells at 37 °C. Cell lysates were prepared, and an immunoprecipitation assay was performed using anti-FLAG M2 beads. The precipitated proteins were analyzed by Western blotting using each antibody as described under “Experimental Procedures.” Upper panel, cell lysates; lower panel, immunoprecipitates. B, quantities of precipitated HA-CD4 or V5-SMS with the 3×FLAG co-receptor were represented as the ratio between the intensities of CD4 and HIV-1 co-receptor (upper panel), and SMS isoforms and HIV-1 co-receptor (lower panel), respectively, in the immunoprecipitation assay. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01; **, p < 0.05; IP, immunoprecipitation. C, HEK cells stably expressing SMS-V5 were transfected with plasmids (HA-CD4 and 3×FLAG-CCR5). Cells were incubated for 10 min with or without HIV-1 EnvJRFL-expressing HEK293 cells at 37 °C. Cell lysates were prepared, and an immunoprecipitation assay was performed using anti-FLAG M2 beads. Left panel, cell lysates; right panel, immunoprecipitates. D, HEK cells stably expressing SMS-V5 were transfected with or without plasmids (HA-CD4 and 3×FLAG-CCR5). Cells were incubated for 10 min with HIV-1 EnvJRFL-expressing HEK293 cells at 37 °C. Cell lysates were prepared, and an immunoprecipitation assay was performed using anti-FLAG M2 or anti-HA beads. Left panel, cell lysates; center panel, immunoprecipitates (anti-FLAG beads); right panel, immunoprecipitates (anti-HA beads). IB, immunoblot.
SMS2 Involvement in HIV-1 Env-induced Pyk2 Activation and F-actin Polymerization
To address the SMS2-dependent mechanism that augments membrane fusion susceptibility, we focused on Pyk2, the nonreceptor tyrosine kinase known to modulate HIV-1 gp120-induced migration of dendritic cells (36). To investigate the roles of Pyk2 signaling, we examined the effects of a pharmacologic Pyk2 inhibitor (36, 37), tyrphostin A9, on HIV-1 Env-mediated membrane fusion using ZS/Sms2 cells as target cells. The Pyk2 inhibitor-treated ZS/Sms2 cells significantly inhibited membrane fusion in a dose-dependent manner compared with vehicle-treated control (Fig. 9A). Nearly 70% inhibition of membrane fusion was observed in 10 μm tyrphostin A9 concentrations. Pyk2 is mainly expressed in hematopoietic cells, neuronal cells, and fibroblasts (38–40). We therefore examined the Pyk2 expression in ZS/Sms2 cells derived from mouse embryonic fibroblasts (target cells) and HEK293 cells (effector cells) by immunoblotting. As expected, the expression of Pyk2 was observed in ZS/Sms2 and not HEK293 cells (Fig. 9B); therefore, the alteration of Pyk2 signaling could be observed in only target cells. Pyk2 phosphorylation peaked at 5–15 min with HIV-1 gp120 stimulation in dendritic cells (34). We therefore decided to examine Pyk2 phosphorylation for 10 and 30 min of incubation with EnvJRFL-expressing HEK293 cells. As shown in Fig. 9C, we observed a transient but significant increase in Pyk2 phosphorylation in ZS/Sms2 and Sms2-H229A cells compared with ZS and ZS/Sms1 cells. For the loading control, the total Pyk2 protein concentration was examined in all samples but was not altered after HIV-1 Env treatment.
FIGURE 9.

SMS2 involvement in HIV-1 Env-mediated Pyk2 activation. A, ZS/Sms2 cells that expressed CD4/CCR5 were incubated with medium including 0.1, 1, or 10 μm tyrphostin A9 in 0.05% DMSO for 30 min, and the cells were harvested. The cell-cell fusion assay was performed by incubating tyrphostin A9-treated target cells with HIV-1 EnvJRFL-expressing HEK293 cells. Fusion susceptibility was determined with luciferase activity using the reporter gene activation assay. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01. B, cell lysates (15 μg of protein/lane) from HEK293 and ZS/Sms2 cells were subjected to Western blotting with the anti-Pyk2 antibody. Western blotting was also performed with the anti-GAPDH antibody for the loading control. IB, immunoblot. C, ZS, ZS/Sms1, and ZS/Sms2 cells that expressed CD4/CCR5 were incubated for 0, 10, or 30 min with HIV-1 EnvJRFL-expressing HEK293 cells. The lysates were analyzed by Western blotting with anti-Pyk2 (upper panel), anti-phosphoryl Pyk2, and Tyr-402 (lower panel) antibodies. For quantitative analysis of protein phosphorylation, the ratio of phosphorylation versus total protein in each lane was obtained by densitometry. The phosphorylation index was determined by calculating the value of this ratio in each lane and presenting the ratio as the fold increase over the control value (unstimulated sample, 1). Values represent the mean ± S.D. from three independent experiments. **, p < 0.05.
Next, we examined whether SMS2 was involved in F-actin polymerization. Pyk2 signaling promotes the activation of the F-actin-binding protein, leukocyte-specific protein 1, which then associates with actin, leading to actin polymerization and chemotaxis in dendritic cells (36). Furthermore, actin polymerization was previously shown to play an essential role in the fusion process during HIV-1 infection, which could act as a driving force for membrane fusion (41). The pretreatment of peripheral blood mononuclear cells with cytochalasin D, a specific inhibitor of actin polymerization, inhibited the entry of HIV-1 (42). To determine the roles of F-actin polymerization, we examined the effects of cytochalasin D on HIV-1 Env-mediated membrane fusion using ZS/Sms2 cells as target cells. As shown in Fig. 10A, cytochalasin D significantly inhibited membrane fusion in a dose-dependent manner.
FIGURE 10.

Actin polymerization was involved in HIV-1 Env-mediated membrane fusion. A, ZS/Sms2 cells that expressed CD4/CCR5 or CD4/CXCR4 were incubated with medium, including 0.25 or 1 μm cytochalasin D in 0.05% DMSO for 1 h, and cells were harvested. The cell-cell fusion assay was performed by incubating cytochalasin D-treated target cells with the corresponding HIV-1 mimetics (HIV-1 EnvJRFL- or EnvNL4-3-expressing HEK293 cells). Fusion susceptibility was determined with luciferase activity using the reporter gene activation assay. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01; **, p < 0.05. B, actin polymerization was augmented by SMS2 at cell-cell contact sites between the effector and target cells. Three types of target cells (CD4- and CCR5-expressing ZS, ZS/Sms1, and ZS/Sms2 cells) were labeled with CMAC (blue) and incubated for 10 min with HIV-1 EnvJRFL- and EGFP-expressing COS7 cells (green). These cells were observed by confocal microscopy after staining with phalloidin-AlexaFluor 546 (red). The arrowheads indicate heterotypic cell contacts. The arrows indicate ZS/Sms2 cells that contacted target cells and were not in a contact. DIC, differential interference contrast. C, F-actin polymerization was quantified as the ratio between the signals from noncontacting to contacting regions at plasma membrane in target cells. Values represent the mean ± S.D. for ZS (n = 87), ZS/Sms1 (n = 89), and ZS/Sms2 (n = 77) cells. *, p < 0.01.
We next examined actin polymerization during HIV-1 EnvJRFL-mediated membrane fusion. Staining with phalloidin-AlexaFluor 546, a probe for F-actin, indicated that actin polymerization was observed in the contact site of HIV-1 mimetics and target cells (Fig. 10B). Actin polymerization was more prominent in the contact site when ZS/Sms2 cells, compared with ZS or ZS/Sms1 cells, were the target cells. The staining intensity of the contact area in ZS/Sms2 cells was ∼1.7-fold higher than that in ZS and ZS/Sms1 cells (Fig. 10, B and C). However, we did not observe significant accumulation of F-actin in ZS/Sms2 cells that contacted target cells or were not in contact (indicated by an arrow in Fig. 10B). Based on these results, we propose that SMS2 may be able to promote actin remodeling via Pyk2 activation, which implies a novel function of SMS2 in membrane fusion.
DISCUSSION
The HIV-1 envelope is composed of phospholipid bilayers derived from the membrane of host cells. Therefore, membrane fusion between the HIV-1 envelope and plasma membranes of target cells is essential for HIV-1 entry. However, very little is known about the role of lipid-metabolizing enzymes in membrane fusion. To examine the roles of lipids and their lipid-metabolizing enzymes, we employed the cell-cell fusion assay using HIV-1 mimetics and lipid-manipulating target cells. In the target cells, the levels of Sms expression and cell surface SM are manipulated by the overexpression and silencing of Sms1 and Sms2 in Sms-deficient cells.
In this study, we found that SMS2, not SMS1, is involved in HIV-1 Env-mediated membrane fusion. The difference of membrane fusion susceptibility between Sms1- and Sms2-expressing cells cannot be attributed to the differences of SM generated by Sms1 and Sms2, because the lipid molecular species of SM in Sms1- and Sms2-expressing ZS cells is similar (43). However, the cellular localizations of SMS1 and SMS2 are different; SMS1 is mainly localized in the Golgi apparatus, whereas SMS2 is localized in both the Golgi apparatus and plasma membrane. Therefore, we hypothesize that the location of SMS2 in the plasma membrane is a critical determinant for augmenting membrane fusion.
We also found no correlation between membrane fusion susceptibility and cell-surface SM levels. Although cell-surface SM levels in ZS/Sms1 cells are higher than those in Sms-deficient ZS cells, these SM levels do not augment membrane fusion. Furthermore, cell-surface SM levels in ZS/Sms2-H229A cells are significantly lower than those in ZS/Sms2 cells, although membrane fusion susceptibility is similar between these cells. However, our results do not completely reject that SM is involved in HIV-1 Env-mediated membrane fusion, because a significant amount of SM is observed on the cell surface of Sms-deficient ZS cells, which might be derived from serum in the culture medium. We presume that this level of SM is enough for membrane fusion, although it cannot form SM-rich domains with which to bind and form pores with lysenin. Our results demonstrate that the enhancement observed in fusion by SMS2 could contribute to SM-dependent membrane fusion. Tafesse et al. (44) reported that the knockdown of either SMS1 or SMS2 reduces SM content and blocks cell growth. Because the addition of exogenous SM does not restore cell growth, the authors suggested that the biological role of SMS may extend beyond formation of SM. Our findings provide support for this idea and further reveal the role of SMS2 in membrane fusion.
In our study, we observed that SMS2 co-localizes and is constitutively associated with the HIV receptor·co-receptor complex in the plasma membrane and with them. However, no significant difference was observed in the association efficiency between the HIV-1 receptor and its co-receptor in the presence or absence of SMS2. These results indicate that SMS2 could be associated with, but not promote, HIV-1 receptor·co-receptor clustering. Furthermore, the association levels of SMS2/CD4/CCR5 are not significantly different with and without HIV-1 Env stimulation. This result is consistent with the constitutive association between CD4 and CCR5 that was detected by co-immunoprecipitation (45). We need to use a sequential BRET/FRET technique to more directly investigate the association of this heterotrimerization (46).
We also found that HIV-1 Env induces Pyk2 phosphorylation at tyrosine residue 402 in Sms2-expressing and catalytically nonactive Sms2-expressing cells, with a peak at 10 min after HIV-1 EnvJRFL-expressing HEK293. Tyr-402 has been known to be the Pyk2 autophosphorylation site and binds to the Src homology 2 domain of Src family protein tyrosine kinases (47). This binding permits the subsequent phosphorylation of other tyrosine residues on Pyk2, leading to an increase in Pyk2 activity (47). Tyrphostin A9 has emerged as the most potent and selective Pyk2 inhibitor among the 51 tyrosine kinase inhibitors tested in the TNF-induced neutrophils (37), and this pharmacologic inhibitor is an effective tool for investigating the role of Pyk2 signaling in HIV-1 Env-mediated membrane fusion. In this study, tyrphostin A9-treated ZS/Sms2 cells significantly inhibited membrane fusion in a dose-dependent manner compared with vehicle-treated control. These results indicate that Pyk2 signaling is necessary for efficient membrane fusion in ZS/Sms2 cells.
Furthermore, we investigated the possibility that SMS2 may regulate the cytoskeleton, which acts as a driving force for membrane fusion. We observed significant inhibition by cytochalasin D, a specific inhibitor of actin polymerization, in HIV-1 Env-mediated membrane fusion. The intensity of actin polymerization in the contact area in ZS/Sms2 cells was higher than those in ZS and ZS/Sms1 cells after 10 min of incubation with HIV-1 EnvJRFL-expressing HEK293 cells. There was no significant increase of F-actin polymerization in ZS/Sms2 cells after 30 min of incubation (data not shown), indicating transient activation of actin polymerization by HIV-1 Env treatment. The time course of actin polymerization was in agreement with that of Pyk2 phosphorylation. These results demonstrate that F-actin polymerization in ZS/Sms2 cells is mediated by Pyk2 signaling.
The role of F-actin polymerization induced by HIV-1 Env remains to be elucidated. The most appealing hypothesis is that it is required to assemble high concentrations of HIV-1 receptor·co-receptor at the plasma membrane to promote viral binding and entry (41). Because it is difficult to express high levels of HIV-1 receptor/co-receptor in ZS cells, we did not observe the CD4/co-receptor capping in detail by confocal microscopy.
The cell-cell fusion assay should be used with other lipid-metabolizing enzymes. We examined the effects of various membrane-bound O-acyltransferases, 1-acylglycerol-3-phosphate O-acyltransferases, and phospholipase A1 in HIV-1 Env-mediated membrane fusion. However, the overexpression or knockdown of these enzymes in HEK293 or COS7 cells did not significantly affect membrane fusion susceptibility (data not shown). Because acyltransferases or phospholipases comprise many isoforms and are redundant in substrates (48), we hypothesized that other isoforms or related proteins, which can act on the same substrate, may be activated/inactivated to maintain lipid homeostasis within the cells. Target gene-manipulated cells, such as ZS/Sms1 and ZS/Sms2 cells in this study, need to be examined to understand the function of the target gene itself using this cell-cell fusion assay.
In summary, our study demonstrates that SMS2 is involved in HIV-1 Env-mediated membrane fusion, regardless of its enzyme activity. Moreover, we propose a model to explain that SMS can augment the membrane fusion susceptibility. SMS2 is associated with the HIV-1 receptor·co-receptor complex in the plasma membrane, which promotes HIV-1 receptor·co-receptor-mediated Pyk2 signaling in response to HIV-1 Env. Pyk2 signaling induces F-actin polymerization at the at the cell-cell contact sites, leading to consequent induction of membrane fusion. These novel findings further our understanding of the role of lipid-metabolic enzymes in membrane fusion.
Acknowledgments
We are grateful to Dr. T. Kobayashi (RIKEN, Japan) and Dr. H. Mitsuya (National Institutes of Health, Bethesda) for donating the EGFP-lysenin expression vector and plasmids, respectively, for the cell-cell fusion assay. We also thank Drs. K. Maeda (National Institutes of Health) and H. Nakata (Kumamoto University, Japan) for their valuable technical advice for the cell-cell fusion assay. HIV-1 (YU2) gp140 (−/GCN4) in pcDNA3.1 was obtained from Dr. Joseph Sodroski through the AIDS Reagent Program, Division of AIDS, NIAID, National Institutes of Health.
This work was supported in part by Grant-in-aid for Young Scientists (B) 25860055 (to Y. H.) and for Scientific Research (C) 24590095 (to A. Y.) from the Japan Society for the Promotion of Science and in part by Ministry of Education, Culture, Sports, and Technology-supported program for the Strategic Research Foundation at Private Universities.
- Env
- envelope protein
- SM
- sphingomyelin
- SMS
- sphingomyelin synthase
- EGFP
- enhanced GFP
- NBD
- 4-nitrobenz-2-oxa-1,3-diazole
- Tat
- trans-activator of transcription.
REFERENCES
- 1. Jahn R., Lang T., Südhof T. C. (2003) Membrane fusion. Cell 112, 519–533 [DOI] [PubMed] [Google Scholar]
- 2. Godlee C., Kaksonen M. (2013) From uncertain beginnings: initiation mechanisms of clathrin-mediated endocytosis. J. Cell Biol. 203, 717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lord C., Ferro-Novick S., Miller E. A. (2013) The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the Golgi. Cold Spring Harbor Perspect. Biol. 5, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Curtis I., Meldolesi J. (2012) Cell surface dynamics–how Rho GTPases orchestrate the interplay between the plasma membrane and the cortical cytoskeleton. J. Cell Sci. 125, 4435–4444 [DOI] [PubMed] [Google Scholar]
- 5. Chen Y. G., Siddhanta A., Austin C. D., Hammond S. M., Sung T. C., Frohman M. A., Morris A. J., Shields D. (1997) Phospholipase D stimulates release of nascent secretory vesicles from the trans-Golgi network. J. Cell Biol. 138, 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ha K. D., Clarke B. A., Brown W. J. (2012) Regulation of the Golgi complex by phospholipid remodeling enzymes. Biochim. Biophys. Acta 1821, 1078–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gallo S. A., Finnegan C. M., Viard M., Raviv Y., Dimitrov A., Rawat S. S., Puri A., Durell S., Blumenthal R. (2003) The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 1614, 36–50 [DOI] [PubMed] [Google Scholar]
- 8. Koot M., van 't Wout A. B., Kootstra N. A., de Goede R. E., Tersmette M., Schuitemaker H. (1996) Relation between changes in cellular load, evolution of viral phenotype, and the clonal composition of virus populations in the course of human immunodeficiency virus type 1 infection. J. Infect. Dis. 173, 349–354 [DOI] [PubMed] [Google Scholar]
- 9. Speck R. F., Wehrly K., Platt E. J., Atchison R. E., Charo I. F., Kabat D., Chesebro B., Goldsmith M. A. (1997) Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71, 7136–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vieira C. R., Munoz-Olaya J. M., Sot J., Jiménez-Baranda S., Izquierdo-Useros N., Abad J. L., Apellániz B., Delgado R., Martinez-Picado J., Alonso A., Casas J., Nieva J. L., Fabriás G., Mañes S., Goñi F. M. (2010) Dihydrosphingomyelin impairs HIV-1 infection by rigidifying liquid-ordered membrane domains. Chem. Biol. 17, 766–775 [DOI] [PubMed] [Google Scholar]
- 11. Finnegan C. M., Rawat S. S., Puri A., Wang J. M., Ruscetti F. W., Blumenthal R. (2004) Ceramide, a target for antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 101, 15452–15457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Finnegan C. M., Rawat S. S., Cho E. H., Guiffre D. L., Lockett S., Merrill A. H., Jr., Blumenthal R. (2007) Sphingomyelinase restricts the lateral diffusion of CD4 and inhibits human immunodeficiency virus fusion. J. Virol. 81, 5294–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiantia S., Kahya N., Ries J., Schwille P. (2006) Effects of ceramide on liquid-ordered domains investigated by simultaneous AFM and FCS. Biophys. J. 90, 4500–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hug P., Lin H. M., Korte T., Xiao X., Dimitrov D. S., Wang J. M., Puri A., Blumenthal R. (2000) Glycosphingolipids promote entry of a broad range of human immunodeficiency virus type 1 isolates into cell lines expressing CD4, CXCR4, and/or CCR5. J. Virol. 74, 6377–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimizu T., Arai S., Imai H., Oishi T., Hirama M., Koito A., Kida Y., Kuwano K. (2004) Glycoglycerolipid from the membranes of Acholeplasma laidlawii binds to human immunodeficiency virus-1 (HIV-1) and accelerates its entry into cells. Curr. Microbiol. 48, 182–188 [DOI] [PubMed] [Google Scholar]
- 16. Huitema K., van den Dikkenberg J., Brouwers J. F., Holthuis J. C. (2004) Identification of a family of animal sphingomyelin synthases. EMBO J. 23, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z., Fan Y., Liu J., Li Y., Huan C., Bui H. H., Kuo M. S., Park T. S., Cao G., Jiang X. C. (2012) Impact of sphingomyelin synthase 1 deficiency on sphingolipid metabolism and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 32, 1577–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yano M., Watanabe K., Yamamoto T., Ikeda K., Senokuchi T., Lu M., Kadomatsu T., Tsukano H., Ikawa M., Okabe M., Yamaoka S., Okazaki T., Umehara H., Gotoh T., Song W. J., Node K., Taguchi R., Yamagata K., Oike Y. (2011) Mitochondrial dysfunction and increased reactive oxygen species impair insulin secretion in sphingomyelin synthase 1-null mice. J. Biol. Chem. 286, 3992–4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mitsutake S., Zama K., Yokota H., Yoshida T., Tanaka M., Mitsui M., Ikawa M., Okabe M., Tanaka Y., Yamashita T., Takemoto H., Okazaki T., Watanabe K., Igarashi Y. (2011) Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J. Biol. Chem. 286, 28544–28555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rucker J., Doranz B. J., Edinger A. L., Long D., Berson J. F., Doms R. W. (1997) Use of a cell-cell fusion assay to study the role of chemokine receptors in human immunodeficiency virus type 1 (HIV-1) entry. Methods Enzymol. 288, 118–133 [DOI] [PubMed] [Google Scholar]
- 21. Jenkinson S., McCoy D. C., Kerner S. A., Ferris R. G., Lawrence W. K., Clay W. C., Condreay J. P., Smith C. D. (2003) Development of a novel high-throughput surrogate assay to measure HIV envelope/CCR5/CD4-mediated viral/cell fusion using BacMam baculovirus technology. J. Biomol. Screen. 8, 463–470 [DOI] [PubMed] [Google Scholar]
- 22. Ji C., Zhang J., Cammack N., Sankuratri S. (2006) Development of a novel dual CCR5-dependent and CXCR4-dependent cell-cell fusion assay system with inducible gp160 expression. J. Biomol. Screen. 11, 65–74 [DOI] [PubMed] [Google Scholar]
- 23. Zama K., Mitsutake S., Watanabe K., Okazaki T., Igarashi Y. (2012) A sensitive cell-based method to screen for selective inhibitors of SMS1 or SMS2 using HPLC and a fluorescent substrate. Chem. Phys. Lipids. 165, 760–768 [DOI] [PubMed] [Google Scholar]
- 24. Abe M., Makino A., Hullin-Matsuda F., Kamijo K., Ohno-Iwashita Y., Hanada K., Mizuno H., Miyawaki A., Kobayashi T. (2012) A role for sphingomyelin-rich lipid domains in the accumulation of phosphatidylinositol-4,5-bisphosphate to the cleavage furrow during cytokinesis. Mol. Cell. Biol. 32, 1396–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maeda K., Das D., Yin P. D., Tsuchiya K., Ogata-Aoki H., Nakata H., Norman R. B., Hackney L. A., Takaoka Y., Mitsuya H. (2008) Involvement of the second extracellular loop and transmembrane residues of CCR5 in inhibitor binding and HIV-1 fusion: insights into the mechanism of allosteric inhibition. J. Mol. Biol. 381, 956–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsuchiya K., Ode H., Hayashida T., Kakizawa J., Sato H., Oka S., Gatanaga H. (2013) Arginine insertion and loss of N-linked glycosylation site in HIV-1 envelope V3 region confer CXCR4-tropism. Sci. Rep. 3, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang X., Wyatt R., Sodroski J. (2001) Improved elicitation of neutralizing antibodies against primary human immunodeficiency viruses by soluble stabilized envelope glycoprotein trimers. J. Virol. 75, 1165–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barrero-Villar M., Cabrero J. R., Gordón-Alonso M., Barroso-González J., Alvarez-Losada S., Muñoz-Fernández M. A., Sánchez-Madrid F., Valenzuela-Fernández A. (2009) Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 122, 103–113 [DOI] [PubMed] [Google Scholar]
- 29. Jiménez-Baranda S., Gómez-Moutón C., Rojas A., Martínez-Prats L., Mira E., Ana Lacalle R., Valencia A., Dimitrov D. S., Viola A., Delgado R., Martínez-A C., Mañes S. (2007) Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 9, 838–846 [DOI] [PubMed] [Google Scholar]
- 30. Florkiewicz R. Z., Rose J. K. (1984) A cell line expressing vesicular stomatitis virus glycoprotein fuses at low pH. Science 225, 721–723 [DOI] [PubMed] [Google Scholar]
- 31. Roberts P. C., Kipperman T., Compans R. W. (1999) Vesicular stomatitis virus G protein acquires pH-independent fusion activity during transport in a polarized endometrial cell line. J. Virol. 73, 10447–10457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., Kabat D. (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72, 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeang C., Varshney S., Wang R., Zhang Y., Ye D., Jiang X. C. (2008) The domain responsible for sphingomyelin synthase (SMS) activity. Biochem. Biophys. Acta 1781, 610–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y., Liu H., Kim B. O., Gattone V. H., Li J., Nath A., Blum J., He J. J. (2004) CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J. Virol. 78, 4120–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arthos J., Cicala C., Martinelli E., Macleod K., Van Ryk D., Wei D., Xiao Z., Veenstra T. D., Conrad T. P., Lempicki R. A., McLaughlin S., Pascuccio M., Gopaul R., McNally J., Cruz C. C., Censoplano N., Chung E., Reitano K. N., Kottilil S., Goode D. J., Fauci A. S. (2008) HIV-1 envelope protein binds to and signals through integrin α4β7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 9, 301–309 [DOI] [PubMed] [Google Scholar]
- 36. Anand A. R., Prasad A., Bradley R. R., Deol Y. S., Nagaraja T., Ren X., Terwilliger E. F., Ganju R. K. (2009) HIV-1 gp120-induced migration of dendritic cells is regulated by a novel kinase cascade involving Pyk2, p38 MAP kinase, and LSP1. Blood 114, 3588–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fuortes M., Melchior M., Han H., Lyon G. J., Nathan C. (1999) Role of the tyrosine kinase pyk2 in the integrin-dependent activation of human neutrophils by TNF. J. Clin. Invest. 104, 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Astier A., Avraham H., Manie S. N. (1997) The related adhesion focal tyrosine kinase is tyrosinephosphorylated after β1-integrin stimulation in B cells and binds to p130cas. J. Biol. Chem. 272, 228–232 [DOI] [PubMed] [Google Scholar]
- 39. Keogh R. J., Houliston R. A., Wheeler-Jones C. P. (2002) Human endothelial Pyk2 is expressed in two isoforms and associates with paxillin and p130Cas. Biochem. Biophys. Res. Commun. 290, 1470–1477 [DOI] [PubMed] [Google Scholar]
- 40. Lee C., Liu Q. H., Tomkowicz B., Yi Y., Freedman B. D., Collman R. G. (2003) Macrophage activation through CCR5- and CXCR4-mediated gp120-elicited signaling pathways. J. Leukocyte Biol. 74, 676–682 [DOI] [PubMed] [Google Scholar]
- 41. Liu Y., Belkina N. V., Shaw S. (2009) HIV Infection of T cells: actin-in and actin-out. Sci. Signal. 2, 1–3 [DOI] [PubMed] [Google Scholar]
- 42. Iyengar S., Hildreth J. E., Schwartz D. H. (1998) Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 72, 5251–5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Asano S., Kitatani K., Taniguchi M., Hashimoto M., Zama K., Mitsutake S., Igarashi Y., Takeya H., Kigawa J., Hayashi A., Umehara H., Okazaki T. (2012) Regulation of cell migration by sphingomyelin synthases:sphingomyelin in lipid rafts decreases responsiveness to signaling by the CXCL12/CXCR4 pathway. Mol. Cell. Biol. 32, 3242–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tafesse F. G., Huitema K., Hermansson M., van der Poel S., van den Dikkenberg J., Uphoff A., Somerharju P., Holthuis J. C. (2007) Both sphingomyelin synthases SMS1 and SMS2 are required for sphingomyelin homeostasis and growth in human HeLa cells. J. Biol. Chem. 282, 17537–17547 [DOI] [PubMed] [Google Scholar]
- 45. Xiao X., Wu L., Stantchev T. S., Feng Y. R., Ugolini S., Chen H., Shen Z., Riley J. L., Broder C. C., Sattentau Q. J., Dimitrov D. S. (1999) Constitutive cell surface association between CD4 and CCR5. Proc. Natl. Acad. Sci. U.S.A. 96, 7496–7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carriba P., Navarro G., Ciruela F., Ferré S., Casadó V., Agnati L., Cortés A., Mallol J., Fuxe K., Canela E. I., Lluís C., Franco R. (2008) Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 5, 727–733 [DOI] [PubMed] [Google Scholar]
- 47. Avraham H., Park S. Y., Schinkmann K., Avraham S. (2000) RAFTK/Pyk2-mediated cellular signalling. Cell. Signal. 12, 123–133 [DOI] [PubMed] [Google Scholar]
- 48. Yamashita A., Hayashi Y., Nemoto-Sasaki Y., Ito M., Oka S., Tanikawa T., Waku K., Sugiura T. (2014) Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid. Res. 53, 18–81 [DOI] [PubMed] [Google Scholar]