

Abstract
The hypothalamic-pituitary-adrenal (HPA) axis is a classic neuroendocrine system. One of the best ways to understand the HPA axis is to appreciate its dynamics in the variety of diseases and syndromes that affect it. Excess glucocorticoid activity can be due to endogenous cortisol overproduction (spontaneous Cushing’s syndrome) or exogenous glucocorticoid therapy (iatrogenic Cushing’s syndrome). Endogenous Cushing’s syndrome can be subdivided into ACTH-dependent and ACTH-independent, the latter of which is usually due to autonomous adrenal overproduction. The former can be due to a pituitary corticotroph tumor (usually benign) or ectopic ACTH production from tumors outside the pituitary; both of these tumor types overexpress the proopiomelanocortin gene. The converse of Cushing’s syndrome is the lack of normal cortisol secretion and is usually due to adrenal destruction (primary adrenal insufficiency) or hypopituitarism (secondary adrenal insufficiency). Secondary adrenal insufficiency can also result from a rapid discontinuation of long-term, pharmacological glucocorticoid therapy because of HPA axis suppression and adrenal atrophy. Finally, mutations in the steroidogenic enzymes of the adrenal cortex can lead to congenital adrenal hyperplasia and an increase in precursor steroids, particularly androgens. When present in utero, this can lead to masculinization of a female fetus. An understanding of the dynamics of the HPA axis is necessary to master the diagnosis and differential diagnosis of pituitary-adrenal diseases. Furthermore, understanding the pathophysiology of the HPA axis gives great insight into its normal control.
Introduction—Normal Physiology
Cortisol, the principal glucocorticoid produced by the human adrenal cortex, is integral in the control of most physiological systems. Excess endogenous cortisol or the overuse of exogenous glucocorticoids—Cushing’s syndrome—has wide-ranging and devastating effects on most organ systems, whereas deficiency of cortisol secretion—adrenal insufficiency—can be fatal if untreated. Finally, defects in the steroidogenic pathway leading to congenital adrenal hyperplasia during fetal life can result in dramatic effects on development. Most previous reviews of adrenal disorders have been tailored to a primarily clinical audience, with a few exceptions (92,214). Although there are other disorders of the control of adrenocortical function, this review will focus on these three as a vehicle to understand the physiology and pathophysiology of adrenocortical control.
There are several reviews in this section of Comprehensive Physiology that focus on all aspects of normal function of the adrenal cortex and the hypothalamic-pituitary adrenal (HPA) axis. In this article, we will briefly review normal control where appropriate because this information is necessary for understanding the pathophysiology of the HPA axis.
The control of cortisol secretion is one of the classic examples of a hypothalamic-pituitary-target gland axis (Fig. 1). Basal and stress-inputs to the hypothalamic parvocellular nuclei lead to an increase in the neurocrine factor corticotrophin-releasing hormone (CRH) release into the hypophysical-portal veins. From the point of view of patho-physiology, the most important basal input to CRH secretion is from the circadian rhythm generator in the hypothalamic suprachiasmatic nucleus (117, 214, 225). In individuals with established nocturnal sleep and daytime wakefulness, circulating cortisol concentrations peak between 0600 and 0900 h and have a nadir between 2300 and 0100 h. From a metabolic point of view, the increase in cortisol that starts at about 0400 h helps to maintain plasma glucose (via increases in hepatic gluconeogenesis) until awakening when the overnight fast can be broken (143). Interestingly, there is also a cortisol awakening response. Although its precise function is unclear, it may correlate with general stress reactivity (41, 42).
Figure 1.

The hypothalamic-pituitary-adrenal axis. Inputs from the hypothalamic circadian rhythm generator in the suprachiasmatic nucleus (SCN) and neural stress pathways in the central nervous system (CNS) control the activity of the corticotrophin-releasing hormone (CRH) neuronal cell bodies in the paraventricular nucleus. These neurons are also capable of synthesizing arginine vasopressin (AVP), which can augment the pituitary response to CRH. CRH (and AVP) are released into the portal circulation in capillaries in the median eminence and drain onto the anterior pituitary where they stimulate the pituitary corticotrophs to release adrenocorticotropic hormone (ACTH). ACTH stimulates the zona fasciculata (ZF) and zona reticularis (ZR) via the MC2R (melanocortin 2 receptor, also known as the ACTH receptor). This G-protein coupled receptor increases intracellular cAMP release, which activates StAR-mediated cholesterol transport into the mitochondria (the rate-limiting step of steroidogenesis). Once cholesterol reaches the inner mitochondrial (Mito) membrane, it is acted on by the first steroidogenic enzyme, and then by subsequent sequential enzymes in the smooth endoplasmic reticulum (SER) and Mito with cortisol as an end product (see Fig. 12). Cortisol is released into the plasma compartment where it binds reversibly to corticosteroid-binding globulin (CBG; also known as cortisol-binding globulin). As CBG-bound plasma cortisol enters the capillaries in target tissue, it dissociates from CBG and diffuses into the target cell. In the pituitary and hypothalamus, negative feedback inhibition is exerted with the binding of cortisol to glucocorticoid (GR) and mineralocorticoid (MR) receptors.
CRH acutely stimulates the corticotrophs of the anterior pituitary to increase the release of already stored adrenocorticotrophic hormone (ACTH) into the pituitary venous effluent that, through the inferior petrosal sinuses, drains into the internal jugular veins. CRH also stimulates the synthesis of new ACTH by activating gene transcription for the precursor molecule proopiomelanocortin (POMC) and its post-translational processing to ACTH and other byproducts (see Fig. 10). ACTH stimulates the release of cortisol by binding to the melanocyte type-2 receptor (MC2R) on the cells of the zona fasciculata and zona reticularis in the adrenal cortex (44). This is via a G-protein coupled receptor leading to increases in intracellular cAMP (second messenger) and the liberation of steroidogenic acute regulatory (StAR) protein, which mediates translocation of cholesterol to the inner mitochondrial membrane—the rate limiting step of steroidogenesis (87, 142, 244). After cholesterol makes contact with the first steroidogenic enzyme in the inner mitochondrial membrane—side chain cleavage (P450scc)—and is converted to pregnenolone, the remainder of steroidogenesis occurs in an obligatory fashion with cortisol as the main final product. The steroidogenic pathway will be discussed in more detail in the section on congenital adrenal hyperplasia.
Figure 10.
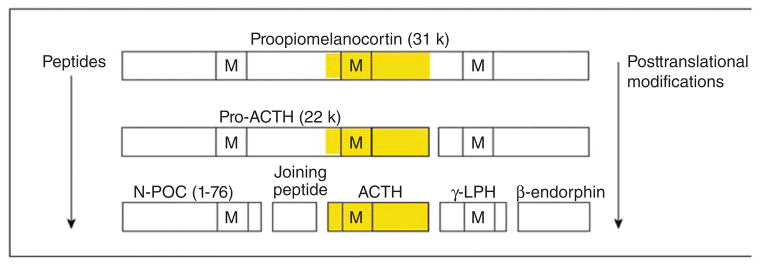
The large protein proopiomelanocortin (POMC) is produced by transcription and translation of the POMC gene. Adrenocorticotropic hormone is then produced by posttranslational processing. Note that other products of POMC can be produced (for example, beta and gamma-lipotropic hormone [LPH], N-terminal POMC fragment [N-POC], and melanocyte-stimulating hormone [M]). Also notice that ACTH contains the sequence of MSH within it. Ectopic ACTH-secreting tumors can perform the same processing but often produce large amounts of precursors (particularly pro-ACTH). From (214) with kind permission of the Annals of Internal Medicine/American College of Physicians.
Once cortisol is released into the blood, it can circulate in the free form (~5–6% of the total plasma cortisol), or bound primarily to two proteins. Like all steroids, it can significantly bind to the low affinity-high capacity protein albumin when cortisol secretion rate is high. It also has a specific carrier—corticosteroid binding globulin (CBG; transcortin)—that is produced by the liver (144,196). This high affinity-low capacity protein is more important at physiological cortisol secretion rates. CBG production can be modulated; in particular, estrogen increases hepatic CBG production, which can confound some of the screening tests described below (171,204). It is the free cortisol in the plasma that can diffuse into the cells of the target organs to exert the biological effects of cortisol shown in Figure 1.
The metabolic clearance of plasma cortisol is a complex process beyond the scope of this clinical review. Briefly, cortisol is conjugated to glucuronide in the liver, and then excreted in the urine (83). However, the inactivation and generation of cortisol in target tissues is an important physiological process described below.
The mechanism of action of cortisol, like other steroid hormones, involves an intracellular receptor and changes in gene transcription and translation (223). The two main receptors of interest are cytoplasmic, and are the glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) (17, 100). At first glance, it would seem that all of the effects in Table 1 should be accounted for by binding of cortisol to the GR. However, although not well appreciated, cortisol has significant affinity for MR. How, then, can aldosterone, the primary adrenocortical mineralocorticoid, control sodium reabsorption and potassium secretion in the distal nephron, since the free cortisol concentration in plasma is higher than free aldosterone (85, 86)? It turns out that the MRs in the distal tubules of the kidney are protected from the action of cortisol by the enzyme 11-beta-hydroxysteroid dehydrogenase type 2 (11BHSD2), which converts the active cortisol into inactive cortisone (63, 85, 86, 243). At physiological concentrations, the direct effect of cortisol on sodium and potassium balance is minimal compared to aldosterone. However, in Cushing’s syndrome, when cortisol secretion is increased, the ability of 11BHSD2 to inactivate cortisol is overwhelmed and cortisol binding to MR leads to significant mineralocorticoid effects (e.g., sodium reabsorption and hypertension) shown in Table 1. Interestingly, plasma cortisone can be reactivated to cortisol by the enzyme 11-beta-hydroxysteroid dehydrogenase type 1 (11BHSD1) found in abundance in the liver (258).
Table 1.
Clinical Features of Cushing’s Syndrome (% Prevalence) and Their Physiological Causes
| Clinical feature | Physiological basis of effecta |
|---|---|
| General | |
| Obesity 90% | Hyperphagia; insulin resistance |
| Hypertension 85% | Sodium retention (via MR) |
| Skin | |
| Plethora 70% | Thinning of skin (connective tissue) |
| Hirsutism 75% | Androgen excess |
| Striae 50% | Thinning of skin (connective tissue) |
| Acne 35% | Androgen and glucocorticoid excess |
| Bruising 35% | Thinning of skin (connective tissue) |
| Musculoskeletal | |
| Osteopenia 80% | Increase in bone resorption |
| Weakness 65% | Catabolic effect on muscle |
| Neuropsychiatric 85% | CNS effects |
| Emotional lability | Cortisol excess |
| Euphoria | Cortisol excess |
| Depression | Cortisol excess |
| Psychosis | Cortisol excess |
| Gonadal dysfunction | Inhibition of gonadotroph function |
| Menstrual disorders 70% | Inhibition of GnRH, LH, and FSH release |
| Impotence, decreased libido 85% | Inhibition of GnRH, LH, and FSH release |
| Metabolic | |
| Glucose intolerance 75% | Insulin resistance |
| Diabetes 20% | Insulin resistance |
| Hyperlipidemia 70% | Increased lipolysis, hepatic steatosis, insulin resistance |
| Polyuria 30% | Increased GFR; suppression of AVP |
| Kidney stones 50% | urine: ↑ calcium, uric acid, oxalate, ↓ citrate |
| Pediatricb | |
| Growth retardation (83%) | Inhibition of GH secretion (see Fig. 2) |
The final and probably most important component of the HPA axis necessary to understand the diagnosis, differential diagnosis, and treatment of adrenal disorders is glucocorticoid negative feedback (130,214). Negative feedback control is a fundamental process in homeostasis—the foundation of physiology. Cortisol can inhibit POMC transcription and ACTH secretion directly and also can indirectly inhibit ACTH secretion by decreasing CRH secretion (117, 130, 152, 214). Glucocorticoids inhibit pituitary POMC expression through binding of GR to a negative promoter on the POMC gene, and also through the action of a variety of transcription factors (124, 227). This negative feedback can be exerted quickly (so-called fast feedback) as well as the better appreciated delayed feedback that takes at least 30 min to 2 h to be expressed (46, 130). It is delayed feedback that is exploited in clinical suppression tests of HPA axis function.
One other physiological phenomenon necessary to understand Cushing’s syndrome, certain forms of adrenal insufficiency, exogenous glucocorticoid therapy, and congenital adrenal hyperplasia is adrenal growth. Like other anterior pituitary hormones, ACTH has potent influences on the growth of its target gland. Simply, ACTH excess leads to adrenocortical hyperplasia whereas ACTH deficiency leads to adrenocortical atrophy. This fact becomes extremely important when weaning patients from long-term glucocorticoid therapy and during recovery from surgery to correct endogenous cortisol excess. When discontinuing exogenous steroid therapy, the HPA axis needs a considerable amount of time to recover—ACTH must increase sufficiently for an adequate amount of time to induce the restoration of adrenocortical volume (93,119). The approach to weaning a patient from exogenous steroid therapy requires a thorough understanding of the normal function of the HPA axis and is described later in this review (111, 221).
Cushing’s Syndrome
Definitions
Cushing’s syndrome is generally defined as a state of glucocorticoid excess. It can be caused by endogenous production of cortisol from the adrenal gland (regardless of the reason) or by synthetic glucocorticoids administered therapeutically for any number of diseases. Endogenous Cushing’s syndrome is a relatively unusual disorder, although it is now clear that its incidence and prevalence is higher than previously thought (99, 254). This is primarily due to the development of simple screening tests that have dramatically improved the detection of the disorder. Appreciation of these screening tests relies on understanding normal physiological function (Fig. 1).
There are two general causes of endogenous hypercortisolism—ACTH-dependent Cushing’s syndrome and ACTH-independent Cushing’s syndrome (77–81, 168). ACTH-dependent adrenal hyperfunction can be caused by excess ACTH secretion by pituitary corticotroph tumors (Cushing’s disease) or ectopic ACTH production, usually from neuroendocrine tumors (114). Endogenous ACTH-independent Cushing’s syndrome is caused by autonomous adrenal overproduction of cortisol, usually due to a benign, solitary adrenocortical adenoma (188).
There are several other more unusual causes of adrenal Cushing’s syndrome such as adrenal carcinoma and a variety of adrenal hyperplastic syndromes (28). Exogenous glucocorticoid therapy can be classified as “ACTH-independent” since, from a physiological point of view, it is similar to the adrenal gland producing cortisol autonomously (without ACTH stimulation).
Glucocorticoids are potent anti-inflammatory and immunosuppressive drugs (239), so are used to treat a wide spectrum of diseases. For the most part, the signs and symptoms of exogenous glucocorticoid excess are the same as endogenous hypercortisolism (111). There may be exceptions, particularly since increases in adrenal androgens can occur with endogenous Cushing’s syndrome (19, 62). Furthermore, there may be the effects of synthetic glucocorticoids that are different from endogenous cortisol, particularly in the central nervous system (160, 279).
Clinical Features of Cushing’s Syndrome
In keeping with the pleiotropic actions of cortisol, the clinical spectrum of Cushing’s syndrome is broad, and establishing a diagnosis based on clinical presentation alone can often be difficult as none of the signs and symptoms are pathognomonic of the syndrome. Table 1 summarizes the varied clinical features of Cushing’s syndrome.
Weight gain is one of the most common clinical findings, and clearly requires overeating—cortisol induces hyperphagia (Table 1) (251). However, obesity is not specific to Cushing’s syndrome and has significant clinical overlap in those with and without the syndrome (18). Some of the signs that more reliably distinguish Cushing’s syndrome from obesity include proximal muscle weakness, easy bruising, and violaceous striae greater than 1 cm in width (175). Growth retardation along with progressive weight gain is a hallmark of Cushing’s syndrome in children (95). Glucocorticoids act directly on long bones in children to arrest the development of the epiphyseal cartilage. Furthermore, suppression of growth hormone secretion and action leads to blunted somatic growth (Fig. 2) (139, 245).
Figure 2.

The mechanisms of glucocorticoid (GC)-induced decreases in growth in children and suppression of growth hormone (GH) in adults. GHRH is growth-hormone release hormone, IGF1 is insulin-like growth factor 1; GnRH is gonadotropin-releasing hormone, LH is luteinizing hormone, and FSH is follicle-stimulating hormone. Adapted from (7) with kind permission from WB Saunders through the Copyright Clearance Center.
Excess glucocorticoids have a catabolic effect on skeletal muscle with increased activity of myofibrillar proteinases and reduced uptake and conversion of amino acids into proteins. This negative nitrogen balance is associated with increased protein wasting, type II muscle fiber atrophy, and significant muscle weakness with predominant involvement of the muscles of the pelvic girdle (157, 219). Similar catabolic effects also occur in the epidermis and underlying connective tissue. This thinning of the skin leads to the development of violaceous striae, easy bruisability, and a plethoric appearance of the face.
Decreased bone mineral density, osteoporosis and related fragility fractures have been reported in 60% and 80% of patients with glucocorticoid excess. The pathogenesis of this bone loss is multifactorial. GRs are present on both osteoblasts and osteoclasts. Excess glucocorticoids impair osteoblastic differentiation and increase apoptosis of both osteoblasts and osteoclasts (273). Increased production of the receptor activator of NF-kappa beta ligand (RANKL), which increases osteoclastogenesis, and decreases osteoprotegrin, a decoy receptor for RANKL that acts to decrease osteoclast differentiation, together lead to increased osteoclastic bone resorption (108). Hypercortisolism suppresses gonadotropin and growth hormone concentration (Fig. 2), further contributing to decreased bone mass. Decreased intestinal and tubular reabsorption of calcium and related normocalcemic hypercalciuria has been reported in up to 85% of patients with Cushing’s syndrome (67). However, its significance in the development of glucocorticoid-induced bone loss is not completely clear. Recently, higher levels of sclerostin, an extra-cellular antagonist of the Wingless/β-catenin (Wnt) signaling pathway, were noted in patients with Cushing’s syndrome compared to healthy individuals (22). However contradictory results were found in another study (266). Therefore, the role of sclerostin in the pathogenesis of bone loss in Cushing’s syndrome needs further study.
Psychiatric disturbances are present in up to 85% patients with Cushing’s syndrome. Depression and irritability are the most common manifestations (51–86%); others include emotional lability, mania, paranoia, acute psychosis, anxiety, and panic attacks (61, 105, 241). Glucocorticoids mediate their effects on brain via both GR and MR. GRs are distributed widely in the central nervous system while MRs are expressed highly in the limbic brain areas such as the hippocampus. MRs in the brain have greater affinity for glucocorticoids than GRs and are almost completely occupied at basal corticosteroid levels. On the other hand, GRs are activated when cortisol levels are higher, as at the peak of the circadian rhythm, during stress and in Cushing’s syndrome (49). Differences in neuropsychiatric effects between endogenous and exogenous glucocorticoids could be due, at least in part, to the fact that most exogenous glucocorticoids do not bind appreciably to MR in the brain.
Hypercortisolemia is associated with a decrease in apparent brain volume, particularly the hippocampus, and related impairment in learning, cognition and short-term memory (27, 240). The mechanism underlying structural atrophy is not fully understood but is thought to include reversible atrophy of the dendritic processes, inhibition of neurogenesis, and increased susceptibility to cell injury and death due to increased levels of excitatory amino acid neurotransmitters such as glutamate (160).
Chronic glucocorticoid excess is associated with dyslipidemia and glucose intolerance. Increased hepatic gluconeogenesis, peripheral insulin resistance, and direct suppression of insulin release together contribute to the development of impaired glucose tolerance in 30% and 60% of patients and overt diabetes in 20% and 50% of patients (200). This reported incidence is likely an underestimate as many patients with normal fasting glucose may have underlying glucose intolerance and not all patients undergo glucose tolerance tests. Dyslipidemia observed in patients with Cushing’s syndrome is similar to the metabolic syndrome with an increase in very low density lipoprotein (VLDL) and low density lipoprotein (LDL) and a decrease in high density lipoprotein levels. This is thought to be related to the direct and indirect effect of cortisol to increase lipolysis, VLDL synthesis, fatty accumulation in the liver, and peripheral insulin resistance (14).
In addition to dyslipidemia and glucose intolerance, hypertension (related to upregulation of the renin-angiotensin system and the mineralocorticoid effects of cortisol described earlier) (232) and a hypercoagulable state [related to increased synthesis of clotting factors like fibrinogen as well as plasminogen activator inhibitor type-1, an inhibitor of the fibrinolytic system (265)] lead to an additional increase in cardiovascular risk that may not return to baseline even after remission of hypercortisolemia.
Screening tests for endogenous hypercortisolism
As outlined above, Cushing’s syndrome is a relatively unusual disease. However, aspects of the Cushingoid phenotype are very common, so it is vital that a robust screening test be used to identify the rare patients with true Cushing’s syndrome from the large number of patients with the clinical features shown in Table 1. The screening test cannot be too complicated or costly or it will not be practical. However, it has to have acceptable sensitivity and specificity to avoid false negative and false positive results leading to missed diagnosis or excessive unnecessary follow-up testing. The three tests outlined below exploit our knowledge of the normal control of the HPA axis and, hence, its pathophysiology. The first step, of course, is to take an extensive drug history to make sure the patient is not using any form of exogenous glucocorticoid therapy (described below).
Late-night cortisol measurement
Figure 1 demonstrates that the circadian rhythm of the HPA axis is a fundamental characteristic such that cortisol peaks upon awakening in the morning and is at its nadir between 2300 and 0100 h (136, 214). It was hypothesized that one of the first derangements that occurs in Cushing’s syndrome regardless of the cause is the failure to achieve a normal late-night circadian nadir (53). In mild ACTH-independent (adrenal) Cushing’s syndrome, the abnormal steroidogenic tissue in the adrenal is secreting cortisol autonomously, so that cortisol secretion is disconnected from the CNS circadian rhythm generator (35,138). The same logic follows with ectopic ACTH secretion. ACTH-secreting pituitary adenomas are still “connected” to the portal vein CRH concentration and often express the CRH receptor (96). However, these corticotroph adenomas are more active and less sensitive to glucocorticoid negative feedback (214). Therefore, although they can exhibit a circadian rhythm, the nadir in cortisol secretion that normally occurs between 2300 and 0100 h is usually not fully achieved (89, 264).
The abnormal circadian cortisol rhythmicity was first exploited by Newell-Price et al. in a comprehensive way; unstressed blood samples at midnight demonstrated that patients with Cushing’s syndrome had increased cortisol compared to controls (170). This was confirmed in a larger cohort by Papanicolaou et al. (192). The practical problem that this blood sampling imposed, however, was the requirement that the late-night blood sample be drawn under unstressed conditions. The patient had to have a blood-drawing catheter placed many hours before sampling, or (in the Newell-Price study) be sleeping when the sample was drawn after admission to an inpatient unit (170). This highlights the physiological principle that any assessment of the HPA axis has to take into account the level of ambient stress in environment of the patient or subject. Although the late-night serum cortisol test performs extremely well, unstressed late-night blood sampling is not practical for endocrinologists to execute routinely and, therefore, is not a practical screening test. For this reason, the test that has become widely accepted is using late-night (bedtime) salivary cortisol (LNSC) as a surrogate for serum free cortisol—the biologically active endpoint of the HPA axis.
Salivary steroid concentrations are in equilibrium with the concentration of free steroids in plasma. Salivary cortisol concentration has been used to assess HPA axis function for several decades. An early study demonstrated the potential for subjects and patients to sample saliva at bedtime (2300–2400 h) at home in a presumably low stress environment (127). Many subsequent studies in a large number of patients have demonstrated that measurement of late night salivary cortisol concentration performs well as a surrogate for plasma free cortisol (32, 33, 203, 208–212, 216, 218, 262). The high sensitivity, specificity, and simplicity of this test has led to it becoming widely available and utilized throughout the world (55, 210, 211).
There are four major potential problems with the assessment of salivary cortisol. One is compliance of the patient in terms of when the saliva sample is obtained. It has been shown that compliance with proper sample timing can be problematic and it is vital that the patient and clinician be carefully educated about this (30, 103). In fact, since salivary cortisol is extensively used in human stress research, this is an experimental as well as a clinical problem.
A second problem is that the normal circadian variation in cortisol can be disrupted in individuals with altered sleep patterns, such as shift workers (272). In this setting, it is difficult to predict the time of the nadir value, and that value may be abnormally increased.
A third problem is the possible effects of stress or excitement to increase serum and hence salivary cortisol. This was shown in a study of 15 healthy subjects who provided a saliva sample 1 h after watching a football game; one individual had an abnormal level, suggesting that individuals should be cautioned to avoid exciting experiences in the evening of the test (216).
The final potential problem is the possibility of contamination of the saliva sample. When salivary cortisol (or for that matter serum cortisol) is assayed by immunoassay, certain synthetic steroids can cross-react in the assay and falsely increase the result. The more common problem, however, is contamination of the saliva sample with widely available nonprescription topical hydrocortisone (authentic cortisol) anti-itch creams and ointments. This will falsely increase salivary cortisol whether measured by immunoassay or the more specific liquid chromatograph tandem mass spectrometry (LCMSMS) methodology (211,212,218). Since plasma cortisol can be converted to cortisone by 11BHSD2 as described above and since the salivary gland expresses 11BHSD2, transfer of cortisone from plasma or its local production in the salivary gland normally results in a ratio of cortisol to cortisone in saliva that is significantly less than 1 (195, 218). Since contamination of the saliva with topical cortisol does not result in an increase in salivary cortisone, it has been shown that measurement of a greatly increased ratio of cortisol to cortisone and a lack of increase in the absolute concentration of cortisone in saliva established that a false positive result is due to contamination (218). Therefore, although we usually first analyze all saliva samples with a cortisol immunoassay because of its better diagnostic performance (211, 212), if the result is very high and/or out of proportion to the clinical findings, the sample is reanalyzed for cortisol and cortisone by LCMSMS (Fig. 3). Finally, many clinicians advocate using a confirmatory screening test (described below) before referring a patient for differential diagnosis (32).
Figure 3.

The use of late-night salivary cortisol (LNSC) for screening patients with suspected Cushing’s syndrome. Note that because Cushing’s syndrome is relatively rare and its phenotype very common, most patients screened will have normal LNSCs and Cushing’s syndrome will be ruled out. Conversely, most patients with true Cushing’s syndrome will have consistently increased LNSCs. Even so, the diagnosis is usually confirmed with urine free cortisol (UFC) measurements and/or the overnight low-dose dexamethasone suppression test (oDST). Occasionally, the LNSCs are discordant as shown or the samples are suspicious for contamination with over-the-counter hydrocortisone creams. In that case, measurement of cortisol and cortisone by liquid chromatography/tandem mass spectrometry (LC-MS/MS) will resolve the problem. Adapted from (211) with kind permission from Springer.
It also is theoretically possible that leakage of blood carrying cortisol bound to CBG into saliva due to bad dental health could be a problem. Interestingly, this has not shown to be the case for salivary cortisol (133).
Urine free cortisol
Cortisol is excreted in the urine in the free form and conjugated to, for example, glucuronides (83). The physiological theory of the measurement of urine free cortisol (UFC) in a 24 h urinary collection is that it will represent an integration of the activity of the HPA axis over that time. This test has been used for decades. Although its sensitivity and specificity are adequate, it does not perform well in the milder forms of Cushing’s syndrome, particularly when LCMSMS is used for measurement (4,132). In comparison to earlier studies, this likely reflects the fact that immunoassays often had cross-reactivity with noncortisol glucocorticoids that are also produced in excess in Cushing’s syndrome. With the newer structurally based assays, this cross-reactivity is not present, and the “UFC” value is less. There are several factors and drugs that can falsely increase UFC, even when measured by LCMSMS or HPLC (73, 161, 175). As shown in Figure 3, UFC can be useful in the confirmation of the diagnosis of Cushing’s syndrome by increased LNSC, before proceeding with differential diagnosis.
Low-dose dexamethasone suppression test
As described above, one of the hallmarks of the normal HPA axis is cortisol negative feedback inhibition of CRH and ACTH secretion. Dexamethasone, a potent GR agonist with a long half-life, has been used for 50 years to test negative feedback sensitivity. In regards to Cushing’s disease (pituitary ACTH-secreting adenomas), the theory is that corticotroph cells in an adenoma have decreased feedback sensitivity, because they can maintain ACTH hypersecretion in the face of increased endogenous cortisol (70, 151). Thus, if just the right concentration of plasma dexamethasone were achieved, normal corticotrophs will fully suppress ACTH, and therefore, cortisol secretion, whereas patients with corticotroph adenomas will not (177). Since ectopic ACTH-secreting neuroendocrine tumors were never normal corticotrophs, they should not be inhibited by dexamethasone. Finally, as adrenal adenomas are not dependent on ACTH, a GR agonist would not suppress their cortisol secretion. The low-dose dexam-ethasone suppression test (DST) is often abnormal in patients with adrenal incidentalomas (adrenal masses incidentally discovered during abdominal imaging for other reasons) who have minimal signs and symptoms of Cushing’s syndrome (174). Additional abnormal screening tests are needed before a diagnosis of Cushing’s syndrome can be established (125).
There are two forms of the low-dose oral dexamethasone suppression test currently in use that exploit these physiological concepts (82, 175). One is the 1 mg overnight test (oDST) in which dexamethasone is given at 2300 h and a serum or plasma cortisol is measured at 0800 h the next morning. The other is the 2 day 2-mg low-dose DST (LDDST) in which divided doses are given over several days, and serum or plasma cortisol is measured six hours after the last dose.
Although the theory of the low-dose dexamethasone suppression test is appealing, it has several theoretical and empirical problems that require an understanding of normal HPA axis physiology to appreciate.
The test assumes that dexamethasone absorption, volume of distribution, and metabolism is similar in all patients—a “one size [dose] fits all” approach. This is obviously not a defendable assumption since gastrointestinal function, body weight and composition (and hence blood volume and volume of distribution), and hepatic and renal function (that affect metabolism) can vary dramatically. To counteract this problem, several approaches have been used. Different doses of dexamethasone have been advocated (112), but, in practice, most clinicians still use the 1 mg oDST (175). In addition, some advocate measuring plasma dexamethasone levels in all subjects in the morning cortisol blood sample to verify adequate glucocorticoid negative feedback (172). Measurement of serum dexamethasone is not a standard test in many laboratories, acceptable ranges of plasma dexamethasone have not been validated, and it adds to the expense of the low-dose DST. If dexamethasone levels in the blood are found to be inadequate, then a positive test (i.e., failure to suppress) would have to be repeated with a higher dose of dexamethasone. One study suggested that patients taking multiple medications are more likely to have a clinically inappropriate response to the oDST (263). Dexamethasone levels were not measured, but the implication is that other medications influenced its metabolism.
The test assumes that all corticotroph adenomas have significantly decreased sensitivity to dexamethasone (glucocorticoid negative feedback agonist). This has been proven incorrect since up to 7% of corticotroph adenomas suppress cortisol with the low-dose DST (82).
The test assumes that all ectopic ACTH-secreting tumors are insensitive to dexamethasone. This has been proven incorrect since some of these neuroendocrine tumors do express the GR and dexamethasone inhibits their ACTH secretion (114).
The test assumes that cortisol-secreting adenomas cannot be inhibited by dexamethasone since they are not ACTH-dependent. Although this assumption is probably the safest in this list of drawbacks, the adrenal cortex does express the GR, and it is at least theoretically possible that dexamethasone could directly alter cortisol secretion from adrenal tumors (150, 226).
These issues cause a significant number of false negative and false positive results with the low-dose dexamethasone suppression test (18,82). Despite this, it is still a useful test as long as the clinician is aware of the caveats outlined above.
Stimulation tests
The desmopressin (DDAVP) test may be useful to distinguish patients with Cushing’s disease from those without pathologic hypercortisolism (98, 253). The basic theory of using DDAVP stimulation testing is that corticotroph tumors will still express the vasopressin V1b receptor and will hyper-respond to provocative stimulation despite ambient hypercortisolemic negative feedback (13). Although this test is usually not required, it may help to resolve the diagnosis in patients without definitive screening test results (256).
Combined testing
The general concept of combined testing (whether simultaneous or sequential) is that the drawbacks of one test will be overcome by the strengths of another, particular if they interrogate different aspects of the HPA axis control system. The combined LDDST-oCRH test operates on the assumption that corticotroph adenomas will still respond to CRH when suppressed with lower doses of dexamethasone, whereas normal corticotrophs will not (280). Although this test is expensive and difficult to execute properly, it can be useful, although there are significant caveats. In particular, it seems to be very sensitive to confounding by concomitant medications that alter the metabolism of dexamethasone (175, 263). Another combined approach is the oDST and LNSC test. Since the oDST interrogates glucocorticoid negative feedback sensitivity and the LNSC interrogates abnormalities in the HPA axis circadian rhythm (two different pathophysiological features of Cushing’s disease), combining them can improve the sensitivity and specificity of either test (20, 37, 38).
A final comment on the diagnosis of Cushing’s syndrome
It should be evident from the previous discussion that knowledge of normal physiological processes is vital in the appropriate choice and interpretation of screening tests for endogenous Cushing’s syndrome. A special mention should be made concerning the diagnosis of an enigmatic subtype of this already protean syndrome—cyclical Cushing’s syndrome. This is characterized by waxing and waning of cortisol secretion that can be unpredictable in terms of rhythm. Obviously, if hypercortisolism is not present during clinical screening, the tests will be negative for Cushing’s syndrome despite the fact that the patient is affected. One approach that may work is waiting for the symptoms to increase or return, and then use one of the screening tests listed above. Multiple LNSC measurements over an extended period have been used to identify patients with cyclical Cushing’s syndrome (5, 164).
Differentiation of ACTH-dependent and ACTH-independent Cushing’s syndrome
Once the diagnosis of endogenous glucocorticoid excess (Cushing’s syndrome) is established, the first step in the differential diagnostic algorithm is usually distinguishing ACTH-dependent (Cushing’s disease or ectopic ACTH) and ACTH-independent (adrenal) Cushing’s syndrome (Fig. 4).
Figure 4.

The physiological basis for the approach to the differential diagnosis of Cushing’s syndrome. CT is computed tomography radiography and MRI is magnetic resonance imaging. Once the diagnosis is established (see Fig. 3), measurement of a suppressed plasma level of adrenocorticotropic hormone (ACTH) identifies ACTH-independent (adrenal) Cushing’s syndrome. *Adrenal computed tomography (CT) is then performed, and a more detailed analysis is needed to differentiate among the subtypes of adrenal Cushing’s syndrome. The most challenging problem is the differential diagnosis of ACTH-dependent Cushing’s syndrome. The high-dose dexamethasone suppression test is no longer recommended. If the results of magnetic resonance imaging (MRI) of the pituitary show a mass > 6 mm, referral to a neurosurgeon is appropriate. If not, bilateral inferior petrosal sinus sampling with administration of corticotropin-releasing hormone (CRH) is performed. This method reliably distinguishes pituitary Cushing’s disease from occult ectopic ACTH syndrome. For a more thorough discussion, see text. From (214) with kind permission of the Annals of Internal Medicine/American College of Physicians.
Plasma ACTH measurements
As shown in Figure 4, the measurement of plasma ACTH is critical in the differentiation of ACTH-independent and ACTH-dependent Cushing’s syndrome. The original clinical plasma ACTH measurements were by radioimmunoassay, with all of its inherent problems. In particular, its lack of sensitivity at the critical ACTH concentrations of <20 pg/mL was a problem as was its cross-reactivity with other POMC posttranslational products (74, 88, 146, 213). The two-site immunometric assay has overcome most of these problems with the added benefit of avoiding the need for radioactive tracers. The physiological theory behind the usefulness of measuring plasma ACTH to differentiate ACTH-dependent and independent Cushing’s syndrome is somewhat obvious. However, its explanation helps to illuminate some subtleties of the physiological control of the HPA axis.
Figure 5 shows plasma ACTH values in the three subtypes of Cushing’s syndrome. When measured at the circadian peak (around 0800 h), it is common to find a plasma ACTH concentration within the reference range in patients with Cushing’s disease (ACTH-secreting pituitary corticotroph adenoma). Although there is significant overlap between the two forms of ACTH-dependent Cushing’s syndrome (Cushing’s disease and ectopic ACTH), extraordinarily high plasma ACTH levels (>500 pg/mL) are usually only found in ectopic ACTH. Finally, plasma ACTH is usually suppressed in ACTH-independent Cushing’s syndrome (adrenal tumor) due to cortisol negative feedback suppression of the normal pituitary corticotrophs.
Figure 5.

Plasma ACTH concentrations in patients with established ACTH-dependent Cushing’s syndrome (Cushing’s disease [pituitary corticotroph adenomas] and ectopic ACTH) and ACTH-independent Cushing’s syndrome (Adrenal tumor). Note that plasma ACTH is often within the reference range (blue shading) in Cushing’s disease and that, on average, patients with ectopic ACTH have very high plasma ACTH. To convert to pmol/L, multiply pg/mL by 0.2202.
How can plasma ACTH be in the reference range in Cushing’s disease if this is an ACTH-dependent form of hypercortisolemia? This is a common source of confusion and highlights an important physiological concept that requires an understanding of the normal feedback control of ACTH that can be difficult for the novice to appreciate. Remember that cortisol excess normally suppresses ACTH secretion via negative feedback (Fig. 1) as in patients with adrenal tumors shown in Figure 5. The recognition that plasma ACTH is not suppressed below the reference range in patients with Cushing’s disease (Fig. 5) demonstrates that that it is inappropriately normal and that tumoral ACTH excess is the cause of the hypercortisolism. If anterior pituitary corticotroph function were normal, negative feedback would have fully suppressed ACTH secretion as in the patients with ACTH-independent Cushing’s syndrome (adrenal tumor in Fig. 5).
As with all laboratory tests, the plasma ACTH immunometric assay is not infallible. There is significant variability between immunometric assay methods particularly around and below the critical lower end of the reference range shown in Figure 5 (250). Furthermore, there is the rare patient, particularly with ectopic ACTH, who makes biologically active fragments of POMC that are not detected in the highly specific ACTH immunometric assay (74,215) Additionally, some patients with ACTH-independent (adrenal) causes of Cushing’s syndrome have mild hypercortisolism that is not sufficient to fully suppress ACTH levels (115, 169). Despite rare misleading results, the ACTH immunometric assay has revolutionized the diagnostic strategy for the evaluation of Cushing’s syndrome.
Other tests to distinguish ACTH-dependent and ACTH-independent Cushing’s syndrome
For the vast majority of patients, the measurement of plasma ACTH as shown in the algorithm in Figure 4 is sufficient to distinguish the ACTH-dependent vs. independent causes of the adrenal hypersecretion of cortisol. Occasionally, there is a patient with mild Cushing’s disease or mild adrenal Cushing’s syndrome whose plasma ACTH concentration is not definitive in establishing the diagnosis (168). From a physiological point of view, the best test to evaluate these patients is the oCRH stimulation test (or perhaps the desmopressin stimulation test) (106, 129). The physiological concept is that abnormal corticotrophs in mild Cushing’s disease will still respond to stimulation by exogenous oCRH (and perhaps desmopressin), whereas the normal corticotrophs will be somewhat suppressed from mild cortisol excess due to a small adrenal tumor and, therefore, will not respond to provocative stimulation.
Treatment of ACTH-independent (adrenal) Cushing’s syndrome
Once an adrenal tumor has been localized by abdominal CT (Fig. 4), the optimal therapy is surgical removal (281). As described for discontinuation of glucocorticoid therapy in exogenous Cushing’s later in this review, postsurgical secondary adrenal insufficiency will ensue due to long-term suppression of the HPA axis and glucocorticoid therapy must be given.
A brief description of approaches to the differential diagnosis of Cushing’s syndrome and subsequent treatment
From a physiological point of view, it would seem fairly simple to distinguish patients with pituitary tumors (Cushing’s disease) from those with ectopic ACTH-secreting tumors using medical imaging techniques. If the patient has a known nonpituitary neoplasm with the potential to secrete ACTH (e.g., small cell lung cancer), it is likely that the neoplasm is the source of ACTH. However, most patients with ACTH-dependent Cushing’s syndrome have an occult (i.e., hidden) tumor. Pituitary adenomas can be very small and be missed by even the most advanced magnetic resonance scanning techniques described below (123,194,278). Furthermore, ectopic ACTH-secreting neuroendocrine tumors (e.g., bronchial carcinoids) can be small (< 1 cm) and difficult to identify in imaging studies. Therefore, the differential diagnosis (i.e., localization) of ACTH-secreting tumors is one of the most challenging problems in clinical endocrinology (3, 77–80, 114). Table 2 shows the different approaches that have been used. The goal here is not to delve into a long exposition on the clinical subtleties of this testing, which is usually reserved for highly experienced clinical endocrinologists. The purpose is to use this list to explore the physiology and pathophysiology of the HPA axis.
Table 2.
Methods that Have Been Used to Differentiate Cushing’s Disease (Pituitary Tumor) from Occult Ectopic ACTH
|
In actuality, these are usually only performed to look for an ectopic tumor (after a pituitary tumor has been ruled out).
For the most part, HDDST has been supplanted by the tests listed above it.
Imaging
Despite the fact that the pituitary MRI is normal in up to 50% of patients with Cushing’s disease, it should be obtained in all patients with ACTH-dependent Cushing’s syndrome. Corticotroph adenomas are hypodense on T1 SE weighted MRI and usually do not enhance after gadopentate dimeglumine (Gd-DPTA) contrast administration. Use of newer MRI techniques like the spoiled gradient recalled acquisition in a steady state improves tumor detection from 49% to 80% (194). However, incidental pituitary lesions up to 6 mm in size are detected in 10% of individuals without identifiable pituitary disease, limiting the ability of MRI to distinguish between a pituitary and ectopic source of ACTH (102). Despite this limitation, the presence of a larger mass (> 6–10 mm) strongly suggests Cushing’s disease and may obviate the need for invasive testing.
Identifying the source of ACTH in ectopic Cushing’s syndrome can be challenging. Nuclear imaging studies along with CT and MRI of the chest, abdomen, and neck facilitate localization and are described in more detail later in this review.
Inferior petrosal sinus sampling for ACTH with provocative stimulation and subsequent surgical therapy
This technique represents a major advance in the differentiation of ACTH-secreting pituitary tumors (usually small adenomas) and occult ectopic ACTH-secreting tumors (59,76,185). The physiological principle behind inferior petrosal sinus sampling (IPSS) is intuitive and illustrates classic physiological concepts (Fig. 6). If the pituitary is the primary source of ACTH excess, the concentration of ACTH in the pituitary venous effluent should be higher than in the peripheral circulation (P) (such as the inferior vena cava). Because of the ambient cortisol excess, the normal corticotrophs will be suppressed. Furthermore, oCRH is injected to stimulate the corticotroph tumor to secrete even more ACTH to maximize the gradient between the petrosal sinuses and a peripheral (e.g., inferior vena cava) sample. If the primary source of ACTH is ectopic, the ambient hypercortisolism will suppress pituitary corticotroph function so that, even under the rare circumstance in which the ectopic tumor is stimulated by oCRH, there will not be a significant gradient between the petrosal sinuses and the vena cava (Fig. 6).
Figure 6.

Summary of inferior petrosal sinus (IPS) sampling in the differential diagnosis of ACTH-dependent Cushing’s syndrome. IPS:P is the ratio of plasma ACTH concentration between an inferior petrosal sinus sample and a sample from a peripheral vein (usually the inferior vena cava) drawn simultaneously. Adapted from (77) with permission from John Wiley and Sons.
There are a few other subtleties of petrosal sinus sampling that are worth briefly mentioning. Even the most experienced neurosurgeon occasionally cannot find the ACTH-secreting microadenoma during exploration of the pituitary (6, 149, 202). Based on a small group of patients, it was proposed that a dominant IPS ACTH to nondominant IPS ACTH ratio > 1.4 might suggest that the tumor is on the dominant side of the pituitary (right vs. left) (184). However, in a larger study from the same group, this strategy correctly predicted the side of the tumor in only 69% of 501 patients (278). Thus, while this approach can be helpful, it is not good enough to reliably depend on.
Furthermore, the pituitary circulation can occasionally have anomalous venous drainage and/or anatomic deviations from normal (57,249) that lead to false negative IPSS results. Potential false negative results can be identified by peak dominant IPSS ACTH values <300 to 400 pg/mL, probably reflecting such anatomic deviation in drainage (278). Additionally, measuring prolactin in the IPS samples can verify the fidelity of the catheterization and venous drainage (75,233). Because prolactin has a very high pituitary secretion rate, it can be a marker for proper venous drainage and catheterization.
This discussion brings up another extremely important question with a physiological message. Once the source of the ACTH-secreting tumor is localized, the optimal treatment is surgical removal. If the IPS:P ratio is >3 after administration of oCRH, a neurosurgeon specializing in pituitary surgery attempts to remove the adenoma while leaving the remainder of the pituitary intact. Although it is beyond the scope of this article to describe how clinicians localize ectopic tumors, surgical removal is the optimal treatment.
So what happens to the patient once the ACTH-secreting tumor is removed, regardless of its location? Remember that the normal pituitary corticotrophs have been functionally suppressed because of cortisol negative feedback. It may take months for the corticotrophs to return to normal function. If the ACTH-secreting tumor is successfully removed, ACTH secretion will dramatically decrease because the remaining normal corticotrophs in the pituitary remain suppressed for some time. As a result, the adrenal gland will be almost immediately deprived of corticotrophic stimulation, and the cortisol levels will plummet to lethally low levels (postsurgical secondary adrenal insufficiency described below). This illustrates again how knowledge of normal physiology informs clinical medicine. These patients must be treated with exogenous glucocorticoid therapy until the suppressed pituitary corticotrophs recover. There is no clear consensus among endocrinologists on the exact dosing and management of glucocorticoid therapy after surgical cure. Our practice has been to initiate replacement glucocorticoids based on body surface area (10–12 mg/m2) and decrease the dose according to postoperative weight loss and recovery of the hypothalamic-pituitary-adrenal axis as ascertained by a 250 μg ACTH stimulation test (described later in this review) performed every 3 months. Other clinicians have used an empiric weekly or monthly tapering regimen with an ACTH stimulation test performed every 6 months (148). There is no clear evidence whether one approach is superior to the other with respect to recovery time and patient tolerability. Although in most cases the hypothalamic-pituitary-adrenal axis recovers during the first year after surgery, occasionally exogenous glucocorticoid support may be required for up to 3 years (148). The same can be said for weaning patients from long-term glucocorticoid therapy (described later in this review) or after removal of adrenal tumors secreting cortisol (54, 93).
High-dose dexamethasone suppression test
The physiological concept behind this test is that since ACTH-secreting pituitary tumors arise clonally from normal corticotrophs, they still may express the GR. On the other hand, ectopic ACTH-secreting tumors were never normal corticotrophs and should theoretically not be inhibited by GR agonist (i.e., dexamethasone). If sufficient GR agonist is given, it is conceivable that pituitary tumors will suppress and ectopic tumors will not. Unfortunately, these theoretical assumptions are not accurate.
As shown in Table 3 (logistic models 5 and 6), the diagnostic accuracy of the high-dose dexamethasone suppression test (HDDST) is poor (<80%) and is currently not recommended unless IPSS is not available (16). This is particularly so since (a) about 90% of patients with ACTH-dependent Cushing’s syndrome will have a pituitary cause and (b) routine clinical characteristics are actually more predictive so that HDDST does not improve on that accuracy (logistic models 1 and 2 compared to 3 and 4 in Table 3). One would be better off just sending all patients to pituitary surgery with the small chance that an unnecessary surgery would be performed on a patient with ectopic ACTH secretion. Like with most tests of endocrine function, there are certain practicalities such as the inability to perform IPSS that may result in use of combinations of several other tests such as peripheral CRH stimulation testing, pituitary MRI, and the HDDST (169).
Table 3.
Logistic Regression Modeling of Probability of Cushing’s Disease Based on Pathophysiological Findings, Other Variables, and the High-Dose Dexamethasone Suppression Test (HDDST)
| Model no. | Variables | Sensitivity | Specificity | Diagnostic accuracy |
|---|---|---|---|---|
| 1 | Age, sex, duration, hypokalemia, urinary free cortisol, plasma ACTH, suppression by ≤50%a | 100 | 80 | 96 |
| 2 | Age, sex, duration, hypokalemia, urinary free cortisol, plasma ACTH, % suppressionb | 98 | 80 | 94 |
| 3 | Age, sex, duration, hypokalemia, urine free cortisol, plasma ACTH | 98 | 78 | 93 |
| 4 | Duration, hypokalemia, plasma ACTH | 98 | 67 | 91 |
| 5 | Suppression by ≥50%a | 79 | 67 | 77 |
| 6 | % Suppressionb | 93 | 27 | 77 |
Suppression of cortisol after HDDST;
Suppression of cortisol (after HDDST) as a continuous variable.
Adapted from (16).
Clinical characteristics
It is clear from Figure 5 that many patients with ectopic ACTH have extremely high plasma ACTH levels that lead to prodigious hypercortisolism. These tumors tend to be unregulated and insensitive to glucocorticoid negative feedback such that ACTH, and hence cortisol secretion is unrestrained (77). As stated above, there is too much overlap between Cushing’s disease and ectopic ACTH to depend solely on plasma ACTH to differentiate the two, and clinical characteristics can be very revealing in the differential diagnosis of ACTH-dependent Cushing’s syndrome (Table 3) (114, 116).
This provides us with a great opportunity to review the physiological and pathophysiological effects of cortisol excess that can be helpful in differentiating Cushing’s disease from ectopic ACTH secretion (Table 3). Moreover, Cushing’s disease is much more common in women than men whereas there does not appear to be a gender difference in ectopic ACTH. While not all ectopic ACTH patients are older, the presence of Cushing’s syndrome after the reproductive years should prompt its consideration. As described earlier in this review, hypokalemia is primarily due to the ability of very high levels of cortisol to overwhelm the protective effects of 11BHSD2, bind to the MR in the distal nephron, and so increase potassium excretion (260). As mentioned already, very high plasma ACTH (Fig. 5), and therefore, high urinary free cortisol excretion rate, is also more common in patients with ectopic ACTH. Therefore, an older male patient with a rapid onset of symptoms, hypokalemia, and very high urinary cortisol excretion is more likely to have tumoral ectopic ACTH secretion compared to a younger woman with a more insidious onset, milder symptoms, normokalemia, and slightly elevated cortisol excretion.
Localization of ectopic ACTH-secreting tumors and subsequent treatment
We have already described how inferior petrosal sinus sampling and pituitary imaging can localize pituitary tumors. By contrast, selective venous sampling of every organ as a potential source of ACTH to look for occult ACTH-secreting ectopic tumors is not feasible. This gives us a chance to review a classic physiological concept—the Fick principle—that states that blood flow is proportional to the difference in concentration of a substance in the arterial and venous blood of an individual organ. Restated, for the same secretion rate of a substance (like ACTH), the A-V concentration difference will be smaller in organs with higher blood flows and very high in small organs with small blood flow as a percentage of the cardiac output (131). This is one reason why inferior petrosal sinus sampling works well in localizing ACTH-secreting tumors to the pituitary (60,76,113,185). Pituitary blood flow is a very small percentage of the cerebral blood flow, which, by the Fick principle, will lead to a large pituitary A-V concentration difference in ACTH when the pituitary is the source of virtually all of the circulating ACTH. With admixture of other cephalic venous drainage mixing with petrosal sinus effluent in the jugular vein, the gradient of ACTH in the jugular vein compared to the inferior vena cava is considerably less and not nearly as reliable as samples taken closer to the pituitary venous outflow (113).
Anatomical (CT and MRI) and functional (somatostatin scintigraphy and PET scans) imaging studies form the basis of localization of the source of ACTH in ectopic Cushing’s syndrome. Most of these tumors are intrathoracic (70–85%) and therefore it is logical that initial imaging studies should focus on the thorax with additional studies performed as needed. Given the small size of these tumors, as many as 50% of them may remain undetected on conventional CT and MRI scans. Improved imaging techniques including thinner (1 to 2 mm) sections on CT scan, and 3 Tesla MRI may improve tumor detection but can lead to false positive results (282). Functional imaging studies rely on specific physiological properties of these neuroendocrine tumor cells and can help improve accuracy when used along with anatomical studies. Many of the neuroendocrine tumors express somatostatin (sst) receptors on their cell surface and may be identified via somatostatin receptor scintigraphy. The ability of scintigraphy to localize these tumors depends on the type (sst 1–5) and degree of sst receptor expression, size of the tumor and the dose (6–18 mCi pentetreotide) of the radio-pharmaceutical (137, 145). In vitro studies have shown that glucocorticoids selectively downregulate sst2, and to a lesser degree sst5, receptor expression in human neuroendocrine cells (47, 193). This phenomenon was recently described in vivo as well in two patients with ACTH-secreting pulmonary carcinoids where mifepristone treatment was followed by a change in the octreotide scan status and correct tumor localization suggesting an effect of elevated functional glucocorticoid levels on tumoral sst2 receptor expression (48). This effect of antiglucocorticoid or cortisol lowering therapy on tumoral sst receptor expression and in improving the diagnostic ability of the octreotide scan to detect these tumors needs to be further tested in larger studies.
Studies have also shown that many neuroendocrine tumors take up and decarboxylate L-3,4 dihydroxyphenylalanine and (11C) 5-hydroxytryptamine (5-HT), allowing for visualization via PET scans (128, 186, 282). Given that most occult tumors are slow-growing and have low metabolic activity, fluorodeoxyglucose-PET scan has limited utility in tumor localization and is mainly useful in defining the extent of metastatic disease (189).
Because no single imaging modality identifies all tumors, correlation of different imaging studies is needed for optimal diagnostic accuracy (282). In our experience, non-IPSS venous sampling does not provide any additional information than that obtained by imaging studies and is not necessary in the evaluation of ectopic Cushing’s syndrome (58, 114). We therefore recommend that the initial evaluation of a patient with occult Cushing’s syndrome should include thin-slice thoracic CT and MRI along with standard dose (6mCi) pentetreotide scintigraphy. This is based on the fact that intrathoracic tumors are the most likely cause of ectopic ACTH secretion.
Once the ectopic source of ACTH is discovered, surgical removal is the optimal treatment, following the same logic as for pituitary corticotroph tumors described above. Postoperative glucocorticoid administration is required as in Cushing’s disease because the pituitary corticotrophs have been suppressed by the prodigious hypercortisolism from ectopic ACTH (217). Recovery of the HPA axis and weaning from glucocorticoid therapy again follows the same physiological concepts as described above for recovery of pituitary surgery for Cushing’s disease and from weaning patients with exogenous Cushing’s syndrome described below.
Ectopic CRH
As shown in Figure 1, the hypothalamic neurohormone CRH is the main stimulator of pituitary ACTH secretion in healthy individuals. The actual amount of CRH released into the portal blood per unit time (secretion rate) is small because portal blood flow is very small percentage of cerebral blood flow. Again, by the Fick principle, the concentration of CRH in the portal vein bathing the anterior pituitary will be inversely proportional to the blood flow assuming a constant secretion rate of CRH from hypothalamic nerves terminating in the median eminence. Therefore, the CRH in the systemic circulation does not reflect hypothalamic activity and is essentially background noise compared to the high concentration of CRH in portal blood derived from the paraventricular nucleus. It is theoretically possible that an ectopic tumor can release CRH in sufficient quantities to stimulate the pituitary corticotrophs, thereby causing a biochemical and clinical scenario similar to Cushing’s disease. Although this has been reported, it is extremely rare (231).
Medical treatment of endogenous Cushing’s syndrome
Optimal treatment of endogenous Cushing’s syndrome, regardless of the cause, is surgical resection of the source of glucocorticoid excess (pituitary adenoma, nonpituitary tumor secreting ACTH, or adrenal pathology) as described above. Medical control of hypercortisolemia may be needed when a source cannot be found, while awaiting surgery, when surgery is contraindicated or unsuccessful and while awaiting the effect of radiation treatment. Medical agents to control hyper-cortisolemia include compounds that inhibit steroidogenesis (ketoconazole, metyrapone, mitotane, and etomidate), modulate ACTH release (somatostatin and dopamine agonists) or block glucocorticoid action at its receptor (mifepristone) (69).
The steroidogenesis inhibitors ketoconazole and metyrapone have a rapid onset of action. Ketoconazole inhibits the first step in cortisol biosynthesis (side-chain cleavage) and to a lesser degree 11-beta-hydroxylase and 17,20 desmolase (64,236). It is usually the first-line agent for medical control of hypercortisolism but its use may be limited by gastrointestinal side-effects, hepatocellular dysfunction, and gynecomastia and decreased libido in men (140). It requires an acidic environment for maximal absorption and therefore cannot be used in combination with proton pump inhibitors.
Metyrapone inhibits 11-beta-hydroxylase and has been used as monotherapy in ectopic ACTH secretion and as adjunctive therapy with radiation therapy in Cushing’s disease (187). As a result of inhibition of 11-beta-hydroxylase, increased deoxycorticosterone (DOC) levels can lead to salt retention but rarely hypertension, and increased adrenal androgens can lead to new or worsening hirsutism and acne in women.
Mitotane has a slow onset but long-lasting action and works both as an adrenal steroidogenesis inhibitor and an adrenolytic agent (21). It is teratogenic, not well-tolerated and is primarily used in the treatment of adrenocortical carcinoma.
The parenteral agent etomidate is a substituted imidazole anesthetic agent that inhibits 11-beta-hydroxylase. It can be used for rapid control of hypercortisolemia in hospitalized patients and is the only available agent that can be used in patients who are unable to take medication by mouth (201).
One of the main concerns with all medical agents is overtreatment that can lead to adrenal insufficiency. This leads to two strategies: the first is to block cortisol production to achieve normal levels. The second is to block cortisol secretion completely and give back replacement glucocorticoid. Regardless of the choice, all patients receiving such treatment should be educated in the use of glucocorticoids in emergency.
Corticotroph adenomas can express somatostatin (sst5 and sst2) and dopamine (D2) receptors. Pituitary-directed therapy with pasireotide (a somatostatin analogue) and cabergoline (a dopamine agonist) has recently been shown to be successful in 20% to 40% of patients with Cushing’s disease (43, 90, 199). Pasireotide is more effective when the UFC is less than twice the upper limit of normal and has been associated with glucose intolerance in 73% of patients.
Mifepristone (RU486), a GR antagonist and antiprogestin, has been approved in the United States for control of hyperglycemia secondary to hypercortisolism in patients with Cushing’s syndrome who failed surgery or are not surgical candidates. It can be rapidly effective for some acute complications of Cushing’s syndrome, in particular cortisol-induced psychosis (36). Of note, cortisol levels remain unchanged or may increase during mifepristone treatment and therefore hormonal measurements cannot be used to guide medical therapy or to diagnose adrenal insufficiency.
Combination therapy with different agents may be needed to achieve normal plasma cortisol levels in patients with moderate to severe hypercortisolism and can help minimize drug-related adverse events if lower combined doses are effective. Overall, medical treatment of hypercortisolemia should be tailored to each individual patient according to patient characteristics, potential side-effects, and other pharmacological properties of the drugs (69).
Exogenous (iatrogenic, factitious) Cushing’s syndrome
Patients treated with supraphysiological doses of glucocorticoids for any number of autoimmune and inflammatory diseases can exhibit the signs and symptoms of endogenous Cushing’s syndrome (111). The pathophysiological difference is obvious—they will have suppressed hypothalamic and pituitary function due to glucocorticoid negative feedback and adrenal atrophy as a result. The assumption when performing all of the screening tests for Cushing’s syndrome described above is that the patient does not have exogenous Cushing’s syndrome. It is usually from oral glucocorticoid therapy, but can even result from high-dose inhaled or topical corticos-teroid therapy and can be exacerbated by drugs that interfere with hepatic corticosteroid metabolism (79, 207). The process of weaning these patients from glucocorticoid therapy is described in the section on adrenal insufficiency.
Adrenal Insufficiency
Definitions
The physiological definition of adrenal insufficiency is the inadequate secretion of cortisol from the adrenal gland regardless of the cause (Fig. 7). In primary adrenal insufficiency, the essential pathophysiological problem started with a malfunction of the adrenal cortex (235). In secondary adrenal insufficiency, the essential problem started with decreased ACTH stimulation of the adrenal cortex. However, due to prolonged deprivation of the adrenal cortex of corticotrophic stimulation, the adrenal cortices atrophy. This distinction is often confusing to students and appreciation of this is absolutely critical to understanding the physiology and pathophysiology of the HPA axis.
Figure 7.
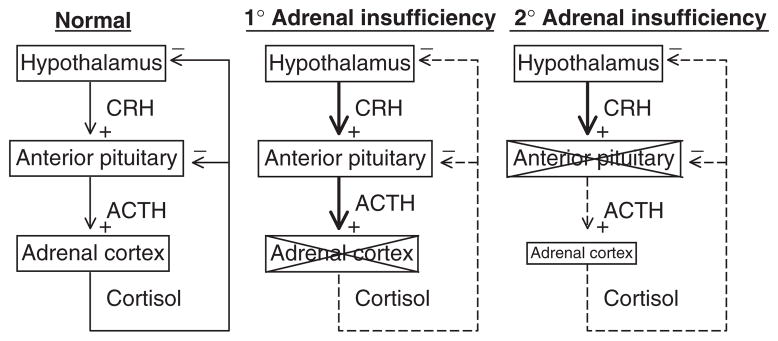
Adrenal insufficiency. In primary adrenal insufficiency, the adrenal cortex is typically destroyed (indicated by an X). This relieves the hypothalamus of cortisol negative feedback such that, presumably, corticotrophin-release hormone (CRH) is increased, although sampling portal vein blood is not possible in humans. The loss of negative feedback at the pituitary leads to a large increase in plasma ACTH. In secondary adrenal insufficiency, adequate ACTH secretion is lost (indicated by an X) resulting in suboptimal plasma ACTH and adrenal atrophy.
Primary adrenal insufficiency
Although this is popularly known as “Addison’s disease,” in actuality that is not a generic term for adrenal insufficiency. A “disease” usually has only one cause, and there are many causes of primary adrenal insufficiency (Table 4). When Thomas Addison first described this disorder in the 19th century, the most common cause was destruction of the adrenal cortex due to tuberculosis (26,52). In the 21st century in the developed parts of the world, autoimmune destruction of the adrenal cortex is more common, although tuberculosis still exists and can still cause primary adrenal insufficiency (65, 135). Either way, the term Addison’s disease is usually reserved for irreversible destruction of the adrenal cortex (like an infection or autoimmune cause). For example, although surgical removal of both adrenal glands will clearly lead to the same symptoms and pathophysiology, that is usually not called Addison’s disease. We have even heard the term Addison’s disease completely misused to describe secondary adrenal insufficiency.
Table 4.
Causes of Adrenal Insufficiency
Primary Adrenal Insufficiency
|
Secondary Adrenal Insufficiency
|
Adapted from (242).
Regardless of the cause, primary adrenal insufficiency leads to a failure to produce adequate amounts of cortisol and has an extremely high morbidity and mortality if left untreated. Also lost is the secretion of adrenal androgens (e.g., DHEA) and mineralocorticoids (aldosterone). Because the adrenal androgens are of low potency, their loss is usually not a major concern. However, the loss of aldosterone secretion does have significant pathophysiological consequences.
Secondary (and tertiary) adrenal insufficiency
As its name implies, secondary adrenal insufficiency is defined as low cortisol secretion due to a loss of normal ACTH input to the adrenal cortex. For the purposes of this review, this is associated with a decrease in function of all of the hormone-producing cells of the anterior pituitary (hypopituitarism or panhypopituitarism) or rarely is due to isolated ACTH deficiency (Table 4). As stated above, deficiency in ACTH for long periods of time results in adrenocortical atrophy. However, it is extremely important to point out that it is typically only the loss of ACTH-dependent adrenocortical function (cortisol and androgens). The zona glomerulosa—the source of aldosterone—is usually not affected in secondary adrenal insufficiency because the renin-angiotensin system remains intact (180). Therefore, although cortisol does have some mineralocorticoid action when it is in excess, adequate aldosterone secretion in secondary adrenal insufficiency is often able to maintain normal sodium and potassium balance, obviating the need for mineralocorticoid replacement therapy (discussed below).
Tertiary adrenal insufficiency can be caused by any process that involves the hypothalamus and interferes with CRH secretion. The most common causes are abrupt cessation of high-dose glucocorticoid therapy or cure of Cushing’s syndrome. High levels of glucocorticoids (endogenous or exogenous) decrease hypothalamic CRH synthesis and secretion and also block the trophic and ACTH-secretagogue actions of CRH on anterior pituitary cells. This leads to decrease in size, and eventually, number of corticotroph cells (198). The CRH stimulation test can be used to differentiate between secondary and tertiary forms of adrenal insufficiency, although there is overlap in the responses (97, 180, 229). In both conditions, the cortisol levels are low at baseline and after CRH administration. There is little or no ACTH response to CRH in secondary adrenal insufficiency while in the tertiary form, there is a delayed and exaggerated increase in ACTH after CRH with a subnormal plasma cortisol response (228, 229). The clinical presentation and treatment of tertiary adrenal insufficiency is similar to secondary adrenal insufficiency and a formal distinction between the two forms is not needed for clinical management.
Clinical features of adrenal insufficiency
The clinical features of adrenal insufficiency depend on the extent of loss of adrenal function and whether mineralocorticoid production is preserved (Table 5). The onset is often gradual and the condition may remain undiagnosed until an illness or stress precipitates an adrenal crisis.
Table 5.
Clinical Features of Adrenal Insufficiency
| Type of adrenal insufficiency | Clinical features | Frequency |
|---|---|---|
| Primary | Hyperpigmentation | 92%–100% |
| Dehydration, salt craving | 12%–20% | |
| Hyperkalemia | 64% | |
| Adrenal calcification | ||
| Vitiligo | 8%–20% | |
| Secondary or tertiary | Signs of other pituitary hormone deficiencies | |
| Delayed puberty | ||
| Headaches, visual field deficits | ||
| Both | Gastrointestinal symptoms (nausea, vomiting, abdominal pain, and diarrhea alternating with constipation) | 56%–92% |
| Hyponatremia | 88% | |
| Hypoglycemia (more common in secondary AI) | ||
| Orthostatic hypotension | 56%–90% | |
| Anorexia, weight loss | 73%–100% | |
| Fatigue, generalized malaise | 95%–100% | |
| Amenorrhea | 25% | |
| Decreased libido, loss of axillary, or pubic hair in women | ||
| Depression | 20%–40% | |
| Auricular cartilage calcification | 5% | |
| Normocytic anemia | 13%–40% | |
| Eosinophilia | 17% | |
| Hypercalcemia | 5% |
The most common symptoms in any cause of adrenal insufficiency are related to glucocorticoid insufficiency and include general malaise, weakness, fatigue, anorexia, weight loss, nausea, vomiting, abdominal pain, or diarrhea that alternates with constipation. Hyponatremia is observed in 85% to 90% of patients and is related to both mineralocorticoid deficiency and increased vasopressin action due to cortisol deficiency, as cortisol directly inhibits neurohypophysial vasopressin secretion (179, 191, 205, 206). Hypoglycemia occurs more commonly in infants and children with primary adrenal insufficiency, in isolated ACTH deficiency, and in patients with type 1 diabetes mellitus who develop adrenal insufficiency (31, 141). It may occur rarely in other adults, usually in the setting of infection, fever or alcohol intake.
Additional clinical features found in primary adrenal insufficiency reflect mineralocorticoid deficiency and hypovolemia, and include postural dizziness, hypotension/orthostasis, dehydration, and hyperkalemia. Salt craving, sometimes with massive ingestion of salt or salty food items like pickle juice and beef jerky, is a distinctive feature of primary adrenal insufficiency related to aldosterone deficiency.
Hyperpigmentation is a distinctive feature of chronic primary adrenal insufficiency and is present in nearly all patients. This results from an increased production of pro-opiomelanocortin, a pro-hormone which is then cleaved into ACTH and melanocyte-stimulating hormone (MSH) during posttranslational processing (see Fig. 10). In vitro, ACTH and alpha-MSH have been noted to be equally potent stimulators of melanogenesis (261). These melanocortins act in a paracrine/autocrine manner and regulate melanogenesis by binding to the human melanocortin-1 receptor (121, 248). Although the relative etiologic roles of ACTH and MSH in hyperpigmentation in primary adrenal insufficiency have not been well studied, it is likely to be a combination of increases in both plasma ACTH and MSH (1,252). Incidentally, hyper-pigmentation is usually not observed in ACTH-dependent Cushing’s syndrome unless plasma ACTH concentrations are extremely high, as can occur in ectopic ACTH syndrome (Fig. 5). In both primary adrenal insufficiency and Cushing’s syndrome, the resulting hyperpigmentation is more prominent in areas exposed to light (face, neck, and dorsal surface of hands), areas exposed to friction (elbows, knees, knuckles, and waist), and palmar creases. Buccal, nipple, vaginal, and anal mucosal pigmentation may also be seen. Scars acquired during the period of untreated adrenal insufficiency become permanently pigmented and nails may shows bands of hyper-pigmentation.
The clinical features of secondary or tertiary adrenal insufficiency are similar to those of primary adrenal insufficiency with a few exceptions. Hyperpigmentation is not present, as ACTH levels are not increased. Hyperkalemia and dehydration do not occur and hypotension is less prominent because the renin-angiotensin-aldosterone system is intact. However, glucocorticoids do potentiate the effects of catecholamines on the vasculature, so that some patients with secondary adrenal insufficiency may have relative hypotension (8).
Hyponatremia and water retention can occur related to an inappropriate secretion or action of vasopressin due to cortisol deficiency (179). Clinical manifestations of a pituitary or hypothalamic lesion, such as deficiency of other pituitary hormones, headaches or visual deficits may be present.
Screening tests for adrenal insufficiency
As experienced teachers, we have found that this is one of the most difficult concepts for students and trainees to understand. The devastating consequences of missing the diagnosis are so great that one needs only a small index of suspicion to look for this disorder. The screening test has to be relatively easy to execute and have a very low false negative rate. Once again, understanding the physiology of the adrenal cortex is of immense help in understanding the approach outlined before.
Unlike cortisol excess that can be detected using a late-night serum or saliva sample, it is very difficult to assess the lack of cortisol secretion in ambulatory patients. That is, one could hypothesize that, from a physiological point of view, if cortisol is increased at night in patients with Cushing’s syndrome, plasma or salivary cortisol should be low in the morning in patients with adrenal insufficiency. Unfortunately, the variability in morning cortisol levels in healthy subjects makes measuring basal plasma or salivary cortisol levels in the morning suboptimal for the detection of adrenal insufficiency (97,101,210). Therefore, it is routine to use the synthetic ACTH (1–24) (cosyntropin, synacthen) stimulation test to evaluate patients for cortisol deficiency.
The ACTH stimulation test
Although there are a variety of clinical protocols for the ACTH stimulation test, we will describe the simplest as they all illustrate the important pathophysiological principles. The test is usually performed in the morning (around 0800 h) at the circadian peak of the HPA axis. The injection of ACTH (1–24) is used to evaluate the integrity of cortisol secretory capacity.
Standard Dose
The simplest test is (a) draw a baseline sample for cortisol (and sometimes aldosterone and ACTH), (b) give a bolus injection of 250 μg of ACTH (1–24), and then (c) draw a blood sample for cortisol (and sometimes aldosterone) 30 min later (51). Some advocate also drawing a sample at 60 min after ACTH (1–24) injection (237). Regardless, an inadequate cortisol response to this pharmacological dose of ACTH (1–24) is prima facie evidence that the maximal cortisol secretory capacity of the adrenal cortex is diminished. Interestingly, this does not detect subtle abnormalities and a large volume of the adrenal cortex has to be lost before the test is reliably subnormal (173). A measurement of the aldosterone response to ACTH is helpful to document miner-alocorticoid deficiency (51,110). Finally, we usually measure plasma ACTH in the basal (preinjection) sample to differentiate primary and secondary adrenal insufficiency (see below). Although it would seem more logical to measure the cortisol response to ACTH (1–24) first, and then baseline plasma ACTH only if the cortisol response is abnormal, it is usually more convenient for the patient to do this testing at one visit.
The logic behind the ACTH stimulation test in primary adrenal insufficiency is intuitively obvious. However, why would the adrenal response to ACTH (1–24) be decreased in secondary adrenal insufficiency? Because, as described above, deprivation of the adrenal cortex of endogenous ACTH stimulation leads to adrenocortical atrophy such that, when stimulated with a pharmacological dose of ACTH (1–24), the response will be subnormal. Therefore, the ACTH stimulation test is useful for screening for both primary and established secondary adrenal insufficiency. The test may not be abnormal in early secondary adrenal insufficiency because it takes time for the adrenal to atrophy and lose sensitivity to a pharmacological dose of ACTH (45, 97, 107).
There are generally accepted cut-offs for an adequate peak serum cortisol response to the high dose of ACTH that rules out adrenal insufficiency—most advocate 18 μg/dL (500 nmol/L) (97). As described below, there are a variety of factors (such as CBG concentration) that can confound these cutoffs, not to mention variations in assay performance. Therefore, there is an art to interpreting these tests particularly when the cortisol response is at or near the cut-off to make the diagnosis of adrenal insufficiency, and the clinical scenario must always be considered.
Low-dose
The standard 250 μg dose of ACTH (1–24) is clearly pharmacological and increases plasma ACTH to levels not usually achieved from endogenous pituitary secretion, even in many patients with primary adrenal insufficiency (94, 159, 215). Because of this, it was thought that this huge dose of ACTH might lead to falsely normal testing in patients with more subtle losses of adrenal function. For that reason, a bolus low dose (1–2 μg) test was developed (9, 50, 259). From a physiological point of view, the lower dose of ACTH makes sense because plasma ACTH concentration would be increased to more physiological levels and on the steepest part of the ACTH-cortisol dose-response curve. However, there are issues with this test that have rendered it diagnostically suboptimal. First, the standard 250 μg vial of ACTH (1–24) has to be diluted in an extremely accurate way, because a ±5% difference in the concentration of ACTH (1–24) injected will lead to a large enough difference in plasma ACTH on the steepest part of the ACTH-cortisol dose response curve to lead to an unacceptable variability of the cortisol response (181). Second, we have observed that the aldosterone response to low-dose testing is not reliable. Third, the injection technique can greatly influence the plasma ACTH levels achieved and hence add to the variability of the cortisol response (165,270). Fourthly, the ACTH injection is given as a bolus. For the high dose test, this is not an issue since plasma ACTH is increased to pharmacological levels and remains so for a long time after injection (178, 215). However, for the low dose test, a bolus injection leads to a very variable concentration of plasma ACTH in the critical portion of the ACTH-cortisol dose-response curve. In fact, from a physiological point of view, the best test would be a continuous infusion of a low-dose of ACTH to achieve steady state plasma ACTH concentrations (178, 222). However, this is logistically difficult for clinicians to execute and is rarely done. Finally, because the concentration of plasma ACTH achieved is optimally in the middle of the ACTH-cortisol dose response curve, any change in the Km (shifting in the dose-response curve to the left or right) will clearly confound the results. For these and other reasons, the low-dose ACTH stimulation test has not achieved its goal of minimizing false-negative responses and has, rather, increased the number of falsely low (abnormal) results (false positives) (84, 247).
Insulin-induced hypoglycemia (insulin tolerance test)
This method to evaluate the integrity of the HPA axis, although not practical, is an excellent way to summarize the physiology of the HPA axis (Fig. 8). The general concept is that insulin injection in a fasting patient with normal insulin sensitivity will lead to significant hypoglycemia (271). This is a classic stress that leads to a dramatic increase in plasma ACTH and serum cortisol in healthy subjects. In patients with a loss of adrenocortical function (for whatever reason), the cortisol response will be inadequate (56). Notice in Figure 8 that in hypopituitarism (secondary adrenal insufficiency), there still can be a small, measurable ACTH response because the few remaining normal corticotrophs are highly active due to decreased cortisol negative feedback. Also notice that there is a significant, albeit subnormal, cortisol response in secondary adrenal insufficiency, presumably because the adrenal gland in this patient is very sensitive to small changes in ACTH. The insulin tolerance test (ITT) is particularly useful for interrogating the integrity the hypothalamic-pituitary unit since hypoglycemia leads to an increase in hypothalamic CRH and pituitary ACTH release. Although many acknowledge the ITT as the “gold standard” test for secondary adrenal insufficiency, it is a dangerous test with significant risk of neuroglycopenia (109, 120). For that reason, it is not typically performed by most clinical facilities.
Figure 8.

Typical plasma glucose, ACTH, and cortisol response to insulin-induced hypoglycemia in a healthy subject and a patient with secondary adrenal insufficiency (hypopituitarism). Notice there is a small increase in ACTH and cortisol in this patient with secondary adrenal insufficiency indicating there are still a few remaining functioning pituitary corticotrophs. To convert glucose to mmol/L, multiply mg/100 ml by 0.056; to convert ACTH to pmol/L, multiply pg/mL by 0.2202. To convert cortisol to nmol/L, multiply μg/dL by 27.59.
CRH testing
This was described earlier in this review. The physiological concept is simple and straightforward—patients with secondary adrenal insufficiency will have diminished pituitary corticotroph function and will have a diminished response to injection of synthetic ovine CRH (229). Because of the expense of ovine CRH and the overlap in responses between the groups, this test is usually only performed if measuring the plasma ACTH concentration and the cortisol response during ACTH stimulation test do not provide definitive results (173). It is also important to point out that human CRH is used outside the United States, and that oCRH seems to be more potent at least due to its longer half-life and different pharmacokinetics (176, 230).
Differentiation of endogenous primary and secondary adrenal insufficiency
Although this has been alluded to in the previous section, it is a good place to review this from a physiological point of view. Remember that in classic primary adrenal insufficiency (destruction of the adrenal cortex), all adrenocortical function is lost including cortisol, aldosterone, and androgens, whereas in secondary adrenal insufficiency, aldosterone secretion is often intact due to the control of the zona glomerulosa by the renin-angiotensin system. From symptoms alone (see Table 5), one might expect the water and electrolyte disturbances to be less severe in secondary adrenal insufficiency (180). As a result, patients with secondary adrenal insufficiency can have a more insidious presentation.
In most patients, measurement of plasma ACTH concentration in the morning is sufficient to differentiate primary and secondary adrenal insufficiency (Fig. 9). Because of the loss of glucocorticoid negative feedback and the presence of normal pituitary function, patients with primary adrenal insufficiency have increased plasma ACTH concentration, sometimes to extremely high levels (Fig. 9). This again illustrates an interesting aspect of ACTH biochemistry as described previously. Because ACTH (1–39) contains within it the sequence for alpha-melanocyte stimulating hormone (αMSH) (Fig. 10), very high levels of ACTH (1–39) and increases in MSH itself in patients with primary adrenal insufficiency can lead to hyperpigmentation (Table 5) (1, 66, 252, 261).
Figure 9.

Plasma ACTH in patients with untreated primary and secondary adrenal insufficiency, and in patients on chronic pharmacological glucocorticoid therapy. Notice that plasma ACTH is often within the reference range (blue shading) in patients with secondary adrenal insufficiency. To convert to pmol/L, multiply pg/mL by 0.2202.
Notice in Figure 9 that plasma ACTH concentration can be within the reference range in patients with secondary adrenal insufficiency. This is the other side of the same coin described earlier as to why plasma ACTH can be in the reference range in patients with ACTH-dependent Cushing’s syndrome (Fig. 5). If the patient has clinically significant loss of adrenal function due to a lack of normal ACTH input, how can plasma ACTH be within the reference range? The logic is as before. If pituitary function were normal, the plasma ACTH concentration would be greatly increased as in primary adrenal insufficiency. Thus, plasma ACTH can be “inappropriately normal” or “inappropriately not increased” in secondary adrenal insufficiency (97). The plasma ACTH levels, although in the reference range, are not sufficient to restore adrenocortical size and function. This is a very common general finding in patients with hypopituitarism: LH and FSH can be in the reference range in hypogonadotropic (secondary) hypogonadism and TSH can be in the reference range in secondary hypothyroidism (197, 234). This is an extremely difficult concept for trainees to appreciate, but it makes perfect sense from a physiological point of view.
Occasionally, the plasma ACTH concentrations and cortisol response to ACTH (1–24) are not definitive. In that case, as described above, an oCRH test, metyrapone stimulation test, or an ITT may be helpful. Metyrapone inhibits 11-beta-hydroxylase in the steroidogenic pathway (see Fig. 12), which decreases cortisol secretion and increases 11-deoxycortisol, the precursor to cortisol in the steroidogenic pathway. Traditionally, one could evaluate the loss of cortisol negative feedback by measuring the magnitude of the increase in plasma ACTH and/or 11-deoxycortisol (the substrate for 11-beta-hydroxylase) (24, 72). However, at present it is cumbersome to obtain metyrapone in the United States, so that this test is only rarely done.
Figure 12.

The normal human steroidogenic pathway. Under normal conditions, the human adrenal cortex produces only small amounts of estrone, estradiol, testosterone, and adrenostenediol. The main adrenal secretory products are aldosterone (produced in the zona glomerulosa), and cortisol, dehydroepiandrosterone (DHEA), and androstenedione (produced in the zonae fasciculata and reticularis). StAR, steroidogenic acute regulatory protein; P450scc, side-chain cleavage, HSD, hydroxysteroid dehydrogenase; P450c17, 17-hydroxylase; POR, P450 oxoreductase; P450c21, 21-hydroxylase; P450c11, 11-hydroxylase; P450aro, aromatase; 5α-red, 5-alpha-reductase; DHT, dihydrotestosterone. Reproduced from (118) with kind permission from Elsevier.
Treatment of adrenal insufficiency
The general approach to the treatment of adrenal insufficiency, regardless of the cause, is to replace the missing cortisol in as physiological a pattern as possible. Because cortisol is normally secreted in a circadian rhythm, it is most common to give cortisol (hydrocortisone) by mouth in a circadian pattern. For example, patients can take 10 mg of hydrocortisone on awakening and then 5 mg in the afternoon (166). The obvious problem with this is that cortisol normally starts to increase well before awakening (e.g., at 0400 h), and most patients do not want to wake up that early to take their morning dose. As a result, a time-release hydrocortisone capsule has been developed that can be taken at bedtime and starts to release hydrocortisone a few hours before awakening (268). This preparation is currently available in Europe as Plenadren®. Another preparation, Chronocort®, is in clinical trials in the United States. The increased cost of these preparations will probably slow their widespread use.
Some advocate the use of other synthetic forms of glucocorticoids such as prednisone or dexamethasone. Because these drugs have long half-lives, it is difficult to achieve a physiological and circadian replacement regimen and this can lead to overreplacement (68, 246). An additional drawback with dexamethasone is that the standard dose formulations in the pharmacopeia do not allow for fine-tuning.
It is often necessary to replace endogenous aldosterone in primary adrenal insufficiency. Aldosterone pills are not available; rather, the mineralocorticoid agonist 9-αfluorocortisol (fludrocortisone) is used (255). Patients with secondary adrenal insufficiency usually do not require mineralocorticoid replacement (again, because the renin-angiotensin-aldosterone system is intact).
Monitoring patients with adrenal insufficiency for under-or overreplacement of glucocorticoid (and fludrocortisone, if given) is critical and requires an appreciation of physiological control. The goal of treatment in adrenal insufficiency is to use the lowest glucocorticoid dose possible that relieves symptoms while avoiding glucocorticoid excess. The choice of replacement dose is somewhat of an art and empirical, and often requires adjustment based as a variety of factors including body weight (154). As with all oral drugs, initial doses are predicated on normal gastrointestinal absorption. The replacement dose may need to be increased if the patient has gastrointestinal disease or surgery resulting in malabsorption (154, 257).
The dose can be adjusted if there are persistent signs and symptoms of glucocorticoid deficiency while monitoring for weight gain, facial plethora, and other signs of glucocorticoid excess. Long-acting glucocorticoids, like prednisone or dex-amethasone, are more likely to be associated with increased bone loss and are not the preferred agents for glucocorticoid replacement (68). Increasing the glucocorticoid dose in an attempt to normalize ACTH levels in patients with primary adrenal insufficiency can lead to overtreatment and is not recommended. ACTH levels remain elevated in patients with primary adrenal insufficiency; the underlying reason is not clear but may represent the nonphysiologic nature of the replacement administration schedule. Realizing the importance of a proper circadian rhythm of cortisol to restore physiological function, some physicians advocate the use of normative cortisol day curves to assess the adequacy of glucocorticoid dosing (154). However, clinical assessment usually works equally well (12) and timed serum cortisol measurements are not needed for routine monitoring in the majority of patients.
The adequacy of mineralocorticoid replacement can be assessed by monitoring of blood pressure and electrolytes. Postural dizziness, salt craving, orthostatic hypotension, hyponatremia, and hyperkalemia reflect underreplacement. Tiredness and lassitude, without clear-cut hypotension, also may reflect underreplacement, and the adequacy of mineralocorticoid dosage should be assessed before increasing the glucocorticoid dose. Measurement of plasma renin activity also can be useful, especially in newly diagnosed patients until they are on a stable dose of fludrocortisone, with the goal to keep levels in the upper normal range (182, 183). It is important to note that mineralocorticoid replacement may need to be increased in hot weather due to increased salt loss in perspiration, especially in patients who are exposed to ambient temperatures (10). This should be accompanied by liberal salt intake, especially when the patient is exercising. Hypertension, edema, and hypokalemia may be signs of mineralocorticoid overreplacement. If hypertension develops, the fludrocortisone dose can be decreased gradually with close monitoring of electrolytes, but it can never be completely stopped.
Iatrogenic adrenal insufficiency
This is an extremely common and dangerous phenomenon that illustrates an extremely important physiological concept that was mentioned at the beginning of this review. When a patient with long-standing adrenal Cushing’s syndrome is treated by removal of the adrenal gland containing the tumor, the contralateral adrenal gland is atrophic due to long-standing suppression of pituitary ACTH secretion by the endogenous hypercortisolism (negative feedback). The same is true for patients receiving exogenous glucocorticoid therapy (Fig. 9). Although the patient may be Cushingoid during pharmacological glucocorticoid therapy, secondary adrenal insufficiency will rapidly ensue if the glucocorticoid therapy is abruptly stopped. This is due to the long-term suppression of the endogenous HPA axis. Patients receiving glucocorticoid therapy for extended periods of time are counseled extensively on this fact. As a result, patients have to be slowly weaned from chronic glucocorticoid therapy (111, 221).
Figure 11 shows an example of a profile of plasma ACTH and cortisol during weaning from glucocorticoid therapy. Notice first that both plasma ACTH and cortisol are suppressed when weaning starts. As the dose of glucocorticoid therapy is tapered, the hypothalamic-pituitary unit starts to recover. As a result, plasma ACTH begins to increase and starts the process of “waking up” the atrophied adrenal cortex. As plasma ACTH continues to increase, adrenal function starts to recover leading to an increase in plasma cortisol. Notice that plasma ACTH overshoots the reference range which leads to a complete recovery of the axis, in this case in about a year after starting the weaning process. The duration of this process is very variable, but probably correlates to the dose and the amount of time the patient had received glucocorticoid therapy (93, 119, 147). There are many practical aspects of this process that are not relevant for this article focusing on physiological mechanisms.
Figure 11.

Pattern of the recovery of ACTH from the pituitary and cortisol from the adrenal after discontinuation of chronic pharmacological glucocorticoid therapy. From (71) with kind permission of McGraw Hill.
Glucocorticoid resistance
Syndromes of glucocorticoid resistance are rare and can present a diagnostic challenge to clinicians (40). They illustrate the importance of understanding the physiology of cortisol negative feedback in clinical medicine. These disorders usually present with high circulating cortisol and inappropriately normal or increased ACTH levels because the hypothalamus and pituitary corticotrophs “sense” glucocorticoid negative feedback through the same steroid hormone receptors as those that respond to cortisol in peripheral tissue (see Fig. 1). Therefore, biochemically, patients with glucocorticoid resistance need to be distinguished from Cushing’s syndrome. Resistance to glucocorticoids can be categorized depending on the location of resistance (generalized, peripheral, or localized). A complete review of this topic is beyond the scope of this article. Here we briefly review the alterations in normal endocrine physiology seen in generalized glucocorticoid resistance.
Generalized glucocorticoid resistance is a familial GR-mediated disorder and is characterized by hypercortisolism in the absence of Cushingoid features (39,269). Mutations in the alpha form of the GR are the most common cause of this disorder and lead to abnormal binding of cortisol to the receptor or abnormal binding of the hormone-receptor complex to DNA, with subsequent decreased effect compared to that of a normal GR. As a result, circulating cortisol levels are less able to suppress ACTH and CRH production, causing partial resistance of the hypothalamus and pituitary to negative feedback. In response, the HPA axis is reset at a higher ACTH set point to achieve normal negative feedback. The subsequent increase in ACTH leads to increased production of DOC, cortisol, and adrenal androgens. The peripheral tissues are relatively resistant to cortisol as well such that Cushingoid features do not develop despite elevated cortisol levels. In contrast, sensitivity to mineralocorticoids and androgens is preserved. At high concentrations, DOC and cortisol can activate the MR in the renal distal tubule (as described earlier in this review). These patients, therefore, can present with signs and symptoms of mineralocorticoid (hypertension and hypokalemia). Because of adrenal androgen excess, hirsutism, acne and menstrual irregularity in women can be present (156). However, the clinical features can be quite variable.
In addition to the absence of a Cushingoid phenotype, preserved diurnal variation in cortisol levels, an increase in cortisol following hypoglycemic stress and a family history of hypercortisolism can help distinguish glucocorticoid resistance from Cushing’s syndrome. Evaluation of linear growth in children also can help make this distinction. Accelerated growth with advanced bone age (due to androgen excess) is suggestive of glucocorticoid resistance while growth retardation along with weight gain is indicative of Cushing’s syndrome (95, 155).
Treatment is reserved for patients with clinically significant disease and includes the use of long-acting glucocorticoids, and antagonists to androgens and/or mineralocorticoids. Patients with mild resistance and minimal symptoms only need intermittent monitoring to ensure that they remain clinically unaffected (267).
Congenital Adrenal Hyperplasia
As the name implies, this syndrome is usually discovered at birth and is characterized by a hyperplastic adrenal cortex. The etiology of this fascinating disorder is a very enlightening way to teach about the steroidogenic pathway, glucocorticoid negative feedback, and control of adrenal growth, as well as to describe the overall effects of steroid hormones in fetal development. Although congenital adrenal hyperplasia (CAH) can express itself in adolescence or adulthood—so-called “nonclassic” CAH, this section will focus on in utero (fetal) expression that is apparent at delivery of the affected newborn (classic CAH). The sine qua non of this disorder is the decreased functional expression of a critical component of adrenocortical steroidogenic pathway usually due to a partially inactivating mutation of a critical steroidogenic enzyme (162). To understand this, we must first describe the physiology of the normal steroidogenic pathway (Fig. 12).
Steroidogenic pathway
Steroidogenesis in the adrenal cortex (as well as the gonads) starts with cholesterol translocation from the cytoplasm to the inner membrane of the mitochondria where the first steroidogenic enzyme is located. The presence of cytoplasmic StAR protein is necessary for this rate-limiting process (122, 142, 244). Thereafter, steroidogenesis proceeds in an unregulated way—the amount of each steroid product produced is determined primarily by the amount of steroidogenic enzyme activity present. The mineralocorticoid pathway producing aldosterone occurs in the zona glomerulosa and is regulated primarily by angiotensin II and potassium and, acutely, by ACTH (2, 277).
The glucocorticoid (cortisol), mineralocorticoid (aldosterone), and adrenal androgen synthetic pathways are the focus of the pathophysiology of CAH. ACTH binds to its receptor, the MC2R, which is a G-protein coupled receptor that increases adenylyl cyclase activity and cytoplasmic cAMP concentration (see Fig. 1). Increased cAMP then leads to an activation of StAR protein and an increase in cholesterol transport into the mitochondria. The sequential steps thereafter leading to cortisol, aldosterone, DHEA, and androstenedione are shown in Figure 12. The adrenal reticularis zone is also capable of producing small amounts of estrogens and testosterone (104).
General physiological concept of cah
The overall scheme explaining CAH from a physiological point of view is shown in Figure 13. Assume that a fetus with an XX genotype (female) has a partial mutation in, for example, P450c21 shown in Figure 12. It is important to emphasize that this cannot be a completely inactivating mutation or no cortisol would be produced, which would be lethal. However, if, for example, a mutated P450c21 enzyme retained 10% of its normal activity, adequate cortisol for survival could be produced if the substrate for P450c21 (17-α-OH-progesterone) were increased tenfold and the number of steroidogenic cells (i.e., adrenal volume) were increased dramatically. This is exactly what happens in the fetus with CAH.
Figure 13.
Mechanism of virilization in female fetuses with congenital adrenal hyperplasia. An enzyme defect (usually partial; in this case to P450c21) in the steroidogenic pathway leads to decreased production of cortisol and a shift of precursors into the adrenal androgen pathway. Because cortisol negative feedback is decreased, ACTH release from the fetal pituitary gland increases. Although cortisol can eventually be normalized, it is at the expense of ACTH-stimulated adrenal hypertrophy and excess fetal adrenal androgen production. Adapted from (276) with kind permission from McGraw-Hill.
The inadequate cortisol production in the fetus provides subnormal negative feedback inhibition of the fetal hypothalamus and pituitary. This leads to an increase in fetal plasma ACTH in an attempt to normalize fetal cortisol (Fig. 1). The increase in fetal plasma ACTH increases StAR-mediated cholesterol transport into the mitochondria to acutely increase steroidogenesis in the fetal adrenal gland. In the long term, the increased ACTH leads to hyperplasia of the fetal adrenal cortex with a dramatic increase in size of the fetal adrenal gland. A consequence of this increased activity in the early steroidogenic pathway is an increase in the production of precursors for adrenal androgens (e.g., 17-α-OH-progesterone) upstream from the enzyme deficiency. This will lead to steroid precursor “spill-over” into the androgen pathway resulting in enormous increases in, for example, DHEA. Whereas DHEA is a relatively weak androgen under usual circumstances, the enormous increase in DHEA in an XX fetus can lead to virilization of the highly steroid sensitive genitalia during fetal development. As a result, an XX-fetus can be born with ambiguous genitalia—that is, it is unclear to the observer whether the newborn is a phenotypic girl or boy. If this occurs in a normally virilized XY fetus, it is typically not apparent at birth because the normal fetal testes produce testosterone, which is a much more potent androgen than DHEA. However, CAH can lead to the adrenogenital syndrome in boys in childhood; they are overly virilized due to adrenal androgens because testosterone secretion is typically low in boys from infancy until puberty (275).
All of the components of the steroidogenic pathway shown in Figure 12 have been shown to have inactivating mutations (162), although mutations in P450c21 are the most common by far. Important for this discussion is the notion that the symptoms, steroid profile, and treatment protocol can vary depending on which enzyme is mutated, and the residual amount of cortisol and aldosterone produced. This is an elegant example of how understanding the physiology and biochemistry of adrenal steroidogenesis informs the clinical diagnosis and treatment of diseases resulting from relatively common mutations.
Brief comment about treatment of the affected fetus in utero
If a woman has already given birth to an infant with CAH and, therefore, is known to carry one of the mutations described below, or if there is a pertinent family history, or the parents are known to be gene carriers, it is possible to make a diagnosis in utero as well as treat the fetus via the mother. The clinical details of this are not relevant to this article, but a general statement reveals the importance of understanding normal physiology to appreciate the clinical consequences of this disorder. In general, the fetal genotype (XX vs. XY) and presence of a mutation can be evaluated in the fetus by chorionic villus sampling (29, 220). If an XX fetus is discovered to carry one of the mutations described below, the overall goal is to reduce fetal ACTH secretion thus preventing adrenal hyperplasia, androgen excess, and virilization, while supplying adequate glucocorticoid activity to the developing fetus. The simplest treatment in this scenario is administration of low-doses of oral dexamethasone—the glucocorticoid agonist—to the pregnant woman (167, 238). The dexamethasone crosses the placenta from the maternal circulation into the fetal circulation and suppresses fetal ACTH secretion by negative feedback. Furthermore, dexamethasone (as opposed to hydrocortisone) is used because it is not metabolized by 11BHSD2 which is highly expressed in the placenta (23). In women with a history of previously giving birth to a CAH infant, glucocorticoid therapy may be started as soon as pregnancy is discovered to prevent abnormal genital development early in gestation. The dexamethasone is then discontinued if the genetic testing reveals a male, or an unaffected female. There may be some subtle long-term negative effects on the adult who was treated as a fetus, and there is a risk of causing Cushing’s syndrome in the mother. As a result, the strategy is controversial and a recent guideline suggested that this be provided only within the context of an IRB-reviewed clinical trial (163, 167, 190, 238).
Subtypes of congenital adrenal hyperplasia (Table 6)
Table 6.
Congenital Adrenal Hyperplasia
| Disorder | Affected gene and chromosome | Signs and symptoms |
|---|---|---|
| 21-Hydroxylase deficiency (classic) | CYP21 | Glucocorticoid deficiency |
| 6p21.3 | Mineralocorticoid deficiency (salt-wasting) | |
| Adrenal androgen excess (Ambiguous genitalia in females; Postnatal virilization in males) | ||
| 11-Beta-hydroxylase deficiency | CYP11B1 | Hypertension |
| 8q24.3 | Glucocorticoid deficiency | |
| Mineralocorticoid deficiency (salt-wasting) | ||
| Adrenal androgen excess (ambiguous genitalia in females; Postnatal virilization in males and females) | ||
| 3-Beta-hydroxysteroid dehydrogenase deficiency (classic) | HSD3B2 | Adrenal androgen excess (ambiguous genitalia) |
| 1p13.1 | Disordered puberty (precocious adrenarche) | |
| Cortisol deficiency (with adequate corticosterone) | ||
| 17-Alpha-hydroxylase | CYP17 | Ambiguous genitalia in males |
| 17–20 Lyase deficiency | 10q24.3 | Sexual infantilism |
| Hypertension | ||
| Cortisol deficiency |
As described above, virtually any catalytic component in the steroidogenic pathway may decrease its expression due to a mutation, and lead to CAH. Also as stated above, the most common by far is an inactivating mutation in P450c21 (21-hydroxylase deficiency). One can anticipate from Figure 12 that this will lead to a deficiency in the synthesis of cortisol and aldosterone leading to (a) increased ACTH (due to loss of cortisol negative feedback) and adrenocortical hyperplasia and (b) hyponatremia and hyperkalemia (due to a loss of aldosterone action at the distal nephron) and resultant hyperreninemia. The spillover to the androgen pathway can lead to ambiguous genitalia in XX newborns and postnatal virilization in both genders. Postnatal treatment, not surprisingly, is administration of glucocorticoid and mineralocorticoid replacement, the former resulting in suppression of ACTH and preventing androgen excess. Surgical treatment of the ambiguous genitalia is often required.
A brief comment on the diagnostic testing of 21-hydroxylase deficiency does help explain the basic physiology. In most cases, measuring an increased baseline 17-hydroxyprogestosterone (substrate for 21-hydroxylase enzyme; Fig. 12) is sufficient to make the diagnosis (238). Occasionally, ACTH stimulation is necessary to amplify the analysis of the steroidogenic intermediates shown in Figure 12. Genotyping is usually performed only in patients in whom the diagnosis is equivocal or if the patient will be referred for genetic counseling (238).
An inactivating mutation in P450c11 (11β-hydroxylase deficiency) often leads to hypertension due to increased precursors (synthesized in the zona fasciculata) with mineralocorticoid activity (e.g., 11-deoxycorticosterone) (274). Because the synthesis of aldosterone in the zona glomerulosa is catalyzed by a different isoform (CYP11B2), the ability to synthesize aldosterone is theoretically normal. However, the long-term suppression of renin secretion from the kidney due to the presence of increased mineralocorticoids from the zona fasciculata may result in hyponatremia when glucocorticoid replacement is given, leading to the need for mineralocorticoid administration. The hyperandrogenemia and ambiguous genitalia follows the same logic as 21-hydroxylase deficiency.
A mutation in 3β-HSD which encodes a noncytochrome P450 enzyme—3β-hydroxysteroid dehydrogenase—leads to decreased production of cortisol, aldosterone, androgens and estrogens (283). Cortisol deficiency leads to increased ACTH secretion and accumulation of various steroid precursors (17-alpha-hydroxypregnenolone, DHEA, and DHEA sulfate). Most patients present as neonates or in early infancy with symptoms of glucocorticoid and mineralocorticoid deficiency. Female infants may have mild virilization of their external genitalia due to high DHEA levels (158). Males present with varying degrees of failure of normal genital development ranging from hypospadias to male pseudohermaphroditism (224). Treatment involves replacing all deficient hormones.
A mutation in P450c17 leads to 17-hydroxylase/17,20 lyase deficiency (25, 91). Decreases in cortisol production due to 17-hydroxylase deficiency lead to increased ACTH secretion and accumulation of the 11-deoxysteroids including corticosterone, 11-deoxycorticosterone and 18-hydroxy-deoxycorticosterone. Impairment of 17,20 lyase activity leads to complete androgen and estrogen deficiency. These patients, therefore, present usually at the time of expected puberty with hypertension, hypokalemia and hypogonadism. Glucocorticoid deficiency may or may not be present depending on the degree of cortisol secretory defect. Female patients present with amenorrhea and absence of secondary sexual characteristics. Male patients usually have complete male pseudohermaphroditism and are often raised as girls with the disorder being recognized only when evaluation is sought for lack of pubertal development. Therapy is based on glucocorticoid replacement at doses sufficient to decrease ACTH production and normalize 11-deoxycorticosterone levels while avoiding glucocorticoid excess.
Finally, there are truly rare forms of CAH due to mutations in StAR (126) and POR (P450 oxoreductase) that are too unusual to discuss in this article focusing on physiological mechanisms.
Conclusion
Understanding clinical disorders requires a thorough understanding of basic physiology; this is particularly true in endocrinology. For the most part, most disorders of the hypothalamic-pituitary-adrenal axis can be explained by the understanding of negative feedback—one of the fundamental processes in homeostasis. The persistently increased secretion of cortisol from the adrenal gland demonstrates resistance to suppression of ACTH secretion by negative feedback, autonomous, non-ACTH-dependent production of cortisol from the adrenal gland, or resistance to the action of cortisol. Understanding negative feedback also explains the development of secondary adrenal insufficiency after removal of the source of excess ACTH or cortisol as well as the recovery of the HPA axis from exogenous glucocorticoid therapy. The signs and symptoms of Cushing’s syndrome, adrenal insufficiency, and glucocorticoid resistance require an understanding of the normal effects of cortisol via the activation of the glucocorticoid and mineralocorticoid receptors, and the effects of adrenal androgens via activation of the androgen receptors. Finally, the effects of enzyme mutations in the steroidogenic pathway elegantly illustrate the feedback control of the HPA axis, ACTH-mediated adrenocortical growth, and the effects of androgen excess on the development of the genitalia in the fetus.
Acknowledgments
The authors thank Catherine Warner for her editorial assistance.
Footnotes
This article is a U.S. government work and is in the public domain in the U.S.A.
References
- 1.Abe K, Nicholson WE, Liddle GW, Orth DN, Island DP. Normal and abnormal regulation of β-MSH in man. J Clin Inves. 1969;48:1580–1585. doi: 10.1172/JCI106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilera G. Factors controlling steroid biosynthesis in the zona glomerulosa of the adrenal. J Steroid Biochem Mol Biol. 1993;45:147–151. doi: 10.1016/0960-0760(93)90134-i. [DOI] [PubMed] [Google Scholar]
- 3.Alexandraki KI, Grossman AB. The ectopic ACTH syndrome. Rev Endocr Metab Disord. 2010;11:117–126. doi: 10.1007/s11154-010-9139-z. [DOI] [PubMed] [Google Scholar]
- 4.Alexandraki KI, Grossman AB. Is urinary free cortisol of value in the diagnosis of Cushing’s syndrome? Curr Opin Endocrinol Diabetes Obes. 2011;18:259–263. doi: 10.1097/MED.0b013e3283487193. [DOI] [PubMed] [Google Scholar]
- 5.Alexandraki KI, Kaltsas GA, Isidori AM, Akker SA, Drake WM, Chew SL, Monson JP, Besser GM, Grossman AB. The prevalence and characteristic features of cyclicity and variability in Cushing’s disease. Eur J Endocrinol. 2009;160:1011–1018. doi: 10.1530/EJE-09-0046. [DOI] [PubMed] [Google Scholar]
- 6.Alexandraki KI, Kaltsas GA, Isidori AM, Storr HL, Afshar F, Sabin I, Akker SA, Chew SL, Drake WM, Monson JP, Besser GM, Grossman AB. Long-term remission and recurrence rates in Cushing’s disease: Predictive factors in a single-centre study. Eur J Endocrinol. 2013;168:639–648. doi: 10.1530/EJE-12-0921. [DOI] [PubMed] [Google Scholar]
- 7.Allen DB. Growth suppression by glucocorticoid therapy. Endocrinol Metab Clin North Am. 1996;25:699–717. doi: 10.1016/s0889-8529(05)70348-0. [DOI] [PubMed] [Google Scholar]
- 8.Altura BM, Altura BT. Effects of local anesthetics, antihistamines, and glucocorticoids on peripheral blood flow and vascular smooth muscle. Anesthesiology. 1974;41:197–214. doi: 10.1097/00000542-197408000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Ambrosi B, Barbetta L, Re T, Passini E, Faglia G. The one microgram adrenocorticotropin test in the assessment of hypothalamic-pituitary-adrenal function. Eur J Endocrinol. 1998;139:575–579. doi: 10.1530/eje.0.1390575. [DOI] [PubMed] [Google Scholar]
- 10.Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab. 2009;94:1059–1067. doi: 10.1210/jc.2009-0032. [DOI] [PubMed] [Google Scholar]
- 11.Arlt W. Disorders of the Adrenal Cortex. In: Longo Dan L, Fauci Anthony S, Kasper Dennis L, Hauser Stephen L, Larry Jameson J, Loscalzo Joseph., editors. Harrison’s Online, Internal Medicine. 18. Chapter 342. McGraw-Hill; New York: 2012. [Google Scholar]
- 12.Arlt W, Rosenthal C, Hahner S, Allolio B. Quality of glucocorticoid replacement in adrenal insufficiency: Clinical assessment vs. timed serum cortisol measurements. Clin Endocrinol (Oxf) 2006;64:384–389. doi: 10.1111/j.1365-2265.2006.02473.x. [DOI] [PubMed] [Google Scholar]
- 13.Arnaldi G, de Keyzer Y, Gasc JM, Clauser E, Bertagna X. Vasopressin receptors modulate the pharmacological phenotypes of Cushing’s syndrome. Endocr Res. 1998;24:807–816. doi: 10.3109/07435809809032691. [DOI] [PubMed] [Google Scholar]
- 14.Arnaldi G, Scandali VM, Trementino L, Cardinaletti M, Appolloni G, Boscaro M. Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology. 2010;92 (Suppl 1):86–90. doi: 10.1159/000314213. [DOI] [PubMed] [Google Scholar]
- 15.Aron DC, Findling JW, Tyrrell JB. Glucocorticoids and adrenal androgens. In: Gardner DG, Shoback D, editors. Greenspan’s Basic and Clinical Endocrinology. McGraw-Hill; New York: 2007. [Google Scholar]
- 16.Aron DC, Raff H, Findling JW. Effectiveness versus efficacy: The limited value in clinical practice of high dose dexamethasone suppression testing in the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1997;82:1780–1785. doi: 10.1210/jcem.82.6.3991. [DOI] [PubMed] [Google Scholar]
- 17.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- 18.Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK. Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J Clin Endocrinol Metab. 2009;94:3857–3864. doi: 10.1210/jc.2008-2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbetta L, Dall’Asta C, Re T, Colombo P, Travaglini P, Ambrosi B. Androgen secretion in ectopic ACTH syndrome and in Cushing’s disease: Modifications before and after surgery. Horm Metab Res. 2001;33:596–601. doi: 10.1055/s-2001-17906. [DOI] [PubMed] [Google Scholar]
- 20.Barrou Z, Guiban D, Maroufi A, Fournier C, Dugue MA, Luton JP, Thomopoulos P. Overnight dexamethasone suppression test: Comparison of plasma and salivary cortisol measurement for the screening of Cushing’s syndrome. Eur J Endocrinol. 1996;134:93–96. doi: 10.1530/eje.0.1340093. [DOI] [PubMed] [Google Scholar]
- 21.Baudry C, Coste J, Bou KR, Silvera S, Guignat L, Guibourdenche J, Abbas H, Legmann P, Bertagna X, Bertherat J. Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol. 2012;167:473–481. doi: 10.1530/EJE-12-0358. [DOI] [PubMed] [Google Scholar]
- 22.Belaya ZE, Rozhinskaya LY, Melnichenko GA, Solodovnikov AG, Dragunova NV, Iljin AV, Dzeranova LK, Dedov II. Serum extracellular secreted antagonists of the canonical Wnt/beta-catenin signaling pathway in patients with Cushing’s syndrome. Osteoporos Int. 2013;24:2191–2199. doi: 10.1007/s00198-013-2268-y. [DOI] [PubMed] [Google Scholar]
- 23.Benediktsson R, Calder AA, Edwards CRW, Seckl JR. Placental 11β-hydroxysteroid dehydrogenase: A key regulator of fetal glucocorticoid exposure. Clin Endocrinol. 1997;46:161–166. doi: 10.1046/j.1365-2265.1997.1230939.x. [DOI] [PubMed] [Google Scholar]
- 24.Berneis K, Staub JJ, Gessler A, Meier C, Girard J, Muller B. Combined stimulation of adrenocorticotropin and compound-S by single dose metyrapone test as an outpatient procedure to assess hypothalamic-pituitary-adrenal function. J Clin Endocrinol Metab. 2002;87:5470–5475. doi: 10.1210/jc.2001-011959. [DOI] [PubMed] [Google Scholar]
- 25.Biglieri EG. 17 alpha-Hydroxylase deficiency: 1963–1966. J Clin Endocrinol Metab. 1997;82:48–50. doi: 10.1210/jcem.82.1.3653. [DOI] [PubMed] [Google Scholar]
- 26.Bishop PM. The history of the discovery of Addison’s disease. Proc R Soc Med. 1950;43:35–42. [PMC free article] [PubMed] [Google Scholar]
- 27.Bourdeau I, Bard C, Noel B, Leclerc I, Cordeau MP, Belair M, Lesage J, Lafontaine L, Lacroix A. Loss of brain volume in endogenous Cushing’s syndrome and its reversibility after correction of hypercortisolism. J Clin Endocrinol Metab. 2002;87:1949–1954. doi: 10.1210/jcem.87.5.8493. [DOI] [PubMed] [Google Scholar]
- 28.Bourdeau I, Lampron A, Costa MH, Tadjine M, Lacroix A. Adrenocorticotropic hormone-independent Cushing’s syndrome. Curr Opin Endocrinol Diabetes Obes. 2007;14:219–225. doi: 10.1097/MED.0b013e32814db842. [DOI] [PubMed] [Google Scholar]
- 29.Brambati B, Tului L. Chorionic villus sampling and amniocentesis. Curr Opin Obstet Gynecol. 2005;17:197–201. doi: 10.1097/01.gco.0000162191.22091.e9. [DOI] [PubMed] [Google Scholar]
- 30.Broderick JE, Arnold D, Kudielka BM, Kirschbaum C. Salivary cortisol sampling compliance: Comparison of patients and healthy volunteers. Psychoneuroendocrinology. 2004;29:636–650. doi: 10.1016/S0306-4530(03)00093-3. [DOI] [PubMed] [Google Scholar]
- 31.Burke CW. Adrenocortical insufficiency. Clin Endocrinol Metab. 1985;14:947–976. doi: 10.1016/s0300-595x(85)80084-0. [DOI] [PubMed] [Google Scholar]
- 32.Carroll T, Raff H, Findling JW. Late-night salivary cortisol measurement in the diagnosis of Cushing’s syndrome. Nat Clin Pract Endocrinol Metab. 2008;4:344–350. doi: 10.1038/ncpendmet0837. [DOI] [PubMed] [Google Scholar]
- 33.Carroll T, Raff H, Findling JW. Late-night salivary cortisol for the diagnosis of Cushing syndrome: A meta-analysis. Endocr Pract. 2009;15:335–342. doi: 10.4158/EP09023OR. [DOI] [PubMed] [Google Scholar]
- 34.Carroll TB, Aron DC, Findling JW, Tyrrell B. Glucocorticoids and adrenal androgens. In: Gardner DG, Shoback D, editors. Access Medicine online; Greenspan’s Basic & Clinical Endocrinology. 9. Chapter 9. McGraw-Hill Education; New York: 2011. [Google Scholar]
- 35.Carroll TB, Findling JW. Cushing’s syndrome of nonpituitary causes. Curr Opin Endocrinol Diabetes Obes. 2009;16:308–315. doi: 10.1097/MED.0b013e32832d8950. [DOI] [PubMed] [Google Scholar]
- 36.Castinetti F, Fassnacht M, Johanssen S, Terzolo M, Bouchard P, Chanson P, Do CC, Morange I, Pico A, Ouzounian S, Young J, Hahner S, Brue T, Allolio B, Conte-Devolx B. Merits and pitfalls of mifepristone in Cushing’s syndrome. Eur J Endocrinol. 2009;160:1003–1010. doi: 10.1530/EJE-09-0098. [DOI] [PubMed] [Google Scholar]
- 37.Castro M, Elias LL, Elias PC, Moreira AC. A dose-response study of salivary cortisol after dexamethasone suppression test in Cushing’s disease and its potential use in the differential diagnosis of Cushing’s syndrome. Clin Endocrinol (Oxf) 2003;59:800–805. doi: 10.1046/j.1365-2265.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 38.Castro M, Elias PC, Quidute AR, Halah FP, Moreira AC. Out-patient screening for Cushing’s syndrome: The sensitivity of the combination of circadian rhythm and overnight dexamethasone suppression salivary cortisol tests. J Clin Endocrinol Metab. 1999;84:878–882. doi: 10.1210/jcem.84.3.5521. [DOI] [PubMed] [Google Scholar]
- 39.Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: Clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572. doi: 10.1210/jc.2008-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chrousos GP, tera-Wadleigh SD, Karl M. Syndromes of glucocorticoid resistance. Ann Intern Med. 1993;119:1113–1124. doi: 10.7326/0003-4819-119-11-199312010-00009. [DOI] [PubMed] [Google Scholar]
- 41.Clow A, Hucklebridge F, Stalder T, Evans P, Thorn L. The cortisol awakening response: More than a measure of HPA axis function. Neurosci Biobehav Rev. 2010;35:97–103. doi: 10.1016/j.neubiorev.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Clow A, Hucklebridge F, Thorn L. The cortisol awakening response in context. Int Rev Neurobiol. 2010;93:153–175. doi: 10.1016/S0074-7742(10)93007-9. [DOI] [PubMed] [Google Scholar]
- 43.Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Schoenherr U, Mills D, Salgado LR, Biller BM. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med. 2012;366:914–924. doi: 10.1056/NEJMoa1105743. [DOI] [PubMed] [Google Scholar]
- 44.Cone RD, Mountjoy KG. Molecular genetics of the ACTH and melanocyte-stimulating hormone receptors. Trends Endocrinol Metab. 1993;4:242–247. doi: 10.1016/1043-2760(93)90129-3. [DOI] [PubMed] [Google Scholar]
- 45.Cunningham SK, Moore A, McKenna TJ. Normal cortisol response to corticotropin in patients with secondary adrenal failure. Arch Intern Med. 1983;143:2276–2279. [PubMed] [Google Scholar]
- 46.Dayanithi G, Antoni FA. Rapid as well as delayed inhibitory effects of glucocorticoid hormones on pituitary adrenocorticotropic hormone release are mediated by type II glucocorticoid receptors and require newly synthesized messenger ribonucleic acid as well as protein. Endocrinology. 1989;125:308–313. doi: 10.1210/endo-125-1-308. [DOI] [PubMed] [Google Scholar]
- 47.de Bruin C, Feelders RA, Waaijers AM, van Koetsveld PM, Sprij-Mooij DM, Lamberts SWJ, Hofland LJ. Differential regulation of human dopamine D2 and somatostatin receptor subtype expression by glucocorticoids in vitro. J Mol Endocrinol. 2009;42:47–56. doi: 10.1677/JME-08-0110. [DOI] [PubMed] [Google Scholar]
- 48.de Bruin C, Hofland LJ, Nieman LK, van Koetsveld PM, Waaijers AM, Sprij-Mooij DM, van Essen M, Lamberts SW, de Herder WW, Feelders RA. Mifepristone effects on tumor somatostatin receptor expression in two patients with Cushing’s syndrome due to ectopic adrenocorticotropin secretion. J Clin Endocrinol Metab. 2012;97:455–62. doi: 10.1210/jc.2011-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 50.Dickstein G, Shechner C, Nicholson WE, Rosner I, Shen-Orr Z, Adawi F, Lahav M. Adrenocorticotropin stimulation test: Effects of basal cortisol level, time of day, and suggested new sensitive low dose test. J Clin Endocrinol Metab. 1991;72:773–778. doi: 10.1210/jcem-72-4-773. [DOI] [PubMed] [Google Scholar]
- 51.Dluhy RG, Himathongkam T, Greenfield M. Rapid ACTH test with plasma aldosterone levels. Improved diagnostic discrimination. Ann Intern Med. 1974;80:693–696. doi: 10.7326/0003-4819-80-6-693. [DOI] [PubMed] [Google Scholar]
- 52.Dodds C. Thomas Addison and after. Guys Hosp Rep. 1960;109:227–239. [PubMed] [Google Scholar]
- 53.Doe RP, Vennes JA, Flink EB. Diurnal variation of 17-hydroxycorticosteroids, sodium, potassium, magnesium and creatinine in normal subjects and in cases of treated adrenal insufficiency and Cushing’s syndrome. J Clin Endocrinol Metab. 1960;20:253–265. doi: 10.1210/jcem-20-2-253. [DOI] [PubMed] [Google Scholar]
- 54.Doherty GM, Nieman LK, Cutler GB, Jr, Chrousos GP, Norton JA. Time to recovery of the hypothalamic-pituitary-adrenal axis after curative resection of adrenal tumors in patients with Cushing’s syndrome. Surgery. 1990;108:1085–1090. [PubMed] [Google Scholar]
- 55.Doi M, Sekizawa N, Tani Y, Tsuchiya K, Kouyama R, Tateno T, Izumiyama H, Yoshimoto T, Hirata Y. Late-night salivary cortisol as a screening test for the diagnosis of Cushing’s syndrome in Japan. Endocr J. 2008;55:121–126. doi: 10.1507/endocrj.k07e-023. [DOI] [PubMed] [Google Scholar]
- 56.Donald RA. Plasma immunoreactive corticotrophin and cortisol response to insulin hypoglycemia in normal subjects and patients with pituitary disease. J Clin Endocrinol Metab. 1971;32:225–231. doi: 10.1210/jcem-32-2-225. [DOI] [PubMed] [Google Scholar]
- 57.Doppman JL, Chang R, Oldfield EH, Chrousos G, Stratakis CA, Nieman LK. The hypoplastic inferior petrosal sinus: A potential source of false-negative results in petrosal sampling for Cushing’s disease. J Clin Endocrinol Metab. 1999;84:533–540. doi: 10.1210/jcem.84.2.5475. [DOI] [PubMed] [Google Scholar]
- 58.Doppman JL, Nieman L, Miller DL, Pass HI, Chang R, Cutler GB, Jr, Schaaf M, Chrousos GP, Norton JA, Ziessman HA. Ectopic adrenocorticotropic hormone syndrome: Localization studies in 28 patients. Radiology. 1989;172:115–124. doi: 10.1148/radiology.172.1.2544919. [DOI] [PubMed] [Google Scholar]
- 59.Doppman JL, Oldfield E, Krudy AG, Chrousos GP, Schulte HM, Schaaf M, Loriaux DL. Petrosal sinus sampling for Cushing syndrome: Anatomical and technical considerations. Work in progress. Radiology. 1984;150:99–103. doi: 10.1148/radiology.150.1.6316418. [DOI] [PubMed] [Google Scholar]
- 60.Doppman JL, Oldfield EH, Nieman LK. Bilateral sampling of the internal jugular vein to distinguish between mechanisms of adrenocorticotropic hormone-dependent Cushing syndrome. Ann Intern Med. 1998;128:33–36. doi: 10.7326/0003-4819-128-1-199801010-00006. [DOI] [PubMed] [Google Scholar]
- 61.Dorn LD, Burgess ES, Dubbert B, Simpson SE, Friedman T, Kling M, Gold PW, Chrousos GP. Psychopathology in patients with endogenous Cushing’s syndrome: ‘Atypical’ or melancholic features. Clin Endocrinol (Oxf) 1995;43:433–442. doi: 10.1111/j.1365-2265.1995.tb02614.x. [DOI] [PubMed] [Google Scholar]
- 62.Dupuis CC, Storr HL, Perry LA, Ho JT, Ahmed L, Ong KK, Dunger DB, Monson JP, Grossman AB, Besser GM, Savage MO. Abnormal puberty in paediatric Cushing’s disease: Relationship with adrenal androgen, sex hormone binding globulin and gonadotrophin concentrations. Clin Endocrinol (Oxf) 2007;66:838–843. doi: 10.1111/j.1365-2265.2007.02822.x. [DOI] [PubMed] [Google Scholar]
- 63.Edwards CR, Stewart PM, Burt D, Brett L, McIntyre MA, Sutanto WS, de Kloet ER, Monder C. Localisation of 11 beta-hydroxysteroid dehydrogenase—Tissue specific protector of the mineralocorticoid receptor. Lancet. 1988;2:986–989. doi: 10.1016/s0140-6736(88)90742-8. [DOI] [PubMed] [Google Scholar]
- 64.Engelhardt D, Weber MM. Therapy of Cushing’s syndrome with steroid biosynthesis inhibitors. J Steroid Biochem Mol Biol. 1994;49:261–267. doi: 10.1016/0960-0760(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 65.Erichsen MM, Lovas K, Skinningsrud B, Wolff AB, Undlien DE, Svartberg J, Fougner KJ, Berg TJ, Bollerslev J, Mella B, Carlson JA, Erlich H, Husebye ES. Clinical, immunological, and genetic features of autoim-mune primary adrenal insufficiency: Observations from a Norwegian registry. J Clin Endocrinol Metab. 2009;94:4882–4890. doi: 10.1210/jc.2009-1368. [DOI] [PubMed] [Google Scholar]
- 66.Erickson QL, Faleski EJ, Koops MK, Elston DM. Addison’s disease: The potentially life-threatening tan. Cutis. 2000;66:72–74. [PubMed] [Google Scholar]
- 67.Faggiano A, Pivonello R, Melis D, Filippella M, Di SC, Petretta M, Lombardi G, Colao A. Nephrolithiasis in Cushing’s disease: Prevalence, etiopathogenesis, and modification after disease cure. J Clin Endocrinol Metab. 2003;88:2076–2080. doi: 10.1210/jc.2002-021494. [DOI] [PubMed] [Google Scholar]
- 68.Falhammar H, Filipsson NH, Wedell A, Brismar K, Thoren M. Bone mineral density, bone markers, and fractures in adult males with congenital adrenal hyperplasia. Eur J Endocrinol. 2013;168:331–341. doi: 10.1530/EJE-12-0865. [DOI] [PubMed] [Google Scholar]
- 69.Feelders RA, Hofland LJ. Medical treatment of Cushing’s disease. J Clin Endocrinol Metab. 2013;98:425–438. doi: 10.1210/jc.2012-3126. [DOI] [PubMed] [Google Scholar]
- 70.Fehm HL, Voigt KH. Pathophysiology of Cushing’s disease. Pathobiol Annu. 1979;9:225–255. [PubMed] [Google Scholar]
- 71.Felig P, Frohman L. Endocrinology and Metabolism 4/e. McGraw-Hill; New York: 2001. p. 627. Figure 14–11. [Google Scholar]
- 72.Fiad TM, Kirby JM, Cunningham SK, McKenna TJ. The overnight single-dose metyrapone test is a simple and reliable index of the hypothalamic-pituitary-adrenal axis. Clin Endocrinol (Oxf) 1994;40:603–609. doi: 10.1111/j.1365-2265.1994.tb03011.x. [DOI] [PubMed] [Google Scholar]
- 73.Findling JW, Pinkstaff SM, Shaker JL, Raff H, Nelson JC. Pseudohypercortisoluria: Spurious elevation of urinary cortisol due to carbamazepine. The Endocrinologist. 1998;8:51–54. [Google Scholar]
- 74.Findling JW, Engeland WC, Raff H. The use of immunoradiometric assay for the measurement of ACTH in human plasma. Trends Endocrinol Metab. 1990;1:283–287. doi: 10.1016/1043-2760(90)90064-a. [DOI] [PubMed] [Google Scholar]
- 75.Findling JW, Kehoe ME, Raff H. Identification of patients with Cushing’s disease with negative pituitary adrenocorticotropin gradients during inferior petrosal sinus sampling: Prolactin as an index of pituitary venous effluent. J Clin Endocrinol Metab. 2004;89:6005–6009. doi: 10.1210/jc.2004-1378. [DOI] [PubMed] [Google Scholar]
- 76.Findling JW, Kehoe ME, Shaker JL, Raff H. Routine inferior petrosal sinus sampling in the differential diagnosis of adrenocorticotropin (ACTH)-dependent Cushing’s syndrome: Early recognition of the occult ectopic ACTH syndrome. J Clin Endocrinol Metab. 1991;73:408–413. doi: 10.1210/jcem-73-2-408. [DOI] [PubMed] [Google Scholar]
- 77.Findling JW, Raff H. Endocrine Tumors. Cambridge: Blackwell Scientific Publications; 1993. Ectopic ACTH; pp. 554–566. [Google Scholar]
- 78.Findling JW, Raff H. Newer diagnostic techniques and problems in Cushing’s disease. Endocrinol Metab Clin North Am. 1999;28:191–210. doi: 10.1016/s0889-8529(05)70063-3. [DOI] [PubMed] [Google Scholar]
- 79.Findling JW, Raff H. Diagnosis and differential diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2001;30:729–747. doi: 10.1016/s0889-8529(05)70209-7. [DOI] [PubMed] [Google Scholar]
- 80.Findling JW, Raff H. Screening and diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2005;34:385–38x. doi: 10.1016/j.ecl.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Findling JW, Raff H. Cushing’s syndrome: Important issues in diagnosis and management. J Clin Endocrinol Metab. 2006;91:3746–3753. doi: 10.1210/jc.2006-0997. [DOI] [PubMed] [Google Scholar]
- 82.Findling JW, Raff H, Aron DC. The low-dose dexamethasone suppression test: A reevaluation in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2004;89:1222–1226. doi: 10.1210/jc.2003-030207. [DOI] [PubMed] [Google Scholar]
- 83.Finken MJ, Andrews RC, Andrew R, Walker BR. Cortisol metabolism in healthy young adults: Sexual dimorphism in activities of A-ring reductases, but not 11beta-hydroxysteroid dehydrogenases. J Clin Endocrinol Metab. 1999;84:3316–3321. doi: 10.1210/jcem.84.9.6009. [DOI] [PubMed] [Google Scholar]
- 84.Fleseriu M, Gassner M, Yedinak C, Chicea L, Delashaw JB, Jr, Loriaux DL. Normal hypothalamic-pituitary-adrenal axis by high-dose cosyntropin testing in patients with abnormal response to low-dose cosyntropin stimulation: A retrospective review. Endocr Pract. 2010;16:64–70. doi: 10.4158/EP09153.OR. [DOI] [PubMed] [Google Scholar]
- 85.Funder JW. Aldosterone and mineralocorticoid receptors: A personal reflection. Mol Cell Endocrinol. 2012;350:146–150. doi: 10.1016/j.mce.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 86.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- 87.Gallo-Payet N, Payet MD. Mechanism of action of ACTH: Beyond cAMP. Microsc Res Tech. 2003;61:275–287. doi: 10.1002/jemt.10337. [DOI] [PubMed] [Google Scholar]
- 88.Gibson S, Pollock A, Littley M, Shalet S, White A. Advantages of IRMA over RIA in the measurement of ACTH. Ann Clin Biochem. 1989;26 (Pt 6):500–507. doi: 10.1177/000456328902600608. [DOI] [PubMed] [Google Scholar]
- 89.Glass AR, Zavadil AP, III, Halberg F, Cornelissen G, Schaaf M. Circadian rhythm of serum cortisol in Cushing’s disease. J Clin Endocrinol Metab. 1984;59:161–165. doi: 10.1210/jcem-59-1-161. [DOI] [PubMed] [Google Scholar]
- 90.Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol. 2010;163:709–716. doi: 10.1530/EJE-10-0382. [DOI] [PubMed] [Google Scholar]
- 91.Goldsmith O, Solomon DH, Horton R. Hypogonadism and mineralocorticoid excess. The 17-hydroxylase deficiency syndrome. N Engl J Med. 1967;277:673–677. doi: 10.1056/NEJM196709282771302. [DOI] [PubMed] [Google Scholar]
- 92.Gorman LS. The adrenal gland: Common disease states and suspected new applications. Clin Lab Sci. 2013;26:118–125. [PubMed] [Google Scholar]
- 93.Graber AL, Ney RL, Nicholson WE, Island DP, Liddle GW. Natural history of pituitary-adrenal recovery following long-term suppression with corticosteroids. Trans Assoc Am Phys. 1964;77:296–306. [PubMed] [Google Scholar]
- 94.Graybeal ML, Fang VS. Physiological dosing of exogenous ACTH. Acta Endocrinol (Copenh) 1985;108:401–406. doi: 10.1530/acta.0.1080401. [DOI] [PubMed] [Google Scholar]
- 95.Greening JE, Storr HL, McKenzie SA, Davies KM, Martin L, Grossman AB, Savage MO. Linear growth and body mass index in pediatric patients with Cushing’s disease or simple obesity. J Endocrinol Invest. 2006;29:885–887. doi: 10.1007/BF03349191. [DOI] [PubMed] [Google Scholar]
- 96.Grino M, Guillaume V, Boudouresque F, Margioris AN, Grisoli F, Jaquet P, Oliver C, Conte-Devolx B. Characterization of corticotropin-releasing hormone receptors on human pituitary corticotroph adenomas and their correlation with endogenous glucocorticoids. J Clin Endocrinol Metab. 1988;67:279–283. doi: 10.1210/jcem-67-2-279. [DOI] [PubMed] [Google Scholar]
- 97.Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab. 1994;79:923–931. doi: 10.1210/jcem.79.4.7962298. [DOI] [PubMed] [Google Scholar]
- 98.Grossman A. New uses for an old peptide: Desmopressin and Cushing’s syndrome. Clin Endocrinol (Oxf) 1993;38:461–462. doi: 10.1111/j.1365-2265.1993.tb00340.x. [DOI] [PubMed] [Google Scholar]
- 99.Guaraldi F, Salvatori R. Cushing syndrome: Maybe not so uncommon of an endocrine disease. J Am Board Fam Med. 2012;25:199–208. doi: 10.3122/jabfm.2012.02.110227. [DOI] [PubMed] [Google Scholar]
- 100.Gustafsson JA, Carlstedt-Duke J, Poellinger L, Okret S, Wikstrom AC, Bronnegard M, Gillner M, Dong Y, Fuxe K, Cintra A. Biochemistry, molecular biology, and physiology of the glucocorticoid receptor. Endocr Rev. 1987;8:185–234. doi: 10.1210/edrv-8-2-185. [DOI] [PubMed] [Google Scholar]
- 101.Hagg E, Asplund K, Lithner F. Value of basal plasma cortisol assays in the assessment of pituitary-adrenal insufficiency. Clin Endocrinol (Oxf) 1987;26:221–226. doi: 10.1111/j.1365-2265.1987.tb00780.x. [DOI] [PubMed] [Google Scholar]
- 102.Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: Occult adenomas in the general population. Ann Intern Med. 1994;120:817–820. doi: 10.7326/0003-4819-120-10-199405150-00001. [DOI] [PubMed] [Google Scholar]
- 103.Hanrahan K, McCarthy AM, Kleiber C, Lutgendorf S, Tsalikian E. Strategies for salivary cortisol collection and analysis in research with children. Appl Nurs Res. 2006;19:95–101. doi: 10.1016/j.apnr.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Hanukoglu I. Steroidogenic enzymes: Structure, function, and role in regulation of steroid hormone biosynthesis. J Steroid Biochem Mol Biol. 1992;43:779–804. doi: 10.1016/0960-0760(92)90307-5. [DOI] [PubMed] [Google Scholar]
- 105.Haskett RF. Diagnostic categorization of psychiatric disturbance in Cushing’s syndrome. Am J Psychiatry. 1985;142:911–916. doi: 10.1176/ajp.142.8.911. [DOI] [PubMed] [Google Scholar]
- 106.Hermus AR, Pieters GF, Pesman GJ, Smals AG, Benraad TJ, Kloppenborg PW. The corticotropin-releasing-hormone test versus the high-dose dexamethasone test in the differential diagnosis of Cushing’s syndrome. Lancet. 1986;2:540–544. doi: 10.1016/s0140-6736(86)90113-3. [DOI] [PubMed] [Google Scholar]
- 107.Hjortrup A, Kehlet H, Lindholm J, Stentoft P. Value of the 30-minute adrenocorticotropin (ACTH) test in demonstrating hypothalamic-pituitary-adrenocortical insufficiency after acute ACTH deprivation. J Clin Endocrinol Metab. 1983;57:668–670. doi: 10.1210/jcem-57-3-668. [DOI] [PubMed] [Google Scholar]
- 108.Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, Khosla S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: Potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140:4382–4389. doi: 10.1210/endo.140.10.7034. [DOI] [PubMed] [Google Scholar]
- 109.Holmer H, Link K, Erfurth EM. Risk for severe hypoglycaemia with unawareness in GH-deficient patients during the insulin tolerance test. Clin Endocrinol (Oxf) 2006;64:136–140. doi: 10.1111/j.1365-2265.2006.02436.x. [DOI] [PubMed] [Google Scholar]
- 110.Holst JP, Soldin SJ, Tractenberg RE, Guo T, Kundra P, Verbalis JG, Jonklaas J. Use of steroid profiles in determining the cause of adrenal insufficiency. Steroids. 2007;72:71–84. doi: 10.1016/j.steroids.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am. 2005;34:371–84. ix. doi: 10.1016/j.ecl.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 112.Huizenga NATM, Koper JW, De Lange P, Pols HAP, Stolk RP, Grobbee DE, de jong FH, Lamberts SWJ. Interperson variability but intraperson stability of baseline plasma cortisol concentrations, and its relation to feedback sensitivity of the hypothalamo-pituitary-adrenal axis to a low dose of dexamethasone in elderly individuals. J Clin Endocrinol Metab. 1998;83:47–54. doi: 10.1210/jcem.83.1.4498. [DOI] [PubMed] [Google Scholar]
- 113.Ilias I, Chang R, Pacak K, Oldfield EH, Wesley R, Doppman J, Nieman LK. Jugular venous sampling: An alternative to petrosal sinus sampling for the diagnostic evaluation of adrenocorticotropic hormone-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2004;89:3795–3800. doi: 10.1210/jc.2003-032014. [DOI] [PubMed] [Google Scholar]
- 114.Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: Twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90:4955–4962. doi: 10.1210/jc.2004-2527. [DOI] [PubMed] [Google Scholar]
- 115.Invitti C, Pecori GF, de MM, Cavagnini F. Diagnosis and management of Cushing’s syndrome: Results of an Italian multicentre study. Study Group of the Italian Society of Endocrinology on the Pathophysiology of the Hypothalamic-Pituitary-Adrenal Axis. J Clin Endocrinol Metab. 1999;84:440–448. doi: 10.1210/jcem.84.2.5465. [DOI] [PubMed] [Google Scholar]
- 116.Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newell-Price J, Reznek RH, Jenkins PJ, Monson JP, Grossman AB, Besser GM. The ectopic adrenocorticotropin syndrome: Clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91:371–377. doi: 10.1210/jc.2005-1542. [DOI] [PubMed] [Google Scholar]
- 117.Jacobson L. Hypothalamic-pituitary-adrenocortical axis regulation. Endocrinol Metab Clin North Am. 2005;34:271–92. vii. doi: 10.1016/j.ecl.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 118.Jameson JL, DeGroot LJ. Endocrinology Adult and Pediatric. Vol. 2005. Saunders, Elsevier; Philadelphia: 2010. Figure 119.9. [Google Scholar]
- 119.Jamilloux Y, Liozon E, Pugnet G, Nadalon S, Heang LK, Dumonteil S, Gondran G, Fauchais AL, Vidal E. Recovery of adrenal function after long-term glucocorticoid therapy for giant cell arteritis: A cohort study. PLoS One. 2013;8:e68713. doi: 10.1371/journal.pone.0068713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones SL, Trainer PJ, Perry L, Wass JA, Bessser GM, Grossman A. An audit of the insulin tolerance test in adult subjects in an acute investigation unit over one year. Clin Endocrinol (Oxf) 1994;41:123–128. doi: 10.1111/j.1365-2265.1994.tb03793.x. [DOI] [PubMed] [Google Scholar]
- 121.Kadekaro AL, Kanto H, Kavanagh R, Abdel-Malek Z. Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann N Y Acad Sci. 2003;994:359–65. doi: 10.1111/j.1749-6632.2003.tb03200.x. [DOI] [PubMed] [Google Scholar]
- 122.Kallen CB, Arakane F, Christenson LK, Watari H, Devoto L, Strauss JF., III Unveiling the mechanism of action and regulation of the steroidogenic acute regulatory protein. Mol Cell Endocrinol. 1998;145:39–45. doi: 10.1016/s0303-7207(98)00167-1. [DOI] [PubMed] [Google Scholar]
- 123.Kasaliwal R, Sankhe SS, Lila AR, Budyal SR, Jagtap VS, Sarathi V, Kakade H, Bandgar T, Menon PS, Shah NS. Volume interpolated 3D-spoiled gradient echo sequence is better than dynamic contrast spin echo sequence for MRI detection of corticotropin secreting pituitary microadenomas. Clin Endocrinol (Oxf) 2013;78:825–830. doi: 10.1111/cen.12069. [DOI] [PubMed] [Google Scholar]
- 124.Kassela O, Herrlicha P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 125.Katabami T, Obi R, Shirai N, Naito S, Saito N. Discrepancies in results of low-and high-dose dexamethasone suppression tests for diagnosing preclinical Cushing’s syndrome. Endocr J. 2005;52:463–469. doi: 10.1507/endocrj.52.463. [DOI] [PubMed] [Google Scholar]
- 126.Katsumata N, Kawada Y, Yamamoto Y, Noda M, Nimura A, Horikawa R, Tanaka T. A novel compound heterozygous mutation in the steroidogenic acute regulatory protein gene in a patient with congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab. 1999;84:3983–3987. doi: 10.1210/jcem.84.11.6118. [DOI] [PubMed] [Google Scholar]
- 127.Katz FH, Shannon IL. Parotid fluid cortisol and cortisone. J Clin Invest. 1969;48:848–855. doi: 10.1172/JCI106042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kauhanen S, Seppanen M, Ovaska J, Minn H, Bergman J, Korsoff P, Salmela P, Saltevo J, Sane T, Valimaki M, Nuutila P. The clinical value of [18F]fluorodihydroxyphenylalanine positron emission tomography in primary diagnosis, staging, and restaging of neuroendocrine tumors. Endocr Relat Cancer. 2009;16:255–265. doi: 10.1677/ERC-08-0229. [DOI] [PubMed] [Google Scholar]
- 129.Kaye TB, Crapo L. The Cushing syndrome: An update on diagnostic tests. Ann Intern Med. 1990;112:434–444. doi: 10.7326/0003-4819-76-3-112-6-434. [DOI] [PubMed] [Google Scholar]
- 130.Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- 131.Kety SS, Schmidt CF. The nitrous oxide method for the quantitative determination of cerebral blood flow in man: Theory, procedure and normal values. J Clin Invest. 1948;27:476–483. doi: 10.1172/JCI101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kidambi S, Raff H, Findling JW. Limitations of nocturnal salivary cortisol and urine free cortisol in the diagnosis of mild Cushing’s syndrome. Eur J Endocrinol. 2007;157:725–731. doi: 10.1530/EJE-07-0424. [DOI] [PubMed] [Google Scholar]
- 133.Kivlighan KT, Granger DA, Schwartz EB, Nelson V, Curran M, Shirtcliff EA. Quantifying blood leakage into the oral mucosa and its effects on the measurement of cortisol, dehydroepiandrosterone, and testosterone in saliva. Horm Behav. 2004;46:39–46. doi: 10.1016/j.yhbeh.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 134.Kliegman RM, Stanton BF, St Geme JW, Schor NF, Behrman RE. Nelson Textbook of Pediatrics. 19. Chapter 570. Philadelphia: Saunders-Elsevier; 2011. pp. 1930–1939. [Google Scholar]
- 135.Kong MF, Jeffcoate W. Eighty-six cases of Addison’s disease. Clin Endocrinol (Oxf) 1994;41:757–761. doi: 10.1111/j.1365-2265.1994.tb02790.x. [DOI] [PubMed] [Google Scholar]
- 136.Krieger DT, Allen W, Rizzo F, Krieger HP. Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab. 1971;32:266–284. doi: 10.1210/jcem-32-2-266. [DOI] [PubMed] [Google Scholar]
- 137.Kwekkeboom DJ, Krenning EP, Bakker WH, Oei HY, Kooij PP, Lamberts SW. Somatostatin analogue scintigraphy in carcinoid tumours. Eur J Nucl Med. 1993;20:283–292. doi: 10.1007/BF00169802. [DOI] [PubMed] [Google Scholar]
- 138.Lacroix A, Bourdeau I. Bilateral adrenal Cushing’s syndrome: Macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin North Am. 2005;34:441–58. x. doi: 10.1016/j.ecl.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 139.Lebrethon MC, Grossman AB, Afshar F, Plowman PN, Besser GM, Savage MO. Linear growth and final height after treatment for Cushing’s disease in childhood. J Clin Endocrinol Metab. 2000;85:3262–3265. doi: 10.1210/jcem.85.9.6817. [DOI] [PubMed] [Google Scholar]
- 140.Lewis JH, Zimmerman HJ, Benson GD, Ishak KG. Hepatic injury associated with ketoconazole therapy. Analysis of 33 cases. Gastroenterology. 1984;86:503–513. [PubMed] [Google Scholar]
- 141.Likhari T, Magzoub S, Griffiths MJ, Buch HN, Gama R. Screening for Addison’s disease in patients with type 1 diabetes mellitus and recurrent hypoglycaemia. Postgrad Med J. 2007;83:420–421. doi: 10.1136/pgmj.2007.058321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin D, Sugawara T, Strauss JF, III, Clark BJ, Stocco DM, Saenger P, Rogol A, Miller WL. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828–1831. doi: 10.1126/science.7892608. [DOI] [PubMed] [Google Scholar]
- 143.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lin HY, Muller YA, Hammond GL. Molecular and structural basis of steroid hormone binding and release from corticosteroid-binding globulin. Mol Cell Endocrinol. 2010;316:3–12. doi: 10.1016/j.mce.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 145.Lindsay JR, Nieman LK. Differential diagnosis and imaging in Cushing’s syndrome. Endocrinol Metab Clin North Am. 2005;34:403–21. x. doi: 10.1016/j.ecl.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 146.Lindsay JR, Shanmugam VK, Oldfield EH, Remaley AT, Nieman LK. A comparison of immunometric and radioimmunoassay measurement of ACTH for the differential diagnosis of Cushing’s syndrome. J Endocrinol Invest. 2006;29:983–988. doi: 10.1007/BF03349211. [DOI] [PubMed] [Google Scholar]
- 147.Livanou T, Ferriman D, James VH. Recovery of hypothalamo-pituitary-adrenal function after corticosteroid therapy. Lancet. 1967;2:856–859. doi: 10.1016/s0140-6736(67)92592-5. [DOI] [PubMed] [Google Scholar]
- 148.Lodish M, Dunn SV, Sinaii N, Keil MF, Stratakis CA. Recovery of the hypothalamic-pituitary-adrenal axis in children and adolescents after surgical cure of Cushing’s disease. J Clin Endocrinol Metab. 2012;97:1483–1491. doi: 10.1210/jc.2011-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lonser RR, Wind JJ, Nieman LK, Weil RJ, Devroom HL, Oldfield EH. Outcome of surgical treatment of 200 children with Cushing’s disease. J Clin Endocrinol Metab. 2013;98:892–901. doi: 10.1210/jc.2012-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Loose DS, Do YS, Chen TL, Feldman D. Demonstration of glucocorticoid receptors in the adrenal cortex: Evidence for a direct dexamethasone suppressive effect on the rat adrenal gland. Endocrinology. 1980;107:137–146. doi: 10.1210/endo-107-1-137. [DOI] [PubMed] [Google Scholar]
- 151.Ludecke DK, Westphal M, Schabet M, Hollt V. In vitro secretion of ACTH, beta-endorphin and beta-lipotropin in Cushing’s disease and Nelson’s syndrome. Horm Res. 1980;13:259–279. doi: 10.1159/000179294. [DOI] [PubMed] [Google Scholar]
- 152.Lundblad JR, Roberts JL. Regulation of proopiomelanocortin gene expression in pituitary. Endocr Rev. 1988;9:135–158. doi: 10.1210/edrv-9-1-135. [DOI] [PubMed] [Google Scholar]
- 153.Magiakou MA, Mastorakos G, Oldfield EH, Gomez MT, Doppman JL, Cutler GB, Jr, Nieman LK, Chrousos GP. Cushing’s syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med. 1994;331:629–636. doi: 10.1056/NEJM199409083311002. [DOI] [PubMed] [Google Scholar]
- 154.Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, Tucker GT, Ross RJ. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol (Oxf) 2004;61:367–375. doi: 10.1111/j.1365-2265.2004.02106.x. [DOI] [PubMed] [Google Scholar]
- 155.Malchoff CD, Javier EC, Malchoff DM, Martin T, Rogol A, Brandon D, Loriaux DL, Reardon GE. Primary cortisol resistance presenting as isosexual precocity. J Clin Endocrinol Metab. 1990;70:503–507. doi: 10.1210/jcem-70-2-503. [DOI] [PubMed] [Google Scholar]
- 156.Malchoff CD, Malchoff DM. Glucocorticoid resistance and hypersensitivity. Endocrinol Metab Clin North Am. 2005;34:315–26. viii. doi: 10.1016/j.ecl.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 157.Mandel S. Steroid myopathy. Insidious cause of muscle weakness. Postgrad Med. 1982;72:207–5. doi: 10.1080/00325481.1982.11716260. [DOI] [PubMed] [Google Scholar]
- 158.Marui S, Castro M, Latronico AC, Elias LL, Arnhold IJ, Moreira AC, Mendonca BB. Mutations in the type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol (Oxf) 2000;52:67–75. doi: 10.1046/j.1365-2265.2000.00873.x. [DOI] [PubMed] [Google Scholar]
- 159.Mayenknecht J, Diederich S, Bahr V, Plockinger U, Oelkers W. Comparison of low and high dose corticotropin stimulation tests in patients with pituitary disease. J Clin Endocrinol Metab. 1998;83:1558–1562. doi: 10.1210/jcem.83.5.4831. [DOI] [PubMed] [Google Scholar]
- 160.McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 161.Meikle AW, Findling J, Kushnir MM, Rockwood AL, Nelson GJ, Terry AH. Pseudo-Cushing syndrome caused by fenofibrate interference with urinary cortisol assayed by high-performance liquid chromatography. J Clin Endocrinol Metab. 2003;88:3521–3524. doi: 10.1210/jc.2003-030234. [DOI] [PubMed] [Google Scholar]
- 162.Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–2136. doi: 10.1016/S0140-6736(05)66736-0. [DOI] [PubMed] [Google Scholar]
- 163.Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: Risks outweigh benefits. Am J Obstet Gynecol. 2013;208:354–359. doi: 10.1016/j.ajog.2012.10.885. [DOI] [PubMed] [Google Scholar]
- 164.Mosnier-Pudar H, Thomopoulos P, Bertagna X, Fournier C, Guiban D, Luton JP. Long-distance and long-term follow-up of a patient with intermittent Cushing’s disease by salivary cortisol measurements. Eur J Endocrinol. 1995;133:313–316. doi: 10.1530/eje.0.1330313. [DOI] [PubMed] [Google Scholar]
- 165.Murphy H, Livesey J, Espiner EA, Donald RA. The low dose ACTH test–a further word of caution. J Clin Endocrinol Metab. 1998;83:712–713. doi: 10.1210/jc.83.2.712-a. [DOI] [PubMed] [Google Scholar]
- 166.Neary N, Nieman L. Adrenal insufficiency: Etiology, diagnosis and treatment. Curr Opin Endocrinol Diabetes Obes. 2010;17:217–223. doi: 10.1097/MED.0b013e328338f608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.New MI, Carlson A, Obeid J, Marshall I, Cabrera MS, Goseco A, Lin-Su K, Putnam AS, Wei JQ, Wilson RC. Prenatal diagnosis for congenital adrenal hyperplasia in 532 pregnancies. J Clin Endocrinol Metab. 2001;86:5651–5657. doi: 10.1210/jcem.86.12.8072. [DOI] [PubMed] [Google Scholar]
- 168.Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605–1617. doi: 10.1016/S0140-6736(06)68699-6. [DOI] [PubMed] [Google Scholar]
- 169.Newell-Price J, Trainer P, Besser M, Grossman A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647–672. doi: 10.1210/edrv.19.5.0346. [DOI] [PubMed] [Google Scholar]
- 170.Newell-Price J, Trainer P, Perry L, Wass J, Grossman A, Besser M. A single sleeping midnight cortisol has 100% sensitivity for the diagnosis of Cushing’s syndrome. Clin Endocrinol (Oxf) 1995;43:545–550. doi: 10.1111/j.1365-2265.1995.tb02918.x. [DOI] [PubMed] [Google Scholar]
- 171.Nickelsen T, Lissner W, Schoffling K. The dexamethasone suppression test and long-term contraceptive treatment: Measurement of ACTH or salivary cortisol does not improve the reliability of the test. Exp Clin Endocrinol. 1989;94:275–280. doi: 10.1055/s-0029-1210910. [DOI] [PubMed] [Google Scholar]
- 172.Nieman LK. Diagnostic tests for Cushing’s syndrome. Ann N Y Acad Sci. 2002;970:112–118. doi: 10.1111/j.1749-6632.2002.tb04417.x. [DOI] [PubMed] [Google Scholar]
- 173.Nieman LK. Dynamic evaluation of adrenal hypofunction. J Endocrinol Invest. 2003;26:74–82. [PubMed] [Google Scholar]
- 174.Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab. 2010;95:4106–4113. doi: 10.1210/jc.2010-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing’s syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526–1540. doi: 10.1210/jc.2008-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nieman LK, Loriaux DL. Corticotropin-releasing hormone: Clinical applications. Annu Rev Med. 1989;40:331–339. doi: 10.1146/annurev.me.40.020189.001555. [DOI] [PubMed] [Google Scholar]
- 177.Nugent CA, Nichols T, Tyler FH. Diagnosis of cushing’s syndrome; single dose dexamethasone suppression test. Arch Intern Med. 1965;116:172–176. doi: 10.1001/archinte.1965.03870020012006. [DOI] [PubMed] [Google Scholar]
- 178.Nye EJ, Grice JE, Hockings GI, Strakosch CR, Crosbie GV, Walters MM, Jackson RV. Comparison of adrenocorticotropin (ACTH) stimulation tests and insulin hypoglycemia in normal humans: Low dose, standard high dose, and 8-hour ACTH-(1–24) infusion tests. J Clin Endocrinol Metab. 1999;84:3648–3655. doi: 10.1210/jcem.84.10.6062. [DOI] [PubMed] [Google Scholar]
- 179.Oelkers W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med. 1989;321:492–496. doi: 10.1056/NEJM198908243210802. [DOI] [PubMed] [Google Scholar]
- 180.Oelkers W. Adrenal insufficiency. N Engl J Med. 1996;335:1206–1212. doi: 10.1056/NEJM199610173351607. [DOI] [PubMed] [Google Scholar]
- 181.Oelkers W, Boelke T, Bahr V. Dose-response relationships between plasma adrenocorticotropin (ACTH), cortisol, aldosterone, and 18-hydroxycorticosterone after injection of ACTH-(1–39) or human corticotropin-releasing hormone in man. J Clin Endocrinol Metab. 1988;66:181–186. doi: 10.1210/jcem-66-1-181. [DOI] [PubMed] [Google Scholar]
- 182.Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab. 1992;75:259–264. doi: 10.1210/jcem.75.1.1320051. [DOI] [PubMed] [Google Scholar]
- 183.Oelkers W, L’age M. Control of mineralocorticoid substitution in Addison’s disease by plasma renin measurement. Klin Wochenschr. 1976;54:607–612. doi: 10.1007/BF01469025. [DOI] [PubMed] [Google Scholar]
- 184.Oldfield EH, Chrousos GP, Schulte HM, Schaaf M, McKeever PE, Krudy AG, Cutler GB, Jr, Loriaux DL, Doppman JL. Preoperative lateralization of ACTH-secreting pituitary microadenomas by bilateral and simultaneous inferior petrosal venous sinus sampling. N Engl J Med. 1985;312:100–103. doi: 10.1056/NEJM198501103120207. [DOI] [PubMed] [Google Scholar]
- 185.Oldfield EH, Doppman JL, Nieman LK, Chrousos GP, Miller DL, Katz DA, Cutler GB, Jr, Loriaux DL. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N Engl J Med. 1991;325:897–905. doi: 10.1056/NEJM199109263251301. [DOI] [PubMed] [Google Scholar]
- 186.Orlefors H, Sundin A, Garske U, Juhlin C, Oberg K, Skogseid B, Langstrom B, Bergstrom M, Eriksson B. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392–3400. doi: 10.1210/jc.2004-1938. [DOI] [PubMed] [Google Scholar]
- 187.Orth DN. Metyrapone is useful only as adjunctive therapy in Cushing’s disease. Ann Intern Med. 1978;89:128–130. doi: 10.7326/0003-4819-89-1-128. [DOI] [PubMed] [Google Scholar]
- 188.Orth DN, Liddle GW. Results of treatment in 108 patients with Cushing’s syndrome. N Engl J Med. 1971;285:243–247. doi: 10.1056/NEJM197107292850501. [DOI] [PubMed] [Google Scholar]
- 189.Pacak K, Ilias I, Chen CC, Carrasquillo JA, Whatley M, Nieman LK. The role of [(18)F]fluorodeoxyglucose positron emission tomography and [(111)In]-diethylenetriaminepentaacetate-D-Phe-pentetreotide scintigraphy in the localization of ectopic adrenocorticotropin-secreting tumors causing Cushing’s syndrome. J Clin Endocrinol Metab. 2004;89:2214–2221. doi: 10.1210/jc.2003-031812. [DOI] [PubMed] [Google Scholar]
- 190.Pang S, Clark AT, Freeman LC, Dolan LM, Immken L, Mueller OT, Stiff D, Shulman DI. Maternal side effects of prenatal dexamethasone therapy for fetal congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1992;75:249–253. doi: 10.1210/jcem.75.1.1619017. [DOI] [PubMed] [Google Scholar]
- 191.Papanek PE, Sladek CD, Raff H. Corticosterone inhibition of osmotically stimulated vasopressin from hypothalamic-neurohypophysial explants. Am J Physiol. 1997;272:R158–R162. doi: 10.1152/ajpregu.1997.272.1.R158. [DOI] [PubMed] [Google Scholar]
- 192.Papanicolaou DA, Yanovski JA, Cutler GB, Jr, Chrousos GP, Nieman LK. A single midnight serum cortisol measurement distinguishes Cushing’s syndrome from pseudo-Cushing states. J Clin Endocrinol Metab. 1998;83:1163–1167. doi: 10.1210/jcem.83.4.4733. [DOI] [PubMed] [Google Scholar]
- 193.Park S, Kamegai J, Kineman RD. Role of glucocorticoids in the regulation of pituitary somatostatin receptor subtype (sst1-sst5) mRNA levels: Evidence for direct and somatostatin-mediated effects. Neuroendocrinology. 2003;78:163–75. doi: 10.1159/000072798. [DOI] [PubMed] [Google Scholar]
- 194.Patronas N, Bulakbasi N, Stratakis CA, Lafferty A, Oldfield EH, Doppman J, Nieman LK. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003;88:1565–1569. doi: 10.1210/jc.2002-021438. [DOI] [PubMed] [Google Scholar]
- 195.Perogamvros I, Keevil BG, Ray DW, Trainer PJ. Salivary cortisone is a potential biomarker for serum free cortisol. J Clin Endocrinol Metab. 2010;95:4951–4958. doi: 10.1210/jc.2010-1215. [DOI] [PubMed] [Google Scholar]
- 196.Perogamvros I, Ray DW, Trainer PJ. Regulation of cortisol bioavailability—Effects on hormone measurement and action. Nat Rev Endocrinol. 2012;8:717–727. doi: 10.1038/nrendo.2012.134. [DOI] [PubMed] [Google Scholar]
- 197.Persani L. Clinical review: Central hypothyroidism: Pathogenic, diagnostic, and therapeutic challenges. J Clin Endocrinol Metab. 2012;97:3068–3078. doi: 10.1210/jc.2012-1616. [DOI] [PubMed] [Google Scholar]
- 198.Phifer RF, Orth DN, Spicer SS. Specific demonstration of the human hypophyseal adrenocortico-melanotropic (ACTH-MSH) cell. J Clin Endocrinol Metab. 1974;39:684–692. doi: 10.1210/jcem-39-4-684. [DOI] [PubMed] [Google Scholar]
- 199.Pivonello R, De Martino MC, Cappabianca P, De LM, Faggiano A, Lombardi G, Hofland LJ, Lamberts SW, Colao A. The medical treatment of Cushing’s disease: Effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab. 2009;94:223–230. doi: 10.1210/jc.2008-1533. [DOI] [PubMed] [Google Scholar]
- 200.Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, Lombardi G, Colao A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010;92(Suppl 1):77–81. doi: 10.1159/000314319. [DOI] [PubMed] [Google Scholar]
- 201.Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: A review. Eur J Endocrinol. 2012;167:137–143. doi: 10.1530/EJE-12-0274. [DOI] [PubMed] [Google Scholar]
- 202.Prevedello DM, Pouratian N, Sherman J, Jane JA, Jr, Vance ML, Lopes MB, Laws ER., Jr Management of Cushing’s disease: Outcome in patients with microadenoma detected on pituitary magnetic resonance imaging. J Neurosurg. 2008;109:751–759. doi: 10.3171/JNS/2008/109/10/0751. [DOI] [PubMed] [Google Scholar]
- 203.Putignano P, Dubini A, Toja P, Invitti C, Bonfanti S, Redaelli G, Zappulli D, Cavagnini F. Salivary cortisol measurement in normal-weight, obese and anorexic women: Comparison with plasma cortisol. Eur J Endocrinol. 2001;145:165–171. doi: 10.1530/eje.0.1450165. [DOI] [PubMed] [Google Scholar]
- 204.Qureshi AC, Bahri A, Breen LA, Barnes SC, Powrie JK, Thomas SM, Carroll PV. The influence of the route of oestrogen administration on serum levels of cortisol-binding globulin and total cortisol. Clin Endocrinol (Oxf) 2007;66:632–635. doi: 10.1111/j.1365-2265.2007.02784.x. [DOI] [PubMed] [Google Scholar]
- 205.Raff H. Glucocorticoid inhibition of neurohypophysial vasopressin secretion. Am J Physiol. 1987;252:R635–R644. doi: 10.1152/ajpregu.1987.252.4.R635. [DOI] [PubMed] [Google Scholar]
- 206.Raff H. Interactions between neurohypophysial hormones and the ACTH-adrenocortical axis. Ann N Y Acad Sci. 1993;689:411–425. doi: 10.1111/j.1749-6632.1993.tb55564.x. [DOI] [PubMed] [Google Scholar]
- 207.Raff H. Suppression of the hypothalamic-pituitary-adrenal axis and other systemic effects of inhaled corticosteroids in asthma. The Endocrinologist. 1998;8:9–14. [Google Scholar]
- 208.Raff H. Salivary cortisol: A useful measurement in the diagnosis of Cushing’s syndrome and the evaluation of the hypothalamic-pituitary adrenal axis. The Endocrinologist. 2000;10:9–17. [Google Scholar]
- 209.Raff H. The role of salivary cortisol determinations in the diagnosis of Cushing’s syndrome. Curr Opin Endocrinol Diabetes. 2004;11:271–275. [Google Scholar]
- 210.Raff H. Utility of salivary cortisol measurements in Cushing’s syndrome and adrenal insufficiency. J Clin Endocrinol Metab. 2009;94:3647–3655. doi: 10.1210/jc.2009-1166. [DOI] [PubMed] [Google Scholar]
- 211.Raff H. Cushing’s syndrome: Diagnosis and surveillance using salivary cortisol. Pituitary. 2012;15:64–70. doi: 10.1007/s11102-011-0333-0. [DOI] [PubMed] [Google Scholar]
- 212.Raff H. Update on late-night salivary cortisol for the diagnosis of Cushing’s syndrome: Methodological considerations. Endocrine. 2013;44:346–349. doi: 10.1007/s12020-013-0013-0. [DOI] [PubMed] [Google Scholar]
- 213.Raff H, Findling JW. A new immunoradiometric assay for corticotropin evaluated in normal subjects and patients with Cushing’s syndrome. Clin Chem. 1989;35:596–600. [PubMed] [Google Scholar]
- 214.Raff H, Findling JW. A physiologic approach to diagnosis of the Cushing syndrome. Ann Intern Med. 2003;138:980–991. doi: 10.7326/0003-4819-138-12-200306170-00010. [DOI] [PubMed] [Google Scholar]
- 215.Raff H, Findling JW, Wong J. Short loop adrenocorticotropin (ACTH) feedback after ACTH-(1–24) injection in man is an artifact of the immunoradiometric assay. J Clin Endocrinol Metab. 1989;69:678–680. doi: 10.1210/jcem-69-3-678. [DOI] [PubMed] [Google Scholar]
- 216.Raff H, Raff JL, Findling JW. Late-night salivary cortisol as a screening test for Cushing’s syndrome. J Clin Endocrinol Metab. 1998;83:2681–2686. doi: 10.1210/jcem.83.8.4936. [DOI] [PubMed] [Google Scholar]
- 217.Raff H, Shaker JL, Seifert PE, Werner PH, Hazelrigg SR, Findling JW. Intraoperative measurement of adrenocorticotropin (ACTH) during removal of ACTH-secreting bronchial carcinoid tumors. J Clin Endocrinol Metab. 1995;80:1036–1039. doi: 10.1210/jcem.80.3.7883819. [DOI] [PubMed] [Google Scholar]
- 218.Raff H, Singh RJ. Measurement of late-night salivary cortisol and cortisone by LC-MS/MS to assess preanalytical sample contamination with topical hydrocortisone. Clin Chem. 2012;58:947–948. doi: 10.1373/clinchem.2012.182717. [DOI] [PubMed] [Google Scholar]
- 219.Rebuffe-Scrive M, Krotkiewski M, Elfverson J, Bjorntorp P. Muscle and adipose tissue morphology and metabolism in Cushing’s syndrome. J Clin Endocrinol Metab. 1988;67:1122–1128. doi: 10.1210/jcem-67-6-1122. [DOI] [PubMed] [Google Scholar]
- 220.Rhoads GG, Jackson LG, Schlesselman SE, de la Cruz FF, Desnick RJ, Golbus MS, Ledbetter DH, Lubs HA, Mahoney MJ, Pergament E, Simpson JL, Carpenter RJ, Elias S, Ginsberg NA, Goldberg JD, Hobbins JC, Lynch L, Shiono PH, Wapner RJ, Zachary JM. The safety and efficacy of chorionic villus sampling for early prenatal diagnosis of cytogenetic abnormalities. N Engl J Med. 1989;320:609–617. doi: 10.1056/NEJM198903093201001. [DOI] [PubMed] [Google Scholar]
- 221.Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am. 2002;31:751–778. doi: 10.1016/s0889-8529(02)00008-7. [DOI] [PubMed] [Google Scholar]
- 222.Rose LI, Williams GH, Jagger PI, Lauler DP. The 48-hour adrenocorticotrophin infusion test for adrenocortical insufficiency. Ann Intern Med. 1970;73:49–54. doi: 10.7326/0003-4819-73-1-49. [DOI] [PubMed] [Google Scholar]
- 223.Rousseau GG. Fifty years ago: The quest for steroid hormone receptors. Mol Cell Endocrinol. 2013;375:10–13. doi: 10.1016/j.mce.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 224.Russell AJ, Wallace AM, Forest MG, Donaldson MD, Edwards CR, Sutcliffe RG. Mutation in the human gene for 3 beta-hydroxysteroid dehydrogenase type II leading to male pseudohermaphroditism without salt loss. J Mol Endocrinol. 1994;12:225–237. doi: 10.1677/jme.0.0120225. [DOI] [PubMed] [Google Scholar]
- 225.Sage D, Maurel D, Bosler O. Involvement of the suprachiasmatic nucleus in diurnal ACTH and corticosterone responsiveness to stress. Am J Physiol Endocrinol Metab. 2001;280:E260–E269. doi: 10.1152/ajpendo.2001.280.2.E260. [DOI] [PubMed] [Google Scholar]
- 226.Saito E, Ichikawa Y, Homma M. Direct inhibitory effect of dexamethasone on steroidogenesis of human adrenal in vivo. J Clin Endocrinol Metab. 1979;48:861–863. doi: 10.1210/jcem-48-5-861. [DOI] [PubMed] [Google Scholar]
- 227.Schaaf MJM, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83:37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 228.Schlaghecke R, Kornely E, Santen RT, Ridderskamp P. The effect of long-term glucocorticoid therapy on pituitary-adrenal responses to exogenous corticotropin-releasing hormone. N Engl J Med. 1992;326:226–230. doi: 10.1056/NEJM199201233260403. [DOI] [PubMed] [Google Scholar]
- 229.Schulte HM, Chrousos GP, Avgerinos P, Oldfield EH, Gold PW, Cutler GB, Jr, Loriaux DL. The corticotropin-releasing hormone stimulation test: A possible aid in the evaluation of patients with adrenal insufficiency. J Clin Endocrinol Metab. 1984;58:1064–1067. doi: 10.1210/jcem-58-6-1064. [DOI] [PubMed] [Google Scholar]
- 230.Schürmeyer TH, Schulte HM, Avgerinos PC, Tomai TP, Loriaux DL, Gold PW, Chrousos GP. Pharmacology of ovine and human CRH. Horm Metab Res. 1987;16:24–30. [PubMed] [Google Scholar]
- 231.Shahani S, Nudelman RJ, Nalini R, Kim HS, Samson SL. Ectopic corticotropin-releasing hormone (CRH) syndrome from metastatic small cell carcinoma: A case report and review of the literature. Diagn Pathol. 2010;5:56. doi: 10.1186/1746-1596-5-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sharma ST, Nieman LK. Cushing’s syndrome: All variants, detection, and treatment. Endocrinol Metab Clin North Am. 2011;40:379–391. doi: 10.1016/j.ecl.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sharma ST, Raff H, Nieman LK. Prolactin as a marker of successful catheterization during IPSS in patients with ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2011;96:3687–3694. doi: 10.1210/jc.2011-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Silveira LF, Latronico AC. Approach to the patient with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013;98:1781–1788. doi: 10.1210/jc.2012-3550. [DOI] [PubMed] [Google Scholar]
- 235.Simpson SL. Addison’s disease. Br Med J. 1950;2:1164–1166. doi: 10.1136/bmj.2.4689.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sonino N. The use of ketoconazole as an inhibitor of steroid production. N Engl J Med. 1987;317:812–818. doi: 10.1056/NEJM198709243171307. [DOI] [PubMed] [Google Scholar]
- 237.Speckart PF, Nicoloff JT, Bethune JE. Screening for adrenocortical insufficiency with cosyntropin (synthetic ACTH) Arch Intern Med. 1971;128:761–763. [PubMed] [Google Scholar]
- 238.Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HF, Miller WL, Montori VM, Oberfield SE, Ritzen M, White PC. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133–4160. doi: 10.1210/jc.2009-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4:525–533. doi: 10.1038/ncprheum0898. [DOI] [PubMed] [Google Scholar]
- 240.Starkman MN, Gebarski SS, Berent S, Schteingart DE. Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing’s syndrome. Biol Psychiatry. 1992;32:756–765. doi: 10.1016/0006-3223(92)90079-f. [DOI] [PubMed] [Google Scholar]
- 241.Starkman MN, Schteingart DE. Neuropsychiatric manifestations of patients with Cushing’s syndrome. Relationship to cortisol and adrenocorticotropic hormone levels. Arch Intern Med. 1981;141:215–219. [PubMed] [Google Scholar]
- 242.Stewart PM. The Adrenal Cortex. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams Textbook of Endocrinology. Chapter 14. Philadelphia: Saunders Elsevier; 2008. [Google Scholar]
- 243.Stewart PM, Whorwood CB, Walker BR. Steroid hormones and hypertension: The cortisolcortisone shuttle. Steroids. 1993;58:614–620. doi: 10.1016/0039-128x(93)90104-u. [DOI] [PubMed] [Google Scholar]
- 244.Stocco DM, Clark BJ. Regulation of the acute production of steroids in steroidogenic cells. Endocr Rev. 1996;17:221–244. doi: 10.1210/edrv-17-3-221. [DOI] [PubMed] [Google Scholar]
- 245.Stratakis CA. Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am. 2012;41:793–803. doi: 10.1016/j.ecl.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Suliman AM, Freaney R, Smith TP, McBrinn Y, Murray B, McKenna TJ. The impact of different glucocorticoid replacement schedules on bone turnover and insulin sensitivity in patients with adrenal insufficiency. Clin Endocrinol (Oxf) 2003;59:380–387. doi: 10.1046/j.1365-2265.2003.01860.x. [DOI] [PubMed] [Google Scholar]
- 247.Suliman AM, Smith TP, Labib M, Fiad TM, McKenna TJ. The low-dose ACTH test does not provide a useful assessment of the hypothalamic-pituitary-adrenal axis in secondary adrenal insufficiency. Clin Endocrinol (Oxf) 2002;56:533–539. doi: 10.1046/j.1365-2265.2002.01509.x. [DOI] [PubMed] [Google Scholar]
- 248.Suzuki I, Cone RD, Im S, Nordlund J, Abdel-Malek ZA. Binding of melanotropic hormones to the melanocortin receptor MC1R on human melanocytes stimulates proliferation and melanogenesis. Endocrinology. 1996;137:1627–33. doi: 10.1210/endo.137.5.8612494. [DOI] [PubMed] [Google Scholar]
- 249.Swearingen B, Katznelson L, Miller K, Grinspoon S, Waltman A, Dorer DJ, Klibanski A, Biller BM. Diagnostic errors after inferior petrosal sinus sampling. J Clin Endocrinol Metab. 2004;89:3752–3763. doi: 10.1210/jc.2003-032249. [DOI] [PubMed] [Google Scholar]
- 250.Talbot JA, Kane JW, White A. Analytical and clinical aspects of adrenocorticotrophin determination. Ann Clin Biochem. 2003;40:453–471. doi: 10.1258/000456303322326371. [DOI] [PubMed] [Google Scholar]
- 251.Tataranni PA, Larson DE, Snitker S, Young JB, Flatt JP, Ravussin E. Effects of glucocorticoids on energy metabolism and food intake in humans. Am J Physiol. 1996;271:E317–E325. doi: 10.1152/ajpendo.1996.271.2.E317. [DOI] [PubMed] [Google Scholar]
- 252.Ten S, New M, Maclaren N. Addison’s disease 2001. J Clin Endocrinol Metab. 2001;86:2909–2922. doi: 10.1210/jcem.86.7.7636. [DOI] [PubMed] [Google Scholar]
- 253.Terzolo M, Reimondo G, Ali A, Borretta G, Cesario F, Pia A, Paccotti P, Angeli A. The limited value of the desmopressin test in the diagnostic approach to Cushing’s syndrome. Clin Endocrinol (Oxf) 2001;54:609–616. doi: 10.1046/j.1365-2265.2001.01260.x. [DOI] [PubMed] [Google Scholar]
- 254.Terzolo M, Reimondo G, Chiodini I, Castello R, Giordano R, Ciccarelli E, Limone P, Crivellaro C, Martinelli I, Montini M, Disoteo O, Ambrosi B, Lanzi R, Arosio M, Senni S, Balestrieri A, Solaroli E, Madeo B, De GR, Strollo F, Battista R, Scorsone A, Giagulli VA, Collura D, Scillitani A, Cozzi R, Faustini-Fustini M, Pia A, Rinaldi R, Allasino B, Peraga G, Tassone F, Garofalo P, Papini E, Borretta G. Screening of Cushing’s syndrome in outpatients with type 2 diabetes: Results of a prospective multicentric study in Italy. J Clin Endocrinol Metab. 2012;97:3467–3475. doi: 10.1210/jc.2012-1323. [DOI] [PubMed] [Google Scholar]
- 255.Thompson DG, Mason AS, Goodwin FJ. Mineralocorticoid replacement in Addison’s disease. Clin Endocrinol (Oxf) 1979;10:499–506. doi: 10.1111/j.1365-2265.1979.tb02107.x. [DOI] [PubMed] [Google Scholar]
- 256.Tirabassi G, Faloia E, Papa R, Furlani G, Boscaro M, Arnaldi G. Use of the desmopressin test in the differential diagnosis of pseudo-Cushing state from Cushing’s disease. J Clin Endocrinol Metab. 2010;95:1115–1122. doi: 10.1210/jc.2009-1146. [DOI] [PubMed] [Google Scholar]
- 257.Titus R, Kastenmeier A, Otterson MF. Consequences of gastrointestinal surgery on drug absorption. Nutr Clin Pract. 2013;28:429–436. doi: 10.1177/0884533613490740. [DOI] [PubMed] [Google Scholar]
- 258.Tomlinson JW, Stewart PM. Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract Res Clin Endocrinol Metab. 2001;15:61–78. doi: 10.1053/beem.2000.0119. [DOI] [PubMed] [Google Scholar]
- 259.Tordjman K, Jaffe A, Grazas N, Apter C, Stern N. The role of the low dose (1 microgram) adrenocorticotropin test in the evaluation of patients with pituitary diseases. J Clin Endocrinol Metab. 1995;80:1301–1305. doi: 10.1210/jcem.80.4.7714104. [DOI] [PubMed] [Google Scholar]
- 260.Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: A series of 58 cases. Ann N Y Acad Sci. 2002;970:134–144. doi: 10.1111/j.1749-6632.2002.tb04419.x. [DOI] [PubMed] [Google Scholar]
- 261.Tsatmali M, Ancans J, Yukitake J, Thody AJ. Skin POMC peptides: Their actions at the human MC-1 receptor and roles in the tanning response. Pigment Cell Res. 2000;13(Suppl 8):125–129. doi: 10.1034/j.1600-0749.13.s8.22.x. [DOI] [PubMed] [Google Scholar]
- 262.Tunn S, Mollmann H, Barth J, Derendorf H, Krieg M. Simultaneous measurement of cortisol in serum and saliva after different forms of cortisol administration. Clin Chem. 1992;38:1491–1494. [PubMed] [Google Scholar]
- 263.Valassi E, Swearingen B, Lee H, Nachtigall LB, Donoho DA, Klibanski A, Biller BM. Concomitant medication use can confound interpretation of the combined dexamethasone-corticotropin releasing hormone test in Cushing’s syndrome. J Clin Endocrinol Metab. 2009;94:4851–4859. doi: 10.1210/jc.2009-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Van Cauter E, Refetoff S. Evidence for two subtypes of Cushing’s disease based on the analysis of episodic cortisol secretion. N Engl J Med. 1985;312:1343–1349. doi: 10.1056/NEJM198505233122102. [DOI] [PubMed] [Google Scholar]
- 265.van der Pas R, Leebeek FW, Hofland LJ, de Herder WW, Feelders RA. Hypercoagulability in Cushing’s syndrome: Prevalence, pathogenesis and treatment. Clin Endocrinol (Oxf) 2013;78:481–488. doi: 10.1111/cen.12094. [DOI] [PubMed] [Google Scholar]
- 266.van Lierop AH, van der Eerden AW, Hamdy NA, Hermus AR, den HM, Papapoulos SE. Circulating sclerostin levels are decreased in patients with endogenous hypercortisolism and increase after treatment. J Clin Endocrinol Metab. 2012;97:E1953–E1957. doi: 10.1210/jc.2012-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.van Rossum EF, Lamberts SW. Glucocorticoid resistance syndrome: A diagnostic and therapeutic approach. Best Pract Res Clin Endocrinol Metab. 2006;20:611–626. doi: 10.1016/j.beem.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 268.Verma S, Vanryzin C, Sinaii N, Kim MS, Nieman LK, Ravindran S, Calis KA, Arlt W, Ross RJ, Merke DP. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2010;72:441–447. doi: 10.1111/j.1365-2265.2009.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Vingerhoeds AC, Thijssen JH, Schwarz F. Spontaneous hypercortisolism without Cushing’s syndrome. J Clin Endocrinol Metab. 1976;43:1128–1133. doi: 10.1210/jcem-43-5-1128. [DOI] [PubMed] [Google Scholar]
- 270.Wade M, Baid S, Calis K, Raff H, Sinaii N, Nieman L. Technical details influence the diagnostic accuracy of the 1 microg ACTH stimulation test. Eur J Endocrinol. 2010;162:109–113. doi: 10.1530/EJE-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Watabe T, Tanaka K, Kumagae M, Itoh S, Takeda F, Morio K, Hasegawa M, Horiuchi T, Miyabe S, Shimizu N. Hormonal responses to insulin-induced hypoglycemia in man. J Clin Endocrinol Metab. 1987;65:1187–1191. doi: 10.1210/jcem-65-6-1187. [DOI] [PubMed] [Google Scholar]
- 272.Weibel L, Spiegel K, Follenius M, Ehrhart J, Brandenberger G. Internal dissociation of the circadian markers of the cortisol rhythm in night workers. Am J Physiol. 1996;270:E608–E613. doi: 10.1152/ajpendo.1996.270.4.E608. [DOI] [PubMed] [Google Scholar]
- 273.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.White PC, Curnow KM, Pascoe L. Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev. 1994;15:421–438. doi: 10.1210/edrv-15-4-421. [DOI] [PubMed] [Google Scholar]
- 275.White PC, Speiser PW. Long-term consequences of childhood-onset congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2002;16:273–288. doi: 10.1053/beem.2002.0198. [DOI] [PubMed] [Google Scholar]
- 276.Widmaier EP, Raff H, Strang KT. Vander’s Human Physiology: The Mechanisms of Body Function. 13. McGraw-Hill; Boston: 2014. p. 609. figure 17.5. [Google Scholar]
- 277.Willenberg HS, Schinner S, Ansurudeen I. New mechanisms to control aldosterone synthesis. Horm Metab Res. 2008;40:435–441. doi: 10.1055/s-2008-1065336. [DOI] [PubMed] [Google Scholar]
- 278.Wind JJ, Lonser RR, Nieman LK, Devroom HL, Chang R, Oldfield EH. The lateralization accuracy of inferior petrosal sinus sampling in 501 patients with Cushing’s disease. J Clin Endocrinol Metab. 2013;98:2285–2293. doi: 10.1210/jc.2012-3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Wolkowitz OM, Burke H, Epel ES, Reus VI. Glucocorticoids. Mood, memory, and mechanisms. Ann N Y Acad Sci. 2009;1179:19–40. doi: 10.1111/j.1749-6632.2009.04980.x. [DOI] [PubMed] [Google Scholar]
- 280.Yanovski JA, Cutler GB, Jr, Chrousos GP, Nieman LK. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test differentiates mild Cushing’s disease from normal physiology. J Clin Endocrinol Metab. 1998;83:348–352. doi: 10.1210/jcem.83.2.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Young WF, Jr, Thompson GB. Laparoscopic adrenalectomy for patients who have Cushing’s syndrome. Endocrinol Metab Clin North Am. 2005;34:489–99. doi: 10.1016/j.ecl.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 282.Zemskova MS, Gundabolu B, Sinaii N, Chen CC, Carrasquillo JA, Whatley M, Chowdhury I, Gharib AM, Nieman LK. Utility of various functional and anatomic imaging modalities for detection of ectopic adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab. 2010;95:1207–1219. doi: 10.1210/jc.2009-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zhang L, Mason JI, Naiki Y, Copeland KC, Castro-Magana M, Gordon-Walker TT, Chang YT, Pang S. Characterization of two novel homozygous missense mutations involving codon 6 and 259 of type II 3beta-hydroxysteroid dehydrogenase (3betaHSD) gene causing, respectively, nonsalt-wasting and salt-wasting 3betaHSD deficiency disorder. J Clin Endocrinol Metab. 2000;85:1678–1685. doi: 10.1210/jcem.85.4.6539. [DOI] [PubMed] [Google Scholar]