

Abstract
Background
Autism spectrum disorders (ASDs) are common and have a strong genetic basis, yet the cause of ∼70–80% ASDs remains unknown. By clinical cytogenetic testing, we identified a family in which two brothers had ASD, mild intellectual disability and a chromosome 22 pericentric inversion, not detected in either parent, indicating de novo mutation with parental germinal mosaicism. We hypothesised that the rearrangement was causative of their ASD and localised the chromosome 22 breakpoints.
Methods
The rearrangement was characterised using fluorescence in situ hybridisation, Southern blotting, inverse PCR and dideoxy-sequencing. Open reading frames and intron/exon boundaries of the two physically disrupted genes identified, TCF20 and TNRC6B, were sequenced in 342 families (260 multiplex and 82 simplex) ascertained by the International Molecular Genetic Study of Autism Consortium (IMGSAC).
Results
IMGSAC family screening identified a de novo missense mutation of TCF20 in a single case and significant association of a different missense mutation of TCF20 with ASD in three further families. Through exome sequencing in another project, we independently identified a de novo frameshifting mutation of TCF20 in a woman with ASD and moderate intellectual disability. We did not identify a significant association of TNRC6B mutations with ASD.
Conclusions
TCF20 encodes a transcriptional coregulator (also termed SPBP) that is structurally and functionally related to RAI1, the critical dosage-sensitive protein implicated in the behavioural phenotypes of the Smith–Magenis and Potocki–Lupski 17p11.2 deletion/duplication syndromes, in which ASD is frequently diagnosed. This study provides the first evidence that mutations in TCF20 are also associated with ASD.
Keywords: Genetics, Molecular genetics, Chromosomal, Clinical genetics, Psychiatry
Introduction
Autism spectrum disorders (ASDs) are common neurodevelopmental conditions characterised by impairments in social communication, the presence of repetitive behaviours and a restricted range of interests; intellectual disability is present in around 50% of people with ASD.1 2 Family and twin studies show that ASDs have a strong genetic basis: at least 5–10% of siblings of children with ASD have an ASD diagnosis themselves.2 Siblings and parents of children with ASD are more likely than controls to show behavioural traits similar to those seen in people with ASD (the broader autism phenotype (BAP)).3 4 Additionally, monozygotic twins are more likely to be concordant for ASD compared with dizygotic twins.5
Many rare mutations and variants have been shown to cause or increase the risk of ASD.6–9 For example, ASD occurs in several clinically defined monogenic and chromosomal disorders (including fragile X, Down, Angelman and Rett syndromes, neurofibromatosis and tuberous sclerosis). No common variants of large effect in ASD have been found10; however, multiple rare variants causing ASD have been identified in research and clinical settings through array comparative genomic hybridisation (CGH) and high-throughput exome and genome sequencing.7–9 11–19 Taking account of genetic causes and other medical/neurodevelopmental conditions, the cause of ASD remains unidentified in ∼70–80% of affected individuals; hence, a substantial proportion of causative loci remains to be identified.6–8
The present study started with the identification of a de novo pericentric inversion of chromosome 22, present in two brothers who both had ASD. Further characterisation of the rearrangement revealed it to be complex, consisting of four separate chromosome 22 breakpoints physically disrupting two genes, TCF20 (encoding transcription factor 20) and TNRC6B (encoding trinucleotide repeat containing 6B), both of which appeared plausible candidates for involvement in ASD. Building on this initial finding, we present additional evidence implicating TCF20 in ASD, based both on the results of resequencing of TCF20 and TNRC6B in samples from the International Molecular Genetic Study of Autism Consortium (IMGSAC) and on the separate identification of an additional TCF20 frameshifting mutation associated with ASD. We propose that precise dosage of TCF20 is important for neurodevelopment, and that functional perturbation of TCF20 confers susceptibility to ASD.
Materials and methods
Patient ascertainment and diagnostic studies
Patients from two unrelated families (#1 and #6) were originally referred for assessment of coexisting craniosynostosis. Ethical approval for research into craniofacial malformations, and the specific ASD studies undertaken in these families, was provided by the Oxfordshire Research Ethics Committee B (C02.143) and the West London Research Ethics Committee (09/H0706/20), and informed consent was obtained. Genetic analyses were performed on DNA and RNA extracted from peripheral blood and lymphoblastoid cell lines. The human genome hg19 sequence release (February 2009) was used for all analyses.
Ascertainment of ASD and control samples
Multiplex and simplex ASD families were identified, collected and assessed by the IMGSAC as previously described.20 21 Ethical approval was obtained for the collection of all data, and written informed consent was obtained from all parents/guardians or, where appropriate, the proband. Parents were administered the Autism Diagnostic Interview-Revised (ADI-R)22 and the Vineland Adaptive Behavior Scales.23 Probands were assessed using the Autism Diagnostic Observation Schedule-Generic (ADOS-G),24 and a medical examination was carried out to exclude cases of known aetiology. IQ was assessed using standardised measures of verbal and performance ability.25 26 Whenever possible, probands were karyotyped and molecular genetic testing for fragile X syndrome was performed. Family history interviews4 were used to investigate BAP behaviours and traits in siblings and parents when possible.
A cohort of 384 UK DNA controls from randomly selected unrelated UK Caucasian blood donors was obtained from the European Collection of Cell Cultures (ECACC) (http://www.hpacultures.org.uk/products/dna/hrcdna/hrcdna.jsp). An additional 432 locally sourced controls were tested in the case of the TCF20 c.4670C>T variant.
Fluorescence in situ hybridisation
Fluorescence in situ hybridisation (FISH) mapping of the chromosome 22 breakpoints in family #1 used BACs and fosmids obtained from the Children's Hospital Oakland Research Institute (CHORI); see table 1 for clone names and locations. Clones were labelled by nick-translation (Abbott Molecular) either with digoxigenin-11-dUTP (Roche) or biotin-16-dUTP (Roche). FISH was carried out following standard procedures. Briefly, the DNA probes were denatured at 75°C for 5 min and preannealed at 37°C for 45 min. Slides were denatured in 70% formamide/2× saline sodium citrate (SSC) at 70°C for 1 min and hybridised in a moist chamber at 37°C overnight. After washes in 50% formamide/1× SSC and 2× SSC at 42°C, the probes were detected with either fluorescein-conjugated antidigoxigenin (Roche) or Cy3-conjugated streptavidin (Sigma). The slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in Vectashield (Vector Laboratories) and analysed on a Cytovision system (Leica).
Table 1.
Clones used for fluorescence in situ hybridization (FISH) analysis in family #1
| Clone name | Genomic location on Chr22 | Position of signal on der(22) | Breakpoint |
|---|---|---|---|
| CTA-150C2 | 39280232-39481326 | Long arm | |
| WI2-1570N6 | 39476065-39520769 | Split short/long arms | C |
| WI2-1013H1 | 39557188-39594983 | Short arm | |
| WI2-2202O13 | 39516811-39555371 | Short arm | |
| WI2-1769B14 | 39587327-39627092 | Short arm | |
| WI2-3097P13 | 39612987-39654267 | Short arm | |
| WI2-1881P6 | 39642520-39684613 | Short arm | |
| WI2-624P20 | 40026816-40067597 | Short arm | |
| WI2-1574G19 | 40631976-40678518 | Short arm | B |
| WI2-1927K3 | 40743240-40784809 | Long arm | B |
| CTA-250D10 | 42252765-42473659 | Long arm | |
| G248P86612G1 | 42600994-42642421 | Split short/long arms | A |
| RP11-241G19 | 42605118-42782007 | Split short/long arms | A |
| G248P84377G7 | 42640176-42679204 | Short arm | |
| RP11-794G14 | 43105492-43331920 | Short arm | |
| RP11-1021O19 | 43972241-44158005 | Short arm | |
| RP11-357F14 | 44543405-44721394 | Short arm | |
| RP11-49A20 | 45141573-45322938 | Short arm | |
| CTA-268H5 | 45574232-45797207 | Short arm | |
| CTA-722E9 | 49795787-49928065 | Short arm | |
| CTA-799F10 | 51078917-51174589 | Short arm |
Array CGH
Array CGH was performed using a human genome-wide 185K oligonucleotide array (Agilent Technologies). Genomic DNA from the inversion patient (II-4, family #1) and from a sex-matched reference were double-digested separately using the restriction endonucleases AluI and RsaI (Promega) and purified using Microcon centrifugal filter devices (Merck Millipore). A total of 1.5 μg of the digested products was differentially labelled by the random priming method using the fluorophores Cy3-dUTP and Cy5-dUTP (Perkin Elmer) and co-hybridised to the array for 48 h at 65°C in a rotating oven. The hybridised arrays were washed and scanned using an Agilent Microarray Scanner. The image data were extracted using Agilent Feature Extraction software V.8.5, and the data analysed using Agilent CGH Analytics software V.3.4 (z-score method setting).
Single-nucleotide polymorphism array hybridisation
Genomic DNA from the inversion patient (II-4, family #1) was analysed using an ∼300K Human CytoSNP-12 BeadChip according to manufacturer's guidelines (Illumina Inc, San Diego, CA). Briefly, ∼200 ng DNA was denatured, amplified, fragmented enzymatically and hybridised to the BeadChips in an Illumina Inc. hybridisation oven at 48°C for 16–24 h. The BeadChips were washed according to the manufacturer's protocol and the hybridised DNA subjected to primer extension with labelled nucleotides prior to detection using fluorescent antibodies. Data were processed using GenomeStudioV2009.2 (Illumina Inc) and analysed using Nexus Discovery Edition v6.1 (BioDiscovery, Hawthorne, California, USA).
Isolation of breakpoints A, B and C on chromosome 22
We obtained BACs and fosmids and performed FISH analysis, initially to identify breakpoint A (table 1). Identification of a split signal using two fosmids localised the breakpoint within ∼35 kb; single-copy probes spanning this region were synthesised and hybridised to Southern blots of patient and control DNA, further refining the breakpoint within ∼1 kb. Three breakpoint-specific primers (TSP1, 5′-GTTTTGGAGCGCCACAAAGCACT-3′; TSP2, 5′-CAAAGCACTCCCATATAAGACGGCG-3′; TSP3, 5′-AGACGGCGAACTTAATATATACATGTTGTG-3′) were combined with redundant primers in nested PCR with the DNA Walking SpeedUp Premix Kit (Seegene). After DNA sequencing to determine the site of the breakpoint and to identify the sequence and location of DNA on the other side of it (breakpoint B), a further primer pair (5′-GATAAATTTTAGCTATTATTATTACCACCTAGAAGCT-3′ and 5′-TTATAGACAAAGGCTAAGGGCAGATG-3′) was designed to confirm the breakpoint by amplifying a 1.5 kb fragment.
To identify breakpoint C, we conducted further FISH and found a split signal with BAC W12-1570N6 (table 1). We screened this ∼44.7 kb region by Southern blot analysis and identified a 15 kb HindIII fragment as likely to span the breakpoint.
Identification of novel TCF20 exon
A comparison of the human and mouse cDNA sequences showed that the mouse Tcf20 transcript contains an extra exon encoding an extended 5′ untranslated region (UTR).27 Correspondingly, comparison of the human and mouse genomic sequence revealed a highly conserved region ∼68.5 kb telomeric of the first annotated exon of TCF20 in the human genome. We isolated total RNA from normal human transformed B-lymphocytes and generated cDNA using random hexamer primers (RevertAid, Fermentas). Following amplification using cDNA as template with primers in the large exon of TCF20 and the conserved region (primer pair 5′-TCCTCCCCCGCCTCGGCTCAG -3′ and 5′-CACTGCTGCCACTACTGCCACCTGTAC-3′), we found the conserved region to be spliced to the previously identified exon 1 of TCF20, indicating that this region represents a previously unannotated exon of human TCF20 (GenBank KF851355).
DNA sequencing of TCF20 and TNRC6B
The entire open reading frames of TCF20 (RefSeq accession: NM_005650.1) and TNRC6B (isoform 1: NM_001162501.1 and isoform 3: NM_001024843.1) were screened in the ASD panel using primers and reaction conditions shown in online supplementary table S1. Fragments were DNA sequenced on the ABI PRISM 3730 DNA sequencer, employing Big Dye Terminator mix V.3.1 (Applied Biosystems). Sequence chromatogram traces were analysed using Mutation Surveyor (Softgenetics) and Sequencher (Gene Codes). We compared the occurrence of variants in a normal control panel of 384 samples by dideoxy sequencing and examined the frequency of each variant in 8600 European American (EA) alleles from the Exome Variant Server (EVS).28 Synonymous and intronic variants were assessed for their potential to affect splicing using the Splice Site Prediction by Neural Network (http://www.fruitfly.org/seq_tools/splice.html), and pathogenicity of missense substitutions was investigated with PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). Nucleotide numbering of variants in cDNA starts at the initiation ATG codon (A=1).
Microsatellite and single-nucleotide polymorphism analysis
The haplotype surrounding the TCF20 c.4670C>T variant identified in three families was investigated by amplifying seven flanking microsatellites (see online supplementary table S2) in proband and parental samples using primers labelled with the fluorophore 6-FAM. Fragments were analysed by capillary electrophoresis on an ABI 3730 containing POP-7 polymer, and peaks were visualised using Gene Mapper V.3.7 (Applied Biosystems). Informative single-nucleotide polymorphisms (SNPs) (see online supplementary table S2) were amplified and sequenced as described above.
Correct biological relationships of samples (and hence, exclusion of non-paternity) were confirmed in all three families with de novo TCF20 mutations (#1, 2 and 6) using at least 10 microsatellites located on different chromosomes.
cDNA analysis
RNA was extracted from a lymphoblastoid cell line using TRIzol/RNeasy (Qiagen) and ∼1 µg used for cDNA synthesis with random hexamers. The region containing the mutation was amplified from the proband's cDNA, an equivalent (-RT) control without addition of reverse transcriptase, and genomic DNA from proband and parents using TCF20 Exon 2.9 primers (see online supplementary table S1), followed by a digestion with BslI and agarose gel electrophoresis.
Results
Chromosome 22 rearrangement associated with ASD
The proband II-4 in family #1 (pedigree, figure 1A) was assessed at the age of 7 months because of an abnormal craniofacial appearance (figure 1C). Plain radiographs and CT of the skull showed fusion of the metopic and coronal sutures and extensive copperbeating suggestive of raised intracranial pressure; the brain appeared structurally normal. Subtotal calvarial remodelling was performed at the age of 1 year. Karyotyping of peripheral lymphocytes revealed a pericentric inversion of chromosome 22, reported as 46,XY,inv(22)(p11?.2-q13?.1). Testing of the family showed the same abnormal karyotype in his older brother, who had no craniofacial dysmorphism (II-2; figure 1B); surprisingly, the karyotypes of both parents (I-1 and I-2), as well as the other two siblings (II-1 and II-3), were normal. During childhood, the two brothers with the inversion (II-2, II-4), but not their siblings or parents, were diagnosed with clinical autism and mild intellectual disability by their local clinicians; subsequently, both individuals met autism criteria during research assessments using ADOS-G (table 2). Array CGH of DNA from the proband was performed using 185K and 300K genome-wide oligonucleotide arrays (see ‘Methods’), but neither revealed any significant gain or loss of material.
Figure 1.
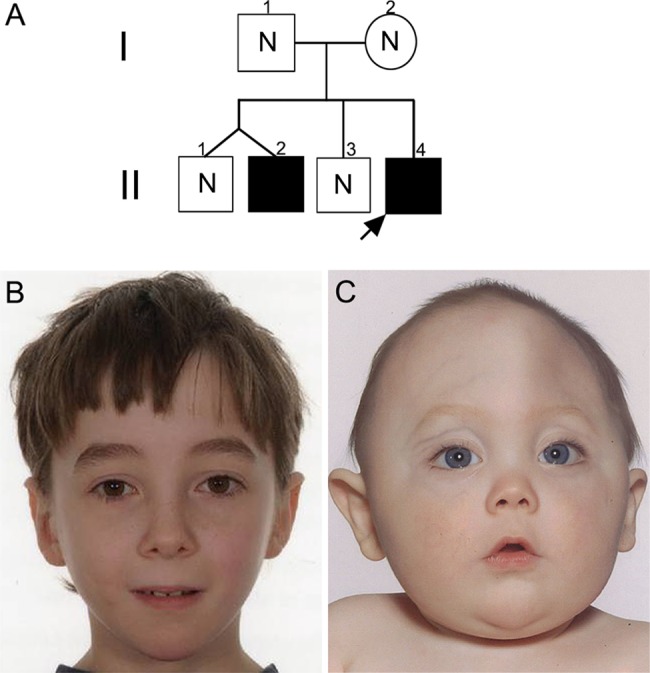
Pedigree of family #1 and facial appearance of individuals heterozygous for chromosome 22 rearrangement. (A) Pedigree showing the immediate family of the proband (arrow). Filled symbols represent individuals shown to carry the rearrangement. N indicates absence of the rearrangement. (B) Normal facial appearance of the proband's older brother II-2, aged 10 years. (C) Facial appearance of the proband aged 10 months showing trigonocephaly associated with hypotelorism and mild exorbitism, caused by premature synostosis of the metopic suture.
Table 2.
Summary results of Autism Diagnostic Observation Schedule-Generic (ADOS-G) and IQ/developmental assessments in subjects with TCF20 mutations
| Family # | Patient ID | TCF20 abnormality | ADOS-G social communication score (age at assessment in years) | IQ/developmental quotient (test, age at assessment in years) |
|---|---|---|---|---|
| 1 | II-4 (proband) | Inversion break intron 1 | 13 (10 years) | Full scale 79, verbal 79, performance 79 (WPPSI-3, 3.5 years) |
| 1 | II-2 (brother) | Inversion break intron 1 | 16 (12 years) | Communication 45, daily living 55, socialisation 44 (VABS, 7 years) |
| 2 | proband | p.K512E | 16 (7 years) | Full scale 120 (WASI, 13 years) |
| 3 | proband | p.P1557L | 11 (8 years) | Performance 100 (Raven's matrices) |
| 4 | proband | p.P1557L | NA | NA |
| 5 | proband | p.P1557L | 11 (10 years) | Performance 80 (Raven's matrices) |
| 5 | brother | p.P1557L | NA | Performance 107 (Raven's matrices) |
| 6 | proband | p.K1173Rfs*5 | 12 (25 years) | Full scale 45, verbal 50, performance 47 (WISC-3, 14 years) |
NA, not available; VABS, Vineland Adaptive behaviour Scales; WASI, Wechsler Abbreviated Scale of Intelligence; WISC, Wechsler Intelligence Scale for Children; WPPSI, Wechsler Preschool and Primary Scale of Intelligence.
To characterise the molecular nature of the pericentric inversion, we performed FISH using multiple BACs and fosmids (table 1). These probes were initially focused on the 22q13.1 band in which the long-arm breakpoint had been tentatively located, but several further rounds of analysis were performed as greater complexity in the rearrangement became apparent (figure 2). The observation of split signals with two fosmids localised one breakpoint (termed breakpoint A) to a ∼37 kb region (figure 2A). Further analysis by Southern blotting with single-copy probes identified breakpoint fragments, initially within a ∼15 kb EcoRI fragment, and subsequently within a 248 bp fragment bordered by StuI and AflIII restriction sites (not shown). PCR primers were designed to amplify across the breakpoint in sequentially nested amplifications with degenerate primers (see ‘Methods’). Surprisingly, DNA sequencing of this amplification product identified the sequence on the centromeric side of the break as originating from a location ∼1.9 Mb centromeric of breakpoint A (figure 2D, bottom right). These sequence data showed contiguity between nucleotides at coordinates at 40 709 620 bp (breakpoint B) and 42 634 698 bp (breakpoint A), adjacent to a short stretch of 5-nucleotide (5′-GACCT-3′) complementarity (figure 2D). Confirming the identification of breakpoint B, clones closely adjacent on either side of this location mapped to opposite arms of the der(22) (figure 2B). This result implied that a third more centromeric break on the long arm (‘breakpoint C’) must have occurred, to which the intermediate segment (B-A) had been joined. This break was localised using FISH to an ∼44.7 kb region within BAC clone W12-1570N6 (figure 2C). Analysis by Southern blotting revealed a HindIII restriction fragment that likely spanned the breakpoint (figure 2D, bottom left), locating the breakpoint to a ∼4 kb region between 39 507 139 and 39 511 083 bp. Figure 2D summarises the structure of the derivative chromosome 22 as concluded from the FISH, Southern blotting and DNA sequencing results. Breakpoint D is predicted to occur in the short arm satellite sequence of chromosome 22 and was not characterised further. Although (as demonstrated by array CGH) there has been no major gain or loss of material at the breakpoints, we found evidence of a small (∼10 kb) duplication at breakpoint A (data not shown) and this may apply to others too, most consistent with the replication-based fork stalling template switching (FoSTeS)-type mechanism for the complex chromosome rearrangement.29
Figure 2.

Structure of the chromosome 22 rearrangement deduced from fluorescence in situ hybridization (FISH) analysis and DNA sequencing. (A–C) Representative FISH analysis and diagrammatic interpretation of structure of the rearranged chromosome (der22), shown in more detail with positions of breakpoints in (D). (A) Signals from RP11-241G19 (green), which spans breakpoint A, and the more distal RP11-49A20 (red) are adjacent on the normal chromosome 22 (arrowhead) but a split green signal is seen near the opposite end of the der22 (arrow). (B) Clones W12-1927K3 (red) and W12-1574G19 (green), which lie on either side of breakpoint B, showing hybridisation together on the normal chromosome 22 (arrowhead) and at opposite ends of the der22 (arrow). C. Single signal with W12-1570N6 on normal chromosome 22 (arrowhead), but split signal on derived 22 (arrow) indicating position of breakpoint C. (D) Ideograms of wt and derived chromosome 22. The order of BAC and fosmid clones employed in figure parts A–C is shown, together with the locations of breakpoints A–C. The 2 Mb region between breakpoints A and B is shown in light red (orientation on the derived chromosome is uncertain). Breakpoint D on the satellite short arm was not further characterised. Below left, map of the 65 kb region that includes breakpoint C, showing the positions and orientations of genes. The Southern blot analysis shows an apparent breakpoint in the patient sample (P) compared with the control (C), localising the breakpoint to the indicated segment (double-ended arrows) of ∼4 kb. Below right, the DNA sequence chromatogram spanning the breakpoints A and B is shown above an alignment of this sequence with the normal sequences at the telomeric and centromeric ends of breakpoints. Arrows indicate positions and numbering of the last intact bases on either side of the translocated region.
Gene content at breakpoints A, B and C and selection of TCF20 and TNRC6B for further analysis
We analysed the three breakpoints on the long arm of chromosome 22 to determine whether they disrupted any genes. Initially breakpoint A appeared to locate within an intergenic region; however, because of sequence homology with the mouse orthologue of TCF20 in which an extra exon is described,27 we predicted the existence of a previously unannotated exon located 5′ of the currently annotated first exon of human TCF20. Primers for cDNA analysis of the corresponding human region were designed (see ‘Methods’); starting with RNA isolated from transformed B lymphocytes, we found this region is indeed spliced to the previously described first exon of TCF20 (see online supplementary figure S1). This novel exon of the human TCF20 transcript encodes an extended 5′ UTR. Therefore, breakpoint A disrupts TCF20 in intron 1 at a position 23.3 kb 5′ of exon 2 (figure 3A). TCF20 encodes a transcriptional coregulator paralogous to RAI1, the causative gene in Potocki–Lupski syndrome (duplication of 17p11.2), which is associated with ASD in ∼90% of cases;30 31 deletions of this region cause Smith–Magenis syndrome, characterised by severe intellectual disability and neurobehavioural problems, including ASD.32 33 Breakpoint B locates within intron 19 of TNRC6B, which encodes a product that stably associates with argonaute proteins required for microRNA-guided mRNA cleavage.34 Breakpoint C does not apparently disrupt any genes, occurring >12 kb telomeric of APOBEC3H and >5 kb centromeric of CBX7 (figure 2D, bottom left).
Figure 3.

TCF20 gene structure, identification of variants in ASD cases and their location within conserved domains. (A) Schematic representation of TCF20, exons are shown to scale with the coding sequence in white and untranslated regions filled in with black. There is an alternative stop codon in the alternatively spliced exon 5. The position of the first coding nucleotide is shown in exon 2, numbers above boxes indicate cDNA numbering at last nucleotides of exon boundaries or last nucleotide of stop codons; numbers in red below lines indicate intron sizes (not to scale). The location of breakpoint A that interrupts TCF20 23350 bp 5′ of exon 2 is also indicated. (B) Pedigrees of five families with variants of TCF20 that are either novel or enriched compared with control samples. Below each pedigree is a chromatogram showing the sequence change together with the amino acids encoded by the change and by adjacent codons. Black symbols indicate individuals with a clinical and research ASD diagnosis, the white symbol indicates people without clinical ASD; where broader autism phenotype data are available this is described in the text; n/a indicates that no DNA was available for analysis. Under each symbol, the status of that individual for the change found in the proband is shown. (C) Diagram representing the TCF20 protein with previously annotated domains: P1-P3, PEST domains; N1-N3, nuclear localisation signals; MD, minimal DNA binding domain; ZNF2, zinc finger domain. The three lines above the protein denote the following domains: TAD, transactivation domain; DBD, DNA binding domain and the ePHD/ADD domain.37 The lower panel shows the positions and conservation of amino acid residues predicted to be substituted in ASD pedigrees. The entire PEST1 and PEST2 sequences are shown with interspecies conservation in mammals, chicken and frog. (D) Analysis of cDNA amplification product compared with genomic (gDNA) from region containing c.3518delA mutation in family #6. Restriction digestion was performed with BslI, yielding product sizes (bp) of 215, 162, 145, 72, 1 in the absence of the mutation and 233, 215, 145, 1 in the presence of the mutation. Lanes numbered as follows: 1, undigested gDNA from proband; 2, mother's gDNA; 3, father's gDNA; 4, proband's gDNA; 5, proband's cDNA and 6, −RT control for proband's cDNA. Note similar relative intensities of mutant and non-mutant fragments in lanes 4 and 5, indicating lack of significant nonsense-mediated decay associated with the frameshifting mutation.
We hypothesised that the ASD present in the two brothers with the complex chromosome 22 rearrangement was most likely due to altered function of one or both of the two physically disrupted genes, TCF20 and TNRC6B. There is no established abnormal phenotype associated with mutations in either of these two genes, or in their murine orthologues, although there are reports of copy number variations (CNVs) that include TNRC6B being linked to ASD (see ‘Discussion’). We therefore proceeded to resequence both genes in the large number of families recruited by IMGSAC.
Resequencing of TCF20 and TNRC6B in the IMGSAC cohort
TCF20 comprises six exons, five of which encode two open reading frames of 5880 and 5814 nucleotides generated by alternative splicing (figure 3A). TNRC6B is alternatively spliced to generate multiple isoforms, including 25 different coding exons. We undertook DNA sequencing of the coding sequences of both genes, including the intron–exon boundaries, in 342 unrelated ASD probands (260 from multiplex pedigrees and 82 from simplex families) from the IMGSAC cohort, and where possible performed parent and sibling studies of the rare variants identified. The occurrence of all variants likely to be functionally relevant (either amino acid altering or predicted to affect splicing) was compared with normal control data as described in ‘Methods’. The results for TCF20 are summarised in table 3 and those for TNRC6B in online supplementary table S3.
Table 3.
Amino acid sequence altering variants of TCF20 found in 342 ASD samples, comparison with controls, and family follow-up
| Nucleotide change | Amino acid change | Number of heterozygous ASD samples/total sequenced† | Number of heterozygous control samples/total sequenced† | Exome Variant Server (EA) expressed as rare/common alleles | Family follow-up | PolyPhen-2 prediction |
|---|---|---|---|---|---|---|
| c.47G>C | p.S16T | 10/331 | 8/353 | 123/8477 | – | Benign (0.015) |
| c.162_167del | p.S55_G56del | 2/331 | 3/353 | 35/8219 | – | n/a |
| c.del966_968 | p.Q322del | 1/336 | 2/354 | 11/8243 | – | n/a |
| c.1213A>G | p.M405V | 63/338 [4] | 61/351 [3] | 788/7812 | – | Benign (0) |
| c.1534A>G | p.K512E | 1/337 | 0/352 | 0 | De novo | Probably damaging (0.970) |
| c.2164A>G | p.S722G | 102/338 [19] | 119/354 [8] | 1797/6803 | – | Benign (0) |
| c.3495G>A | p.M1165I | 1/335 | 0/356 | 11/8589 | – | Benign (0.01) |
| c.4670C>T | p.P1557L | 3/335 | 0/793 | 3/8597 | See figure 3 | Probably damaging (0.963) |
| c.5810C>T | p.P1937L | 1/339 | 0/354 | 2/8598 | Absent in affected sibling; present in unaffected sibling | Probably damaging (0.988) |
| c.5825C>A | p.P1942H | 1/339 | 0/354 | 1/8599 | Absent in affected half-sibling; transmitted by non-shared parent | Possibly damaging (0.634) |
†The number of samples from each panel found to harbour the variant is shown next to the number of samples successfully screened. Numbers in square brackets refer to homozygous changes.
EA, European American.
In TCF20, we identified two common SNPs and eight different rare heterozygous changes (encoding two in-frame deletions and six non-synonymous substitutions), each present in between 1 and 10 ASD probands. Common SNPs did not differ in frequency between cases and controls. Of the rare variants, six were considered unlikely to be causally contributory either because they were present at significant frequency in the EVS (n=4) or an affected sibling did not inherit the variant allele (n=2). The remaining two variants (c.1534A>G and c.4670C>T), identified in one and three different families respectively, were considered potentially pathogenic. The dideoxy sequencing and segregation of these variants is shown in figure 3B, and the positions of the encoded missense changes in the TCF20 protein domain structure and species conservation in figure 3C. In the multiplex ASD family #2, the c.1534A>G transition encodes a likely damaging p.K512E substitution (PolyPhen-2 score 0.97), which had arisen de novo in the proband. This individual had classical Asperger syndrome with good intellectual function (table 2), whereas his cousin had autism, severe intellectual disability and early-onset epilepsy; phenotyping with the family history interview revealed evidence of the BAP in two other family members. Correct biological relationships were confirmed by microsatellite analysis and by haplotype analysis based on a 1M SNP chip (data not shown). The variant was absent in 352 ECACC samples experimentally tested, and not recorded in 6503 samples from the EVS. Amino acid position 512 locates within the PEST1 sequence of TCF20 and is highly conserved in vertebrates (figure 3C); PEST sequences provide targets for proteolytic protein degradation.35
In unrelated ASD probands from three families (singleton families #3 and #4 and multiplex family #5), a c.4670C>T transition encoding p.P1557L (PolyPhen-2 score 0.963) was present (figure 3B). Proline 1557 locates within the PEST2 domain of TCF20 and is highly conserved in vertebrates (figure 3C). The c.4670C>T variant was inherited from the mother (about whom there are no phenotypic data) in family #3 and from the father in families #4 and #5. In family #5, both boys had ASD and average range IQ; the father had evidence of the BAP. The frequency of this substitution in the ASD cohort (3/342 individuals) is significantly higher (Fisher's exact test) than in control populations, based both on our own resequencing data (0/793; p=0.027) and from EVS (3/4,300; p=0.007). Observing that this C>T transition has arisen at a hypermutable CpG site, we analysed the haplotype background on which each variant T allele was present. Using microsatellites and SNPs within a 0.54 Mb region around the substitution that contains no recombination hotspots (defined as ≥10 cM/Mb) according to the International HapMap Consortium (http://hapmap.ncbi.nlm.nih.gov/), we found multiple differences between each of the three haplotypes (table 4), including different alleles in family #5 for SNPs (rs16986035 and rs1548304) that flank the c.4670C>T variant. These data are consistent with the mutation having arisen independently at least twice.
Table 4.
Microsatellite and single-nucleotide polymorphism (SNP) markers in ∼0.5 Mb region surrounding TCF20 to distinguish c.4670C>T haplotypes in families 3, 4 and 5
| Tcms4* 42390888 bp |
Tcfms3 42433133 bp |
Tcfms2 42433855 bp |
Tcfms1 42544517 bp |
rs2899354 42554409 bp |
rs4453786 42563308 bp |
rs16986035 42602139 bp |
c.4670C>T 42606642 bp |
rs5758652 42612408 bp |
rs1548304 42691488 bp |
rs6002674 42694220 bp |
rs11704558 42695148 bp |
rs6002676 42697216 bp |
Tcfms5 42775494 bp |
Tcfms6 42782403 bp |
Tcfms7 42939056 bp |
Tcfms8 43053571 bp |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 3 (father) | 158/162 | 200/202 | 203/203 | 200/214 | C/A | A/G | A/A | C/C | A/G | C/T | C/T | C/C | G/A | 182/184 | 164/166 | 202/202 | 219/223 |
| Family 3 (mother) | 156/156 | 206/206 | 205/205 | 204/206 | C/C | G/G | G/G | C/T | A/A | C/C | T/T | C/T | G/A | 184/184 | 166/166 | 202/206 | 213/223 |
| Family 3 (proband) | 156/162 | 202/206 | 203/205 | 200/206 | C/C | G/G | G/A | C/T | A/A | C/C | T/T | C/C | A/A | 182/184 | 164/166 | 202/206 | 213/223 |
| Family 3 c.4670T haplotype† | 156 | 206 | 205 | 206 | C | G | G | T | A | C | T | C | A | 184 | 166 | 206 | 213 |
| Family 4 (father) | 156/162 | 200/206 | 201/205 | 204/214 | C/A | A/G | G/A | C/T | A/G | C/T | T/T | C/C | A/A | 184/186 | 166/166 | 202/202 | 219/225 |
| Family 4 (mother) | 156/162 | 200/206 | 203/205 | 200/204 | C/C | G/G | A/G | C/C | A/A | C/C | T/T | T/T | G/G | 188/188 | 164/166 | 202/202 | 219/221 |
| Family 4 (proband) | 162/162 | 200/206 | 203/205 | 200/204 | C/C | G/G | G/G | C/T | A/A | C/C | T/T | C/T | G/A | 186/188 | 166/166 | 202/202 | 219/221 |
| Family 4 c.4670T haplotype | 162 | 200/206 | 205 | 204 | C | G | G | T | A | C | T | C | A | 186 | 166 | 202 | 219 |
| Family 5 (mother) | 162/162 | 200/202 | 201/205 | 200/200 | C/C | G/G | G/G | C/C | A/A | T/C | C/T | C/T | G/G | 184/186 | 166/170 | 202/202 | 219/223 |
| Family 5 (father) | 156/162 | 200/206 | 203/205 | 200/204 | C/A | G/A | A/G | C/T | A/G | T/T | T/T | C/C | G/A | 186/186 | 166/168 | 202/202 | 219/221 |
| Family 5 (proband) | 162/162 | 200/206 | 203/205 | 200/204 | C/C | G/G | A/G | C/T | A/A | T/C | T/T | C/T | G/A | 186/186 | 166/170 | 202/202 | 219/219 |
| Family 5 c.4670T haplotype | 162 | 206 | 203 | 204 | C | G | A | T | A | T | T | C | A | 186 | 166 | 202 | 219 |
*See online supplementary table S2 for details of markers.
†The haplotype associated with the c.4670C>T change in each family is shown in bold, markers in each pedigree that differ from those in the other two pedigrees are underlined.
In the case of TNRC6B, we identified 12 different rare non-synonymous changes (encoding 1 frameshift, 1 single amino acid deletion and 10 missense substitutions) each present in 1 or 2 of 335 IMGSAC samples sequenced (see online supplementary table S3). Of these, six were deemed unlikely to be causally contributory because they were previously identified at significant frequency in the EVS (n=4), the mutation was predicted as being functionally benign (n=1) or an affected sibling did not inherit the variant allele (n=1). In the remaining six cases (which include the frameshift and the amino acid deletion), and in contrast to TCF20, none was shown to have arisen de novo or to show a significant frequency difference between cases and controls (although lack of availability of DNA samples from some family members prevented complete analysis). Hence, these data are inconclusive regarding a contribution of TNRC6B mutations to ASD in the IMGSAC cohort.
A de novo truncating mutation of TCF20 in an individual with ASD and intellectual disability
While this work was being undertaken, we coincidentally discovered a further TCF20 mutation during an unrelated project aimed at identifying novel genetic causes of craniosynostosis.36 The exome sequence from a woman with unicoronal synostosis segregating from her mother (family #6) was found to contain a heterozygous one-nucleotide deletion of TCF20 (c.3518delA encoding p.K1173Rfs*5). Analysis of parental samples showed that it had arisen de novo, indicating that it was not causative of the familial craniosynostosis (figure 3D). Clinical case note review revealed that the proband had clinically diagnosed ASD and moderate intellectual disability; she subsequently met autism criteria during a research ADOS (table 2).
To determine whether this TCF20 mutation causes nonsense-mediated mRNA decay, we analysed cDNA obtained from a lymphoblastoid cell line from the proband. Unexpectedly, this showed equal representation of the normal and mutant alleles in the cDNA product (figure 3D), indicating that the mutant mRNA is stable; hence, a truncated protein is expected to be produced in significant quantities.
Discussion
Starting with the clinical observation of the concurrence of a de novo chromosome 22 inversion and ASD phenotype in two male siblings, we have accumulated three lines of evidence supporting a causative association between disruption of TCF20 and ASD, which was not identified by recent exome or genome sequencing studies.14–19 First, the original inversion separates the coding portion of TCF20 from a previously unannotated upstream untranslated exon that is conserved in mice, and therefore likely to have an important function. Second, we identified two de novo mutations of TCF20 (one encoding a missense change in a predicted PEST domain, the other a one-nucleotide deletional frameshift) in individuals with ASD. Third, we identified a significant association of ASD with a likely recurrent missense variant in a second predicted PEST domain of TCF20. Although we do not exclude a contributory role for disruption of TNRC6B to the ASD phenotype in family #1 (indeed, single CNV-based deletion and duplication events in ASD cases that include TNRC6B were previously catalogued),12 13 16 the evidence from our own study is more compelling for the contribution of TCF20, which is the focus of this discussion.
TCF20 (also termed SPBP, SPRE-binding protein) encodes a transcriptional coregulator,37 initially identified by its ability to bind the stromelysin-1 PDGF-responsive element (SPRE) element of the stromelysin-1 (matrix metalloproteinase-3/MMP3) promoter.38 Although widely expressed, TCF20 shows notably increased expression in premigratory neural crest cells39 and in the developing mouse brain at E13.5,40 with specific enrichment in the hippocampus and cerebellum.41 This brain expression pattern is consistent with a role in ASD.42 Significantly, TCF20 contains seven regions with 97% sequence similarity to RAI1,27 mutations and deletions of which underlie Smith–Magenis syndrome.32 The two proteins show an overall 45% similarity and share organisation of several domains such as the three nuclear localisation signals, a C-terminal extended PHD domain and an N-terminal transactivation domain.37 43 There is also striking similarity in the gene structure of RAI1 and TCF20, with over 90% of the coding region of each located in one exon that also contains the start codon, suggesting that TCF20 and RAI1 evolved from a common ancestor by genome duplication.43 A yeast two-hybrid screen with the ZNF2 domain of TCF20 as bait identified RAI-1 as a binding partner, showing that these proteins are able to interact and therefore may also be functionally related.27
To test whether mutations of TCF20 play a wider role in ASDs, we screened the coding sequence in 342 IMGSAC samples. We found two missense mutations of likely functional significance (see figure 3). One of these, p.K512E, had arisen de novo in the proband. Given that a total of 2 018 826 bp were screened in the ASD samples (5903 bp TCF20 coding region in 342 samples) and assuming a germline mutation rate of 1.2×10−8,44 the chance of coincidentally identifying an unrelated de novo variant is ∼0.05. Hence, the de novo nature of the p.K512E mutation favours a causal contribution to ASD, and the high evolutionary conservation of the K512 residue is consistent with this (figure 3C). Of note, a cousin of the proband also had ASD but did not carry the variant (figure 3B), suggesting that there is genetic heterogeneity for ASD causation within this family.
The second TCF20 variant of note is the c.4670C>T (p.P1557L) substitution identified in three ASD individuals from 335 successfully screened for this amplicon. In each case, the variant was present in one of the parents without ASD; no BAP data were available for two parents. In the multiplex family (#5), it was inherited from a father with evidence of BAP and also present in both an affected brother and a half-sister without ASD but for whom no BAP data were available. Haplotype analysis of the individuals carrying the c.4670C>T transition in the three families strongly supports an independent origin in family 5 compared with the other two families (table 4), compatible with the notion that the p.P1557L substitution confers selective disadvantage but is maintained at a low level in the population by recurrent mutation at the CpG dinucleotide.
Of note, the two ASD-associated missense changes in TCF20 each locates within a different PEST domain. PEST sequences are so-called because of enrichment in proline (P), glutamic acid (E), serine (S) and threonine (T) and are common in proteins that are rapidly degraded in eukaryotic cells35 and interact with Cul3, a subunit of a Cullin-RING ubiquitin E3 ligase complex that polyubiquitinates proteins.45 Loss of PEST motifs occurs, for example, in NOTCH1 and NOTCH2 mutations that characterise T-cell acute lymphoblastic leukaemia46 and Hajdu–Cheney syndrome,47 respectively. Hence, these observations suggest that the ASD-associated mutations might stabilise the protein rather than causing a haploinsufficiency. Alternatively, the p.P1557L substitution might affect the nucleosome-binding activity associated with this region of the TCF20 protein.43
The final piece of evidence linking TCF20 with ASD came serendipitously, while studying the genomic origins of craniosynostosis in an unrelated study. Exome sequencing of family #6 revealed a heterozygous mononucleotide frameshifting mutation of TCF20 in a woman with craniosynostosis, a phenotype that was also present in her mother. Given that the mother was of at least average range IQ, the ASD and moderate intellectual disability in her daughter were unexpected and were not thought to be directly related to the coincident craniosynostosis. In the context of our other findings, the de novo TCF20 mutation now provides a plausible explanation for the proband's phenotype. Although it might be expected that this mutation would lead to haploinsufficiency, cDNA analysis showed that the mutant message is stable (figure 3D). The more C-terminal PEST domain would be absent in the translated product, which could, as in the case of the missense mutations, stabilise the protein against degradation.
In summary, we propose that TCF20 mutations constitute a newly identified contributor to ASD that was not highlighted by recent genome-wide screens.12–19 TCF20 mutations may also contribute to intellectual disability, although not all individuals with mutations had his phenotype (table 2). Interestingly none of the mutations presented here predicts simple haploinsufficiency; this may explain why deletions of TCF20 have not been observed in previous extensive CNV screens of ASD. Rather, the pathophysiological mechanism may involve the persistence or misexpression of TCF20 in critical tissues or timepoints: this possibility should be addressed in future functional studies. Of particular interest in this regard, both underdose and overdose of the paralogous RAI1 protein cause overlapping neurological symptoms, suggesting that RAI1 gene dosage is critical in specific neurodevelopmental pathways.48 Given the likely functional overlap between TCF20 and RAI1, our observations provide strong support for further investigation of the normal functions of TCF20 in neurodevelopment and the role of mutations in ASD.
A recent meta-analysis of genome-wide association studies in schizophrenia49 identified a significant association with a SNP (rs6002655) lying within an intron of TCF20. This raises the possibility that variation in TCF20/SPBP function may impact neuropsychiatric disorders additional to ASD.
Supplementary Material
Acknowledgments
This work was funded by the Wellcome Trust (Core Award 090532/Z/09/Z to GM, EVV and SJLK, programme grant 078666 to AOMW, project grant 093329 to AOMW and SRFT), the Newlife Foundation for Disabled Children (10-11/04 to AOMW and SRFT), Royal College of Surgeons of England with support from the Rosetrees Trust and Blond-McIndoe Research Foundation (Research Fellowship to VPS) and the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health's National Institute for Health Research (NIHR) Biomedical Research Centres funding scheme (SJLK, JRP and AOMW). The views expressed in this publication are those of the authors and not necessarily those of the Department of Health.
Footnotes
Collaborators: International Molecular Genetic Study of Autism Consortium (IMGSAC).
Contributors: Performed experiments: CB, DL, ATP, JG, GM, RN, VPS and EVV. Analysed data and performed bioinformatics analysis: SJM and SJLK. Supervised experiments: SRFT, EVV, VJB, SJLK and AOMW. Referred patients: SAW, International Molecular Genetic Study of Autism Consortium (IMGSAC), JRP and AOMW. Analysed data and drafted manuscript: CB, DL, ATP, JRP and AOMW. All authors approved manuscript.
Competing interests: None.
Ethics approval: West London Research Ethics Committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, Charman T. Prevalence of disorders of the autism spectrum in a population cohort of children in south thames: the Special Needs and Autism Project (SNAP). The Lancet 2006;368:210–15. [DOI] [PubMed] [Google Scholar]
- 2.Parr JR. Autism spectrum disorders. In: Seal A, Robinson G, Kelly AM, Williams J, eds. Children with neurodevelopmental disabilities: the essential guide to assessment and management. London, UK: MacKeith Press, 2013:367–84. [Google Scholar]
- 3.Pickles A, Parr JR, Rutter ML, De Jonge MV, Wallace S, Le Couteur AS, van Engeland H, Wittemeyer K, McConachie H, Roge B, Mantoulan C, Pedersen L, Isager T, Poustka F, Bolte S, Bolton P, Weisblatt E, Green J, Papanikolaou K, Bailey AJ. New interview and observation measures of the broader autism phenotype: impressions of interviewee measure. J Autism Dev Disord 2013;43:2082–89. [DOI] [PubMed] [Google Scholar]
- 4.De Jonge M, Parr JR, Rutter ML, Wallace S, Kemner C, Bailey AJ, van Engeland H, Pickles A. New interview and observation measures of the Broader Autism Phenotype: Group differentiation. J Autism Dev Disord 2014, in press. [DOI] [PubMed] [Google Scholar]
- 5.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995;25:63–77. [DOI] [PubMed] [Google Scholar]
- 6.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 2008;9:341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 2011;1380:42–77. [DOI] [PubMed] [Google Scholar]
- 8.Carter MT, Scherer SW. Autism spectrum disorder in the genetics clinic: a review. Clin Genet 2013;83:399–407. [DOI] [PubMed] [Google Scholar]
- 9.Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, Vorstman JA, Thompson A, Regan R, Pilorge M, Pellecchia G, Pagnamenta AT, Oliveira B, Marshall CR, Magalhaes TR, Lowe JK, Howe JL, Griswold AJ, Gilbert J, Duketis E, Dombroski BA, De Jonge MV, Cuccaro ML, Crawford EL, Correia CT, Conroy J, Conceição IC, Chiocchetti AG, Casey J, Cai G, Cabrol C, Bolshakova N, Bacchelli E, Anney R, Gallinger S, Cotterchio M, Casey G, Zwaigenbaum L, Wittemeyer K, Wing K, Wallace S, van Engeland H, Tryfon A, Thomson S, Soorya L, Roge B, Roberts W, Poustka F, Mouga S, Minshew N, McInnes LA, McGrew SG, Lord C, Leboyer M, Le Couteur AS, Kolevzon A, González PJ, Jacob S, Holt R, Guter SJ, Green J, Green A, Gillberg C, Fernandez B, Duque F, Delorme R, Dawson G, Chaste P, Café C, Brennan S, Bourgeron T, Bolton PF, Bölte S, Bernier R, Baird G, Bailey AJ, Anagnostou E, Almeida J, Wijsman EM, Vieland VJ, Vicente AM, Schellenberg GD, Pericak-Vance MA, Paterson AD, Parr JR, Oliveira G, Nurnberger JI, Monaco AP, Maestrini E, Klauck SM, Hakonarson H, Haines JL, Geschwind DH, Freitag CM, Folstein SE, Ennis S, Coon H, Battaglia A, Szatmari P, Sutcliffe JS, Hallmayer J, Gill M, Cook EH, Buxbaum JD, Devlin B, Gallagher L, Betancur C, Scherer SW. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 2014;94:667–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Casey J, Conroy J, Correia C, Corsello C, Crawford EL, de Jonge M, Delorme R, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Gilbert J, Gillberg C, Glessner JT, Green A, Green J, Guter SJ, Heron EA, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Jacob S, Kenny GP, Kim C, Kolevzon A, Kustanovich V, Lajonchere CM, Lamb JA, Law-Smith M, Leboyer M, Le Couteur A, Leventhal BL, Liu XQ, Lombard F, Lord C, Lotspeich L, Lund SC, Magalhaes TR, Mantoulan C, McDougle CJ, Melhem NM, Merikangas A, Minshew NJ, Mirza GK, Munson J, Noakes C, Nygren G, Papanikolaou K, Pagnamenta AT, Parrini B, Paton T, Pickles A, Posey DJ, Poustka F, Ragoussis J, Regan R, Roberts W, Roeder K, Roge B, Rutter ML, Schlitt S, Shah N, Sheffield VC, Soorya L, Sousa I, Stoppioni V, Sykes N, Tancredi R, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Vorstman J, Wallace S, Wing K, Wittemeyer K, Wood S, Zurawiecki D, Zwaigenbaum L, Bailey AJ, Battaglia A, Cantor RM, Coon H, Cuccaro ML, Dawson G, Ennis S, Freitag CM, Geschwind DH, Haines JL, Klauck SM, McMahon WM, Maestrini E, Miller J, Monaco AP, Nelson SF, Nurnberger JI, Jr, Oliveira G, Parr JR, Pericak-Vance MA, Piven J, Schellenberg GD, Scherer SW, Vicente AM, Wassink TH, Wijsman EM, Betancur C, Buxbaum JD, Cook EH, Gallagher L, Gill M, Hallmayer J, Paterson AD, Sutcliffe JS, Szatmari P, Vieland VJ, Hakonarson H, Devlin B. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet 2012;21:4781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger JI, Jr, Paterson AD, Pericak-Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, Betancur C. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010;466:368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, Buja A, Krieger A, Yoon S, Troge J, Rodgers L, Iossifov I, Wigler M. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011;70:886–97. [DOI] [PubMed] [Google Scholar]
- 13.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O'Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Günel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tischfield JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH, Jr, Geschwind D, Roeder K, Devlin B, State MW. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011;70:863–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron 2012;74:285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH, Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012;485:242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012;485:246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012;485:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, Ju J, Mei J, Shi Y, He M, Wang G, Liang J, Wang Z, Cao D, Carter MT, Chrysler C, Drmic IE, Howe JL, Lau L, Marshall CR, Merico D, Nalpathamkalam T, Thiruvahindrapuram B, Thompson A, Uddin M, Walker S, Luo J, Anagnostou E, Zwaigenbaum L, Ring RH, Wang J, Lajonchere C, Wang J, Shih A, Szatmari P, Yang H, Dawson G, Li Y, Scherer SW. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet 2013;93:249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, D'Gama AM, Lim ET, Sanders SJ, Mochida GH, Partlow JN, Sunu CM, Felie JM, Rodriguez J, Nasir RH, Ware J, Joseph RM, Hill RS, Kwan BY, Al-Saffar M, Mukaddes NM, Hashmi A, Balkhy S, Gascon GG, Hisama FM, LeClair E, Poduri A, Oner O, Al-Saad S, Al-Awadi SA, Bastaki L, Ben-Omran T, Teebi AS, Al-Gazali L, Eapen V, Stevens CR, Rappaport L, Gabriel SB, Markianos K, State MW, Greenberg ME, Taniguchi H, Braverman NE, Morrow EM, Walsh CA. Using whole-exome sequencing to identify inherited causes of autism. Neuron 2013;77:259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Molecular Genetic Study of Autism C. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet 1998;7:571–78. [DOI] [PubMed] [Google Scholar]
- 21.International Molecular Genetic Study of Autism C. A genomewide screen for autism: Strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet 2001;69:570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lord C, Rutter M, Le Couteur A. Autism diagnostic interview-revised: a revised version of a diagnostic interviews for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 1994;24:659–85. [DOI] [PubMed] [Google Scholar]
- 23.Sparrow S, Balla D, Cicchetti D. Vineland adaptive behaviour scales. Circle Pines, MN: American Guidance Service, 1984. [Google Scholar]
- 24.Lord C, Risi S, Lambrecht L, Cook EH, Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 2000;30:205–23. [PubMed] [Google Scholar]
- 25.Raven J, Raven JC, Court JH. Manual for Raven's progressive matrices and vocabulary scales. Section 2: the coloured progressive matrices. San Antonio, TX: Harcourt assessment, 1998. [Google Scholar]
- 26.Dunn LM, Dunn LM, Whetton C, Burley J. The British picture vocabulary scale: 2nd edition—BPVS II. Windsor: NFER-Nelson, 1997. [Google Scholar]
- 27.Rekdal C, Sjøttem E, Johansen T. The nuclear factor SPBP contains different functional domains and stimulates the activity of various transcriptional activators. J Biol Chem 2000;275:40288–300. [DOI] [PubMed] [Google Scholar]
- 28.Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, Broad GO, Seattle GO, on behalf of the NHLBI Exome Sequencing Project. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012;337:64–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 2009;41:849–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioural phenotypes in a mouse model of dup(17)(p11.2p11.2). J Clin Invest 2006;116:3035–-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet 2007;80:633–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet 2003;33:466–8. [DOI] [PubMed] [Google Scholar]
- 33.Laje G, Morse R, Richter W, Ball J, Pao M, Smith AC. Autism spectrum features in Smith-Magenis syndrome. Am J Med Genet C Semin Med Genet 2010;154C:456–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baillat D, Shiekhattar R. Functional dissection of the human TNRC6 (GW182-related) family of proteins. Mol Cell Biol 2009;29:4144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci 1996;21:267–71. [PubMed] [Google Scholar]
- 36.Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JAC, Hoogeboom AJM, Brady AF, Jeelani NO, Lynch SA, Mulliken JB, Murray DJ, Phipps JM, Sweeney E, Tomkins SE, Wilson LC, Bennett S, Cornall RJ, Broxholme J, Kanapin A, 500 Whole-Genome Sequences (WGS500) Consortium. Johnson D, Wall SA, van der Spek PJ, Mathijssen IMJ, Maxson RE, Twigg SRF, Wilkie AOM. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet 2013;45:304–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darvekar S, Johnsen SS, Eriksen AB, Johansen T, Sjøttem E. Identification of two independent nucleosome-binding domains in the transcriptional co-activator SPBP. Biochem J 2012;442:65–75. [DOI] [PubMed] [Google Scholar]
- 38.Sanz L, Moscat J, Diaz-Meco MT. Molecular characterization of a novel transcription factor that controls stromelysin expression. Mol Cell Biol 1995;15:3164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams MS, Gammill LS, Bronner-Fraser M. Discovery of transcription factors and other candidate regulators of neural crest development. Dev Dyn 2008;237:1021–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray PA, Fu H, Luo P, Zhao Q, Yu J, Ferrari A, Tenzen T, Yuk D, Tsung EF, Cai Z, Alberta JA, Cheng L, Liu Y, Stenman JM, Valerius MT, Billings N, Kim HA, Greenberg ME, McMahon AP, Rowitch DH, Stiles CD, Ma Q. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 2004;306:2255–57. [DOI] [PubMed] [Google Scholar]
- 41.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007;445:168–76. [DOI] [PubMed] [Google Scholar]
- 42.Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci 2008;31:137–45. [DOI] [PubMed] [Google Scholar]
- 43.Darvekar S, Rekdal C, Johansen T, Sjøttem E. A phylogenetic study of SPBP and RAI1: evolutionary conservation of chromatin binding modules. PLoS ONE 2013;8:e78907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father's age to disease risk. Nature 2012;488:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xing H, Hong Y, Sarge KD. PEST sequences mediate heat shock factor 2 turnover by interacting with the Cul3 subunit of the Cul3-RING ubiquitin ligase. Cell Stress Chaperones 2010;15:301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weng AP, Ferrando AA, Lee W, Morris JP, IV, Silverman LB, Sanchex-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell actute lymphoblastic leukemia. Science 2004;306:269–71. [DOI] [PubMed] [Google Scholar]
- 47.Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK, Kim KH, Pauli RM, Aftimos S, Stewart H, Kim CA, Holder-Espinasse M, Robertson SP, Drake WM, Trembath RC. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet 2011;43:303–05. [DOI] [PubMed] [Google Scholar]
- 48.Cao L, Molina J, Abad C, Carmona-Mora P, Cárdenas Oyarzo A, Young JI, Walz K. Correct developmental expression level of Rai1 in forebrain neurons is required for control of body weight, activity levels and learning and memory. Hum Mol Genet 2014;23:1771–82. [DOI] [PubMed] [Google Scholar]
- 49.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.