

Key Points
Bone marrow failure can derive from germline mutation of the telomere protein TPP1 that is involved in recruiting telomerase to telomeres.
The observed mutation disrupts recruitment of telomerase to telomeres and segregates with short telomere length and disease phenotype.
Abstract
Telomerase is a ribonucleoprotein enzyme that is necessary for overcoming telomere shortening in human germ and stem cells. Mutations in telomerase or other telomere-maintenance proteins can lead to diseases characterized by depletion of hematopoietic stem cells and bone marrow failure (BMF). Telomerase localization to telomeres requires an interaction with a region on the surface of the telomere-binding protein TPP1 known as the TEL patch. Here, we identify a family with aplastic anemia and other related hematopoietic disorders in which a 1-amino-acid deletion in the TEL patch of TPP1 (ΔK170) segregates with disease. All family members carrying this mutation, but not those with wild-type TPP1, have short telomeres. When introduced into 293T cells, TPP1 with the ΔK170 mutation is able to localize to telomeres but fails to recruit telomerase to telomeres, supporting a causal relationship between this TPP1 mutation and bone marrow disorders. ACD/TPP1 is thus a newly identified telomere-related gene in which mutations cause aplastic anemia and related BMF disorders.
Introduction
Telomeres are the repetitive DNA–protein complexes at the ends of linear chromosomes, which shorten with every cell division in human somatic cells.1 Telomere shortening eventually leads to the activation of a DNA damage response and cellular senescence or apoptosis.2-4 Germ cells, stem cells, and cells in the developing human embryo overcome telomere shortening by expression of the ribonucleoprotein enzyme telomerase.5-8 Telomerase catalyzes addition of telomeric DNA repeats to chromosome ends by reverse transcription of a template sequence within its integral RNA subunit (hTR in humans9). The core active human telomerase complex consists of hTR, the reverse-transcriptase protein subunit (hTERT),10 and the RNA-binding protein dyskerin.11,12 Telomerase interacts with many other proteins during its biogenesis and transport to the telomere, such as the protein TCAB1 (also known as WDR79 and WRAP53), which is responsible for trafficking telomerase to nuclear Cajal bodies13,14 and from there to the telomere.15
Individuals who inherit extremely short telomeres are at risk of developing one of a group of disorders collectively termed “short telomere syndromes.”16,17 The first of these diseases linked to short telomeres was dyskeratosis congenita (DC),11 defined by a mucocutaneous triad of abnormal skin pigmentation, oral leukoplakia, and nail dystrophy and characterized by progressive bone marrow failure due to depletion of functional hematopoietic progenitor and stem cells.18 Patients with DC, or its more severe variants Revesz syndrome and Hoyeraal-Hreidarsson syndrome, harbor deleterious mutations in the genes encoding dyskerin,19 hTR,20 hTERT,21 TCAB1,22 the dyskerin-associated proteins Nop1023 and Nhp2,24 or the telomere-binding proteins TIN2,25,26 CTC1,27,28 and RTel1.29-32 Patients with acquired or familial aplastic anemia, but lacking the other clinical features of DC, can also carry short telomeres and mutations in the genes encoding hTR33 or hTERT.21,34
The telomere-binding protein TPP1 (encoded by the gene adrenocortical dysplasia homolog [mouse] [ACD] and previously known as PTOP, PIP1, or TINT2) is critical for telomere stability and length regulation.35-38 TPP1 binds to the telomere via interaction with TIN2 and in turn tethers the single-stranded DNA binding protein Pot1 to telomeres.36-38 TPP1 is required for Pot1 to perform its role in protecting telomeres from being recognized as DNA damage.39-41 The Pot1/TPP1 heterodimer also has a separate role in promoting the processivity of telomerase, ie, its ability to synthesize long tracts of telomeric DNA.42 A region on the surface of TPP1 known as the TEL patch mediates this interaction with telomerase and is necessary for the recruitment of telomerase to telomeres.43-46
Here, we identify a family with 3 generations of individuals with short telomeres and aplastic anemia or other related blood disorders, all of whom carry a heterozygous mutation in the ACD/TPP1 gene. This mutation results in an in-frame deletion of an amino acid in the TEL patch of TPP1, and we demonstrate that the mutated TPP1 protein is able to localize to telomeres but fails to recruit telomerase to telomeres. These data support a causal relationship between this TPP1 mutation and disease in this family.
Methods
Subjects
The proband presented to Children’s Hospital Westmead, with severe thrombocytopenia and macrocytosis and has been diagnosed with aplastic anemia. Her family history includes other cases of bone marrow failure (BMF) as well as oral carcinoma and leukemia (Figure 1A and Table 1). Peripheral blood DNA was available from the proband, her parents, and her maternal grandparents. Informed consent was obtained from all participating individuals, in accordance with the Declaration of Helsinki, and the studies were approved by the Human Research Ethics Committee of the Sydney Children’s Hospitals Network.
Figure 1.
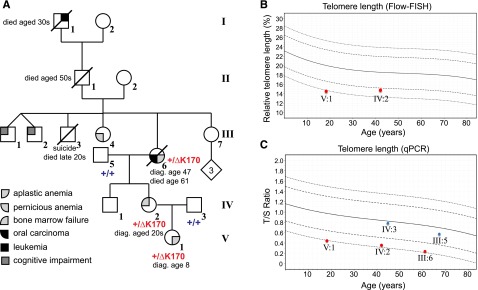
ACD/TPP1 mutation and telomere length in family with history of anemia and cancer. (A) Pedigree and clinical diagnoses of family. The genotypes of the proband and 4 of her family members are shown (+/+ for homozygous WT and +/ΔK170 for heterozygous deletion). Numbering of individuals corresponds with numbering in supplemental Table 1. (B-C) Telomere lengths of 5 family members were measured by Flow-FISH (B) or qPCR (C). Red symbols indicate individuals with the ΔK170 mutation, and blue symbols indicate WT individuals. Curves represent the 1st, 10th, 50th, 90th, and 99th percentiles of telomere lengths in ∼240 healthy individuals. diag., diagnosed.
Table 1.
Hematologic features of affected family members
| Age (y) | Hb (g/L) | MCV (fL) | Neutrophils (× 109/L) | Platelets (× 109/L) | HbF (%) | Bone marrow | BM cytogenetics | Diagnosis | Outcome | |
|---|---|---|---|---|---|---|---|---|---|---|
| Proband V:1 | 8 | 70 | 121 | 0.6 | 18 | 31 | Aplastic | Normal | Severe aplastic anemia | Alive, transfusion dependent |
| Mother IV:2 | 33 | 126 | 120 | 1.7 | 33 | 3.5 | Hypocellular | Normal | BMF, mild | Alive, stable hematology |
| Maternal g/m III:6 | 55 | 104 | 117 | 1.6 | 102 | N/A | Hypocellular | N/A | Aplastic anemia, oral carcinoma | Deceased (61 y) |
g/m, grandmother; Hb, hemoglobin; HbF, fetal hemoglobin; MCV, mean corpuscular volume; N/A, not available.
Whole-exome sequencing and bioinformatics
The procedure was described elsewhere.47 Briefly, exonic regions were captured using Agilent SureSelect Human All Exon kit, and pair-end sequencing was carried out on Illumina HiSeq 2000 machines. The Illumina pipeline at default settings was applied to process raw image files for base calling and generating raw sequencing read files in fastq format. Subsequently, 2 independent analysis workflows were used to perform sequencing read alignment, variant calling, and variant annotation (supplemental Table 1, available on the Blood Web site). In the first pipeline, Burrows-Wheeler alignment was used to align fastq files to the human reference genome (University of California, Santa Cruz [UCSC] hg19).48 Then, variants were called using the Genome Analysis Tool Kit (GATK, version 1.4)49 followed by functional annotation with Annovar50 and SnpEff.51 In the second pipeline, alignment to the human reference genome (UCSC hg19) was performed by Short Oligonucleotide Analysis Package (SOAP, version 2.21).52 We used SOAPsnp (version 1.05)53 to detect single-nucleotide variants (SNVs) and GATK to call insertion-deletions (indels). Finally, annotated by BGI’s self-developed scripts, the variants were grouped into different categories.
Multiple filtering steps and Mendelian genetic analysis
Under an autosomal-dominant inheritance, we applied the following mutational filtering steps in both pipelines described above. We first selected heterozygous variants shared by all 3 affected individuals and required that those variants should not be found in the proband’s unaffected grandfather. Then, we eliminated variants that met either one of the following criteria: (1) out of exonic regions, (2) synonymous changes, and (3) with frequency >0.5% in dbSNP135,54 1000 Genomes Project (n = 1092 genotyped samples),55 HapMap Project (n = 1301 genotyped samples),56 NHLBI Exome Sequencing Project (n = 6500 exomes) (http://evs.gs.washington.edu/EVS/), CAG-CHOP (n = 334 exomes), or BGI (n = 1414 control exomes) databases. We paid special attention to variants close to splicing sites and frameshift indels. In the next step, we considered evolutionary conservation and filtered the nonconserved regions with PhyloP57 value <0.95, then retained variants with “deleterious/damaging” prediction of pathogenicity by PolyPhen58 and SIFT.59 Finally, we took into account biological and clinical relevance of the identified variants (supplemental Table 2).
PCR and Sanger sequencing
The deletion in ACD/TPP1 (RefSeq: NM_001082486.1) identified through whole-exome sequencing (WES) was confirmed by Sanger sequencing of DNA from peripheral blood of all available family members, including the 3 affected individuals and clinically unaffected father and maternal grandfather. The primers TPP1-SeqF and TPP1-SeqR (supplemental Table 3) were used for polymerase chain reactions (PCRs) as reported previously.60 Purified PCR products were sequenced by the Australian Genome Research Facility (AGRF Ltd., Sydney, Australia). The sequencing traces were analyzed using BioEdit Sequence Alignment Editor.61
Patient telomere length analysis (Flow-FISH)
Telomere flow-fluorescence in situ hybridization (Flow-FISH) was performed with a published protocol.62 Approximately 10 mL of lithium-heparin peripheral blood was collected, and mononuclear cells were isolated by Ficoll density centrifugation (1077 Ficoll Histopaque; Sigma-Aldrich). Duplicate samples of 2 × 106 cells were used for FISH analysis. A known cell line with long telomeres (human tetraploid T-cell lymphoblastic line CCRF-CEM, GM03671C; Coriell Institute for Medical Research, Camden, NJ) served as an internal reference standard for telomere length. Equal numbers of CCRF-CEM cells were mixed with patient cells prior to hybridization with a fluorescein isothiocyanate–conjugated (CCCTAA)3 peptide nucleic acid probe (Panagene, Daejeon, Korea) at 0.3 μg/mL. Unstained duplicate tubes were run in parallel to determine autofluorescence. After denaturation, hybridization was performed at 4°C overnight, and excess telomere probe was removed with washing. Flow cytometry was performed on a FACS CANTO II (BD Biosciences, San Jose, CA) instrument and data displayed and analyzed with BD FACSDiva software (BD Biosciences). Calculation of relative telomere length of the patient’s mononuclear cells was performed by comparing the fluorescence of these cells with the tetraploid CCRF-CEM cell line and expressed as a percentage. Values obtained from 240 healthy individuals show the normal percentiles for different age groups (Figure 1B).
Patient telomere length analysis (qPCR)
Genomic DNA from peripheral blood samples was used in telomere-length analysis by quantitative PCR (qPCR), performed on the Rotor-Gene Q (Qiagen), using Rotor-Gene SYBR Green PCR Master Mix (Qiagen). The assay was similar to that previously reported.63,64 Briefly, telomeric (T) DNA was amplified using Tel1b and Tel2b primers (supplemental Table 3), whereas the single copy gene (S), used to normalize the amount of input DNA, was amplified using the HBG1 and HBG2 primers (supplemental Table 3). A standard curve was used to convert the cycle threshold into nanograms of DNA and relative telomere length calculated as the T/S ratio from the mean of triplicate samples. Values obtained from 240 healthy individuals show the normal percentiles for different age groups.
Detection of endogenous TPP1 messenger RNA
RNA was extracted from cells using the RNeasy Mini Kit (Qiagen), followed by DNA digestion with DNase I (Invitrogen). Complementary DNA synthesis was performed with the SuperScript III First Strand Synthesis System for RT-PCR (Invitrogen). To detect expression of the mutant allele, a 400-bp region of ACD/TPP1 spanning the mutation was amplified by PCR using primers TPP1-Fw1 and TPP1-Rv1 (supplemental Table 3). Amplification was performed using Platinum Taq DNA Polymerase (Life Technologies) in an Eppendorf Mastercycler gradient at 95°C for 5 minutes, followed by 36 cycles of 95°C for 30 seconds, 60°C for 45 seconds, and 72°C for 45 seconds. An aliquot of the PCR products was visualized with 1% agarose gel electrophoresis and the remainder purified using a Qiagen PCR Purification Kit and digested with the restriction enzyme XmnI, followed by 1% agarose gel electrophoresis.
For quantitative reverse-transcription PCR (qRT-PCR) of endogenous TPP1 messenger RNA (mRNA) levels, the primers targeting the 5′ untranslated region (UTR) of TPP1 (ie, specific for expression of the endogenous gene) were TPP1-Fw2 and TPP1-Rv2 (supplemental Table 3). Glyceraldehyde-3-phosphate dehydrogenase was used as the internal control (supplemental Table 3). qRT-PCR was performed using SYBR Green Master Mix (Invitrogen) in a LightCycler 96 (Roche) with 40 cycles of 95°C for 30 seconds, 60°C for 45 seconds, and 72°C for 45 seconds.
Site-directed mutagenesis of TPP1 expression plasmid
An expression construct for mutant ΔK170 of TPP1 was constructed in pCMV6-Myc-DDK vector containing wild-type (WT) TPP1 (ACD) (RC204381; Origene) as the template. The deletion of lysine 170 was achieved using the Stratagene QuikChange II XL site-directed mutagenesis kit.
Cell culture, transfections, and cell-cycle synchronizations
Human embryonic kidney 293T cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (vol/vol) fetal bovine serum in a humidified 37°C incubator with 5% CO2. Cells were transfected with 1 µg each of expression constructs of WT and mutant ΔK170 TPP1 using X-tremeGENE 9 (Roche) for 9 hours. After medium change, 90 pmol of silencing RNA (siRNA) targeting the 5′ UTR of TPP1 (Hs_ACD_9 FlexiTube siRNA, Qiagen) was transfected with RNAiMax (Life Technologies) for 24 hours. All Stars Negative Control siRNA (Qiagen) was used as the negative control. Cells were then synchronized at G1/S phase by adding 2 mM thymidine (Sigma-Aldrich) for 15 hours, releasing for 9 hours, and adding 0.5 µg/mL aphidicolin (Sigma-Aldrich) for 16 hours. Cells were harvested 3 hours after release and confirmed to be in mid-S phase by analysis of DNA content by flow cytometry on a FACSDiva (Becton Dickinson).
FISH of hTR and telomeric DNA
Simultaneous FISH against hTR and telomeric DNA was performed as described previously.15 Briefly, harvested synchronized 293T cells were cytospun onto slides, fixed with formaldehyde, and permeabilized. Slides were incubated with 5 Alexa Fluor 488–labeled anti-hTR oligonucleotides (Sigma-Aldrich; supplemental Table 3) and a Texas red–labeled telomere probe (Sigma-Aldrich; supplemental Table 3) in FISH hybridization buffer.15 FISH staining was visualized on a Zeiss AxioImager M1 microscope with a Plan-Apochromat 63× oil objective (numerical aperture 1.4) and an AxioCam MR digital camera (Carl Zeiss) with consistent exposure times between experiments. For presentation purposes, pixel intensity histograms were adjusted in ZEN (Carl Zeiss), equally across all figure panels, and images were cropped in Adobe Photoshop. Colocalizing hTR and telomere foci were manually counted in 100 cells per treatment, and data were analyzed by Student t test for pairwise comparison.
Chromatin immunoprecipitation (ChIP) of telomeric DNA
Telomeric ChIP was conducted as described previously.15 Briefly, nuclei from synchronized cells were crosslinked with formaldehyde. Nuclei were lysed and sonicated, and the chromatin was clarified by centrifugation. An aliquot of chromatin was used as an “input” sample for normalization of recovered chromatin, and the remaining chromatin was incubated with anti-Myc antibody (Myc-tag [9B11] mouse monoclonal antibody, #2276S; Cell Signaling Technology). Samples were immunoprecipitated and DNA purified using Qiagen PCR Purification Kit and applied to Hybond XL membrane (GE Healthcare Life Sciences). The membrane was hybridized overnight with a (CCCTAA)3 telomeric DNA oligonucleotide probe end labeled with [γ-32P]ATP. The amount of DNA recovered was visualized on a Typhoon TRIO Imager (GE Healthcare Life Sciences) and quantified with ImageQuant (GE Healthcare Life Sciences). Standard curves based on the signal of the input samples were used to normalize the amount of telomeric DNA recovered.
Cell lysis and western blot
Harvested synchronized cells were lysed using 4× lithium dodecyl sulfate buffer (106 mM Tris-HCl, 141 mM Tris-base, 2% lithium dodecyl sulfate, 40% glycerol, 0.075% SERVA Blue G50, and 0.025% phenol red) containing 2% β-mercaptoethanol (Sigma-Aldrich) and Benzonase nuclease (0.5 U/μL; Novagen). The cells extracts were incubated at 68°C for 10 min prior to loading 70 000 cell equivalents onto precast NuPAGE 4% to 12% Bis-Tris gels (Life Technologies) and electrophoresis with MES buffer (Life Technologies) for 50 minutes at 200 V. The proteins were transferred onto Hybond ECL membrane (GE Healthcare Life Sciences) at 100 V for 60 minutes at 4°C with transfer buffer (25 mM Tris-base and 192 mM Glycine). The membrane was washed and blocked with 5% skim milk in TBS buffer (15 mM Tris-HCl [Sigma], 4.6 mM Tris-base, and 137 mM NaCl) and 0.1% Tween-20 (TBST) for 1 hour at room temperature. TPP1 antibody (α-ACD MaxPAB, H00065057-B01P, Abnova) was added to 5% skim milk in TBST (1:2500) and incubated with the membrane overnight at 4°C. The membrane was washed 5 times for 5 minutes with TBST and incubated with secondary antibody (Dako goat anti-mouse; 1:5000) conjugated to horseradish peroxidase in 5% skim milk-TBST for 1 hour and then washed as before. Horseradish peroxidase signal was detected using Supersignal West Pico Chemiluminescent Substrate (#34080; Pierce) and visualized with a FujiFilm LAS 4000 Imager.
Results
Disease phenotype of proband and family
The 18-year-old proband (Figure 1A V:1), her mother, and her maternal grandmother presented with BMF of varying severity, and their decreasing ages of presentation in successive generations suggested disease anticipation (Table 1). The proband is the only child of nonconsanguineous white parents. She presented at 8 years of age with increasing pancytopenia, having been previously healthy with normal growth and development. Pancytopenia was associated with marked elevation in fetal hemoglobin (31%) and macrocytosis (Table 1). The bone marrow was markedly hypocellular without morphological dysplasia or cytogenetic abnormality, consistent with aplastic anemia. There was no associated facial dysmorphism or skeletal, cardiac, or urogenital tract anomalies. There were also no mucocutaneous manifestations, and none developed over the next 10 years. The screen for Fanconi anemia was negative, with no increase in chromosomal breakages on exposure to mitomycin C and diepoxybutane. Immunosuppressive therapy (4 courses of anti-thymocyte globulin and corticosteroids) was trialed without response. She responded to androgen therapy (oxymetholone) between the ages of 9 and 14 years, maintaining hemoglobin between 100 and 120 g/L, neutrophils 1.0 to 2.0 × 109/L, and platelets 35 to 50 × 109/L. However, androgen therapy was complicated by virilization, leading to variable compliance, and it was ceased at 14 years of age allowing normal pubertal progression; pancytopenia promptly recurred, and the proband became transfusion dependent for erythrocytes and platelets.
Although the proband’s mother (Figure 1A, IV:2) was diagnosed with myelodysplasia in her early 20s following investigations for thrombocytopenia, she most likely has mild BMF (Table 1). She has remained asymptomatic without disease progression or malignant transformation into her mid-40s. She has 1 unaffected healthy male sibling.
The proband’s maternal grandmother (Figure 1A, III:6) was investigated for mild macrocytic anemia with variable thrombocytopenia associated with hypocellular bone marrow at 47 years of age. A diagnosis of possible aplastic anemia/myelodysplasia was made, and she was monitored without therapy as cytopenia was mild and there were minimal symptoms. Earlier blood counts are not available, but parameters at 55 years are shown in Table 1. She developed carcinoma of the tongue at 59 years of age despite having been a lifelong nonsmoker and teetotaler; this was treated with surgery and local radiotherapy only. Over the ensuing 12 months, there was increasing pancytopenia requiring erythrocyte transfusions. Her death at 61 years was attributed to aplastic anemia. This patient’s paternal grandfather died of leukemia in his 30s; there were, however, no clinical or hematologic data available on this individual.
The proband was screened for mutations in DC genes with autosomal-dominant inheritance at the age of 14 years, and no mutations were detected in TERC, TERT, and TINF2. She was later retested using WES (performed by the Dyskeratosis Congenita Registry, London), and no significant variants in any of the 9 known DC genes were detected. A blood sample referred in 2012 to Repeat Diagnostics (Vancouver, BC, Canada) for telomere-length testing showed an average telomere length of less than the 1st percentile (lymphocytes and granulocytes) by Flow-FISH, supporting the diagnosis of DC or a related disorder of telomere length in the proband (J.T. and Repeat Diagnostics, unpublished data). The proband and her mother were retested when an in-house Flow-FISH assay was available; mononuclear telomere length was less than the 1st percentile for the proband and around the 5th percentile for her mother (Figure 1B). qPCR analysis of peripheral blood telomere length63 confirmed these findings and revealed short telomeres in the affected grandmother, but not in the unaffected father or grandfather (Figure 1C). Thus, short telomeres segregate with disease phenotype in this family.
Genomic variants identified by WES and bioinformatics
We performed WES on the proband (V:1), her mother (IV:2), and her maternal grandparents (III:5 and III:6) (Figure 1A), with each individual’s exome covered at least 62 times (supplemental Table 1). On average, ∼44 000 SNVs and ∼7000 small indels were called for each exome by pipeline 1 (see “Methods”), whereas ∼87 000 SNVs and ∼6000 indels were called by pipeline 2. After filtering, over 16 000 SNVs and ∼600 indels were retained as qualified for further analysis in pipeline 1 and over 17 000 SNVs and ∼700 indels were left in pipeline 2. We followed an autosomal-dominant inheritance, and each pipeline generated a list of candidate variants (supplemental Table 2). We noticed the overlapping variant of a 3-bp deletion in gene ACD, which encodes the human TIN2- and Pot1-interacting protein (TPP1); this variant could only be found in the 3 affected samples and is absent from any public databases (supplemental Table 2). Sanger sequencing confirmed the heterozygous 3-bp deletion (c.499_501del; p.Lys170del) in the genome of the affected individuals, including the proband, her mother, and her maternal grandmother (Figure 2A). The deletion is predicted to result in an in-frame deletion of amino acid lysine 170 (K170) of the TPP1 protein. The proband’s unaffected father and maternal grandfather were homozygous WT, demonstrating that the mutation segregates perfectly with both telomere length and disease phenotype.
Figure 2.

Genomic sequencing and mRNA expression of the ΔK170 mutant allele. (A) Sanger sequencing validates segregation with disease of a 3-bp deletion in the ACD gene, encoding the protein TPP1. The deletion (c.499_501del; p.Lys170del) was detected in the proband, mother, and maternal grandmother. The deletion was not present in the father and maternal grandfather. (B) RT-PCR of a 400-bp region across the ΔK170 region of TPP1 (left). RNA was isolated from fibroblasts of the proband (ΔK170) and the cell line HCT116 as a positive control for WT TPP1 expression (WT). Plasmids encoding WT or ΔK170 TPP1 sequence (pWT and pΔK170) were amplified as positive controls. XmnI digestion of the PCR products from the left panel (right). WT sequence is digested into 2 pieces of 212 and 188 bp, whereas the ΔK170 sequence remains uncut.
Expression of the mutant allele in fibroblasts derived from the proband was confirmed by RT-PCR (Figure 2B). The primers used for this PCR generate a product of 400 bp spanning the mutated region, and deletion of the codon encoding K170 removes an XmnI restriction site in the center of the PCR product, enabling us to distinguish mutant from WT mRNA. A cell line with WT ACD/TPP1 produced a band of 400 bp that was cleaved in two with Xmn1, whereas approximately half of the RT-PCR product from the patient’s cells was not digested with this enzyme (Figure 2B). This demonstrates that the WT and ΔK170 TPP1 alleles are expressed equally at the mRNA level in cells from the patient.
TPP1 mutant ΔK170 localizes to telomeres in human cells
Lysine 170 is localized on the surface of the TPP1 protein known as the TEL patch, between 2 amino acids shown to be vital for binding to telomerase, recruitment of telomerase to telomeres, and telomere length maintenance.44 Substitution of K170 with alanine resulted in a modest reduction in the ability of telomerase to colocalize with TPP1.45 To determine whether deletion of this residue could be causing short telomeres and disease in the family of interest, we analyzed the functional impact of deletion of K170 in immortal human cells in culture.
Nearby mutations in TPP1 are known to be “separation of function” mutations, because they disrupt telomerase interactions without affecting the ability of TPP1 to localize to and protect telomeres.44 To test the hypothesis that ΔK170 TPP1 could localize to telomeres as efficiently as WT TPP1, we transiently transfected plasmids encoding Myc-tagged versions of both proteins into the human embryonic kidney cell line 293T, in the presence of siRNA directed against the 5′ UTR of TPP1 to reduce competition from endogenous TPP1. Western blot analysis of cells harvested 3 days later confirmed equal expression and stability of WT and ΔK170 TPP1 (Figure 3A), and quantitative RT-PCR confirmed knockdown of the endogenous gene (Figure 3B). We then carried out ChIP using an anti-Myc antibody, measuring the amount of recovered telomeric DNA as a measure of TPP1 presence at the telomere (Figure 3C). When normalized against the starting amount of telomeric DNA (“input”), the amounts recovered with WT and ΔK170 TPP1 were equal (Figure 3D). This indicates that WT and ΔK170 TPP1 load onto telomeres at the same density.
Figure 3.

TPP1 with ΔK170 mutation localizes to telomeres. (A) Western blot of WT or ΔK170 TPP1, overexpressed in 293T cells in the presence of siRNA against endogenous TPP1. Vector: empty plasmid transfection only. (B) Levels of endogenous TPP1 mRNA, measured by qRT-PCR using primers in the 5′ UTR of the ACD/TPP1 gene. 293T cells were transfected with plasmids encoding WT or ΔK170 TPP1, followed by transfection with siRNA targeting the 5′ UTR of ACD/TPP1 or control siRNA. Vector: empty plasmid transfection only. (C) Telomere ChIP of myc-tagged, siRNA-resistant TPP1, expressed in 293T cells in the presence of siRNA to reduce endogenous TPP1 levels. Shown are 5%, 10%, and 20% of the input chromatin used in each immunoprecipitation (top). Immunoprecipitation with an anti-myc antibody, in duplicate (bottom). Blot was probed for telomeric DNA. (D) Quantitation of the amount of telomeric DNA recovered as a percentage of input, in three independent transfections and ChIP experiments (data shown as mean ± standard error of the mean).
TPP1 mutant ΔK170 fails to recruit telomerase to telomeres
To test the hypothesis that ΔK170 TPP1 is unable to recruit telomerase to telomeres despite its telomeric localization, we analyzed the presence of telomerase at the telomere using FISH against hTR and telomeres simultaneously.15 This was performed in immortal 293T cells expressing endogenous levels of telomerase, because we have found that telomerase overexpression can perturb some telomerase-recruitment pathways.15 The transfected 293T cells were synchronized in S phase using thymidine and aphidicolin prior to FISH (supplemental Figure 1). As expected, knockdown of endogenous TPP1 reduced the level of telomerase recruitment to telomeres,43 and this was rescued by expression of siRNA-resistant WT TPP1, confirming that the observed recruitment defect was due to lack of TPP1 (Figure 4). Importantly, the levels of endogenous TPP1 were reduced by ∼50% by our siRNA (Figure 3B), demonstrating that a telomerase-recruitment defect is observable even in the presence of remaining WT protein, as would be the case in the heterozygous patients. As predicted, expression of siRNA-resistant ΔK170 TPP1 was completely unable to rescue telomerase recruitment to telomeres (Figure 4).
Figure 4.
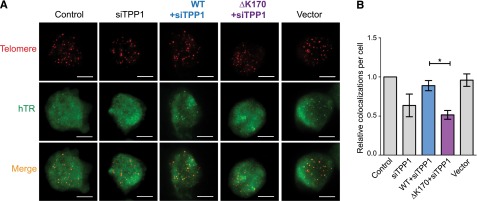
TPP1 with ΔK170 mutation does not recruit telomerase to telomeres. (A) FISH using probes against telomeres and hTR in 293T cells treated with siRNA against endogenous TPP1 and expressing either WT or mutant TPP1 or a vector-only control. (B) Quantitation of the number of hTR/telomere colocalizations in 3 independent transfections and FISH experiments (100 cells [300-1000 colocalizations] counted for each sample in each experiment; data shown as mean ± standard error of the mean; control had 7.8 ± 1.2 colocalizations; *P = .024).
Discussion
The role of the TEL patch of TPP1 in mediating the interaction of this protein with telomerase is well established.44,45 This interaction is necessary for the recruitment of telomerase to telomeres but dispensable for the function of TPP1 in protecting telomeres.44 Our data confirm that this surface of TPP1 is vital for telomerase recruitment and provide evidence that interference with this pathway through TPP1 mutation can lead to telomere shortening in humans. This conclusion is compellingly supported by the finding that mutations in hTERT linked to the short telomere disease idiopathic pulmonary fibrosis have been shown to abrogate interaction with TPP1.45
These data collectively support the hypothesis that a defect in TPP1 renders telomerase unable to maintain telomeres during development and hematopoiesis in the affected family members, leading to short telomeres and progressive BMF and eventual aplastic anemia. The increasing severity and younger age of onset of symptoms over several generations of this family is consistent with the phenomenon of genetic anticipation, leading to more severe disease in each successive generation.65 Curiously, however, telomere lengths did not continue to shorten over the 3 generations of affected females, relative to expected telomere lengths for age (Figure 1C). This is consistent with a previous observation of shorter telomeres in the offspring of fathers, but not mothers, with hTERT mutations,66 suggesting an intriguing paternal influence on telomere length.
In addition to hematologic disorders, the clinical history of this family includes oral carcinoma and leukemia. This is supportive of a link between telomere attrition and increased risk of cancer67 and is consistent with previous reports of leukemia and other malignancies, including oral carcinoma, in families with aplastic anemia due to mutations in telomerase components.68,69 Families with inherited idiopathic pulmonary fibrosis, short telomeres, and hTERT mutations also frequently contain members with leukemia and other malignancies.66,70,71 It is likely that inherited short telomeres can lead to chromosome fusions and genetic instability, which predisposes to malignancy.
This study represents the first identification of ACD/TPP1 as a disease-causing gene in humans, bringing the number of genes linked to BMF disorders to 10. Seven of these genes encode proteins or RNA that are either part of the telomerase complex (hTR, hTERT, dyskerin, Nop10, and Nhp2) or involved in telomerase transport to the telomere (TCAB1 and TPP1), and the remaining 3 proteins (TIN2, CTC1, and RTel1) are known to be involved in telomere protection. This strongly supports the causative role of telomere shortening or deprotection in the etiology of bone marrow disorders in humans.
Acknowledgments
The authors are grateful to the patient and her family for their willing involvement in this study. The authors thank Dr Monica Bessler and her laboratory for their generous assistance in setting up the telomere length Flow-FISH assay and Prof Inderjeet Dokal and Dr Tom Vulliamy for sequencing of known DC-associated genes.
This work was supported by an Institutional Development Award to the Center for Applied Genomics from The Children’s Hospital of Philadelphia, an Australian Awards Scholarship (M.K.), a Cancer Institute New South Wales (NSW) Career Development Fellowship (H.A.P.), a scholarship to A.A.-O. provided by the Academic and Training Affairs at King Faisal Specialist Hospital and Research Center and the Ministry of Higher Education (Riyadh, Saudi Arabia), project support from the Kids Cancer Alliance (NSW), a National Health and Medical Research Council postgraduate medical/dental scholarship (P.M.B.), program grant PG11-08 from Cancer Council NSW (R.R.), and a Cancer Institute NSW Career Development and Support Fellowship and project grant RG10-02 from Cancer Council NSW (T.M.B).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.G., J.L., B.K., Y.C., L.T., X.X., and H.H. designed and performed WES and bioinformatic analysis; M.K. and H.A.P. designed, performed, and analyzed experiments; J.T. collected clinical data and samples; T.K. and P.M.B. performed patient telomere-length analysis; A.A.-O. performed Sanger sequencing; H.A.P., R.R.R., J.C., and T.M.B. planned the study, organized the research, and contributed to the interpretation of results; T.M.B. wrote the manuscript; and all of the authors edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tracy M. Bryan, Children’s Medical Research Institute, 214 Hawkesbury Rd, Westmead, NSW 2145, Australia; e-mail: tbryan@cmri.org.au; and Hakon Hakonarson, The Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, Philadelphia, PA 19104; e-mail: hakonarson@e-mail.chop.edu.
References
- 1.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 2.Allsopp RC, Vaziri H, Patterson C, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992;89(21):10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Counter CM, Avilion AA, LeFeuvre CE, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11(5):1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 5.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 6.Hiyama K, Hirai Y, Kyoizumi S, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol. 1995;155(8):3711–3715. [PubMed] [Google Scholar]
- 7.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18(2):173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Hiyama E, Hiyama K. Telomere and telomerase in stem cells. Br J Cancer. 2007;96(7):1020–1024. doi: 10.1038/sj.bjc.6603671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng J, Funk WD, Wang SS, et al. The RNA component of human telomerase. Science. 1995;269(5228):1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura TM, Morin GB, Chapman KB, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277(5328):955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402(6761):551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 12.Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR. Protein composition of catalytically active human telomerase from immortal cells. Science. 2007;315(5820):1850–1853. doi: 10.1126/science.1138596. [DOI] [PubMed] [Google Scholar]
- 13.Venteicher AS, Abreu EB, Meng Z, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009;323(5914):644–648. doi: 10.1126/science.1165357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tycowski KT, Shu MD, Kukoyi A, Steitz JA. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell. 2009;34(1):47–57. doi: 10.1016/j.molcel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stern JL, Zyner KG, Pickett HA, Cohen SB, Bryan TM. Telomerase recruitment requires both TCAB1 and Cajal bodies independently. Mol Cell Biol. 2012;32(13):2384–2395. doi: 10.1128/MCB.00379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13(10):693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–764. doi: 10.1097/GIM.0b013e3181f415b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldman FD, Aubert G, Klingelhutz AJ, et al. Characterization of primitive hematopoietic cells from patients with dyskeratosis congenita. Blood. 2008;111(9):4523–4531. doi: 10.1182/blood-2007-10-120204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19(1):32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 20.Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413(6854):432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 21.Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005;34(3):257–263. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011;25(1):11–16. doi: 10.1101/gad.2006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16(13):1619–1629. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA. 2008;105(23):8073–8078. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82(2):501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112(9):3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson BH, Kasher PR, Mayer J, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44(3):338–342. doi: 10.1038/ng.1084. [DOI] [PubMed] [Google Scholar]
- 28.Keller RB, Gagne KE, Usmani GN, et al. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer. 2012;59(2):311–314. doi: 10.1002/pbc.24193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballew BJ, Yeager M, Jacobs K, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132(4):473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Guen T, Jullien L, Touzot F, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22(16):3239–3249. doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- 32.Deng Z, Glousker G, Molczan A, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA. 2013;110(36):E3408–E3416. doi: 10.1073/pnas.1300600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359(9324):2168–2170. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352(14):1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 35.Keegan CE, Hutz JE, Else T, et al. Urogenital and caudal dysgenesis in adrenocortical dysplasia (acd) mice is caused by a splicing mutation in a novel telomeric regulator. Hum Mol Genet. 2005;14(1):113–123. doi: 10.1093/hmg/ddi011. [DOI] [PubMed] [Google Scholar]
- 36.Liu D, Safari A, O’Connor MS, et al. PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol. 2004;6(7):673–680. doi: 10.1038/ncb1142. [DOI] [PubMed] [Google Scholar]
- 37.Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18(14):1649–1654. doi: 10.1101/gad.1215404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houghtaling BR, Cuttonaro L, Chang W, Smith S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol. 2004;14(18):1621–1631. doi: 10.1016/j.cub.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 39.Xin H, Liu D, Wan M, et al. TPP1 is a homologue of ciliate TEBP-β and interacts with POT1 to recruit telomerase. Nature. 2007;445(7127):559–562. doi: 10.1038/nature05469. [DOI] [PubMed] [Google Scholar]
- 40.Hockemeyer D, Palm W, Else T, et al. Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat Struct Mol Biol. 2007;14(8):754–761. doi: 10.1038/nsmb1270. [DOI] [PubMed] [Google Scholar]
- 41.Barrientos KS, Kendellen MF, Freibaum BD, Armbruster BN, Etheridge KT, Counter CM. Distinct functions of POT1 at telomeres. Mol Cell Biol. 2008;28(17):5251–5264. doi: 10.1128/MCB.00048-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang F, Podell ER, Zaug AJ, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445(7127):506–510. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- 43.Abreu E, Aritonovska E, Reichenbach P, et al. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30(12):2971–2982. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012;492(7428):285–289. doi: 10.1038/nature11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, Artandi SE. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012;150(3):481–494. doi: 10.1016/j.cell.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sexton AN, Youmans DT, Collins K. Specificity requirements for human telomere protein interaction with telomerase holoenzyme. J Biol Chem. 2012;287(41):34455–34464. doi: 10.1074/jbc.M112.394767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo Y, Prokudin I, Yu C, et al. Advantage of whole exome sequencing over allele-specific and targeted segment sequencing, in detection of novel TULP1 mutation in Leber congenital amaurosis [published online ahead of print February 19, 2014]. Ophthalmic Genet. doi: 10.3109/13816810.2014.886269. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cingolani P, Platts A, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 53.Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19(6):1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. 1000 Genomes Project Consortium, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56-65. [DOI] [PMC free article] [PubMed]
- 56. International HapMap 3 Consortium, Altshuler DM, Gibbs RA, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52-58. [DOI] [PMC free article] [PubMed]
- 57.Pertea M, Pertea GM, Salzberg SL. Detection of lineage-specific evolutionary changes among primate species. BMC Bioinformatics. 2011;12:274. doi: 10.1186/1471-2105-12-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foley AR, Menezes MP, Pandraud A, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137(Pt 1):44–56. doi: 10.1093/brain/awt315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 62.Knudson M, Kulkarni S, Ballas ZK, Bessler M, Goldman F. Association of immune abnormalities with telomere shortening in autosomal-dominant dyskeratosis congenita. Blood. 2005;105(2):682–688. doi: 10.1182/blood-2004-04-1673. [DOI] [PubMed] [Google Scholar]
- 63.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30(10):e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Risques RA, Vaughan TL, Li X, et al. Leukocyte telomere length predicts cancer risk in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2007;16(12):2649–2655. doi: 10.1158/1055-9965.EPI-07-0624. [DOI] [PubMed] [Google Scholar]
- 65.Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA. 2005;102(44):15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diaz de Leon A, Cronkhite JT, Katzenstein AL, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE. 2010;5(5):e10680. doi: 10.1371/journal.pone.0010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361(24):2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Calado RT, Regal JA, Hills M, et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc Natl Acad Sci USA. 2009;106(4):1187–1192. doi: 10.1073/pnas.0807057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marrone A, Sokhal P, Walne A, et al. Functional characterization of novel telomerase RNA (TERC) mutations in patients with diverse clinical and pathological presentations. Haematologica. 2007;92(8):1013–1020. doi: 10.3324/haematol.11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104(18):7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gansner JM, Rosas IO, Ebert BL. Pulmonary fibrosis, bone marrow failure, and telomerase mutation. N Engl J Med. 2012;366(16):1551–1553. doi: 10.1056/NEJMc1200999. [DOI] [PubMed] [Google Scholar]