

Abstract
Inner mitochondrial membrane peptidase 2-like (IMMP2L) protein is a mitochondrial inner membrane peptidase that cleaves the signal peptide sequences of cytochrome c1 (CYC1) and mitochondrial glycerol phosphate dehydrogenase (GPD2). Immp2l mutant mice show infertility and early signs of aging. It is unclear whether mitochondrial respiratory deficiency underlies this phenotype. Here we show that the intermediate forms of GPD2 and CYC1 have normal expression levels and enzymatic function in Immp2l mutants. Mitochondrial respiration is not diminished in isolated mitochondria and cells from mutant mice. Our data suggest that respiratory deficiency is not the cause of the observed Immp2l mutant phenotypes.
Keywords: Immp2l, Cytochrome c1, GPD2, Complex III, Mitochondrial respiration
Abbreviations: IMMP2L, inner mitochondrial membrane peptidase 2-like; CS, citrate synthase; CYC1, cytochrome c1; ECAR, extracellular acidification rate; GAPDH, glyceraldehyde 3-phosphate dehydrogenase (GAPDH); GPD2, mitochondrial glycerol phosphate dehydrogenase; OCR, oxygen consumption rate; VDAC1, voltage-dependent anion channel 1; complex III, ubiquinol-cytochrome-c reductase; complex I:NADH, ubiquinone oxidoreductase; complex II, succinate-ubiquinone oxidoreductase
Graphical abstract
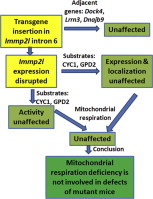
Highlights
-
•
Expression of IMMP2L substrates CYC1 and GPD2 is not affected in Immp2l mutant mice.
-
•
Mitochondria of mutant mice have normal complex III and GPD2 activities.
-
•
Mitochondrial respiration of mutant mice is not diminished.
Introduction
Cytochrome c1 (CYC1) and mitochondrial glycerol phosphate dehydrogenase (GPD2) are mitochondrial inner membrane proteins that need bipartite signal sequences to guide them to their final destination: the mitochondrial-targeting signal sequence and the mitochondrial inner membrane-targeting signal sequence. The mitochondrial targeting signal sequence guides the precursor proteins to the mitochondrial matrix, and is then cleaved by mitochondrial processing peptidase in the matrix. The mitochondrial inner membrane-targeting signal sequence guides the intermediate proteins to the inner membrane, and is finally cleaved by mitochondrial inner membrane peptidase. Yeast mitochondrial inner membrane peptidase has one noncatalytic subunit Som1p [1], and two catalytic subunits Imp1p and Imp2p, each with non-overlapping substrates. Imp1p cleaves the mitochondrial inner membrane-targeting signal sequences from cytochrome c oxidase subunit 2 (Cox2p), cytochrome b2 (Cyb2p), NADH-cytochrome b5 reductase (Mcr1p) and mitochondrial glycerol-3-phosphate dehydrogenase (Gut2p) [2–5], whereas Imp2p cleaves the signal sequence from cytochrome c1 [6].
Mammalian IMMP1L and IMMP2L are homologs of yeast Imp1p and Imp2p [7,8]. Mouse IMMP2L has two substrates, CYC1 and GPD2 [9]. However, in yeast, Gut2p (a homolog of GPD2) is a substrate for Imp1p [5]. Substrates for mammalian IMMP1L have not been described, although mammalian DIABLO/SMAC was suggested as an IMMP1L substrate [8]. Yeast Imp1p substrate Cyb2p lacks a mammalian ortholog. Yeast Imp1p substrates Cox2p and Mcr1p have mammalian orthologs, but appear to lack a typical cleavable signal peptide sequence [10].
Immp2l mutant mice show a series of abnormalities in different organs, which can be classified as either age- dependent or -independent. Age-independent phenotypes include infertility and reduced food intake, which are proposed to be caused by superoxide negation of nitric oxide [9,11]. Age-dependent phenotypes include spermatogenic damage in male mice [12], bladder dysfunction [13], and ataxia in old mutant mice but not in age-matched normal control mice [14]. These phenotypes are proposed to be caused by chronic oxidative stress resulting from increased mitochondrial superoxide generation. While the oxidative stress hypothesis is consistent with the observed phenotypes, it remains unclear whether the phenotypes are caused by functional deficiency of IMMP2L substrates.
CYC1 is one of the 11 subunits of complex III (ubiquinol-cytochrome-c reductase) [15], which is located on the mitochondrial inner membrane involved in mitochondrial electron transfer. Ubiquinol from complex I (NADH:ubiquinone oxidoreductase) and complex II are oxidized by mitochondrial complex III to reduce cytochrome c, where CYC1 is the subunit transferring the electron to cytochrome c [16]. In yeast, the intermediate form of Imp2p substrate Cyt1p (i-Cyt1p, with the inner membrane targeting signal sequence uncleaved) has normal localization in the mitochondria and is respiratory competent [6]. However, the N-terminal signal peptide sequences of yeast Cyt1p and its mouse homolog CYC1 show little homology, and the entire mature sequences are only 58% identical. In addition, yeast and mouse complex III have different numbers of subunits [17]. Furthermore, patients with CYC1 missense point mutations showed reduced complex III activity and insulin-responsive hyperglycemia [18]. Thus, it remains unclear whether the enzymatic function of the intermediate form of CYC1 (i-CYC1) is affected in Immp2l mutant mice.
GPD2, another IMMP2L substrate, catalyzes the conversion of glycerol-3-phosphate to dihydroxyacetone phosphate, reducing the enzyme-bound FAD. Together with the cytosolic glycerol phosphate dehydrogenase (GPD1), they form the glycerol phosphate shuttle, which uses the interconversion of glycerol-3-phosphate and dihydroxyacetone phosphate to transfer reducing equivalents into mitochondria, resulting in the reoxidation of NADH formed during glycolysis. It also remains unclear whether the enzymatic activity of GPD2 is impaired in Immp2l mutant mice due to the presence of uncleaved signal sequences.
The present study was designed to address questions regarding the enzymatic activities of IMMP2L substrates in Immp2l mutant mice. We find that CYC1 and GPD2 are functional in Immp2l mutant mice and conclude that mitochondrial respiration deficiency is not the cause of the observed phenotypes.
Materials and methods
Animals
The development and characterization of the Immp2l mutant mice has been described previously [9]. Mice were housed in the pathogen-free animal facility of Wake Forest University Health Sciences. Experiments were conducted in accordance with the National Research Council publication Guide for Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee of Wake Forest University Health Sciences. Mice were kept in microisolator cages with 12-h light/dark cycles and were fed ad libitum. Genotypes of the mice were determined by coat color. Normal homozygous mice (+/+) were albino due to the FVB background. Heterozygotes (+/−) were slightly pigmented due to the expression of tyrosinase from one copy of the transgene. Homozygous mutant mice (−/−) were darkly pigmented due to the expression of tyrosinase from both copies of the transgene. Mice were fed a chow diet (Prolab, RMH3000).
Quantitative RT-PCR analysis of gene expression
Total RNA was extracted from hypothalamic tissues with Turbo DNase (Ambion) to eliminate DNA contamination, and reverse transcribed as described previously [19]. Primer sequences for mouse Dock4, Lrrn3, Dnajb9, Immp2l exons 3–6 and Immp2l exons 3–7 have been described previously [9]. Pbip gene was used as the internal control, and primer sequences were tcgtctttggactctttggaa (forward primer) and agcgctcaccatagatgctc (reverse primer). RNA from 4 control and 4 mutant mice were compared. Each sample was assayed three times.
Preparation of mitochondrial and cytosolic proteins from mouse tissues
Testicular and brown fat tissues were excised from 2 to 3-month-old mice. Mitochondrial and cytosolic proteins were prepared as described previously [20]. Protease inhibitors (0.5 mM PMSF and 1× Complete Protease Inhibitor Cocktail from Roche) were included to prevent protein degradation.
SDS-PAGE and Western blot analyses
Tissue extracts were prepared from 2 to 3-month-old mice in RIPA buffer including protease inhibitors. Mitochondrial, cytosolic proteins, and tissue extracts were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with anti β-actin (Sigma, 1:5000), anti-GAPDH (Abcam, 1:500), anti-CYC1 (ProteinTech Group, 1:1000), anti-GPD2 (Abnova, 1:500), anti-VDAC1 (Abcam, 1:2000). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Pierce. Chemiluminescent reagents from Pierce were used to visualize the protein signals under the LAS-3000 system from Fujifilm. The Integrated Density function from ImageJ software was used to quantify the expression of individual proteins after normalization by β-actin or GAPDH (for cytosolic proteins or total tissue extracts) and VDAC1 (for mitochondrial proteins).
Oxygen consumption rate (OCR) of isolated mitochondria
Whole hind limb skeletal muscle from a control and an Immp2l−/− mouse (2–3 months) was used for mitochondria isolation, following the protocols described previously [21–23]. Mitochondrial protein concentrations were determined using the BCA protein assay kit (Thermo Scientific, Rockford, IL). Mitochondrial OCR was measured using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Biosciences, Billerica, MA). 10 mM succinate was used as the substrate with the addition of 2 µM rotenone; 5 µg mitochondrial protein was loaded in each well. Three to four replicates of each sample were analyzed for the mean and SD. Following an initial 7 min of equilibration, ADP (2 mM), oligomycin (2 µM), FCCP [carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone, 6 µM] and antimycin (2 µM) were added successively to achieve the final concentration as given in parentheses.
Mitochondrial citrate synthase (CS) activity
CS activity of isolated muscle mitochondria was assayed with the kit from Sigma (Cat no. CS0720), according to the manufacturer's instructions. Assays were performed at 25 °C in a 96-well plate with 4 µg mitochondrial protein per well. Formation of 5-thio-2-nitrobenzoic acid was monitored with a SpectraMax M5 Microplate Reader (Molecular Devices, Sunnyvale, CA) at 412 nm. Before and after the addition of oxaloacetate, absorbance was monitored for 3 min. The difference of the slopes was calculated to obtain the CS specific activity. CS activity was calculated based on the TNB extinction coefficient of 13.6 mM−1 cm−1 at 412 nm. The pathlength of 0.553 cm was adopted for the 200 µl reaction in the 96-well plate.
2.7. Mitochondrial complex III activity
Mitochondrial complex III activity was assayed as described previously [24]. To prepare decylubiquinol, 10 mg decylubiquinone (Sigma, D7911) dissolved in 400 µl of nitrogen-saturated hexane was mixed with 400 µl of 1.15 M sodium dithionite, and vortexed until colorless. The organic phase was collected, and the decylubiquinol was recovered by evaporating the hexane under nitrogen. The decylubiquinol was dissolved in 1 ml ethanol (acidified with 10 mM HCl) and stored in aliquots at −80 °C. Oxidized cytochrome c was from Sigma (C3131). 300 µl reaction mixture (15 µg mitochondrial protein and 60 µM cytochrome c) was added to a cuvette. After the addition of 3 µl decylubiquinol (stock concentration 15 mM, final concentration about 150 µM), reduction of cytochrome c was monitored at 550 nm once every second for 1 min with a SpectraMax M5 Microplate Reader (the chamber temperature was set at 30 °C). The assay was repeated with the addition of 1 µg Antimycin A (Sigma, A8674). Antimycin A-sensitive activity was calculated for the complex III activity. The extinction coefficient of cytochrome c is 21 mM−1 cm−1.
Mitochondrial GPD2 activity
Isolated mitochondria from mouse skeletal muscle were frozen and thawed. GPD2 activity was determined by methods described by Dawson et al. [25] with the addition of complexes I, II, and III inhibitors as described by Orr et al. [26]. The assay was run in a 96-well plate, and each well contained 40 µg total mitochondrial protein in a total volume of 200 µl. The reaction mixture contained 50 mM KH2PO4–NaOH buffer (pH 7.6), 25 mM-l-3-glycerophosphate, 50 µM DCPIP (2,6-dichlorophenolindophenol, Sigma), 4 µM rotenone, 2.5 µM antimycin A, and 1 mM malonate. The assay was started by the addition of mitochondrial proteins, and the decrease in absorption at 600 nm was recorded by a SpectraMax M5 Microplate Reader (Molecular Devices, Sunnyvale, CA) pre-warmed to 37 °C. The progress curve was linear during the first 3–5 min; data from the first 3 min were used to determine the reaction velocity. The reduction of absorption at 600 nm in the absence of mitochondrial protein was negligible. Extinction coefficient of DCPIP at 600 nm was taken to be 21 mM−1 cm−1.
Skeletal muscle myoblasts isolation, myotube differentiation, and oxygen consumption analysis
Myoblasts were isolated from two pairs of young control and mutant mice (aged 6 months), and three pairs of age-matched old control and mutant mice (aged 22, 25 and 29 months), following the procedures described previously [27] but with modifications. The proliferation medium contained Dulbecco's Modified Eagle Medium (DMEM) supplemented with 20% fetal bovine serum, 10% horse serum, 1% chicken embryo extract, and 5 ng/ml bFGF, and the differentiation medium contained 10% horse serum without bFGF. Four to five days after isolation, the myoblasts were collected and filtered through a cell strainer to remove debris. The cells were further re-suspended in proliferation medium, seeded in V7 cell culture plates (Seahorse Biosciences, Billerica, MA) (coated with 1:60 diluted Matrigel) at a concentration of 104 cells/well. After 3–4 days of proliferation, the medium was changed to differentiation medium for another one to two days. The cells were then analyzed with a Seahorse XF24 Extracellular Flux Analyzer as described previously [28]. Five to 11 replicates of each sample were analyzed for the mean and SD. Cells were incubated in Seahorse XF assay media with 5.56 mM d-Glucose, 1 mM sodium pyruvate, and 2 mM glutamax-1 added then pH titrated to 7.4 using NaOH. Following an initial 36 min of equilibration, oligomycin (0.75 µM), FCCP (1.0 µM), and antimycin (1.0 µM) plus rotenone (1 µM) were added successively to achieve the final concentrations (shown in parentheses). OCR (oxygen consumption rate) and extracellular acidification rate (ECAR) were normalized to total protein obtained using a BCA kit after Seahorse analysis.
Statistical analysis
Two-tailed Student's t-tests were performed to compare means of two groups. Tukey's post-tests following analysis of variance (ANOVA) were performed for means of more than two groups. Paired t-tests were also performed to compare mitochondrial GPD2 activities. For measurements with more than one variant, two-way ANOVA was performed followed by Bonferroni post-tests. P <0.05 was accepted as statistically significant.
Results
Immp2l gene but not adjacent genes are affected in Immp2l−/− mice
In Immp2l mutant mice, a transgenic insertion in intron 6 causes a 50 kb deletion in this 250 kb-intron [9] (Fig. 1A). The result is the disruption of normal expression of Immp2l mRNA after exon 6. Immp2l transcripts 5′ to exon 7 can be detected, but full-length Immp2l mRNA is undetectable. Three genes, Dock4, Lrrn3, and Dnajb9, are near this affected region, and Lrrn3 is within intron 4 of Immp2l. Although mRNA of all the three genes was detected by non-quantitative RT-PCR [9], it is unclear whether their expression level could be affected. Here we used quantitative RT-PCR to further examine whether the expression levels of the adjacent genes are affected, which could not be answered by non-quantitative RT-PCR. Since Lrrn3 is mainly expressed in the brain [29], brain expression of the three genes in Immp2l mutant mice was compared by quantitative RT-PCR (qRT-PCR). No significant differences were observed in the expression levels of the genes between control and mutant mice (Fig. 1). Consistent with our previous detection of truncated but not full-length Immp2l transcripts in RT-PCR analysis, Immp2l primers spanning exons 3 and 6 (Immp2lF and Immp2lM) detected similar expression between control and mutant mice in qRT-PCR (Fig. 1B). Immp2l primers spanning exons 3 and 7 (Immp2lF and Immp2lR) could only detect specific amplification from normal control mice (threshold cycle Ct=29 cycles), but not from mutant mice (Ct>37 cycles).
Fig. 1.
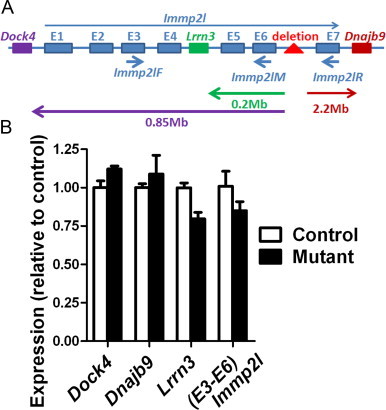
Expression of Immp2l but not other nearby genes is affected in Immp2l mutant mice. (A) Genes in the affected region. E1–E7 are the seven exons of Immp2l gene. The red triangle in intron 6 indicates the location of the foreign DNA insertion and the 50-kb deletion. Short arrows indicate the location of the primers used for qRT-PCR. Long arrows indicate the distances between the deletion and the adjacent genes. The diagram was not drawn to scale. (B) qRT-PCR comparison of expression of genes in the same region as Immp2l. Four control and three mutant mice were analyzed. Means±SEM are presented. No significant differences were found by ANOVA. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
IMMP2L substrate proteins have normal expression and compartmentalization in Immp2l−/− mice
Immp2l is expressed in all tissues, and its mutation affected CYC1 processing in all tissues examined in our previous study [9]. Mitochondrial and cytoplasmic proteins were prepared from mutant mice to determine whether the inability to cleave the signal sequence of CYC1 and GPD2 (the two known IMMP2L substrates) could affect the compartmentalization and expression of these two proteins. GPD2 has relatively high expression in brown adipose tissue [30]. We prepared mitochondrial and cytosolic proteins from brown adipose tissue of control and mutant mice; GPD2 was found in the mitochondrial but not the cytosolic fractions (Fig. 2A). CYC1 is widely expressed in all tissues. We examined CYC1 in the testis, an organ affected in Immp2l−/− mice [9,12]. Like GPD2, it was also detected in the mitochondrial but not the cytosolic fractions (Fig. 2A), demonstrating that the proteins are properly translocated to the mitochondria. This is consistent with observations in yeast, where the intermediate form of Cyt1p (i-Cyt1p) is correctly localized in the mitochondria of IMP2 (the Immp2l homolog) mutant yeast [6]. Compared with voltage-dependent anion channel 1 (VDAC1), a mitochondrial outer membrane protein, IMMP2L substrates showed similar expression between control and mutant mice, in the tissues examined (testis and muscle for CYC1, brown adipose tissue for GPD2, Fig. 2A and B). These data showed that loss of IMMP2L activity does not affect the expression or the compartmentalization of CYC1 and GPD2.
Fig. 2.
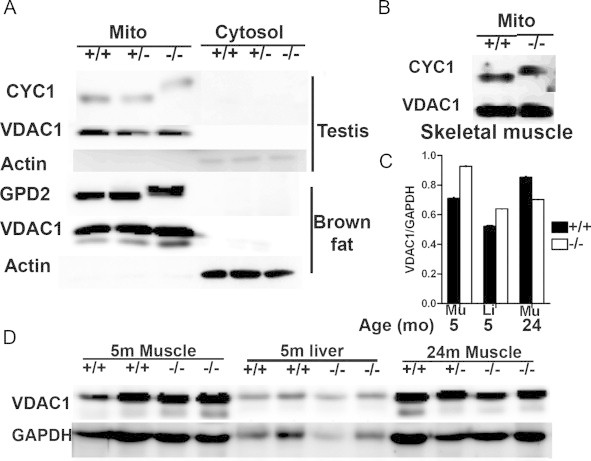
Expression of IMMP2L substrates in Immp2l mutant mice. (A) CYC1 and GPD2 expression determined by Western blot analyses in 3–5-month-old mutant mice. VDAC1 and β-actin were used as mitochondrial and cytosolic markers, respectively. (B) CYC1 expression in muscle mitochondria from 3 to 5-month-old mutant mice. (C) Densitometry comparison of VDAC1 expression in muscle and liver of control and mutant mice. Data were derived from panel “D”. Means±SEM are presented. Mu: skeletal muscle; Li: Liver. (D) Western blot analyses of VDAC1 expression between control and mutant mice using whole tissue lysates. GAPDH was used as a loading control.
A previous study reported no differences in qPCR analyses of mitochondrial DNA content between testicular cells of control and mutant mice [12], suggesting that the Immp2l mutation does not affect total mitochondrial mass of testicular cells. Western blot was performed to compare mitochondrial content in more tissues from control and mutant mice. We analyzed VDAC1 expression relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a cytosol protein, in muscle and liver tissue lysates from 5-month-old mice. The VDAC1/GAPDH density ratios in mutant mice were similar to those of control mice, indicating no mitochondrial mass deficiency in young mutants (Fig. 2C and D). Ratios also were similar between control and mutant mice at the age of 24 months.
Mitochondria of mutant mice have normal complex III activity
CYC1 is one of the three redox centers of mitochondrial complex III. Skeletal muscle is a common source of mitochondria for mitochondrial complex activity analysis. We wondered whether the intermediate form of CYC1 (i-CYC1) could compromise the ubiquinol:cytochrome c oxidoreductase activity of mitochondria from mutant mice. The size of CYC1 in muscle mitochondria from mutant mice (Fig. 2B) confirmed that it is an intermediate form (i-CYC1). We found no differences in CS and complex III activities of muscle mitochondria from control and mutant mice (Fig. 3A). The ratios of complex III/CS were also similar (Fig. 3B). Cells from CYC1 mutant patients show reduced complex III activity [18]. Normal complex III activity of mitochondria from mutant mice suggests that i-CYC1 protein is enzymatically active.
Fig. 3.
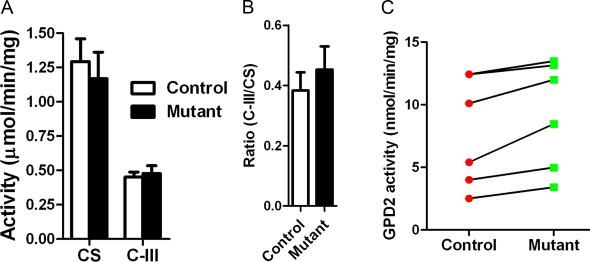
Normal complex III and CS activities of mutant mice. (A) Complex III and CS activities of control and mutant mice. Means±SEM of six mice (3–5 months) are presented. Complex III activity was expressed as the amount cytochrome c reduced in µmol/min/mg. CS activity was expressed as the amount of 5-thio-2-nitrobenzoic acid (TNB) formed in µmol/min/mg. Differences were not statistically significant between control and mutant mice by ANOVA. (B) Similar complex III/CS activity ratios between controls and mutants. No statistically significant differences were found by t-test (p=0.49). (C) GPD2 activity of mitochondria from control and mutant mice. Measurements were done with the same mitochondrial preparations used for complex III activity assay. Values linked by lines were from mitochondria preparations from littermates sacrificed on the same dates.
Mitochondria of mutant mice have normal GPD2 activity
No difference was observed in GPD2 activity in frozen/thawed muscle mitochondria between control and mutant mice (7.81±1.79 nmol/min/mg, N=6 from control mice; 9.25 ±1.76 nmol/min/mg, N =6 for mutant mice; p=0.58 by t-test). Mitochondrial samples were isolated on different days from one control and one mutant mouse (littermates) for experiments requiring fresh mitochondria. We noticed that GPD2 activity shows relatively large intra-group variations. This could be caused by the fact that mice of the same genotype were sacrificed at different dates for tissue collection. Supporting our reasoning, tissues from control and mutant mice sacrificed at the same days had similar values. When compared by paired t-test, mitochondria from mutant mice showed even higher GPD2 activity (p =0.0109, paired t-test). The data exclude GPD2 enzymatic deficiency in mutant mice.
Mitochondrial respiration of mutant mice is not diminished
Isolated mitochondria from skeletal muscle of 3–5-month-old control and mutant mice were analyzed for their oxidative phosphorylation. Mitochondria from control and mutant mice had similar oxygen consumption rates (OCRs) at all states: when only substrate is present (state 2); when respiration is stimulated by addition of ADP to stimulate ATP synthesis or uncoupling reagent FCCP, allowing the inward flow of protons across the inner membrane without ATP synthesis, and when respiration is inhibited by ATP synthase inhibitor oligomycin or complex III inhibitor antimycin A (Fig. 4). No defects of oxidative phosphorylation were noted in mitochondria isolated from mutant mice.
Fig. 4.
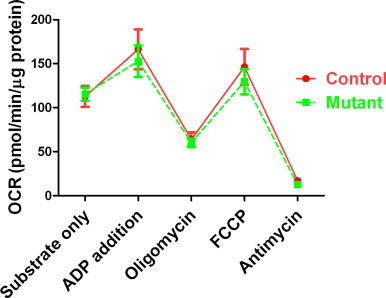
Normal mitochondrial respiration of Immp2l mutant mice. Mean±SEM of 5 pairs are presented. No significant difference was found by ANOVA.
Respiration and glycolysis of myoblast-derived skeletal muscle myotubes from mutant mice are normal
Myoblasts were isolated from skeletal muscles of control and mutant mice and differentiated into myotubes. The OCR (reflecting mitochondrial respiration) and extracellular acidification rate (ECAR, reflecting glycolysis) were compared between myotubes differentiated from myoblasts of control and mutant mice. Differentiated myotubes from 6-month-old mutant mice had similar OCR and ECAR traces as myotubes from their age-matched controls (Fig. 5A and B). Cell numbers were confirmed as similar by determining protein concentrations after the Seahorse reading. The data suggest that cells from control and mutant mice have similar rates of mitochondrial respiration and glycolysis. Similar observations were made in differentiated myotubes from 22–29-month-old control and mutant mice (Fig. 5C and D).
Fig. 5.
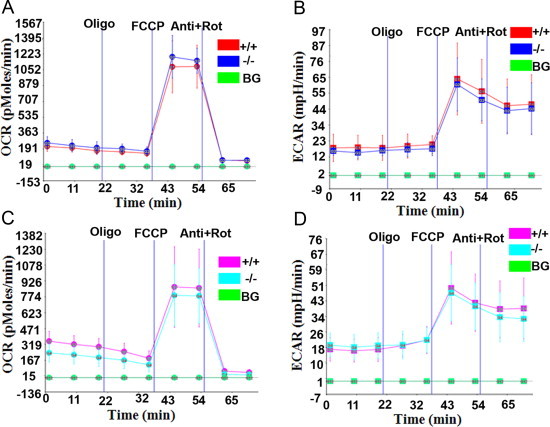
Respiration and glycolysis of in vitro differentiated myotubes. (A and B) Representative OCR (A) and ECAR (B) traces of myotubes from two pairs of 6-month-old control and mutant littermates. Data shown are means±SD of 5 replicates. (C and D) OCR (C) and ECAR (D) traces of myotubes from old mice. Traces were representative of three pairs of control and mutant littermates (aged 22, 25, and 29 months respectively). Presented were means±SD of 6 replicates from mice of 22 months. Oligo: oligomycin; anti: antimycin; FCCP: carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; Rot: rotenone; and BG: no cell background.
Discussion
CYC1 and GPD2 are two known substrates for IMMP2L, a peptidase on the mitochondrial inner membrane that cleaves the space-sorting signal sequences from the precursors of its substrates. The signal peptide sequences of CYC1 and GPD2, also mitochondrial inner membrane proteins, remain unprocessed in Immp2l mutant mice [9]. Immp2l mutant mice show normal embryonic and postnatal development, but exhibit abnormalities in multiple systems, including infertility and a 30% reduced food intake [9,11]. In addition, some abnormalities, including bladder dysfunction [13], ataxia [14], and spermatogenic impairment [12], develop as mice age. Based on our observation of increased mitochondrial superoxide generation in the mutant mice, we proposed that mitochondrial oxidative stress underlies these phenotypes. However, whether CYC1 and GPD2 activity deficiency also contributes to the observed mutant phenotype is not clear.
We confirmed that Immp2l expression is affected due to disruption of exon 7, the portion of Immp2l mRNA 3′ to the transgene insertion site. The adjacent genes, Dock4, Lrrn3, and Dnajb9, are not affected in the mutants. Dock4 and Dnajb9 knockout mice are unavailable; Lrrn3 knockout mice have increased body fat and enhanced glucose tolerance (http://www.informatics.jax.org/marker/MGI:106036). These phenotypes are different from those of Immp2l mutant mice, suggesting that the Lrrn3 gene does not contribute to the phenotype of Immp2l mutant mice.
CYC1 and GPD2 were both observed in the mitochondria but not the cytosol in mutant mice. In addition, CYC1 and GPD2 expression were not affected in the mutants. Thus, IMMP2L deficiency does not affect the expression of its substrates. Furthermore, mitochondria from mutant mice had normal complex III activity (CYC1 is a subunit of complex III) and higher than normal GPD2 activity. The intermediate forms of CYC1 and GPD2 are enzymatically functional, consistent with the observation that yeast i-cyt1p (a CYC1 homolog) is respiratory competent [6]. Furthermore, myofibers from mutant mice showed both normal oxygen consumption (an indication of mitochondrial respiration) and normal extracellular acidification rates (an indication of glycolysis). Considering our previous findings that mutant mice have normal complex I and complex II activities and cellular ATP levels [9], we conclude that the phenotype of Immp2l mutant mice is unlikely to be caused by mitochondrial respiratory deficiency. Most analyses were done with tissues from young mice. We think that this is appropriate since the primary cause must also be observed when young. We agree that examining old tissues may provide more information about the combined effects of the primary defects and age. However, using 2-year-old mice for these experiments is impractical considering the cost and time needed to generate the mice.
We used myofibers and skeletal muscle-derived mitochondria for respirometry analyses because these are common sources of materials for such analyses. Similar analyses using other tissues, especially those most affected in the mutants (e.g. the testis and the brain), could reinforce our argument. CYC1 from muscle lysates of mutant mice has larger than normal size, demonstrating that IMMP2L activity is lost in muscle tissues. We cannot examine the size of GPD2 in muscle lysates because of its relatively low expression in skeletal muscle. However, it is unlikely that GPD2 processing in muscle does not depend on IMMP2L activity. Since the intermediate forms of CYC1 and GPD2 are functional in skeletal muscle, it is logical that they are functional in other tissues.
This work suggests that respiratory deficiency is unlikely the cause of phenotypes observed in Immp2l mutant mice. Our previous works linked the abnormalities found in mutant mice to oxidative stress, either through superoxide negation of nitric oxide [9,11], or through detrimental effects of increased levels of reactive oxygen species [12–14]. Supporting our argument, we observed increased superoxide generation in mitochondria from mutant mice when using endogenous substrates [9]. In addition, multiple oxidative indices (carbonyl, 4-Hydroxynonenal) demonstrated oxidative stress in tissues from mutant mice [12,14]. Finally, testes from mutant mice have significantly increased catalase expression [12], consistent with increased superoxide generation and thus hydrogen peroxide levels.
Interestingly, complex III (of which CYC1 is a subunit) and GPD2 can generate superoxide as a byproduct [26,31–35]. Mutations in three of the 11 complex III subunits have been described in patients, but the pathological mechanism is unclear (MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org, 2013). GPD2 haploinsufficiency was described in a patient with nonsyndromic mental retardation, but whether GPD2 was the cause is unclear [36]. However, conditional knockout of a gene coding for the Rieske iron–sulfur protein (a complex III subunit) in mice causes oxidative stress in the brain [37], confirming that perturbance of complex III can cause oxidative stress. We propose that retention of the signal sequences on CYC1 and/or GPD2 does not affect their ability for electron transfer, but increases the leakage of electrons to oxygen during electron transfer. It remains unknown how electron leakage is increased and whether CYC1 or GPD2 is the greater contributor to increased superoxide generation. We also cannot exclude the contribution of possible unidentified IMMP2L substrates.
In conclusion, the present work demonstrates that Immp2l mutant mice have no mitochondrial respiratory deficit, suggesting that respiratory deficiency is unlikely to be the cause of the observed Immp2l mutant phenotypes.
Acknowledgments
The authors thank Dr. Martin Brand (The Buck Institute for Research on Aging) for constructive discussions and Ms. Karen Klein (Translational Science Institute, Wake Forest University Health Sciences) for editing the manuscript. This work was partially supported by the National Institutes of Health (R01HD058058 to B. L.).
References
- 1.Esser K., Pratje E., Michaelis G. SOM 1, a small new gene required for mitochondrial inner membrane peptidase function in Saccharomyces cerevisiae. Molecular and General Genetics. 1996;252:437–445. doi: 10.1007/BF02173009. 8879245 [DOI] [PubMed] [Google Scholar]
- 2.Pratje E., Guiard B. One nuclear gene controls the removal of transient pre-sequences from two yeast proteins: one encoded by the nuclear the other by the mitochondrial genome. EMBO Journal. 1986;5:1313–1317. doi: 10.1002/j.1460-2075.1986.tb04361.x. 3015596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahne K., Haucke V., Ramage L., Schatz G. Incomplete arrest in the outer membrane sorts NADH-cytochrome b5 reductase to two different submitochondrial compartments. Cell. 1994;79:829–839. doi: 10.1016/0092-8674(94)90072-8. 8001120 [DOI] [PubMed] [Google Scholar]
- 4.Esser K., Jan P.S., Pratje E., Michaelis G. The mitochondrial IMP peptidase of yeast: functional analysis of domains and identification of Gut2 as a new natural substrate. Molecular Genetics and Genomics. 2004;271:616–626. doi: 10.1007/s00438-004-1011-y. 15118906 [DOI] [PubMed] [Google Scholar]
- 5.Luo W., Fang H., Green N. Substrate specificity of inner membrane peptidase in yeast mitochondria. Molecular Genetics and Genomics. 2006;275:431–436. doi: 10.1007/s00438-006-0099-7. 16450175 [DOI] [PubMed] [Google Scholar]
- 6.Nunnari J., Fox T.D., Walter P. A mitochondrial protease with two catalytic subunits of nonoverlapping specificities. Science (New York, N.Y.) 1993;262:1997–2004. doi: 10.1126/science.8266095. 8266095 [DOI] [PubMed] [Google Scholar]
- 7.Petek E., Windpassinger C., Vincent J.B., Cheung J., Boright A.P., Scherer S.W., Kroisel P.M., Wagner K. Disruption of a novel gene (IMMP2L) by a breakpoint in 7q31 associated with Tourette syndrome. American Journal of Human Genetics. 2001;68:848–858. doi: 10.1086/319523. 11254443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burri L., Strahm Y., Hawkins C.J., Gentle I.E., Puryer M.A., Verhagen A., Callus B., Vaux D., Lithgow T. Mature DIABLO/Smac is produced by the IMP protease complex on the mitochondrial inner membrane. Molecular Biology of the Cell. 2005;16:2926–2933. doi: 10.1091/mbc.E04-12-1086. 15814844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu B., Poirier C., Gaspar T., Gratzke C., Harrison W., Busija D., Matzuk M.M., Andersson K.E., Overbeek P.A., Bishop C.E. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biology of Reproduction. 2008;78:601–610. doi: 10.1095/biolreprod.107.065987. 18094351 [DOI] [PubMed] [Google Scholar]
- 10.Borgese N., Aggujaro D., Carrera P., Pietrini G., Bassetti M. A role for N-myristoylation in protein targeting: NADH-cytochrome b5 reductase requires myristic acid for association with outer mitochondrial but not ER membranes. Journal of Cell Biology. 1996;135:1501–1513. doi: 10.1083/jcb.135.6.1501. 8978818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han C., Zhao Q., Lu B. The role of nitric oxide signaling in food intake; insights from the inner mitochondrial membrane peptidase 2 mutant mice. Redox Biology. 2013;1:498–507. doi: 10.1016/j.redox.2013.10.003. 24251118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George S.K., Jiao Y., Bishop C.E., Lu B. Oxidative stress is involved in age-dependent spermatogenic damage of Immp2l mutant mice. Free Radical Biology and Medicine. 2012;52:2223–2233. doi: 10.1016/j.freeradbiomed.2012.04.003. 22569411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soler R., Füllhase C., Lu B., Bishop C.E., Andersson K.E. Bladder dysfunction in a new mutant mouse model with increased superoxide—lack of nitric oxide? Journal of Urology. 2010;183:780–785. doi: 10.1016/j.juro.2009.09.074. 20022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George S.K., Jiao Y., Bishop C.E., Lu B. Mitochondrial peptidase IMMP2L mutation causes early onset of age-associated disorders and impairs adult stem cell self-renewal. Aging Cell. 2011;10:584–594. doi: 10.1111/j.1474-9726.2011.00686.x. 21332923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.González-Halphen D., Lindorfer M.A., Capaldi R.A. Subunit arrangement in beef heart complex III. Biochemistry. 1988;27:7021–7031. doi: 10.1021/bi00418a053. 2848575 [DOI] [PubMed] [Google Scholar]
- 16.Hunte C., Palsdottir H., Trumpower B.L. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Letters. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. 12788490 [DOI] [PubMed] [Google Scholar]
- 17.Brandt U., Yu L., Yu C.A., Trumpower B.L. The mitochondrial targeting presequence of the Rieske iron–sulfur protein is processed in a single step after insertion into the cytochrome bc1 complex in mammals and retained as a subunit in the complex. Journal of Biological Chemistry. 1993;268:8387–8390. 8386158 [PubMed] [Google Scholar]
- 18.Gaignard P., Menezes M., Schiff M., Bayot A., Rak M., Ogier de Baulny H., Su C.H., Gilleron M., Lombes A., Abida H., Tzagoloff A., Riley L., Cooper S.T., Mina K., Sivadorai P., Davis M.R., Allcock R.J., Kresoje N., Laing N.G., Thorburn D.R., Slama A., Christodoulou J., Rustin P. Mutations in CYC1, encoding cytochrome c1 subunit of respiratory chain complex III, cause insulin-responsive hyperglycemia. American Journal of Human Genetics. 2013;93:384–389. doi: 10.1016/j.ajhg.2013.06.015. 23910460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiao Y., Bishop C.E., Lu B. Mex3c regulates insulin-like growth factor 1 (IGF1) expression and promotes postnatal growth. Molecular Biology of the Cell. 2012;23:1404–1413. doi: 10.1091/mbc.E11-11-0960. 22357625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science (New York, N.Y.) 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. 9027314 [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharya S.K., Thakar J.H., Johnson P.L., Shanklin D.R. Isolation of skeletal muscle mitochondria from hamsters using an ionic medium containing ethylenediaminetetraacetic acid and nagarse. Analytical Biochemistry. 1991;192:344–349. doi: 10.1016/0003-2697(91)90546-6. 1903610 [DOI] [PubMed] [Google Scholar]
- 22.Cadenas S., Echtay K.S., Harper J.A., Jekabsons M.B., Buckingham J.A., Grau E., Abuin A., Chapman H., Clapham J.C., Brand M.D. The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. Journal of Biological Chemistry. 2002;277:2773–2778. doi: 10.1074/jbc.M109736200. 11707458 [DOI] [PubMed] [Google Scholar]
- 23.Chappell J.B., Perry S.V. The respiratory and adenosinetriphosphatase activities of skeletal-muscle mitochondria. Biochemical Journal. 1953;55:586–595. doi: 10.1042/bj0550586. 13115340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krähenbühl S., Talos C., Wiesmann U., Hoppel C.L. Development and evaluation of a spectrophotometric assay for complex III in isolated mitochondria, tissues and fibroblasts from rats and humans. Clinica Chimica Acta: International Journal of Clinical Chemistry. 1994;230:177–187. doi: 10.1016/0009-8981(94)90270-4. 7834868 [DOI] [PubMed] [Google Scholar]
- 25.Dawson A.P., Thorne C.J. Preparation and some properties of l-3-glycerophosphate dehydrogenase from pig brain mitochondria. Biochemical Journal. 1969;111:27–34. doi: 10.1042/bj1110027. 5775687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orr A.L., Quinlan C.L., Perevoshchikova I.V., Brand M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. Journal of Biological Chemistry. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. 23124204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danoviz M.E., Yablonka-Reuveni Z. Skeletal muscle satellite cells: background and methods for isolation and analysis in a primary culture system. Methods in Molecular Biology (Clifton, N.J.) 2012;798:21–52. doi: 10.1007/978-1-61779-343-1_2. 22130829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu M., Neilson A., Swift A.L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., Chomicz S., Ferrick D.A. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. American Journal of Physiology. Cell Physiology. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. 16971499 [DOI] [PubMed] [Google Scholar]
- 29.Taniguchi H., Tohyama M., Takagi T. Cloning and expression of a novel gene for a protein with leucine-rich repeats in the developing mouse nervous system. Brain Research Molecular Brain Research. 1996;36:45–52. doi: 10.1016/0169-328x(95)00243-l. 9011764 [DOI] [PubMed] [Google Scholar]
- 30.Koza R.A., Kozak U.C., Brown L.J., Leiter E.H., MacDonald M.J., Kozak L.P. Sequence and tissue-dependent RNA expression of mouse FAD-linked glycerol-3-phosphate dehydrogenase. Archives of Biochemistry and Biophysics. 1996;336:97–104. doi: 10.1006/abbi.1996.0536. 8951039. [DOI] [PubMed] [Google Scholar]
- 31.Herrero A., Barja G. Sites and mechanisms responsible for the low rate of free radical production of heart mitochondria in the long-lived pigeon. Mechanisms of Ageing and Development. 1997;98:95–111. doi: 10.1016/s0047-6374(97)00076-6. 9379714. [DOI] [PubMed] [Google Scholar]
- 32.Turrens J.F., Alexandre A., Lehninger A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Archives of Biochemistry and Biophysics. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 33.Bolter C.J., Chefurka W. Extramitochondrial release of hydrogen peroxide from insect and mouse liver mitochondria using the respiratory inhibitors phosphine, myxothiazol, and antimycin and spectral analysis of inhibited cytochromes. Archives of Biochemistry and Biophysics. 1990;278:65–72. doi: 10.1016/0003-9861(90)90232-n. 2321971 [DOI] [PubMed] [Google Scholar]
- 34.Drahota Z., Chowdhury S.K., Floryk D., Mrácek T., Wilhelm J., Rauchová H., Lenaz G., Houstek J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. Journal of Bioenergetics and Biomembranes. 2002;34:105–113. doi: 10.1023/a:1015123908918. 12018887 [DOI] [PubMed] [Google Scholar]
- 35.Chen Q., Vazquez E.J., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Production of reactive oxygen species by mitochondria: central role of complex III. Journal of Biological Chemistry. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. 12840017 [DOI] [PubMed] [Google Scholar]
- 36.Daoud H., Gruchy N., Constans J.M., Moussaoui E., Saumureau S., Bayou N., Amy M., Védrine S., Vu P.Y., Rötig A., Laumonnier F., Vourc’h P., Andres C.R., Leporrier N., Briault S. Haploinsufficiency of the GPD2 gene in a patient with nonsyndromic mental retardation. Human Genetics. 2009;124:649–658. doi: 10.1007/s00439-008-0588-3. 19011903 [DOI] [PubMed] [Google Scholar]
- 37.Diaz F., Garcia S., Padgett K.R., Moraes C.T. A defect in the mitochondrial complex III, But not complex IV, triggers early ROS-dependent damage in defined brain regions. Human Molecular Genetics. 2012;21:5066–5077. doi: 10.1093/hmg/dds350. 22914734 [DOI] [PMC free article] [PubMed] [Google Scholar]