

Immunoglobulin G4-related disease (IgG4-RD) is a novel clinical entity characterized by elevated serum IgG4 levels and histopathologic features of storiform fibrosis, obliterative phlebitis, tissue eosinophilia, and infiltration of IgG4-positive plasma cells. Although it can involve almost any organ, IgG4-RD rarely involves the nervous system. The principal neurologic manifestations include hypophysitis and hypertrophic pachymeningitis (HP). In some patients, previously known idiopathic HP may in fact be IgG4-related.
IgG4-related CNS disease often resolves with surgery, steroids, and immunomodulators, considered standard treatment. We report a case with IgG4-related HP refractory to steroids and multiple immunomodulators. We share our diagnostic and treatment approach that may change the concepts of IgG4-related HP.
Case report.
A 41-year-old (currently 52-year-old) Hispanic woman initially presented to our service with headaches and seizures in 2003. Brain MRI revealed dural enhancement along the falx and bilateral frontotemporal convexities (figure, A). Biopsy of the affected dura revealed noncaseating granulomatous inflammation, suggesting neurosarcoidosis. She received a 5-day course of IV methylprednisolone at 1,000 mg/day, with transient clinical improvement.
Figure. Radiologic and histopathologic findings in a patient with IgG4-related intracranial hypertrophic pachymeningitis.

(A–D) Radiologic findings over 10 years. (A) Brain MRI from 2003 with and without contrast: axial T1 postcontrast sequence shows extensive dural enhancement along the falx and bilateral frontotemporal convexities. (B) Axial T1 postcontrast sequence from 2011 shows enhancement in the right maxillary sinus. (C) Before treatment with rituximab: coronal T1 postcontrast sequence from 2013 shows diffuse dural enhancement in both frontotemporal convexities and nodular enhancement in the right suprasellar region that extends into the clivus. (D) After treatment with rituximab: coronal T1 postcontrast sequence from 2014 shows significant reduction of the dural enhancement in both frontotemporal convexities and nodular enhancement/mass effect in the right suprasellar region. (E, F) Histopathologic features of dura from middle cranial fossa biopsy. (E) Dense fibrosis admixed with abundant inflammatory cell infiltration (hematoxylin & eosin staining, ×100). (F) Abundant IgG4-positive plasma cell infiltration (IgG4 staining, ×200).
In the following 10 years, she had clinicoradiologic progression of disease despite treatment. She developed worsening headaches, seizures, anosmia, restricted right eye movements, and right facial pain. Serial MRIs showed progression of the dural enhancement and a new right maxillary mass eroding the maxillary sinus, right inferior orbit, and right cavernous sinus (figure, B). She received multiple courses of high-dose steroids and immunomodulators (azathioprine/methotrexate/hydroxychloroquine). She also underwent surgical resection of the right maxillary sinus mass. In October 2012, based on suspicion of IgG4-RD, serum IgG4 level was tested, which revealed a systemic IgG4 level of 98 mg/dL (reference range, 7–89 mg/dL) with IgG4/IgG ratio of 8% (reference range, 5%–6%). Brain MRI revealed disease progression of the dural enhancement and a new area of nodular enhancement in the right suprasellar region extending into the clivus. She was treated with mycophenolate mofetil (MMF) from October 2012 to August 2013, but it was discontinued due to side effects and lack of disease control. Repeat brain MRI demonstrated worsening of the abovementioned lesions (figure, C). Subjecting the tissue from dura biopsy performed in 2003 to staining for IgG4 antibodies revealed significant inflammatory B-cell infiltration but no evidence of lymphoma; more than 25% of the plasma cells were positive for IgG4 immunostaining (figure, E and F). Based on literature that supported use of rituximab in refractory IgG4-RD, the patient received 2 doses of IV rituximab, 1,000 mg each. On the 1- and 4-month follow-up visits after the second dose of rituximab, her headache, facial pain, and diplopia had resolved and she demonstrated excellent clinical and radiologic response (figure, D). CD19 assay revealed sufficient suppression of CD19 B cells. We plan to repeat CD19 assay every 4 months in future follow-ups. Resuming rituximab will be considered based on her neurologic status and CD19 count. The maintenance treatment consists of oral prednisone, 5 mg/day. Over the course of the patient's care, workup for infection and other autoimmune diseases, including CSF studies, was negative.
Discussion.
In 2001, Hamano et al.1 first reported the association of IgG4 with sclerosing disease in patients with autoimmune pancreatitis. Many conditions, including orbital pseudotumor, retroperitoneal fibrosis, Riedel thyroiditis, Küttner tumor, Mikulicz disease, multifocal fibrosclerosis, and tubulointerstitial nephritis, are now considered to be associated with IgG4-RD because they share the same serologic/histologic features. The pathogenesis of IgG4-RD remains unknown. There is no evidence that IgG4 contributes directly to its pathogenesis.2 Proposed pathogenesis includes molecular mimicry of microbial antigens and an allergic disorder. The response to immunosuppressive therapy supports these proposals.
IgG4-RD mainly occurs in middle-aged and elderly men. Clinical presentation depends on the involved organ. IgG4-RD affecting the nervous system is rare. Principal neurologic manifestations include hypophysitis and HP.
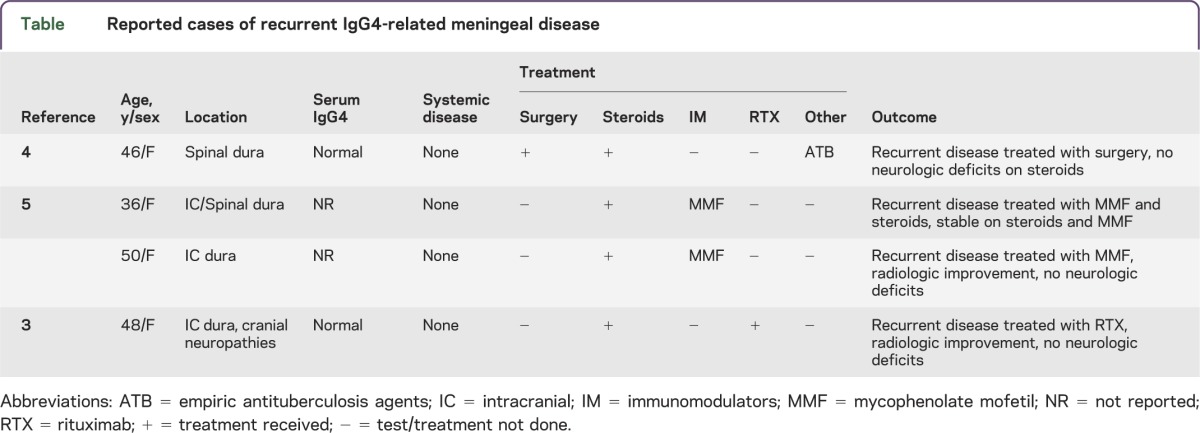
To our knowledge, 20 patients with IgG4-related HP have been reported. Of those, only 4 patients developed recurrent HP (table): one was subsequently treated with rituximab with excellent clinical response3; one underwent surgery followed by oral steroids with resolution of symptoms4; and 2 were treated with MMF, which resulted in stabilized disease after 12 and 18 months, respectively.5 Rituximab was used in 10 patients with steroid/immunomodulator–refractory IgG4-RD not involving the nervous system, reducing serum IgG4 levels that corresponded with clinicoradiologic response, constituting a good indicator of disease activity.6
Table.
Reported cases of recurrent IgG4-related meningeal disease
In our case, the lesions were isolated to the intracranial dura. The disease was refractory to steroids and immunomodulators. Subsequent treatment with rituximab showed clinicoradiologic response. Although rituximab does not cross the blood-brain barrier physiologically, in inflammatory processes within the CNS the barrier might become permeable for monoclonal antibodies,7 so it may represent the next best therapeutic option in steroid/immunomodulator–refractory IgG4-related HP.
Acknowledgments
Acknowledgment: The authors thank Dr. Suzanne Z. Powell for her assistance in IgG4 antibody staining.
Footnotes
Author contributions: Bing Liao: study concept and design. Carlos Kamiya-Matsuoka: study concept and design. Xiang Fang: revision of the manuscript. Robert G. Smith: study supervision.
Study funding: No targeted funding reported.
Disclosure: B. Liao, C. Kamiya-Matsuoka, and X. Fang report no disclosures. R.G. Smith has received research support from the University of Texas system. Go to Neurology.org/nn for full disclosures. The Article Processing Charge was paid by the authors.
References
- 1.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–738 [DOI] [PubMed] [Google Scholar]
- 2.Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol Mech Dis 2014;9:315–347 [DOI] [PubMed] [Google Scholar]
- 3.Shapiro KA, Bove RM, Volpicelli ER, Mallery RM, Stone JH. Relapsing course of immunoglobulin G4-related pachymeningitis. Neurology 2012;79:604–606 [DOI] [PubMed] [Google Scholar]
- 4.Choi SH, Lee SH, Khang SK, Jeon SR. IgG4-related sclerosing pachymeningitis causing spinal cord compression. Neurology 2010;75:1388–1390 [DOI] [PubMed] [Google Scholar]
- 5.Moss HE, Mejico LJ, de la Roza G, Coyne TM, Galetta SL, Liu GT. IgG4-related inflammatory pseudotumor of the central nervous system responsive to mycophenolate mofetil. J Neurol Sci 2012;318:31–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012;91:57–66 [DOI] [PubMed] [Google Scholar]
- 7.Wong ET. Management of central nervous system lymphomas using monoclonal antibodies: challenges and opportunities. Clin Cancer Res 2005;11:7151s–7157s [DOI] [PubMed] [Google Scholar]