

Abstract
Molecular level, mechanistic understanding of the roles of reactive oxygen species (ROS) in a variety of pathological conditions is hindered by the difficulties associated with determining the concentration of various ROS species. Here, we present an approach that converts fold-change in the signal from an intracellular sensor of hydrogen peroxide into changes in absolute concentration. The method uses extracellular additions of peroxide and an improved biochemical measurement of the gradient between extracellular and intracellular peroxide concentrations to calibrate the intracellular sensor. By measuring peroxiredoxin activity, we found that this gradient is 650-fold rather than the 7–10-fold that is widely cited. The resulting calibration is important for understanding the mass-action kinetics of complex networks of redox reactions, and it enables meaningful characterization and comparison of outputs from endogenous peroxide generating tools and therapeutics across studies.
Keywords: Hydrogen peroxide, Peroxiredoxin, Kinetics, HyPer, Quantitative redox biology
Graphical abstract
Highlights
-
•
2-cys peroxiredoxin is the dominant scavenger of H2O2 in mammalian cells.
-
•
H2O2 gradient across the plasma membrane is ~650-fold, 100 times higher than cited in literature.
-
•
We developed an approach that converts fold-change in H2O2 sensor HyPer into change in actual concentration.
-
•
The range of intracellular perturbations associated with the onset of apoptosis is less than 10 nM.
Introduction
Redox reactions underlie a number of signaling pathways within cells, causing a variety of observable cellular responses ranging from homeostasis to proliferation to death [1–4]. Understanding the molecular mechanisms that make one of these responses more likely than another is an active area of research, and such an understanding has far-reaching practical significance since several pathological states are characterized by altered redox biology [5]. As one example of a class of pathological states, cancerous cells are thought to function in the presence of higher levels of oxidative species than noncancerous cells [6]. Redox therapeutics are hypothesized to act by causing the intracellular oxidant level of cancer cells to rise above the upper limit that is compatible with survival [7–9]. For safety, it is important that the perturbation induced by the therapeutic stays within the limits of the defense networks of healthy cells [10]. This hypothesis and approach are inherently quantitative and success would depend on the magnitudes of the changes to oxidant levels in healthy and diseased cells and how the final levels relate to the threshold for survival. However, facile measurement of perturbations to the intracellular levels of oxidants in terms of concentration and as a function of time has been a long-standing challenge in the field.
Hydrogen peroxide is a particularly interesting oxidant because of its stability; its longer lifetime allows it to participate either as a secondary messenger molecule or a lethal oxidant [11]. Most phenotypic and signaling studies of cellular responses to peroxide are done using extracellular addition, either with a bolus or with sustained generation using glucose oxidase [12,13]. However, hydrogen peroxide has limited permeability across the plasma membrane [14]. When peroxide is added to the outside of cells, fast consumption by antioxidant enzymes inside the cell creates a gradient across the plasma membrane, making the intracellular concentration lower than the extracellular concentration. Antunes et al. were the first to move from simply reporting external concentrations to estimating intracellular concentrations by building a compartmental model that estimates the gradient using experimentally determined kinetics of known antioxidants and of the hydrogen peroxide adsorption rate across the membrane [15]. A gradient of ~7- to 10-fold results from their analysis, meaning for a given extracellular concentration, the corresponding change to the intracellular concentration is about 7–10-fold lower. Using this method, changes to intracellular concentrations have been correlated with cellular responses indirectly by observing the effects of extracellular hydrogen peroxide perturbations [13,16].
However, since these publications appeared, other antioxidants have been discovered as important in scavenging hydrogen peroxide [17]. These were not included in the original kinetic calculations and therefore, depending on the magnitude of their contribution, the original gradient estimate may be lower than the actual gradient across the membrane, leading to incorrect intracellular associations for different phenotypes. Furthermore, the study of cellular response to hydrogen peroxide is shifting from using extracellular perturbation to endogenous generators, with the reasoning that endogenous generators better mimic physiological conditions during the action of drugs and for some disease states in terms of the location and kinetics of peroxide generation [12,18–20]. It is currently difficult to assess the hydrogen peroxide concentrations generated by these endogenous tools.
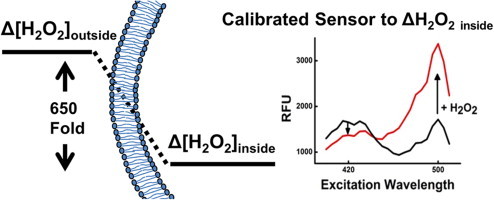
To address this need, we developed a quantification approach, demonstrated here as we correlate fold-change in signal from a genetically encoded peroxide sensor to change in intracellular concentration. HyPer, the particular probe used herein, is derived from the OxyR transcription factor found in bacteria with YFP inserted between two reactive thiol groups to provide a fluorescent readout of OxyR’s oxidation state [21]. The sensor is expressed by the cell and emits a ratiometric fluorescence change upon reaction with hydrogen peroxide only; it is unresponsive to other oxidants. Unlike the readouts in other intracellular peroxide sensing strategies [22], the signal of HyPer is reversible due to the activity of intracellular reductases, and this feature allows peroxide to be monitored as a function of time. To convert fold-change in HyPer’s signal to a change in absolute intracellular concentration, we first added various known amounts of peroxide to the outside of HeLa cells expressing HyPer and measured the resulting fluorescence using methodology established in previous work [23]. Then, we modified the compartmental model from Antunes et al. to include peroxiredoxin, and devised methodology for experimentally determining the kinetic parameters required for calculation of an improved peroxide gradient across the plasma membrane. Using the new gradient, we are able to interpret changes in HyPer fluorescence resulting from the known, external bolus additions in terms of absolute intracellular concentration (Fig. 1a), and the resulting calibration accomplished using extracellular addition of peroxide can be then used to quantify peroxide produced by intracellular generators.
Fig. 1.
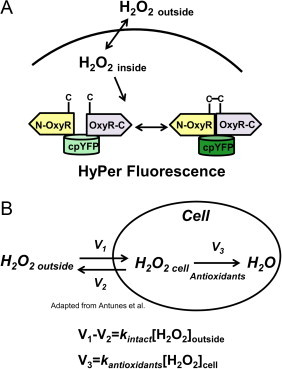
(A) Schematic of a quantification assay for hydrogen peroxide. We measured the gradient of hydrogen peroxide across the plasma membrane of HeLa cells and the fluorescent response of a genetically encoded sensor, HyPer, to known added amounts of extracellular peroxide. Knowledge of the gradient provides a correlation between intracellular concentration and HyPer fold change. (B) Kinetic model of hydrogen peroxide absorption across the plasma membrane and scavenging within the cell. A gradient is established across the plasma membrane due to the rapid consumption of hydrogen peroxide by intracellular antioxidants. The magnitude of this gradient depends on the relative value of the rate constant for intracellular consumption (kantioxidants) and the rate constant for transport across the membrane (kintact).
Results
Determining the hydrogen peroxide gradient across the HeLa biomembrane
We determined the peroxide gradient across the HeLa biomembrane by adapting Antunes’ model (Fig. 1b). Since intracellular concentrations of hydrogen peroxide are generally thought to be sub-micromolar, most of the antioxidant pool is expected to exist in the reduced form, resulting in a pseudo-first order reaction between hydrogen peroxide and the antioxidants [15,24,25]. Thus, at equilibrium, the rate of absorption of hydrogen peroxide across the plasma membrane will equal the rate of consumption of hydrogen peroxide by the antioxidants inside the cell:
| (1) |
where kintact is the pseudo-first order rate constant for absorption of peroxide across the membrane of intact cells and kantioxidants is the sum of pseudo-first-order rate constants for all intracellular antioxidants. Re-arranging these terms yields an equation for the gradient, R, across the plasma membrane
| (2) |
This gradient is independent of peroxide concentration and is only dependent on the first order rate constants of hydrogen peroxide adsorption by the intact cell and by the intracellular antioxidants.
The pseudo-first order rate constants for the rate of adsorption of hydrogen peroxide across the plasma membrane (kintact) and consumption by antioxidants catalase (kcatalase) and glutathione peroxidase (kGPx) in the cytoplasm were determined using methodology similar to that outlined in [15]. To determine kintact, we tracked the extracellular concentration of hydrogen peroxide as a function of time using an absorbance-based horseradish peroxidase assay in place of an oxygen electrode, and we verified the value using steady state generation of peroxide by an extracellular glucose oxidase system (S1). kcatalase and kGPx were determined using the experimental protocol and data analysis methods of the original Antunes paper. We found a kintact of 5.9 ± 0.2 × 10-12 s−1 cell−1 L (N = 6), kcatalase of 8.5 ± 0.3 × 10−13 s−1 cell−1 L (N = 8) and a kGPx 3.4 ± 0.08 × 10−11 s−1 cell−1 L (N = 12) (Fig. 2).
Fig. 2.
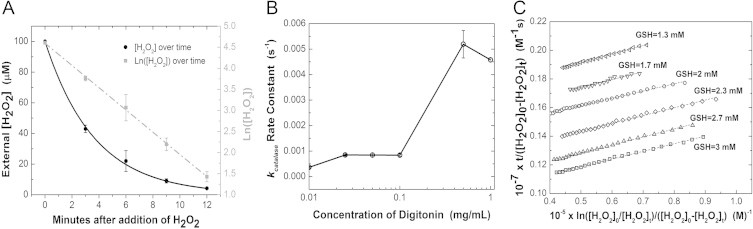
(A) Finding kintact. The rate constant of hydrogen peroxide diffusion across the membrane was determined by adding hydrogen peroxide to a suspension of HeLa cells and measuring the extracellular concentration of peroxide using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) absorbance based assay. (B) Finding kcatalase. HeLa cells were permeabilized with increasing concentrations of digitonin and lysates were incubated with 10 mM H2O2. Depletion of peroxide was measured at 240 nm and the first order rate constant was determined. The plateau before the rise in kinetic activity indicates conditions where the peroxisome is not disrupted. (C) Finding kGPx. HeLa cells lysed with 0.1% Triton were incubated with 1.3–3 mM of GSH, 1.1 U/mL of glutathione reductase, 1 mM NaN3, 0.3 mM NADPH and 35 µM hydrogen peroxide. NADPH depletion, as an indicator of hydrogen peroxide depletion, was measured at 340 nm. Hydrogen peroxide depletion kinetics between 2 and 15 µM were then fitted to an integrated rate equation and kGPx was determined.
Since the time of Antunes’ widely cited gradient estimations, a class of antioxidants called peroxiredoxins (Prx) has been hypothesized to play a major role in scavenging hydrogen peroxide in mammalian cells. Though pseudo-first order rate constants are not yet available in the literature, second order oxidation rate constants the same order of magnitude as that of glutathione peroxidase (GPx) have been reported for purified 2-cysteine forms of the protein [17,26]. The 2-cysteine Prx (2-cys-Prx) that react with peroxide are largely localized in the cytoplasm, and in HeLa cells, cytoplasmic Prx I and II account for over 90% of these isoforms found throughout the cell [27]. Following reaction with peroxide, thioredoxins are responsible for returning oxidized form of Prx to its reduced state. We believe that the contribution of this class of antioxidants cannot be ignored in determining the gradient across the plasma membrane.
One interesting and potentially complicating feature of 2-cys-Prx is that the redox active thiol group can be hyper-oxidized to sulfinic or sulfonic acid groups under sufficient hydrogen peroxide concentrations, rendering it inactive. These species are then very slowly reduced to –SOH by sulfiredoxins (Fig. 3a) [28]. However, hyper-oxidation of 2-cys-Prx has been detected only for cells exposed to high concentrations of extracellular hydrogen peroxide (> 100 µM) and is generally not detected during hydrogen peroxide mediated signaling events [25,29,30]. Below the hyper-oxidation detection threshold, the abundant thioredoxin system in most cell lines rapidly recycles any oxidized 2-cys-Prx, so we can approximate that the majority of the active cytoplasmic 2-cys Prxs are in the reduced form [25,31]. Thus under most in vivo hydrogen peroxide conditions, the Prx scavenging activity can be estimated as pseudo-first order, where the rate constant kprx is a function of the second-order rate constant of the oxidation step, and the total active intracellular 2-cys-Prx concentration, [Prxactive].
Fig. 3.
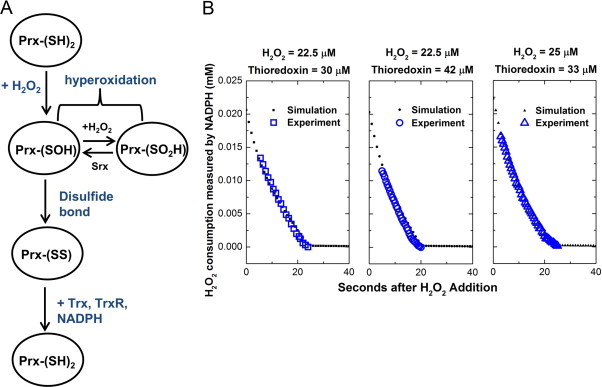
(A) Schematic of 2-cys peroxiredoxin oxidation and reduction cycle through reactions with hydrogen peroxide and thioredoxin. Peroxide oxidizes 2-cys Prx to form Prx-SOH. Prx-SOH can then either react with a second hydrogen peroxide to form a hyper-oxidized state, or it can interact with another Prx to form a disulfide bond, which can then be reduced through the thioredoxin/thioredoxin reductase system. (B) Finding kPrx. HeLa cells lysed with 0.1% Triton were incubated with 30–50 µM yTrx, 2 µM of yTrxR, 1 mM NaN3, 0.3 mM NADPH and 20–25 µM hydrogen peroxide. NADPH depletion, as an indicator of hydrogen peroxide depletion, was measured at 340 nm. The kinetic data were then fitted by minimizing the root mean square deviation to a simulated model of the experimental system. The rate constant kPrx was determined for several peroxide and thioredoxin (Trx) concentrations. For the case of hydrogen peroxide = 22.5 µM and Trx = 30 µM, we obtained a kPrx value of 3.7 × 10−9 s−1 cell−1 L (N = 2). For hydrogen peroxide = 22.5 µM and trx = 42 µM, kPrx was 3.8 × 10−9 s−1 cell−1 L (N = 2), and with hydrogen peroxide = 25 µM and Trx = 33 µM, kPrx was 3.9 × 10−9 s−1 cell−1 L (N = 2). The kPrx value reported in the text is the average of all six trials.
The reaction scheme of 2-cys Prx oxidation, hyperoxidation and reduction is shown below:
| (3) |
| (4) |
or
| (5) |
| (6) |
| (7) |
| (8) |
Writing these reactions as ordinary differential equations yield:
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
We added known initial concentrations of hydrogen peroxide and excess purified thioredoxin/thioredoxin reductase (yTrx/yTrxR) to reduce the 2-cys Prx present in lysates of HeLa cells, and tracked the hydrogen peroxide depletion via NADPH consumption. To determine kprx, we extracted the value of k1*[Prxactive] from our kinetic data of peroxide concentration as a function of time. We did so by fitting the experimental kinetic data to a simulation of the experimental system, built from the system of ODEs above with the appropriate rate constants and known initial conditions, with the parameter to fit being the initial concentration of 2-cys-Prx in the lysate (Prxactive) (Eqs. (9)–(15), SI) [17,32–35]. Fitting using a root-mean square deviation method, we found a kprx of 3.8 ± 0.03 × 10−9 s−1 cell−1 L (N = 6), meaning 2-cys-Prx scavenges peroxide 100 times faster than GPx under most physiological conditions (Fig. 3b). As shown in Fig. S2, it is important to consider hyper-oxidation effects for our experimental set-up since we added a significant peroxide bolus to cell lysate in order to get a measurable kinetic curve.
Substituting the pseudo-first order rate constants for intact cells and for the various antioxidants into Eq. (2), we obtain a gradient of ~650-fold across the plasma membrane. This value is significantly higher than the gradient of 7–10-fold calculated by Antunes et al., and we note that the inclusion of 2-cys-Prx in our calculations is crucial in obtaining the higher value. Typical, non-pathogenic intracellular H2O2 perturbations have been given as between 1 and 700 nM [36]. This widely cited information is based on the gradient calculated by Antunes et al., combined with data in a subsequent paper showing that a steady concentration of less than 5 µM extracellular hydrogen peroxide does not affect Jurkat cell morphology, but a concentration above this threshold causes these cells to undergo apoptosis [16]. Our new gradient suggests that the perturbation to the average intracellular concentration before the onset of cell death is less than 7.5 nM, and for perturbations between 7.5 and 10.5 nM, induction of apoptosis would be expected. Thus, changes to the level of intracellular hydrogen peroxide that lead to the onset of apoptosis appear to be in the low nanomolar range.
Quantifying intracellular [H2O2] with HyPer and the H2O2 membrane gradient
With the improved membrane gradient, we now demonstrate a quantitative approach to calibrate the HyPer sensor such that we can translate its signal output into a change in concentration. We made a standard curve that relates HyPer signals to extracellular peroxide concentrations by adding known boluses of hydrogen peroxide to a suspension of HeLa cells expressing HyPer. We followed the kinetics of HyPer fluorescence over a period of 10 min and extracted the peak ratiometric fluorescence for each bolus addition, and divided that ratio by the baseline ratiometric fluorescence to obtain a fold-change in HyPer response. To account for potential pH effects, buffer without hydrogen peroxide was added to an identical sample of HeLa cells, and fold-change for every hydrogen peroxide bolus is normalized to this control case. The intracellular sensor responded dynamically to 2–7 µM of hydrogen peroxide added externally to a suspension containing 2.5 × 105 cells/mL (Fig. 4). Using our experimentally determined hydrogen peroxide gradient, we can calculate the corresponding intracellular peroxide change for each different external hydrogen peroxide concentration added. This analysis suggests HyPer responds to 3–10 nM changes in intracellular peroxide concentrations (Fig. 4). Now, independent of the method of hydrogen peroxide perturbation, we can measure the intracellular fold-change in HyPer signal and use the standard curve in Fig. 4 to determine the change in terms of an absolute concentration.
Fig. 4.
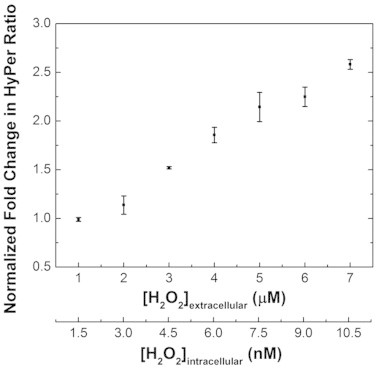
Calibrating HyPer’s ratiometric fluorescent response to hydrogen peroxide. The excitation spectra of cell suspensions containing 2.5 × 105 cells/mL treated with 0–7 µM hydrogen peroxide were measured at 2-min intervals with excitation from 410–510 nm and emission monitored at 545 nm. The peak of the response was noted. Ratiometric responses (Excitation 500 nm/420 nm) were calculated at the peak. This ratio was divided by the baseline ratio before addition of hydrogen peroxide, and normalized to the case where an equal amount of buffer without hydrogen peroxide was added to obtain the normalized fold change values. The error bars for each data point represent 95% confidence intervals. In order to determine the corresponding intracellular hydrogen peroxide concentration for each HyPer response, the extracellular concentration is divided by 650, the estimated gradient of hydrogen peroxide across the plasma membrane.
Discussion
Peroxiredoxins have been proposed as the dominant hydrogen peroxide elimination enzyme in mammalian cells, and our experimental results support this hypothesis as we found that the pseudo-first-order rate constant for the oxidation of Prx by hydrogen peroxide is about 100 times faster than that of GPx. This finding suggests that antioxidant-based therapeutics should focus on targeting Prx isoforms and their reduction partners, thioredoxin and glutathione. Another implication of peroxiredoxin activity is the much higher hydrogen peroxide gradient we measured across the plasma membrane. The new gradient estimates the switch between non-pathological to pathological perturbations to the intracellular concentration of peroxide to be in the low nanomolar range. This lower intracellular concentration means that the rate of cytoplasmic hydrogen peroxide reacting with a protein thiol is even slower than previously thought. Direct action of hydrogen peroxide on its target may require very close proximity of the target to the source of generation, as suggested by the flood-gate model [26,30,37]. Another possibility is that hydrogen peroxide acts on the downstream signaling targets in an indirect manner, through oxidation of more abundant Prx/Gpx antioxidants or conversion to a more reactive radical species such as hydroxyl radical [1,29,38]. Improving our quantitative understanding of these concentration parameters can establish more powerful mathematical models of biological redox systems. Specifically, knowledge of actual intracellular concentrations is important for allowing evaluation of the likelihood of mass–action interactions of various oxidants with their supposed targets, yielding better predictions of fundamental redox processes. Furthermore, concentrations associated with phenotypic responses establish design criteria for future ROS-therapeutics as to how much of a perturbation to the intracellular hydrogen peroxide level is needed to switch from proliferation to cell death [6,7].
Our calibration of an intracellular peroxide sensor using a gradient measurement is an important advance that supports and parallels the development of physiologically relevant intracellular ROS-generators. The gradient can be used for intracellular quantification with a known amount of exogenous addition, but it alone cannot give any concentration information for intracellular generator tools. For this task, an intracellular sensor must be paired with knowledge of the gradient. As Fig. 4 indicates, the response of HyPer to hydrogen peroxide, like that of many popular redox sensors, is non-linear [22]. As a result, it is difficult to interpret the common approach of reporting sensor signal as fold-change, since an increase in signal between a 1.5×-fold change and a 2×-fold change, versus a 2×-fold change and a 2.5×-fold change are not necessary the same. The calibration approach detailed herein will be important in providing real-time verification, visualization and quantification of the perturbations produced by intracellular peroxide generators.
Accuracy in measuring perturbations to the intracellular peroxide concentration depends in part on the choice of sensor. We chose HyPer to demonstrate our quantification approach because it is an intracellular sensor specific for peroxide, and it is reversible and ratio-metric [23]. These characteristics offer an advantage over the dye-based sensors such as DCFH or PG-1 since HyPer measures real-time concentrations rather than a cumulative effect [39], and HyPer provides greater specificity and sensitivity than other fluorescent genetically-encoded sensors such as roGFP [40]. However, using the sensor is not without its caveats. First, the probe is pH sensitive, so proper controls are required to ensure the results are not artifacts of the external environment [41]. We addressed this issue by including a control where buffer without hydrogen peroxide was added to a suspension of HeLa cells, and fold-change for every hydrogen peroxide bolus was normalized to this control case. Secondly, the OxyR domain has a second order rate constant of 105 M−1 s−1 with hydrogen peroxide [42], while 2-cys Prx interacts with hydrogen peroxide with a second order rate constant of 107 M−1 s−1. Thus, in the kinetic competition for reaction with intracellular peroxide, HyPer does not effectively compete with Prx. It follows that in lining up our standard curve with the results of the biochemical gradient measurement, we are slightly over-estimating the intracellular concentration of hydrogen peroxide for each HyPer fold-change. We should note that our quantitative approach generalizes to probes other than HyPer, and accuracy and responsive regions will improve as improved hydrogen peroxide sensors are developed.
The accuracy of this quantification is also dependent on the accuracy of the gradient across the plasma membrane. In fitting our kinetic data for cytoplasmic 2-cys-Prx activity in HeLa cells, where Prx I and II are predominant, we were able to obtain second order rate constants for Prx II but not for Prx I for use in our numerical model. Thus, our experimental data is an average activity of both Prx isoforms in the cytoplasm, but our theoretical fit assumes Prx I have the same rate constants as Prx II. It has been observed that Prx I is less susceptible to hyper-oxidation than Prx II, and is rather inactivated by phosphorylation, however these kinetic rates are unknown and so could not be included in our model [30]. In addition, a recent kinetic model suggests the inclusion of an inhibitor of Prx II as a plausible way to obtain the correct oxidation/reduction kinetics for the antioxidant in red blood cells, and this modeling approach predicts that only a small fraction of the total Prx II protein is active in these cells [43]. Since our kinetic experiments are done under dilute, lysed cell conditions, it is possible that any inhibitor function may be disrupted under these experimental conditions and we over-estimate the concentration of active Prx II. The identity of molecular species that may act as inhibitors and whether this theory applies in HeLa cells remain open questions. Finally, discovery of new antioxidants and targets that react with hydrogen peroxide at a rate similar to that of Prx would further increase the value of the gradient. The model and analysis can easily be updated as more information becomes available. It should be noted that the current calculation as it stands is already a vast improvement over the previous gradient measurement. A combined effort of intracellular quantification and generation tools will allow physiologically relevant and systematic studies of the role of hydrogen peroxide in determining cellular phenotypes. In addition to promoting fundamental, mechanistic understanding of human redox biology, such studies hold promise for determining quantitative design criteria for future ROS-based therapeutics.
Methods and materials
Materials
Hydrogen peroxide, horseradish peroxidase (HRP), neomycin, penicillin–streptomycin, glucose oxidase, catalase, l-glutathione, glutathione reductase, digitonin, triton X-100, were purchased from Sigma (St. Louis, MS, USA). 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) was purchased from Tokyo Chemical Industry Co (Portland, OR, USA). d(+)-Glucose was purchased from Macron Fine Chemicals (Center Valley, PA, USA). NADPH was purchased from Roche (Basel, Switzerland). HeLa cells were a gift from the Wittrup Lab at MIT (Cambridge, MA, USA). The Modified Eagle’s medium (EMEM) and fetal bovine serum were purchased from ATCC (Manassas, VA, USA). Trypsin was purchased from Lonza (Walkerville, MD, USA). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA, USA). A plasmid encoding HyPer-cyto plasmid was purchased from Evrogen (Moscow, Russia). Q HP FF, Phenyl HP and Superdex75 FPLC purification columns were purchased from GE Healthcare (Fairfield, CT, USA). Other common chemicals to make buffer solutions were purchased from VWR International (USA).
Cell culture and transfection
HeLa cells were cultured in EMEM with 10% FBS at 37 °C, 95% air and 5% CO2. A stable cell line of HeLa cells expressing HyPer was created by transfecting HeLa cells with a mixture of plasmid DNA and Lipofectamine for 24 h, and a single colony was gradually selected over a period of 6 weeks using neomycin at 1 mg/mL. The plasmid was maintained in the cell with neomycin at 0.2 mg/mL.
Fluorescence spectroscopy for HyPer readings in intact HeLa cells
HeLa cells expressing HyPer were trypsinized, washed once in phosphate buffer saline (PBS), pH 7.4 and re-suspended in the same buffer at a concentration of 250,000 cells/mL. Excitation spectra of HyPer expressed in cells were recorded using a Tecan M200 microplate reader with excitation at 410–510 nm, 9 nm bandwidth, and emission at 545 nm, 20 nm bandwidth. A baseline excitation spectrum was taken, then a bolus of hydrogen peroxide was added to the suspension and kinetics were followed for a period of 10 min. HyPer’s ratiometric fluorescent response was obtained by dividing the emission reading for excitation at 500 nm by the emission reading for excitation at 420 nm, then dividing the peak of the ratiometric fluorescence by the baseline to obtain a fold change. The fold change data is normalized to a control case where no hydrogen peroxide was added to the system (i.e. an equal volume of buffer only was added).
Consumption of hydrogen peroxide by intact HeLa cells
HeLa cells containing HyPer were grown in 10 cm BD Falcon dishes (VWR International, USA) until confluent. The cells were trypsinized, washed once in PBS, pH 7.4, and re-suspended in PBS with 2 g/L d-glucose at a concentration of 106 cells/mL, 10 mL total. A bolus of hydrogen peroxide was then mixed into the dish, making it a total of 80–100 µM in cell suspension. Every 2 min, a 200 µL sample of the cell suspension was inactivated with 13 µL 5 N HCl to prevent further antioxidant scavenging. The samples were incubated for 10 min in HCl, then centrifuged at 14,000g. 160 µL of the supernatant was removed, mixed with 70 µL of 1 M Kpi (pH 8.0), followed by 50 µL of 2.5 mM of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) solution and 10 µL of 3 mg/mL horse-radish peroxidase. The samples were read at an absorbance wavelength of 405 nm. A standard curve between 0 and 50 µM for hydrogen peroxide was done with each set of data.
Exposure of HeLa cells to steady state levels of hydrogen peroxide
If the first order rate constant of hydrogen peroxide consumption by intact HeLa cells is correct, then we can do a steady state balance between the consumption of hydrogen peroxide and the generation of hydrogen peroxide by a glucose oxidase system [13]. Glucose oxidase was diluted in PBS (pH 7.4) with 2 g/L d(+)-glucose to concentrations of ~5 mU/mL. The rate of hydrogen peroxide generation for the diluted stock was determined using the ABTS assay. Then, 10 µM of hydrogen peroxide mixed with 0.8 million cells/mL and an appropriate amount of glucose oxidase (generates 2.8 µM hydrogen peroxide/min) in the PBS + glucose solution, and the concentration of hydrogen peroxide outside the cell was measured for a period of 60 min using the ABTS assay.
Consumption of hydrogen peroxide by catalase
Trypsinized HeLa cells expressing HyPer (106 cells/mL) were lysed by re-suspending the cells in 50 mM potassium phosphate buffer (pH 7.4) containing digitonin. Digitonin concentration was varied from 0.01 to 1 mg/mL. For each lysate sample, hydrogen peroxide depletion was measured in the presence of 10 mM hydrogen peroxide and kinetics were followed at 37 °C for 5 min at 240 nm (extinction coefficient 43.6 M−1 cm−1). At digitonin concentrations of less 0.1 mg/mL, only the plasma membrane was affected, yielding depletion kinetics due to catalase acting on H2O2 that diffused across an intact peroxisomal membrane.
Consumption of hydrogen peroxide by glutathione peroxidase
HyPer HeLa cells expressing HyPer (5 million cells/mL) were lysed in 50 mM potassium phosphate (pH 7.4), 1 mM DPTA buffer containing 0.1% Triton solution. The lysate mixture was then pre-incubated at 37 °C with a range of 1–3 mM of GSH, 1.1 U/mL glutathione reductase, 0.3 mM NADPH and 1 mM NaN3 to reduce any oxidized GPx for 10 min. Then, a bolus of 35 µM hydrogen peroxide was added to the mixture and NADPH depletion at 340 nm (extinction coefficient 6.22 mM−1 cm−1) was followed for a period of 5 min. For kinetic analysis, the part of the curve corresponding to 2–15 µM hydrogen peroxide remaining was used for fitting to the integrated rate equation shown below:
| (16) |
where t is the time since the addition of a bolus of hydrogen peroxide to the lysate system, k1′, k2′, k3′ are rate constants associated with GPx redox reaction as indicated in SI, and [GPx]total is the total concentration of GPx enzyme. kGpx = k1′*[GPx]total and was obtained by the inverse slope of linear fitting .
Consumption of hydrogen peroxide by 2-cys peroxiredoxin
HeLa cells expressing HyPer (5.8 million/mL) were lysed in 20 mM Hepes (pH 7.4), 1 mM EDTA buffer containing 0.1% Triton solution. The lysate mixture was pre-incubated at 37 °C with a range of 20–50 µM yeast Trx, 2 µM yeast TrxR, 0.4 mM NADPH and 1 mM NaN3 for a period of 10 min. Then, a bolus of hydrogen peroxide between 20 and 25 µM was added to the mixture and NADPH depletion was followed by monitoring absorbance at 340 nm (extinction coefficient 6.22 mM−1 cm−1) for a period of 5 min. For kinetic analysis, the experimental data was fitted to a model simulation using root-mean square deviation and the pseudo-first order rate constant kprx was extracted.
Purification of yeast-thioredoxin and thioredoxin-reductase
Plasmids for yeast Trx/TrxR (pET17b-yTrx1 and pET17b-yTrxR) were a gift from Dr. Sang Won Kang (Ewha Womans University, Seoul, Korea) [32]. To purify the recombinant proteins, Escherichia coli BL21 (DE3) transformed with the plasmids were was grown until OD 0.6 (λ = 600 nm) in 250 mL of Terrific Broth, at which point expression was induced by addition of 1 mM isopropyl-1-thio-B-d-galatoside (IPTG), cells are harvest after shaking for 4 h at 250 rpm. The cells are lysed, on ice, for 20 min, 1 min interval in 10 mL of buffers specified in [32], then spun at 12,000g for 15 min. The supernatant was filtered through a 0.22 µm Acrodisc Syringe Filter (Pall Corporation, USA). For TrxR, the crude extract was loaded onto a Q HP fast-flow anionic exchange FPLC column. The protein was eluted with a linear gradient of 0–500 mM NaCl in 20 mM Tris–HCl (pH 7.5), and the fraction containing yTrxR protein was verified through SDS-PAGE gel and used for a subsequent purification. After dialyzing the fraction against 20 mM Hepes-NaOH (pH 7.0), the protein was equilibrated with 1 M ammonium sulfate (pH 7.0) and loaded onto a Phenyl HP FPLC column. The protein of interest came out in the flow-through before the elution step, and the pool of purified proteins were dialyzed against buffer containing 20 mM Hepes (pH 7.4) and stored at −70 °C until required. For Trx, the crude extract was heat-treated for 30 min at 75 °C and centrifuged to remove denatured protein. The soluble supernatant was loaded onto the Q-HP fast flow anionic exchange and eluted with the same linear NaCl gradient as TrxR. The fractions containing Trx were then loaded onto Superdex 75 gel filtration column equilibrated with 20 mM Tris–HCl, 1 mM EDTA and 150 mM NaCl (pH 7.0). Protein purities for both Trx/TrxR were confirmed through SDS-Page gel and UV spectra (S3). The reducing potential of yTrxR was measured by pre-oxidizing yTrx with equimolar hydrogen peroxide for 10 min, then incubating with yTrxR and NADPH to measure the reduction kinetics [32]. An activity of 2.4 ± 0.3 µmol/min/mg was found, indicating active yTrxR was produced that couples with the yTrx redox state.
Statistical analysis
For kinetics analyses at least three technical replicates and two biological replicates were performed for each rate constant. The experimental values are reported as the mean ± standard error of mean (SEM). For the HyPer standard curve, the data points represent the mean over 4 trials (2 technical and 2 biological replicates), and the error bars are the 95% confidence intervals.
To determine kprx, a parameter sweep from 0.1 to 100, step size 0.1 was performed using Matlab2013, and for each value of kprx, the root mean square deviation (RMSD) is calculated between the simulation kinetic curve and the experimental kinetic curve. The kprx value that gives the minimum of the RMSD function is the estimated kprx. This fitting was performed for various Trx and hydrogen peroxide concentrations, and the overall kprx reported is the mean from all of the fittings
| (17) |
where n is the number of data points, xs is the value of the simulation data, and xe is the value of the experimental data.
Conflicts of interest
No competing financial interests exist.
Acknowledgments
This work was supported by an NSF Graduate Research Fellowship (B.K.H.) (2009085738), a Burroughs Wellcome Fund Career Award at the Scientific Interface (H.D.S.) (1005762) and the Joseph R. Mares Professorship in Chemical Engineering (H.D.S.). We thank Dr. Sang Won Kang (Ewha Womans University, Seoul, Korea) for providing the yTrx/TrxR plasmids, Dr. Dane Wittrup (Massachusetts Institute of Technology, Cambridge, MA) for the HeLa cells, Sohail Ali for assistance with creating the stable HyPer HeLa cell line, Muzhou Wang and members of the Sikes Lab for helpful discussions.
Appendix A. Supplementary information
Supplementary information associated with this article can be found in the online version at 10.1016/j.redox.2014.08.001.
Appendix A. Supplementary informations
Supplementary Fig. S1: (A) Hydrogen peroxide production by glucose oxidase. Glucose oxidase was diluted to concentrations between 3 and 5 mU/mL in in PBS (pH 7.4) + 2 g/L glucose. The production of peroxide by the enzyme was measured using the ABTS assay. (B) A steady-state hydrogen peroxide level obtained using external glucose oxidase as a generator and Hyper HeLa cells as scavengers. HyPer HeLa cells were re-suspended at 0.8 million cells/mL in PBS + 2 g/L glucose. The amount of glucose oxidase producing 2.8 µM hydrogen peroxide/min was added simultaneously with 10 µM of hydrogen peroxide. The extracellular peroxide concentration was monitored for a period of 1 h using the ABTS assay. A constant extracellular concentration verifies the kintact value.
Supplementary Fig. S2: Depletion of hydrogen peroxide due to 2-cys Prx activity in cell lysates with and without hyper-oxidation effects.While hyper-oxidation of 2-cys Prx in intact cells has only been detected for extracellular additions of > 100 µM, it is important to consider hyper-oxidation effects for lower peroxide concentrations when the sample consists of cell lysates rather than intact cells.The kinetics of Prx activity in the experimental system was modeled using a system of ODEs implemented using Matlab (SI). The situation where Prx can undergo hyper-oxidation (solid) versus without hyper-oxidation (dotted) was compared for bolus additions of 20 µM (black) and 35 µM (blue) hydrogen peroxide to a solution of HeLa cell lysate. The possibility of hyper-oxidation changes the kinetics of hydrogen peroxide depletion significantly, and this effect is greater for the 35 µM addition in comparison with the 20 µM addition. From this simulation, we conclude that for both concentrations, hyper-oxidation needs to be taken into account when fitting the Prx kinetic data to a rate equation. Other parameters used in the simulation were Trx = 30 µM, k1*[Prxactive] = 3.8 × 10−9 s−1 cells−1 L and a cell density of 5.8 million cells/mL.
Supplementary Fig. S3: Expression and purification of yTrx and TrxR. After induction with IPTG and expression for 4 h, Trx and TrxR overexpression was detected in the lysate fractions. After chromatographic separations with anionic (Q) and gel filtration (Superdex75) columns using an AKTA FPLC, a single 11 kD fragment for Trx was observed using SDS PAGE. Similarly, TrxR was also purified using Q and Phenyl hydrophobic columns, and a strong band of 35 kD was detectable. The UV-spectra for the purified TrxR showed the correct peaks at 280, 359 and 452 nm.
References
- 1.D’Autréaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews: Molecular Cell Biology. 2007;8:813–824. doi: 10.1038/nrm2256. 17848967 [DOI] [PubMed] [Google Scholar]
- 2.Burdon R.H., Rice-Evans C. Free radicals and the regulation of mammalian cell proliferation. Free Radical Research Communications. 1989;6:345–358. doi: 10.3109/10715768909087918. 2676744 [DOI] [PubMed] [Google Scholar]
- 3.Winterbourn C.C. Reconciling the chemistry and biology of reactive oxygen species. Nature Chemistry and Biology. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 4.Singh M., Sharma H., Singh N. Hydrogen peroxide induces apoptosis in HeLa cells through mitochondrial pathway. Mitochondrion. 2007;7:367–373. doi: 10.1016/j.mito.2007.07.003. 17855174 [DOI] [PubMed] [Google Scholar]
- 5.Lambeth J.D. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radical Biology & Medicine. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. 17602948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurent A., Nicco C., Chéreau C., Goulvestre C., Alexandre J., Alves A., Lévy E., Goldwasser F., Panis Y., Soubrane O., Weill B., Batteux F. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Research. 2005;65:948–956. 15705895 [PubMed] [Google Scholar]
- 7.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature Reviews: Drug Discovery. 2009;8:579–591. doi: 10.1038/nrd2803. 19478820 [DOI] [PubMed] [Google Scholar]
- 8.Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nature Reviews: Drug Discovery. 2013;12:931–947. doi: 10.1038/nrd4002. 24287781 [DOI] [PubMed] [Google Scholar]
- 9.Schumacker P.T. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. 16959608 [DOI] [PubMed] [Google Scholar]
- 10.Raj L., Ide T., Gurkar A.U., Foley M., Schenone M., Li X., Tolliday N.J., Golub T.R., Carr S.A., Shamji A.F., Stern A.M., Mandinova A., Schreiber S.L., Lee S.W. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. 21753854 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Gough D.R., Cotter T.G. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death & Disease. 2011;2:e213. doi: 10.1038/cddis.2011.96. 21975295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forman H.J. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radical Biology & Medicine. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. 17349920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Oliveira-Marques V., Cyrne L., Marinho H.S., Antunes F. A quantitative study of NF-kappaB activation by H2O2: relevance in inflammation and synergy with TNF-alpha. Journal of Immunology. 2007;178:3893–3902. doi: 10.4049/jimmunol.178.6.3893. [DOI] [PubMed] [Google Scholar]
- 14.Miller E.W., Dickinson B.C., Chang C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15681–15686. doi: 10.1073/pnas.1005776107. 20724658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antunes F., Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Letters. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. 10858501 [DOI] [PubMed] [Google Scholar]
- 16.Antunes F., Cadenas E. Cellular titration of apoptosis with steady state concentrations of H(2)O(2): submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radical Biology & Medicine. 2001;30:1008–1018. doi: 10.1016/s0891-5849(01)00493-2. 11316581 [DOI] [PubMed] [Google Scholar]
- 17.Peskin A.V., Low F.M., Paton L.N., Maghzal G.J., Hampton M.B., Winterbourn C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. Journal of Biological Chemistry. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. 17329258 [DOI] [PubMed] [Google Scholar]
- 18.Dickinson B.C., Chang C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature Chemical Biology. 2011;7:504–511. doi: 10.1038/nchembio.607. 21769097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller E.W., Taulet N., Onak C.S., New E.J., Lanselle J.K., Smelick G.S., Chang C.J. Light-activated regulation of cofilin dynamics using a photocaged hydrogen peroxide generator. Journal of the American Chemical Society. 2010;132:17071–17073. doi: 10.1021/ja107783j. 21077675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haskew-Layton R.E., Payappilly J.B., Smirnova N.A., Ma T.C., Chan K.K., Murphy T.H., Guo H., Langley B., Sultana R., Butterfield D.A., Santagata S., Alldred M.J., Gazaryan I.G., Bell G.W., Ginsberg S.D., Ratan R.R. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17385–17390. doi: 10.1073/pnas.1003996107. 20855618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belousov V.V., Fradkov A.F., Lukyanov K.A., Staroverov D.B., Shakhbazov K.S., Terskikh A.V., Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3:281–286. doi: 10.1038/nmeth866. 16554833 [DOI] [PubMed] [Google Scholar]
- 22.Kalyanaraman B., Darley-Usmar V., Davies K.J.A., Dennery P.A., Forman H.J., Grisham M.B., Mann G.E., Moore K., Roberts L.J., Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radical Biology & Medicine. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. 22027063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malinouski M., Zhou Y., Belousov V.V., Hatfield D.L., Gladyshev V.N. Hydrogen peroxide probes directed to different cellular compartments. PloS One. 2011;6:e14564. doi: 10.1371/journal.pone.0014564. 21283738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chance B., Sies H., Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. 37532 [DOI] [PubMed] [Google Scholar]
- 25.Low F.M., Hampton M.B., Peskin A.V., Winterbourn C.C. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109:2611–2617. doi: 10.1182/blood-2006-09-048728. 17105810 [DOI] [PubMed] [Google Scholar]
- 26.Hall A., Karplus P.A., Poole L.B. Typical 2-cys peroxiredoxins: structures, mechanisms and functions. FEBS Journal. 2009;276:2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. 19476488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chae H.Z., Kim H.J., Kang S.W., Rhee S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Research and Clinical Practice. 1999;45:101–112. doi: 10.1016/s0168-8227(99)00037-6. 10588361 [DOI] [PubMed] [Google Scholar]
- 28.Nelson K.J., Parsonage D., Karplus P.A., Poole L.B. Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Methods in Enzymology. 2013;527:21–40. doi: 10.1016/B978-0-12-405882-8.00002-7. 23830624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marinho H.S., Real C., Cyrne L., Soares H., Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biology. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. 24634836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woo H.A., Yim S.H., Shin D.H., Kang D., Yu D.-Y., Rhee S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. 20178744 [DOI] [PubMed] [Google Scholar]
- 31.Hofmann B., Hecht H.J., Flohé L. Peroxiredoxins. Biological Chemistry. 2002;383:347–364. doi: 10.1515/BC.2002.040. 12033427 [DOI] [PubMed] [Google Scholar]
- 32.Kim J.-A., Park S., Kim K., Rhee S.G., Kang S.W. Activity assay of mammalian 2-cys peroxiredoxins using yeast thioredoxin reductase system. Analytical Biochemistry. 2005;338:216–223. doi: 10.1016/j.ab.2004.12.008. 15745741 [DOI] [PubMed] [Google Scholar]
- 33.Arnér E.S.J., Zhong L., Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods in Enzymology. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. 9919525 [DOI] [PubMed] [Google Scholar]
- 34.Peskin A.V., Dickerhof N., Poynton R.A., Paton L.N., Pace P.E., Hampton M.B., Winterbourn C.C. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. Journal of Biological Chemistry. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. 23543738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang T.-S., Jeong W., Woo H.A., Lee S.M., Park S., Rhee S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. Journal of Biological Chemistry. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. 15448164 [DOI] [PubMed] [Google Scholar]
- 36.Stone J.R., Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants & Redox Signaling. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. 16677071 [DOI] [PubMed] [Google Scholar]
- 37.Rhee S.G., Woo H.A., Kil I.S., Bae S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. Journal of Biological Chemistry. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. 22147704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarvis R.M., Hughes S.M., Ledgerwood E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radical Biology & Medicine. 2012;53:1522–1530. doi: 10.1016/j.freeradbiomed.2012.08.001. 22902630 [DOI] [PubMed] [Google Scholar]
- 39.Murphy M.P., Holmgren A., Larsson N.-G., Halliwell B., Chang C.J., Kalyanaraman B., Rhee S.G., Thornalley P.J., Partridge L., Gems D., Nyström T., Belousov V., Schumacker P.T., Winterbourn C.C. Unraveling the biological roles of reactive oxygen species. Cell Metabolism. 2011;13:361–366. doi: 10.1016/j.cmet.2011.03.010. 21459321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukyanov K.A., Belousov V.V. Genetically encoded fluorescent redox sensors. Biochimica et Biophysica Acta. 2014;1840:745–756. doi: 10.1016/j.bbagen.2013.05.030. 23726987 [DOI] [PubMed] [Google Scholar]
- 41.Roma L.P., Duprez J., Takahashi H.K., Gilon P., Wiederkehr A., Jonas J.-C. Dynamic measurements of mitochondrial hydrogen peroxide concentration and glutathione redox state in rat pancreatic β-cells using ratiometric fluorescent proteins: confounding effects of pH with HyPer but not roGFP1. Biochemical Journal. 2012;441:971–978. doi: 10.1042/BJ20111770. 22050124 [DOI] [PubMed] [Google Scholar]
- 42.Kóna J., Brinck T. A combined molecular dynamics simulation and quantum chemical study on the mechanism for activation of the OxyR transcription factor by hydrogen peroxide. Organic & Biomolecular Chemistry. 2006;4:3468–3478. doi: 10.1039/b604602a. 17036142 [DOI] [PubMed] [Google Scholar]
- 43.Benfeitas R., Selvaggio G., Antunes F., Coelho P.M.B.M., Salvador A. Hydrogen peroxide metabolism and sensing in human erythrocytes: a validated kinetic model and reappraisal of the role of peroxiredoxin II. Free Radical Biology & Medicine. 2014;74C:35–49. doi: 10.1016/j.freeradbiomed.2014.06.007. 24952139 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1: (A) Hydrogen peroxide production by glucose oxidase. Glucose oxidase was diluted to concentrations between 3 and 5 mU/mL in in PBS (pH 7.4) + 2 g/L glucose. The production of peroxide by the enzyme was measured using the ABTS assay. (B) A steady-state hydrogen peroxide level obtained using external glucose oxidase as a generator and Hyper HeLa cells as scavengers. HyPer HeLa cells were re-suspended at 0.8 million cells/mL in PBS + 2 g/L glucose. The amount of glucose oxidase producing 2.8 µM hydrogen peroxide/min was added simultaneously with 10 µM of hydrogen peroxide. The extracellular peroxide concentration was monitored for a period of 1 h using the ABTS assay. A constant extracellular concentration verifies the kintact value.
Supplementary Fig. S2: Depletion of hydrogen peroxide due to 2-cys Prx activity in cell lysates with and without hyper-oxidation effects.While hyper-oxidation of 2-cys Prx in intact cells has only been detected for extracellular additions of > 100 µM, it is important to consider hyper-oxidation effects for lower peroxide concentrations when the sample consists of cell lysates rather than intact cells.The kinetics of Prx activity in the experimental system was modeled using a system of ODEs implemented using Matlab (SI). The situation where Prx can undergo hyper-oxidation (solid) versus without hyper-oxidation (dotted) was compared for bolus additions of 20 µM (black) and 35 µM (blue) hydrogen peroxide to a solution of HeLa cell lysate. The possibility of hyper-oxidation changes the kinetics of hydrogen peroxide depletion significantly, and this effect is greater for the 35 µM addition in comparison with the 20 µM addition. From this simulation, we conclude that for both concentrations, hyper-oxidation needs to be taken into account when fitting the Prx kinetic data to a rate equation. Other parameters used in the simulation were Trx = 30 µM, k1*[Prxactive] = 3.8 × 10−9 s−1 cells−1 L and a cell density of 5.8 million cells/mL.
Supplementary Fig. S3: Expression and purification of yTrx and TrxR. After induction with IPTG and expression for 4 h, Trx and TrxR overexpression was detected in the lysate fractions. After chromatographic separations with anionic (Q) and gel filtration (Superdex75) columns using an AKTA FPLC, a single 11 kD fragment for Trx was observed using SDS PAGE. Similarly, TrxR was also purified using Q and Phenyl hydrophobic columns, and a strong band of 35 kD was detectable. The UV-spectra for the purified TrxR showed the correct peaks at 280, 359 and 452 nm.