

Abstract
The Philadelphia chromosome was the first genetic abnormality discovered in cancer (in 1960), and it was found to be consistently associated with CML. The description of the Philadelphia chromosome ushered in a new era in the field of cancer cytogenetics. Accumulating genetic data have been shown to be intimately associated with the diagnosis and prognosis of neoplasms; thus, karyotyping is now considered a mandatory investigation for all newly diagnosed leukemias. The development of FISH in the 1980s overcame many of the drawbacks of assessing the genetic alterations in cancer cells by karyotyping. Karyotyping of cancer cells remains the gold standard since it provides a global analysis of the abnormalities in the entire genome of a single cell. However, subsequent methodological advances in molecular cytogenetics based on the principle of FISH that were initiated in the early 1990s have greatly enhanced the efficiency and accuracy of karyotype analysis by marrying conventional cytogenetics with molecular technologies. In this review, the development, current utilization, and technical pitfalls of both the conventional and molecular cytogenetics approaches used for cancer diagnosis over the past five decades will be discussed.
Keywords: Cancer cytogenetics, FISH, Karyotyping, Molecular cytogenetics
INTRODUCTION
The discovery of a tiny abnormal chromosome, the Philadelphia chromosome, as a hallmark of CML in 1960 by Peter Nowell and David Hungerford marked the first time cancer was shown to result from a specific genetic abnormality [1]. As cytogenetic techniques improved, in 1973, Rowley [2] discovered the existence of a translocation between the long arms of chromosome 9 and 22 in CML. Subsequent work revealed that this translocation resulted in a new fusion protein that was expressed in the cancer cells [3]. Strikingly, the description of the Philadelphia chromosome ushered in a new era of genetic diagnosis. Over the past several decades, accumulating genetic data has been shown to be intimately associated with the diagnosis and prognosis of neoplasms, thereby moving cancer cytogenetics out of research laboratories and into clinical practice. Recognizing the association between specific cytogenetic abnormalities and certain morphologic and clinical features, the World Health Organization has categorized four unique AML subtypes according to cytogenetics [4]. Therefore, conventional cytogenetic analysis is considered mandatory for all newly diagnosed leukemias, owing to its usefulness in diagnosis, classification, and prognostication. FISH can be used to map loci on specific chromosomes [5], detect both numerical and structural chromosomal abnormalities, and reveal cryptic abnormalities. It has overcome many of the drawbacks of karyotyping, such as low specimen cell yield, low mitotic index, poor quality metaphases, and other technical difficulties. Currently, FISH is used as an indispensable tool for the detection of structural rearrangements such as translocations, inversions, insertions, and microdeletions, and for the identification of marker chromosomes and the delineation of chromosome breakpoints [6, 7]. Therefore, FISH has greatly enhanced the efficiency and accuracy of karyotype analysis by bringing together conventional cytogenetics and molecular technologies.
This review summarizes the development, current utilization, and technical pitfalls of the conventional and molecular cytogenetic approaches used in clinical laboratories for cancer diagnosis.
CONVENTIONAL CYTOGENETICS
Chromosome analysis is a simple technique. Under optimal conditions, in most cases of acute leukemia, clonal cytogenetic abnormalities are detected by using this method. Chromosome analysis requires five principal steps: 1) cell culture, 2) harvest of metaphase chromosomes, 3) chromosome preparation, 4) banding and staining using a special protocol, and 5) analysis by light microscopy or karyotype assisted computer analysis (Fig. 1). The discovery that colchicine (or colcemid) pretreatment resulted in mitotic arrest and that treatment of arrested cells with a hypotonic solution improved the yield and quality of metaphases spreads. Therefore, counted and analyzed individual chromosomes in human cells are then possible. Chromosome analysis provides an overview of all chromosomal aberrations in a single tumor cell.
Fig. 1.

Protocol for the preparation of a karyotype from a leukemic patient.
1. Technology considerations
Chromosomal studies of malignancies pose a particular technical challenge. As the results are so unpredictable, there is no a single technique that can be guaranteed to work consistently and reliably. Therefore, every laboratory should adopt a slight variation of the basic protocol. In addition to setting up a bone marrow suspension cell culture of hematologic neoplasms, peripheral blood can also be investigated, if it contains more than 10% circulating blasts or immature cells. In addition, a lymph node biopsy is required, as lymph nodes are the preferred tissue for studies of most lymphomas [8]. The duration of the cell cycle in malignant cells varies greatly among patients; a range of 16 hr to 292 hr was obtained in a series of 37 patients with AML [9]. Therefore, one of the most significant factors in obtaining a successful result is setting up multiple cultures to maximize the chances of obtaining optimal malignant cell divisions: 1) direct harvest of bone marrow cells, 2) overnight culture, and 3) overnight culture with synchronization (by blocking at S-phase of the cell cycle) [10].
High-resolution banding of long chromosomes with good morphology can be achieved by applying synchronization techniques [10]. It enables the identification of subtle structural chromosome aberrations that are commonly found in malignant cells. However, it has been reported that fluorodeoxyuridine synchronization cultures are inferior to short-term cultures for chromosome analysis in ALL [11]. ALL is a frustrating disease for most cytogeneticists, as it has several technical challenges, including frequent poor chromosome morphology, low mitotic index, and samples that have a marked tendency to clot during harvest.
2. Acquired abnormal clones versus constitutional chromosomal anomalies or cytogenetic heteromorphisms
In cases where the cytogenetic analysis reveals only abnormal metaphases, especially in those with a balanced translocation, it may be necessary to rule out a constitutional chromosomal anomaly. In addition, the constitutive heterochromatic regions just below the centromeres of chromosomes 1, 9, and 16 and the telomeric end of the long arm of the Y-chromosome can vary widely in size among individuals and can sometimes appear abnormal (Fig. 2A). Constitutional chromosomal anomalies are best differentiated from differentiate acquired abnormal clones through cytogenetic examination of phytohemagglutinin (PHA)-stimulated lymphocytes from peripheral blood in disease remission. The Cytogenetics Resource Committee (CyRC) of the College of American Pathologists (CAP)/American College of Medical Genetics & Genomics (ACMG) has asked participants not to include heteromorphisms when responding to a challenge of the cytogenetics survey. However, the guidelines for reporting heteromorphisms have not been standardized. Of 223 cytogenetic laboratories, 136 (61%) stated that they would include selected heteromorphism information identified by routine G-banding in a clinical cytogenetic report in a CyRC survey on cytogenetic heteromorphisms [12]. The majority of the clinical cytogenetics surveyed would not include the more common chromosomal variants (such as prominent short arms, large or double satellites, and increased stalk length or double stalks on acrocentric chromosomes, and long arm heterochromatin variations of chromosomes 1, 9, 16, and Y), but would report most pericentric inversions [such as inv(9)(p11q13); Fig. 2B] and other rare heteromorphisms [12].
Fig. 2.

Partial karyotype showing normal variants and chromosome abnormalities of leukemia. (A) Normal variant with 1qh+ (arrow), (B) Normal variant with inv(9)(p11q13) (arrow), (C) dup(1)(q21q32) chromosome with duplication of a 1q21-q32 chromosomal fragment (arrow), (D) trp(1)(q21q32) chromosome with triplication of a 1q21-q32 chromosomal fragment (arrow), (E) del(7)(q) chromosome with terminal deletion ofa7q22-qter chromosomal fragment (arrow), (F) del(5)(q13q33) chromosome with an interstitial deletion of a 5q13-q33 chromosomal fragment (arrow), (G) der(1)t(1;1)(p35;q25) chromosome with loss of a 1p35-pter chromosomal fragment and duplication of a 1q25-qter chromosomal fragment (arrow), (H) i(17)(q10) chromosome with a loss of the whole short arm and duplication of the whole long of chromosome 17 (arrow), (I) t(9;22)(q34;q11.2), a balanced translocation between 9q34 and 22q11.2 (arrows), (J) t(2;9;22)(q37;q34;q11.2), a balanced three-way translocation between 2q37, 9q34, and 22q11.2 (arrows), (K) der(1;7)(q10;p10), a centric fusion of the whole arms of 1q and 7p with a gain of 1q and a loss of 7q (arrow), (L) der(5)ins(5;?)(q13;?) chromosome with an unknown chromosomal fragment inserted into 5q13 (arrow), (M) inv(16)(p13q22) chromosome with G-banding (arrow), (N) inv(16)(p13q22) chromosome with R-banding (arrow).
Constitutional pericentric inversion of chromosome 9 [inv(9)(p11q13)] occurs in 0.8% to 2% of the normal population and has long been considered a normal variant. Whether constitutional inv(9) is a predisposing factor for cancer remains controversial. We were the first group to document a case of an acquired pericentric inv(9) in essential thrombocythemia [13]. Since then, several other hematological malignancies with acquired inv(9) have been reported. However, inv(9) is not over-represented in patients with hematological malignancy; therefore, there is no evidence to suggest that the presence of constitutional inv(9) increases the risk of hematological malignancy [13, 14].
Furthermore, some abnormalities are both acquired and inherited. For example, trisomy 21 is common both as an inherited abnormality in Down syndrome and as an acquired abnormality in AML, MDS, and childhood ALL [15, 16, 17]. Trisomy 21 is the second most common trisomy in AML and MDS after trisomy 8 [18]. Therefore, if trisomy 21 is found in a karyotype, it is necessary to consider both inherited and acquired abnormalities. However, most individuals with Down syndrome have characteristic physical features.
3. Mechanisms that generate chromosome abnormalities
1) Net gain and loss of chromosomal material
In general, gain or loss of chromosomal material results in gene amplification or loss of heterozygosity, respectively. Two main classes of cancer-relevant genes, oncogenes and tumor suppressor genes have been recognized as the major pathogenic targets for cancer-associated karyotypic abnormalities. Numerical chromosome aberrations are detected as the sole clonal change in approximately 15% of all cytogenetically abnormal hematological malignancies, and they show substantial variations in frequency among the various disease subgroups [19]. Despite their relatively frequent occurrence, numerical changes, including single autosomal trisomies, have received less attention than structural changes. There are several reasons for this discrepancy. First, the high degree of correlation between cytogenetic changes and morphology that is observed with structural changes like translocation is lacking for numerical aberrations. Second, the role that numerical changes play in leukemogenesis tends to be obscured. Third, the molecular consequences of whole chromosome gains and losses are not yet clear.
Net gain of chromosomal material may be caused by duplication (Fig. 2C) and triplication (Fig. 2D) of particular chromosomal segments or regions, which may also lead to an unbalanced gene product. Mis-segregation of entire chromosomes in cell division may also result in trisomies or more extensive polysomies [20]. A single autosomal trisomy is a common numerical cytogenetic abnormality in hematological malignancies, and it shows a predilection for myeloid disorders [15, 16, 18, 21]. However, gain of a sex chromosome as the sole acquired abnormality is very rare in hematological malignancies [22]. Interestingly, hand-mirror cell morphology has been described in AML with trisomy 13 [23], particularly in AML-M0 and -M1, and it may not be detected in the more differentiated subtypes of AML [24]. In addition, chromosome gain is nonrandom in childhood ALL, and eight chromosomes (+4, +6, +10, +14, +17, +18, +21, and +X) account for almost 80% of all gains. However, trisomy 4 has also been reported as the sole karyotypic abnormality in ALL [25].
Net loss of chromosomal material may be caused by loss of an entire chromosome (monosomy) [26, 27, 28], deletions of particular chromosomal segments [29] (Fig. 2E, F), unbalanced translocations (Fig. 2G), or isochromosomes (Fig. 2H). Terminal deletions results from a single break in the chromosome arm, with loss of the distal segment (Fig. 2E). Interstitial deletions emerge when two breaks occur within the same chromosome arm and the intervening segment is lost (Fig. 2F).
2) Chromosomal rearrangements
A chromosome rearrangement occurs when a piece of one chromosome breaks off and attaches to another chromosome. A gene fusion can be created when the translocation joins two other separate genes, the occurrence of which is common in cancer. It can be a balanced translocation, unbalanced translocation, insertion, or inversion.
In a balanced translocation, pieces of chromosomes are rearranged, but no genetic material is gained or lost in the cell (Fig. 2I). A balanced translocation can involve more than two chromosomes and form a complex variant translocation [30, 31] (Fig. 2J). In CML, a complex, 3-way variant translocation, involving 3 chromosomes, often occurs as a single-step process with 3 breakpoints and no reciprocal ABL1-BCR fusion [32] (Fig. 2J). Interestingly, a complex 3-way translocation resulting from 4 breakpoints, 2-step process and a reciprocal gene fusion on the third chromosome has been detected in acute promyelocytic leukemia and CML, using dual color dual fusion translocation FISH probes [32, 33].
In an unbalanced translocation, the exchange of chromosome material is unequal, resulting in extra or missing chromosomal fragments [34, 35]. Derivative (1;7)(q10;p10) with an unbalanced whole-arm translocation is a recurrent cytogenetic abnormality in myeloid disorders [36, 37] (Fig. 2K). It had long been regarded as a poor prognostic indicator in MDS and AML [38], until the publication of a large study on the clinicopathological features of myeloid neoplasms with this karyotypic abnormality [39]. It has been proposed that myeloid neoplasms with der(1;7)(q10;p10) may not have a homogeneously favorable clinical behavior compared to MDS, which has known poor-risk cytogenetics [37].
An insertion is a structural rearrangement, in which part of a chromosome is typically interstitially repositioned into a different area of the karyotype (Fig. 2L). The insertion can be cryptic, and at the gene level, we have previously reported a case of childhood CML with a cryptic insertion of BCR at 9q34 and morphologically normal chromosomes 9 and 22 on G-banding [40]. FISH confirmed the presence of the BCR/ABL1 gene fusion on chromosome 9 in metaphase chromosomes. Therefore, in clinical practice, atypical genetic test results should not be interpreted in isolation and should be integrated with information gathered through different genetic studies.
A chromosomal aberration, in which a segment of a chromosome is reversed in orientation but not relocated, is called an inversion. Inversion of chromosome 16 [inv(16)(p13q22)] is the most common chromosomal inversion observed in leukemia (Fig. 2M). It has been detected in approximately 5% of de novo AML cases, which are mostly classified as the M4Eo subtype, and is associated with a relatively favorable outcome. In the AML M4Eo subgroup, inv(16) is much more prevalent (88%) [41]. However, ethnic differences have been reported, including a very low prevalence of inv(16)(p13q22) abnormalities in two Chinese AML cohorts [41, 42]. Interestingly, R-banding is unsuitable for detecting this inv(16)(p13q22) aberration (Fig. 2N) [42], and it is far easier to recognize by G-banding. Therefore, FISH, reverse-transcription (RT)-PCR, and Southern blot analyses are reliable tools for detecting masked inv(16).
MOLECULAR CYTOGENETICS
Molecular cytogenetics involves the use of a series of techniques referred to as FISH, in which DNA probes are labeled with different colored fluorescent tags to visualize one or more specific regions of the genome (Fig. 3). It is used as a rapid, sensitive test for the detection of cryptic or subtle chromosomal changes. Furthermore, it can be used to detect genetic alterations in non-dividing cell populations, and it is a convenient method to support the practice of personalized medicine. However, FISH assays are still hampered by reagent costs, which prevent its adoption by large-scale oncological screening.
Fig. 3.

FISH protocol. It includes sample pretreatment, denaturation of probe and sample, hybridization, post-hybridization washing, and fluorescent signal detection.
1. Methodology
The standard FISH protocol is illustrated in Fig. 3. Briefly, it includes five steps: 1) sample pretreatment, 2) denaturation of probe and sample, 3) hybridization of probe to target cells or metaphase spreads (annealing), 4) post-hybridization washing, and 5) detection using a simple epifluorescence microscope with appropriate filter sets. When a FISH test is initially implemented, the assay performance characteristics assessed should include sensitivity, accuracy, precision, and specificity [43]. The upper cutoff for normal results in a FISH assay can be determined by calculating the 95% confidence interval for probe signal patterns detected in normal control samples that are representative of the sample type to be analyzed.
ACMG sets internationally accepted standards for FISH analysis to ensure that FISH results are clear and interpretable [44]. Furthermore, ongoing monitoring of interobserver reproducibility, accomplished in part by having two laboratory personnel read every case, can help detect changes in assay performance or loss of consistency in applying scoring criteria. The standard FISH nomenclature has been simplified and expanded in the latest edition of the International System for Human Cytogenetic Nomenclature, ISCN 2013 [45]. However, since the use of the full FISH ISCN is likely to make it more difficult for physicians to understand, it is not recommended by the European Myeloma Network [46].
2. Selection of FISH probes for use in oncology
There are three broad types of FISH probes used in clinical genetics laboratories, each with a different application: 1) whole-chromosome painting (WCP) probes for deciphering cytogenetic aberrations [8, 34, 47, 48]; 2) repetitive sequence probes for chromosome enumeration [21, 25, 26, 49, 50, 51]; and 3) locus-specific identifier (LSI) probes for gene fusions [17, 33, 52], gene deletions [35, 53, 54] or duplications [55]. These probes can also be used in various combinations when investigating complex chromosome abnormalities [56, 57].
WCP probes are designed to mark the entire chromosome of interest and are useful for deciphering cytogenetic aberrations that are difficult to resolve on morphological grounds, such as marker chromosomes of uncertain nature or complex changes. However, the use of WCP probes in interphase cells is very limited, as the chromosomes are dispersed in the cell nucleus during interphase and they do not form discrete units.
Centromeric enumeration probes (CEPs) hybridize to alpha (or beta) satellite repeat sequences within the specific centromeric regions of each chromosome and are used for chromosomal enumeration. Interphase FISH analysis using dual color CEPs for chromosomes X and Y is an efficient method to predict relapse or failure of engraftment in patients after sex-mismatched bone marrow transplantation (Fig. 4A) [58].
Fig. 4.

Interphase FISH images. (A) Interphase FISH using dual color centromeric-specific probes for chromosomes X (red) and Y (green) to determine the proportion of donor cells in the peripheral blood of the recipient (XY, arrow; XX, block arrow). (B) Interphase FISH using a dual color dual fusion BCR-ABL1 translocation probe, showing a 2G2R pattern in a normal cell (block arrow) and 1G1R2F in a Philadelphia-positive cell (arrow). (C) Interphase FISH using a dual color breakapart MLL translocation probe, showing an MLL split signal (distal MLL region, arrow; proximal MLL region, block arrow), and indicating MLL gene rearrangement. (D) MYC amplification in a neuroblastoma (arrow) and a normal cell with a 2G2R pattern (block arrow). The MYCN gene is labeled with a green fluorochrome, whereas the centromeric probe for chromosome 2 is labeled with a red fluorochrome.
There are two main LSI FISH probe systems for the detection of gene rearrangements in oncology, dual-color translocation probes and dual-color break-apart probe. The dual-color translocation probes are designed to detect chromosomal translocations involving know partner genes, such as BCR-ABL1 that result from t(9;22)(q34;q11.2) in CML (Fig. 4B). Furthermore, dual-color break-apart probes are useful for detecting chromosomal translocations that involve genes with unknown or multiple translocation partners, such as MLL, ALK, and RARα (Fig. 4C). Break-apart translocation FISH probes conveniently provide important information on the presence of gene rearrangements, although they are unable to identify the specific partner gene. Recently, with the use of long distance inverse-polymerase chain reaction (LDI)-PCR, it has become possible to identify unknown translocation partners and to map breakpoints at the base-pair level. LDI-PCR requires only approximate sequence information for one partner, rendering it ideal for use in combination with FISH to extend and refine cytogenetic breakpoint data [59]. LSI FISH probes have also been used to identify the origin of the amplified DNA that constitutes homogeneously staining regions (HSRs) and double minutes (DMs), which occur in a variety of tumor cells (Fig. 4D) [60].
3. Advances in FISH technology
Numerous methodological advances in molecular cytogenetic technology were initiated in the early 1990s, including comparative genomic hybridization (CGH) [54], spectral karyotyping (SKY) [62], multicolor FISH (MFISH) [63], multicolor banding (mBAND) [64], and array CGH (aCGH) [65]. All of these cytogenetic techniques add colors to the monotonous world of conventional banding. Two multicolor fluorescence technologies have been introduced, MFISH [63] and SKY [62]. These technologies are based on simultaneous hybridization of 24 chromosome-specific composite probes. This technique is suitable for the identification of subtle chromosomal aberrations that include an unidentified chromosome (marker chromosome) and an unbalanced chromosomal translocation. Regarding probe design, these chromosome-painting probes are generated from flow-sorted human chromosomes. Unique chromosome-specific colors are produced by labeling each chromosome library with either a single fluorochrome or specific combinations of multiple fluorochromes.
mBAND has been developed to facilitate the identification of intrachromosomal rearrangements and to map the exact breakpoint by using human overlapping microdissection libraries that are differentially labeled [64]. The color bands have great value for delineating intrachromosomal exchanges, such as inversions, deletions, duplications, and insertions [6].
CGH is based on quantitative dual-color FISH along each chromosome [61]. CGH can be used to detect genetic imbalances in test genomes, and to determine the chromosomal map positions of gains and losses of entire chromosomes or chromosomal subregions present in normal reference metaphase preparations. A distinct advantage of CGH is that tumor DNA is the only requirement for this analysis. Thus, archived, formalin-fixed and paraffin-embedded tissues can be used as well. CGH is useful for cancer research, especially for determining the low mitotic index of malignant cells with poor chromosome morphology and resolution [66, 67, 68].
Tremendous technical advances in cytogenetics have changed the approaches used for clinical diagnostics and research, in particular, the development of array CGH (aCGH) technology for "molecular karyotyping" with a resolution of 100 kb to 1 Mb [65]. aCGH greatly improves the resolution of this technique by substituting the hybridization targets, the metaphase chromosome spread, with genomic segments spotted in an array format. The complexity of the genomic aberrations in most human tumors hampers delineation of the genes that drive the tumorigenic process. Interestingly, cognate mouse models have been shown to recapitulate these genetic alterations with unexpected fidelity [69]. These results indicate that cross-species aCGH analysis is a powerful strategy to identify the responsible genes and assess their oncogenic capacity in the appropriate genetic context [69]. Recently developed genomic microarray methodologies, including aCGH and single-nucleotide polymorphism (SNP)-based arrays, are innovative methods that provide genomic data for multiple neoplastic disorders [70, 71, 72]. In addition, these methods can reveal additional important information about the genetics of specific disorders, such as leukemia with normal cytogenetic and FISH analyses [73, 74]. In 2013, ACMG developed professional standards and guidelines to assist clinical laboratories in the validation, consistent use, and the interpretation and reporting of results from these microarray methodologies [75].
CANCER CYTOGENETICS RESOURCES
1. The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer
The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman) includes a comprehensive database of all published neoplasia-associated karyotypes and their corresponding gene fusions. The available information on chromosome abnormalities in human neoplasias has steadily increased over the past three decades. The data in this database have been manually culled from the literature by Felix Mitelman, Bertil Johansson, and Fredrik Mertens. The total number of tumor cases, in which clonal cytogenetic aberrations have been reported has reached 64,319, and the database was updated with the addition of 2,072 chimeric fusion genes in May 2014 [76].
2. The Atlas of Genetics and Cytogenetics in Oncology and Haematology
The Atlas of Genetics and Cytogenetics in Oncology and Haematology (http://atlasgeneticsoncology.org/) [77], which was established in 1997, is a peer-reviewed, open access, online journal, encyclopedia, and database that is devoted to genes, cytogenetics, and clinical entities in cancer and cancer-prone diseases. Approximately 2,200 authors have contributed to the Atlas so far, making 2,167 review articles available. The Atlas contains peer-reviewed articles on 1,135 genes, 503 leukemia entities, 177 solid tumors, and 104 cancer-prone inherited diseases. It also contains "automated cards" on 8,190 other genes that are potentially implicated in cancer [78].
3. Cytogenetic nomenclature
In the medical genetics community, interpretation and scientific communication is often facilitated by universally accepted nomenclature with precisely defined terms and syntax conventions that minimize complexity and add precision to the process. Cytogenetic nomenclature is based on the reports of an international committee that was established in 1960, known as the International System for Human Cytogenetic Nomenclature (ISCN) [79]. The nomenclature is updated periodically, most recently in 2013 with expanded guidelines for cancer cytogenetics, FISH, and microarray [45]. In cancer cytogenetic, an abnormal cytogenetic clone is defined as a population of cells with the same chromosome complement that is derived from a single progenitor. A clone must have at least two cells with the same aberration if the aberration is a chromosome gain or a structural rearrangement. If the abnormality is loss of a chromosome, the same loss must be present in at least three cells to be accepted as clonal. However, in the current version of the ISCN (2013), two cells with identical losses of one or more chromosomes and the same structural aberration(s) may be considered clonal and newly included [45]. However, whether two cells with the same loss of a single chromosome or one cell with a gain of a single chromosome in a composite karyotype should be counted and included in the size of the clone has not been mentioned in the current nomenclature system.
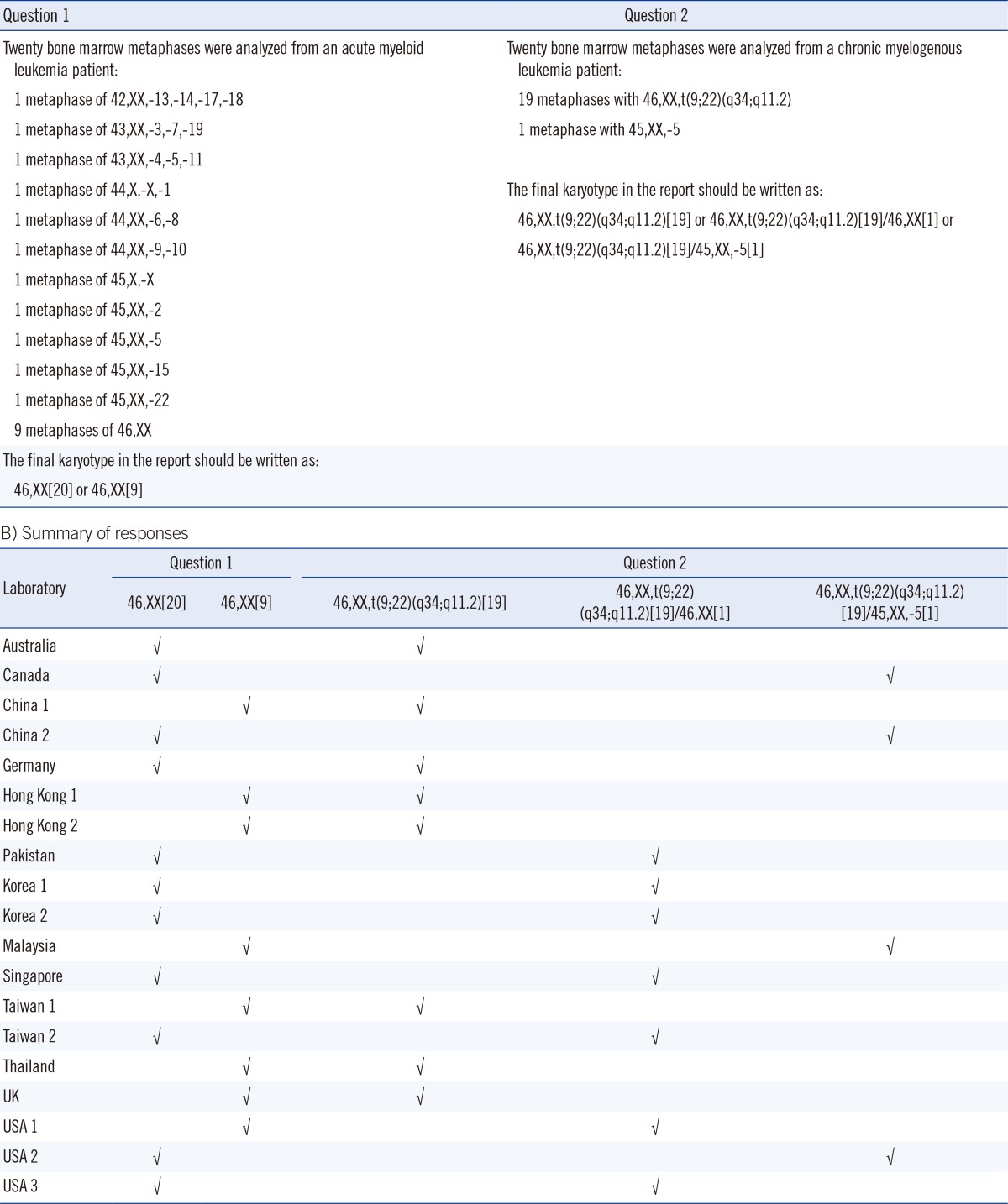
Recently, we conducted a pilot survey of the reporting practices for the random loss or gain of a single chromosome in clinical cytogenetics reports. The questionnaire, which consisted of two questions, was sent to 19 cancer cytogenetic laboratories in 13 counties and cities, and we invited them to report to us using their usual reporting system (Table 1). In question 1, participants were presented with a bone marrow sample from an AML patient with non-clonal (random) loss of chromosome(s), and asked whether these metaphases should be considered as normal and included in the clone size of the normal karyotype. In question 2, participants were presented with a marrow blood sample from a CML patient with 19 metaphases of t(9;22)(q34; q11.2) and one metaphase with only -5. We asked whether the normal metaphase with a loss of a chromosome 5 should be reported in final karyotype in addition to the 19 metaphases of t(9;22)(q34;q11.2). To our surprise, the responses were not unanimous. For question 1, 11 of the 19 laboratories (57.9%) reported 46,XX[20], and the remaining 8 (42.1%) reported 46, XX[9] (Table 1). Therefore, 57.9% of the cytogenetic laboratories assumed that metaphases with non-clonal loss of chromosomes were normal, and they included all these metaphases in the clone size of normal clone. Most laboratory accreditation bodies require the "total number of metaphases analyzed" in the final report. Thus, for the 42.1% of cytogenetic laboratories that reported 46,XX[9], there is a general understanding by the clinicians that the patient has 11 metaphases with non-clonal loss or gain of chromosomes among the 20 metaphases analyzed. For question 2, 8 of the 19 laboratories (42.1%) reported 46, XX,t(9;22)(q34;q11.2) [19] and omitted the only metaphase with -5; 7 laboratories (36.8%) reported 46,XX,t(9;22)(q34;q11.2)[19]/46,XX[1] and counted the only metaphase with -5 as a normal clone; and 4 laboratories (21.1%) reported 46,XX,t(9;22)(q34;q11.2)[19]/45,XX,-5 [1] and simply wrote down the only metaphase with -5 in the final karyotype (Table 1). Taken together, the ISCN standing committee should continue to discuss such discrepancies and make efforts to align the reporting system used by cytogenetic laboratories [80].
Table 1.
Survey on the International System for Human Cytogenetic Nomenclature (ISCN) for non-clonal loss of chromosomes
Interestingly, a review report of the CAP FISH proficiency surveys between 1997 and 2001 showed that syntax errors were far more common than diagnostic errors [81]. In addition, the syntax errors that are common in the proficiency surveys are also common in FISH surveys. Recently, since the full FISH ISCN is likely more difficult for physicians to understand, the use of FISH ISCN is not recommended by the European Myeloma Network [46]. Therefore, continuous improvement of the nomenclature guidelines to address some of these issues and identify the cause of these variations is essential [78, 82].
FUTURE PROSPECTS AND CONCLUDING REMARKS
Over the past five decades, innovative technical advances in the field of cancer cytogenetics have greatly enhanced the detection of chromosomal alterations and have facilitated the research and diagnostic potential of chromosomal studies in malignancies. Karyotyping of a single cell is still the easiest way to understand the relationship between clonal evolution and disease progression. The use of advanced FISH techniques allows for the identification of chromosomal alterations that are unresolved by karyotyping. Recently, DNA microarray technologies have been developed that provide a high-resolution view of the whole genome, which may yield massive amounts of new information on cancer genomics. Strikingly, cancer cytogenetics not only provides key information to improve the care of patients with leukemia and various cancers but also acts as a guide to identify the genes responsible for the development of these neoplastic states.
Ongoing research and development on automated cytogenetics procedures includes: an automated metaphase harvester, automated metaphase spreader, high-throughput slide stainer, high-throughput metaphase finder, automated karyotyper, automated FISH pretreatment processor, automated FISH spot counting, and computerized sample tracking system. However, microscopy is still an essential technique for a final check of the chromosomes. Obviously, cytogenetic laboratories need to obtain more capital investment to keep up with the fast advancing and robotic technologies, since equipment costs are currently high.
The main goal of the FISH laboratory is to identify the techniques that are most useful and informative for a particular study and perform thorough analyses to arrive at an interpretation that is useful for research and diagnostic purposes. The unique characteristics of peptide nucleic acids (PNAs) allow for the use of shorter probes, which combined with the hydrophobic nature of the peptide backbone, enables PNA probes to more easily traverse the hydrophobic core of cell membranes. A staining efficiency of nearly 100% can be achieved by using telomere probes based on PNA [83]. Telomere length can be measured by quantitative FISH in human [84] and other vertebrate cells [85]. Recently, the inherent flexibility of de novo synthesized oligonucleotide libraries was shown to have a powerful advantage that could aid in the visualization of high-resolution fine-scale genomic structure [86].
In the future, targeted therapies will find broader applications in leukemia and cancer. Personalized oncology, in addition to FISH and drug target information for treatment decision making, other aspects include the application of genetic markers for patient risk stratification. This is particularly relevant to CLL [87] and multiple myeloma. Since the neoplastic population in both CLL and multiple myeloma is mitotically inactive, the use of interphase FISH for the detection of genetic abnormalities plays a significant role in the prognostication and risk stratification of these disorders [88]. The identification or selection of malignant cells by morphology, immunophenotyping, or through sorting of plasma cells is required before FISH probes can yield reliable results [89]. In addition, although personalized genomic medicine in the clinic may be very attractive, there is a need for succinct clinicopathological correlation and the rational use of faster, more cost-effective methods among the large array of genomic tests available for drug-target selection. Recently, the power and applicability of whole-genome sequencing for the diagnosis of leukemia patients with cryptic gene fusion has demonstrated, and it hugely affected the clinical management and prognosis of these patients. However, leukemic patients with cryptic gene rearrangements can often be diagnosed without whole-genome analysis - a costly, time consuming, and highly specialized procedure.
Conventional cytogenetics is now complemented by FISH and molecular biology. A FISH-negative cryptic PML/RARα rearrangement detected by long-distance PCR and sequencing analyses has also been reported [90]. Therefore, FISH analysis should be conducted, if the morphologic, cytogenetic, and molecular findings are inconsistent. It is envisaged that efforts made towards the characterization of molecular defects in neoplasms will ultimately be translated into better clinical outcomes for patients. Taken together, the morphologic, karyotyping, FISH, and molecular features should all be considered to obtain accurate diagnoses of malignancies. This highlights the clinical importance of a combined modality approach for the accurate diagnosis and classification of cancers.
Acknowledgements
The author thanks Eden Wan for drawing Figs. 1 and 3, Sophia Ho for expert clerical assistance, Dr. Huifang Huang of Fujian Medical University (China) for providing the R-banded image of inv(16) shown in Fig. 2N, and all of the laboratories who participated in the ICSN nomenclature survey presented in Table 1.
Footnotes
No potential conflicts of interest relevant to this article were reported.
References
- 1.Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497. doi: 10.1126/science.144.3623.1229. [DOI] [PubMed] [Google Scholar]
- 2.Rowley JD. A new consistent chromosomal abnormality in chronic myelogeneous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 3.de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D, et al. A cellular oncogene is translocated to Philadelphia chromosome in chronic myelocytic leukemia. Nature. 1982;300:765–767. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 4.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasm and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 5.Manuelidis L, Langer-Safer PR, Ward DC. High-resolution mapping of satellite DNA using biotin-labeled DNA probes. J Cell Biol. 1982;95:619–625. doi: 10.1083/jcb.95.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan TS, Ma ES. Molecular cytogenetics: an indispensable tool for cancer diagnosis. Chang Gung Med J. 2012;35:96–110. doi: 10.4103/2319-4170.106161. [DOI] [PubMed] [Google Scholar]
- 7.Wan TS, Ma ES. The role of FISH in hematologic cancer. Int J Hematol Oncol. 2012;1:71–86. [Google Scholar]
- 8.Wan TS, Ma SK, Chan GC, Ching LM, Ha SY, Chan LC. Complex cytogenetic abnormalities in T-lymphoblastic lymphoma: resolution by spectral karyotyping. Cancer Genet Cytogenet. 2000;118:24–27. doi: 10.1016/s0165-4608(99)00174-0. [DOI] [PubMed] [Google Scholar]
- 9.Raza A, Maheshwari Y, Preisler HD. Differences in cell cycle characteristics among patients with acute nonlymphocytic leukemia. Blood. 1987;69:1647–1653. [PubMed] [Google Scholar]
- 10.Yunis JJ. Comparative analysis of high-resolution chromosome techniques for leukemic bone marrows. Cancer Genet Cytogenet. 1982;7:43–50. doi: 10.1016/0165-4608(82)90106-6. [DOI] [PubMed] [Google Scholar]
- 11.Garipidou V, Secker-Walker LM. The use of fluorodeoxyuridine synchronization for cytogenetic investigation of acute lymphoblastic leukemia. Cancer Genet Cytogenet. 1991;52:107–111. doi: 10.1016/0165-4608(91)90060-8. [DOI] [PubMed] [Google Scholar]
- 12.Brothman AR, Schneider NR, Saikevych I, Cooley LD, Butler MG, Patil S, et al. Cytogenetic Heteromorphisms. Survey results and reporting practices of Giemsa-band regions that we have pondered for years. Arch Pathol Lab Med. 2006;130:947–949. doi: 10.5858/2006-130-947-CHSRAR. [DOI] [PubMed] [Google Scholar]
- 13.Wan TS, Ma SK, Chan LC. Acquired pericentric inversion of chromosome 9 in essential thrombocythemia. Hum Genet. 2000;106:669–670. doi: 10.1007/s004390050041. [DOI] [PubMed] [Google Scholar]
- 14.Lee SG, Park TS, Lim G, Lee KA, Song J, Choi JR. Constitutional pericentric inversion 9 and hematological disorders: a Korean tertiary institution's experience over eight years. Ann Clin Lab Sci. 2010;40:273–277. [PubMed] [Google Scholar]
- 15.Wan TS, Au WY, Chan JC, Chan LC, Ma SK. Trisomy 21 as the sole acquired karyotypic abnormality in acute myeloid leukemia and myelodysplastic syndrome. Leuk Res. 1999;23:1079–1083. doi: 10.1016/s0145-2126(99)00117-4. [DOI] [PubMed] [Google Scholar]
- 16.Wan TS, Ma SK, Au WY, Liu HS, Chan JC, Chan LC. Trisomy 21 and other chromosomal abnormalities in acute promyelocytic leukemia. Cancer Genet Cytogenet. 2003;140:170–173. doi: 10.1016/s0165-4608(02)00684-2. [DOI] [PubMed] [Google Scholar]
- 17.Ma SK, Wan TS, Cheuk AT, Fung LF, Chan GC, Chan SY, et al. Characterization of additional genetic events in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion: a molecular cytogenetics study. Leukemia. 2001;15:1442–1447. doi: 10.1038/sj.leu.2402202. [DOI] [PubMed] [Google Scholar]
- 18.Ma SK, Wan TS. Single autosomal trisomy in acute myeloid leukemia and myelodysplastic syndrome. Curr Genomics. 2000;1:153–173. [Google Scholar]
- 19.Heim S, Mitelman F. Numerical chromosome aberrations in human neoplasia. Cancer Genet Cytogenet. 1986;22:99–108. doi: 10.1016/0165-4608(86)90169-x. [DOI] [PubMed] [Google Scholar]
- 20.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 21.Ma SK, Wan TS, Au EY, Chan LC. Trisomy 5 in two cases of acute monocytic leukemia with hyperdiploid clones. Leuk Res. 1998;22:961–964. doi: 10.1016/s0145-2126(98)00095-2. [DOI] [PubMed] [Google Scholar]
- 22.Wan TS, Yip SF, Yeung YM, Chan LC, Ma SK. Fatal diffuse alveolar damage complicating acute myeloid leukemia with abnormal eosinophils and trisomy X. Ann Hematol. 2002;81:167–169. doi: 10.1007/s00277-001-0423-6. [DOI] [PubMed] [Google Scholar]
- 23.Mehta AB, Bain BJ, Fitchett M, Shah S, Secker-Walker LM. Trisomy 13 and myeloid malignancy-characteristic blast cell morphology: a United Kingdom Cancer Cytogenetics Group survey. Br J Haematol. 1998;101:749–752. doi: 10.1046/j.1365-2141.1998.00760.x. [DOI] [PubMed] [Google Scholar]
- 24.Ma SK, Wan TS. Blast cell morphology in acute myeloid leukemia with trisomy 13. Leuk Res. 1999;23:767–769. doi: 10.1016/s0145-2126(99)00052-1. [DOI] [PubMed] [Google Scholar]
- 25.Yip SF, Wan TS, Chan LC, Chan GC. Trisomy 4 as sole karyotypic abnormality in acute lymphoblastic leukemia: different clinical features and treatment response between B and T phenotypes? Cancer Genet Cytogenet. 2006;164:94–95. doi: 10.1016/j.cancergencyto.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Au WY, Ma SK, Wan TS, Jim MH, Kwong YL. Subvalvular pulmonary stenosis, demyelination and myelodysplasia with monosomy 7. Leuk Lymphoma. 2002;43:1505–1507. doi: 10.1080/10428190290033530. [DOI] [PubMed] [Google Scholar]
- 27.Au WY, Wan TS, Leung RY, Lie AK. Sequential chronic myelogenous leukemia, B-lineage lymphoma and erythroleukemia with monosomy 7 over 10 years. Leuk Lymphoma. 2012;53:733–735. doi: 10.3109/10428194.2011.628063. [DOI] [PubMed] [Google Scholar]
- 28.Surapolchai P, Ha SY, Chan GC, Lukito J, Wan TS, So CC, et al. Central diabetes insipidus: an unusual complication in a child with juvenile myelomonocytic leukemia and monosomy 7. J Pediatr Hematol Oncol. 2013;35:e84–e87. doi: 10.1097/MPH.0b013e3182580d88. [DOI] [PubMed] [Google Scholar]
- 29.Wan TS, Ma ES, Lam CC, Chan LC, Lee KK, Au WY. Deletion 9q as the sole karyotypic abnormality in myelocytic disorders: a new case of myelodysplastic syndrome and its prognostic implications in acute myelocytic leukemia. Cancer Genet Cytogenet. 2003;145:184–186. doi: 10.1016/s0165-4608(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 30.Wan TS, Chim CS, So CK, Chan LC, Ma SK. Complex variant 15;17 translocations in acute promyelocytic leukemia. A case report and review of three-way translocations. Cancer Genet Cytogenet. 1999;111:139–143. doi: 10.1016/s0165-4608(98)00230-1. [DOI] [PubMed] [Google Scholar]
- 31.So CW, Ma SK, Wan TS, Chan GC, Ha SY, Chan LC. Analysis of MLL-derived transcripts in infant acute monocytic leukemia with a complex translocation (1;11;4)(q21;q23;p16) Cancer Genet Cytogenet. 2000;117:24–27. doi: 10.1016/s0165-4608(99)00136-3. [DOI] [PubMed] [Google Scholar]
- 32.So CC, Wan TS, Yip SF, Chan LC. A dual colour dual fusion fluorescence in situ hybridization study on the genesis of complex variant translocations in chronic myelogenous leukemia. Oncol Rep. 2008;19:1181–1184. [PubMed] [Google Scholar]
- 33.Wan TS, So CC, Hui KC, Yip SF, Ma ES, Chan LC. Diagnostic utility of dual fusion PML/RARα translocation DNA probe (D-FISH) in acute promyelocytic leukemia. Oncol Rep. 2007;17:799–805. [PubMed] [Google Scholar]
- 34.Wan TS, Ma SK, Yip SF, Yeung YM, Chan LC. Molecular characterization of der(15)t(11;15) as a secondary cytogenetic abnormality in acute promyelocytic leukemia with cryptic PML-RARα fusion on chromosome 17q. Cancer Genet Cytogenet. 2000;121:90–93. doi: 10.1016/s0165-4608(00)00234-x. [DOI] [PubMed] [Google Scholar]
- 35.So CC, Wan TS, Ma ES, Chan LC. An unbalanced translocation, der(17)t(1;17)(p13;p11.2), leads to heterozygous loss of TP53 and is associated with clinical evolution in myelodysplastic syndrome. Br J Biomed Sci. 2008;65:36–38. doi: 10.1080/09674845.2008.11978107. [DOI] [PubMed] [Google Scholar]
- 36.Willem P, Pinto M, Bernstein R. Translocation t(1;7) revisited. Report of three further cases and review. Cancer Genet Cytogenet. 1988;36:45–54. doi: 10.1016/0165-4608(88)90074-x. [DOI] [PubMed] [Google Scholar]
- 37.So CC, Ma ES, Wan TS, Yip SF, Chan LC. Clinicopathological features of unbalanced translocation Der(1;7)(q10;p10) in myeloid neoplasms. Leuk Res. 2008;32:1000–1001. doi: 10.1016/j.leukres.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 38.Pedersen B, Nørgaard JM, Pedersen BB, Clausen N, Rasmussen IH, Thorling K. Many unbalanced translocation show duplication of a translocation participant. Clinical and cytogenetic implications in myeloid hematologic malignancies. Am J Hematol. 2000;64:161–169. doi: 10.1002/1096-8652(200007)64:3<161::aid-ajh4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 39.Sanada M, Uike N, Ohyashiki K, Ozawa K, Lili W, Hangaishi A, et al. Unbalanced translocation der(1;7)(q10;p10) defines a unique clinicopathological subgroup of myeloid neoplasms. Leukemia. 2007;21:992–997. doi: 10.1038/sj.leu.2404619. [DOI] [PubMed] [Google Scholar]
- 40.Wan TS, Ma SK, Li CK, Chan LC. Atypical fluorescence in situ hybridization pattern in chronic myeloid leukemia due to cryptic insertion of BCR at 9q34. Leukemia. 2004;18:161–162. doi: 10.1038/sj.leu.2403197. [DOI] [PubMed] [Google Scholar]
- 41.So CC, Wan TS, Chow JL, Hui KC, Choi WW, Lam CC, et al. A single-center cytogenetic study of 629 Chinese patients with de novo acute myeloid leukemia – evidence of major ethnic differences and a high prevalence of acute promyelocytic leukemia in Chinese patients. Cancer Genet. 2011;204:430–438. doi: 10.1016/j.cancergen.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Cheng Y, Wang Y, Wang H, Chen Z, Lou J, Xu H, et al. Cytogenetic profile of de novo acute myeloid leukemia: a study based on 1432 patients in a single institution of China. Leukemia. 2009;23:1801–1806. doi: 10.1038/leu.2009.107. [DOI] [PubMed] [Google Scholar]
- 43.Saxe DF, Persons DL, Wolff DJ, Theil KS Cytogenetics Resource Committee of the College of American Pathologists. Validation of fluorescence in situ hybridization using an analyste-specific reagent for detection of abnormalities involving the mixed lineage leukemia gene. Arch Pathol Lab Med. 2012;136:47–52. doi: 10.5858/arpa.2010-0645-SA. [DOI] [PubMed] [Google Scholar]
- 44.American College of Medical Genetics. Standards and Guidelines for Clinical Genetics Laboratories. https://www.acmg.net/StaticContent/SGs/Section_E_2011.pdf (2009 edition, Revised on Jan 2010)
- 45.Shaffer LG, McGowan-Jordan J, Schmid M, editors. ISCN (2013): An International System for Human Cytogenetic Nomenclature. Basel: S. Karger; 2013. [Google Scholar]
- 46.Ross FM, Avet-Loiseau H, Ameye G, Gutiérrez NC, Liebisch P, O'Connor S, et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica. 2012;97:1272–1277. doi: 10.3324/haematol.2011.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma SK, Wan TS, Au WY, Kwong YL, Chan LC. Atypical chronic myeloid leukemia with der(20)t(17;20)(q21;q13) Cancer Genet Cytogenet. 1999;112:130–133. doi: 10.1016/s0165-4608(98)00265-9. [DOI] [PubMed] [Google Scholar]
- 48.Wan TS, Ma SK, Yip SF, Yeung YM, Chan LC. Two balanced and novel chromosomal translocations in myeloid malignancies.characterization by multiplex fluorescence in situ hybridization. Cancer Genet Cytogenet. 2002;139:52–56. doi: 10.1016/s0165-4608(02)00611-8. [DOI] [PubMed] [Google Scholar]
- 49.Ma SK, Lee AC, Wan TS, Lam CK, Chan LC. Trisomy 8 as a secondary genetic change in acute megakaryoblastic leukemia associated with Down's syndrome. Leukemia. 1999;13:491–492. doi: 10.1038/sj.leu.2401330. [DOI] [PubMed] [Google Scholar]
- 50.Ma SK, Kwong YL, Shek TW, Wan TS, Chow EY, Chan JC, et al. The role of trisomy 8 in the pathogenesis of chronic eosinophilic leukemia. Hum Pathol. 1999;30:864–868. doi: 10.1016/s0046-8177(99)90149-1. [DOI] [PubMed] [Google Scholar]
- 51.Cheung AM, Wan TS, Leung JC, Chan LY, Huang H, Kwong YL, et al. Aldehyde dehydrogenase activity in leukemia blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia. 2007;21:1423–1430. doi: 10.1038/sj.leu.2404721. [DOI] [PubMed] [Google Scholar]
- 52.Meyer C, Kowarz E, Yip SF, Wan TS, Chan TK, Dingermann T, et al. A complex MLL rearrangement identified five years after initial MDS diagnosis results in out-of-frame fusions without progression to acute leukemia. Cancer Genet. 2011;204:557–562. doi: 10.1016/j.cancergen.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Ma SK, Wan TS, Au WY, Fung LF, So CK, Chan LC. Chromosome 11q deletion in myeloid malignacies. Leukemia. 2002;16:953–955. doi: 10.1038/sj.leu.2402442. [DOI] [PubMed] [Google Scholar]
- 54.Cheung AM, Fung TK, Fan AK, Wan TS, Chow HC, Leung JC, et al. Successful engraftment by leukemia initiating cells in adult acute lymphoblastic leukemia after direct intrahepatic injection into unconditioned newborn NOD/SCID mice. Exp Hematol. 2010;38:3–10. doi: 10.1016/j.exphem.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Wan TS, Ma ES, Chen YT. Near-tetraploid acute myeloid leukemia. Br J Haematol. 2011;155:285. doi: 10.1111/j.1365-2141.2011.08774.x. [DOI] [PubMed] [Google Scholar]
- 56.So CC, Yung KH, Chu ML, Wan TS. Diagnostic challenges in a case of B cell lymphoma unclassifiable with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. Int J Hematol. 2013;98:478–482. doi: 10.1007/s12185-013-1414-9. [DOI] [PubMed] [Google Scholar]
- 57.Lee JH, Wan TS, Ha JS. Acute myeloid leukemia with a novel t(8;21) variant: paracentric inversion-associatedins(21;8) Leuk Lymphoma. 2014;55:441–443. doi: 10.3109/10428194.2013.801469. [DOI] [PubMed] [Google Scholar]
- 58.Li YH, Ma SK, Wan TS, Au WY, Fung LF, Leung AY. Lineage-specific differences in telomere length after bone marrow transplantation. Bone Marrow Transplant. 2002;30:475–477. doi: 10.1038/sj.bmt.1703669. [DOI] [PubMed] [Google Scholar]
- 59.Yang JJ, Marschalek R, Meyer C, Park TS. Diagnostic usefulness of genomic breakpoint analysis of various gene rearrangements in acute leukemias: a perspective of long distance- or long distance inverse-PCR-based approaches. Ann Lab Med. 2012;32:316–318. doi: 10.3343/alm.2012.32.4.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wan TS, Ma ES, Chan GC, Chan LC. Investigation of MYCN status in neuroblastoma by fluorescence insitu hybridization. Int J Mol Med. 2004;14:981–987. [PubMed] [Google Scholar]
- 61.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, et al. Compararive genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 62.Schröck E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, et al. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273:494–497. doi: 10.1126/science.273.5274.494. [DOI] [PubMed] [Google Scholar]
- 63.Speicher MR, Gwyn Ballard S, Ward DC. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet. 1996;12:368–375. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- 64.Chudoba I, Plesch A, Lörch T, Lemke J, Claussen U, Senger G. High resolution multicolor-banding: a new technique for refine FISH analysis of human chromosomes. Cytogenet Cell Genet. 1999;84:156–160. doi: 10.1159/000015245. [DOI] [PubMed] [Google Scholar]
- 65.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature Genetics. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 66.Tsao SW, Wong N, Wang X, Liu Y, Wan TS, Fung LF, et al. Nonrandom chromosomal imbalances in human ovarian surface epithelial cells immortalized by HPV16-E6E7 viral oncogenes. Cancer Genet Cytogenet. 2001;130:141–149. doi: 10.1016/s0165-4608(01)00473-3. [DOI] [PubMed] [Google Scholar]
- 67.Hu YC, Lam KY, Law SY, Wan TS, Ma ES, Kwong YL, et al. Establishment, characterization, karyotyping, and comparative genomic hybridization analysis of HKESC-2 and HKESC-3, two newly established human esophageal squamous cell carcinoma cell lines. Cancer Genet Cytogenet. 2002;135:120–127. doi: 10.1016/s0165-4608(01)00580-5. [DOI] [PubMed] [Google Scholar]
- 68.Wong MP, Fung LF, Wang E, Chow WS, Chiu SW, Lam WK, et al. Chromosomal aberrations of primary lung adenocarcinomas in nonsmokers. Cancer. 2003;97:1263–1270. doi: 10.1002/cncr.11183. [DOI] [PubMed] [Google Scholar]
- 69.Peeper D, Berns A. Cross-species oncogenomics in cancer gene identification. Cell. 2006;125:1230–1233. doi: 10.1016/j.cell.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 70.Slovak ML, Bedell V, Hsu YH, Estrine DB, Nowak NJ, Delioukina ML, et al. Molecular karyotypes of Hodgkin and Reed/Sternberg cells at disease onset reveal distinct copy number alterations in chemosensitive versus refractory Hodgkin lymphoma. Clin Cancer Res. 2011;17:3443–3454. doi: 10.1158/1078-0432.CCR-10-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci U S A. 2009;106:12950–12955. doi: 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu L, Slovak ML, Mannoor K, Chen C, Hunger SP, Carroll AJ, et al. Microarray detection of multiple recurring submicroscopic chromosomal aberrations in pediatric T-cell acute lymphoblastic leukemia. Leukemia. 2011;25:1042–1046. doi: 10.1038/leu.2011.33. [DOI] [PubMed] [Google Scholar]
- 73.Kawamata N, Ogawa S, Zimmermann M, Kato M, Sanada M, Hemminki K, et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111:776–784. doi: 10.1182/blood-2007-05-088310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O'Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115:2731–2739. doi: 10.1182/blood-2009-10-201848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cooley LD, Lebo M, Li MM, Slovak ML, Wolff DJ Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders. Genet Med. 2013;15:484–494. doi: 10.1038/gim.2013.49. [DOI] [PubMed] [Google Scholar]
- 76.Mitelman F, Johansson B, Mertens F, editors. Mitelman database of chromosome aberrations in cancer. [Updated on May 2014]. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 77.Atlas of Genetics and Cytogenetics in Oncology and Haematology. [Updated on May 2012]. http://AtlasGeneticsOncology.org. [DOI] [PMC free article] [PubMed]
- 78.Huret JL, Ahmad M, Arsaban M, Bernheim A, Cigna J, Desangles F, et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013;41(Database issue):D920–D924. doi: 10.1093/nar/gks1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaffer LG, Tommerup N, editors. ISCN (2005): an International System for Human Cytogenetic Nomenclature. Basel: S. Karger; 2005. [Google Scholar]
- 80.Mitelman F, Rowley JD. ISCN (2005) is not acceptable for describing clonal evolution in cancer. Genes Chromosomes Cancer. 2007;46:213–214. doi: 10.1002/gcc.20414. [DOI] [PubMed] [Google Scholar]
- 81.Mascarello JT, Cooley LD, Davison K, Dewald GW, Brothman AR, Herrman M, et al. Problems with ISCN FISH Nomenclature make it not practical for use in clinical test reports or cytogenetic databases [corrected] Genet Med. 2003;5:370–377. doi: 10.1097/01.gim.0000086479.80559.ea. [DOI] [PubMed] [Google Scholar]
- 82.Shaffer LG, Slovak ML, Campbell LJ, editors. ISCN (2009): an International System for Human Cytogenetic Nomenclature. Basel: S. Karger; 2009. [Google Scholar]
- 83.Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, et al. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685–691. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- 84.Wan TS, Martens UM, Poon SS, Tsao SW, Chan LC, Lansdorp PM. Absence or low number of telomere repeats at junctions of dicentric chromosomes. Genes Chromosomes Cancer. 1999;24:83–86. doi: 10.1002/(sici)1098-2264(199901)24:1<83::aid-gcc12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 85.Zijlmans JM, Martens UM, Poon SS, Raap AK, Tanke HJ, Ward RK, et al. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci USA. 1997;94:7423–7428. doi: 10.1073/pnas.94.14.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamada NA, Rector LS, Tsang P, Carr E, Scheffer A, Sederberg MC, et al. Visualization of fine-scale genomic structure by oligonucleotide-based high-resolution FISH. Cytogenet Genome Res. 2011;132:248–254. doi: 10.1159/000322717. [DOI] [PubMed] [Google Scholar]
- 87.Stevens-Kroef MJ, van den Berg E, Olde Weghuis D, Geurts van Kessel A, Pfundt R, Linssen-Wiersma M, et al. Identification of prognostic relevant chromosomal abnormalities in chronic lymphocytic leukemia using microarray-based genomic profiling. Mol Cytogenet. 2014;7:3. doi: 10.1186/1755-8166-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- 89.Leung EW, Sin PL, Wan TS. The impact of FICTION on the detection of genetic aberrations in multiple myeloma. J H K Inst Med Lab Sci. 2012;13:1–8. [Google Scholar]
- 90.Kim MJ, Cho SY, Kim MH, Lee JJ, Kang SY, Cho EH, et al. FISH-negative cryptic PML-RARA rearrangement detected by long-distance polymerase chain reaction and sequencing analyses: a case study and review of the literature. Cancer Genet Cytogenet. 2010;203:278–283. doi: 10.1016/j.cancergencyto.2010.08.026. [DOI] [PubMed] [Google Scholar]