

Abstract
Universal stress protein (USP) is a novel target to overcome the tuberculosis resistance. Our present study enlightens the possibilities of some natural polyphenols as an antioxidant for USP. The study has shown some molecular simulations of some selected natural antioxidants with USP. We have considered USP (Rv1636) strain for homology modeling and the selected template was taken for the docking study. Curcumin, catechin, reservetrol has shown ARG 136 (1.8Å) hydrogen bonding and two ionic bonding with carboxyl group of curcumin with LEU 130 (3.3Å) and ASN 144 (3.4Å) respectively. INH was taken for the standard molecule to perform molecular simulation. It showed poor binding interaction with the target, that is, −5.18 kcal, and two hydrogen bonding with SER 140 (1.887Å), ARG 147 (2.064Å) respectively. The study indicates possible new generation curcumin analogue for future therapy to down-regulate USP.
Keywords: Isoniazid, polyphenols, Rv1636, universal stress protein
INTRODUCTION
Mycobacterium latency is responsible for large number of deaths all over the world. Bacterial recrudescence causes disease recurrence and evolution of mutant strains which predisposes the host to suffer undefined resistance toward therapy. A biophysical and biochemical study depicts that universal stress protein (USP) binds with the adenosine triphosphate binding domain of Rv2623 strain.[1] USP was found to proliferate more under oxidative stress and thus induces bacterial cell cycle for further growth. The USP gene is conserved across almost all the bacterial species, however they show immaculate similarity. It encodes either a small USP protein (around 14-15 kDa) or a larger version (around 30 kDa) consisting of two USP domains in tandem or in large proteins where the two peptides are attached as a single functional protein.[2,3] Drumm et al. reported the crystal structure of wild-typeRv2623 at a resolution of 2.9Å thus revealing the 2-fold symmetric dimer form of the protein.[1] The current study depicts that hypothetical in-silico inhibition of USP by natural polyphenols [Figure 1] could be of vital importance and a novel route for targeting multi-drug resistant Mycobacterium tuberculosis species. Docking analysis of all the selected polyphenols with USP (PDB ID: 1TQ8) was carried out and the best compounds were identified on the basis of binding energy, conformational orientation, inhibition constant, etc.
Figure 1.
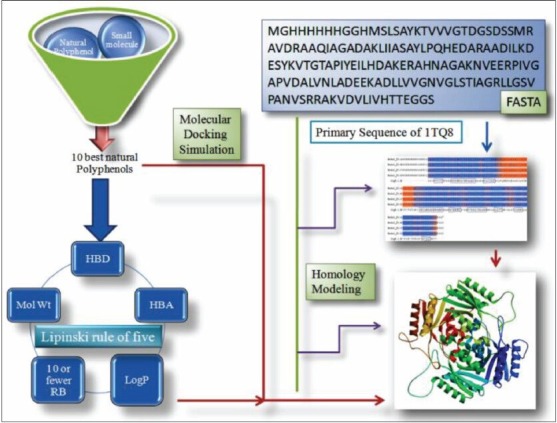
Protocol adopted for identifying polyphenols as universal stress protein inhibitors. Polyphenols from Pubchem server were filtered in terms of their drug likeness and physicochemical properties. Selective ligands were docked to the homology modeled target protein and best activity compounds were identified
MATERIALS AND METHODS
Molecular modelling and docking analysis study was carried out using AutoDock 4.2 MGL Tools (The Scripps Research Institute, USA) and Pymol Molecular Visualization package (Schrödinger).[4,5] Computed Atlas of Surface Topography of Protein (CastP) and FT site open source servers were used to determine the possible binding sites and pocket occupancy of the target protein respectively.[6,7] The polyphenolic ligands were filtered from Pubchem Database.[8] The selected ligands were sketched and converted to their three-dimensional formats using Chemdraw 8.0 and Chemdraw three-dimensional (Cambridge soft. Comp.).[9] SWISS-MODEL server was used to generate the homology model of target protein.[10] BLAST server was used to validate the query sequence of the protein.[11] Procheck server was used to validate the protein by generating Procheck Ramachandran plot (European Bioinformatics Institute, UK).[12] DruLiO tool has been used to calculate the drug-likeness of the molecule set.[13]
Experimental
Homology modeling of universal stress protein
The crystallographic structure of USP was extracted from RCSB Protein Data Bank (www.rcsb.org) identifier (PDB ID: 1TQ8). SWISS-MODEL server was used to model the secondary structure of the protein. FASTA sequences were retrieved from RCSB server to generate the templates. Automated mode was employed in SWISS-MODEL server. This mode automatically selects suitable templates of the query sequence by Blast E-value limit. It has been reported that automated sequence alignment method allows reliable results when the target and templates shared around > 50% identical residues.[13]
Protein preparation
Polar hydrogen atoms were added to the protein. The deletion of the both water molecule and inorganic charges were done to avoid error. Gasteiger charges were computed and added to the macromolecule. Lamarckian Genetic Algorithm was applied for ligand docking. The grid size was set to 54, 48 and 62 along the X-, Y- and Z-axis to recognize the binding site. Spacing was set as 0.381Å. The lowest binding energy conformers were selected out of 20 different conformers for each docking simulation and resultant data was further analyzed. Other miscellaneous parameters were assigned to the default values obtained from the AutoDock 4.2 program.
Ligand preparation
In our present study, polyphenols ligands were extracted from the Pubchem server. The drug likenesses, Lipinski rule of five hypotheses was used to filter the ligands. Best 10 natural polyphenols were selected for the study. Chemdraw 8.0 and Chemdraw three-dimensional were used to prepare all selected ligands. All ligands were converted to protein data bank extension file format (PDBQT). An extended PDB format, termed PDBQT, is used for coordinate files, which includes atomic partial charges and atom types. Torsion angles were calculated to assign the fixable and nonbonded rotations of the molecule.[14]
Pocket validation
The ligand binding region in the macromolecule is often referred as its binding site. The active site is a ligand binding site in the macromolecule with a maximal number of amino acids. Grid generation aids the ligand to specifically recognize its binding region in the receptor. The CastP server (http://sts.bioengr.uic.edu/castp/) was used to validate the receptor active site.
Determination of ligand binding site
All possible binding sites of USP were determined by the open source server called FT site server (http://ftsite.bu.edu/cite). The PDB: 1TQ8 was uploaded into the server and also created the job name for identification of the result.
Molecular docking study
In order to understand the biological activity of a drug molecule and appreciate it as a prototype therapeutic agent, the knowledge of binding selectivity toward its target protein environment is very essential. Docking analysis was performed to understand and correlate their biological efficacy toward the selected binding domain of the USP protein. The AutoDock 4.2-MGL Tools version 1.5.6 software package was utilized to carry out molecular docking analysis. Pymol 1.3 was used to analyze the docking results and bonding interactions.[4,5] Molecular docking simulation was carried out using a described method elsewhere.[15]
RESULTS AND DISCUSSIONS
Homology modeling analysis of universal stress protein
With an identity of 100% and 2.40Å resolution PDB template was identified by the SWISS-MODEL server. The total result contained three distinguished outputs. Sequence similarity, range and coverage of (1tq8.1.B) were found to be 0.59, (15-159), 0.89 respectively. GMQE score and QMEAN4 score were found to be 0.74 and−0.70 respectively [Figure 2] depicts local quality estimation of A, B, C, D chain of USP, which helps to validate the sequence similarity of all homomers of a protein. In our study we used Chain B and Chain D for ligand binding domain. Z-score value and QMEAN value are pointed out in Figure 3. QMEAN scoring function has been used to assess the global and per residue quality. For improved quality, weights of the individual QMEAN terms have been trained specifically for SWISS-MODEL. Top scored model details are tabulated in Table 1. Sequence alignment of 1TQ8.1.B is highlighted in Figure 4. 1TQ8.1.B template structure was used for molecular docking [Figure 5a]. Ramachandran plot was generated to validate the selected protein structure for further study [Figure 5b].
Figure 2.
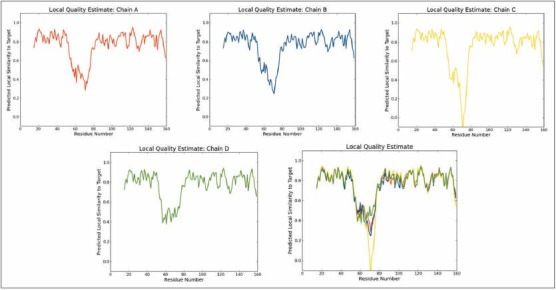
Local quality estimate of A, B, C, D chain of the universal stress protein PDB
Figure 3.
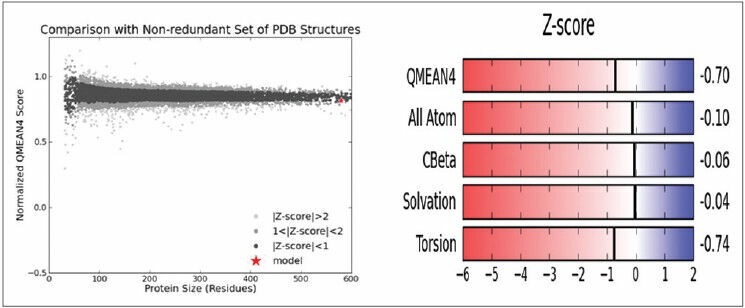
Graphical representation of Z-score value with the nonredundant set of PDB
Table 1.
Homology modeling results of USP
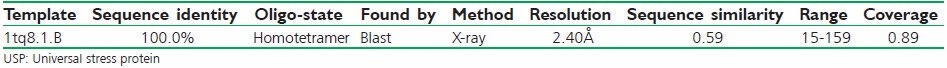
Figure 4.
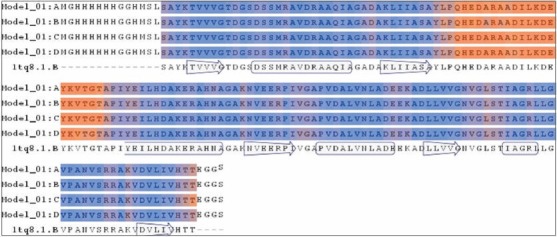
Sequence alignment of 1tq8.1.B template generated by SWISS-MODEL
Figure 5.
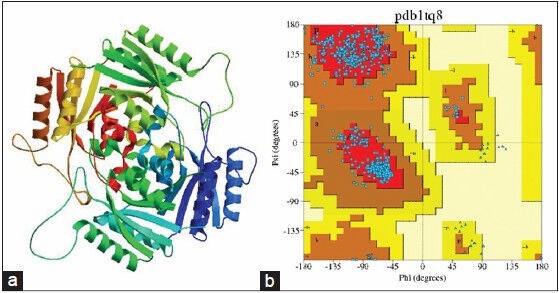
(a) Best scored 1tq8.1.B template protein structure, (b) Ramachandan plot generated by Procheck server
Pocket validation analysis
Computed Atlas of Surface Topography of Protein server (http://sts.bioengr.uic.edu/castp/) helped to recognize the binding pockets into the receptor. They generated several possible pockets by the computation based on the pocket algorithm of the alpha shape theory and its core is the alpha shape API developed in Edelsbrunner's group and the NCSA. They depict the pocket information in respect to the volume and the area of the pocket. The annotated notations were mentioned to identify the pocket region precisely. Pocket information (ID: 30, Job ID: JIDSE10240F) for USP was taken for the preparation of grid.
Ligand binding site analysis
FT site determined the different binding sites of the protein. They have calculated the three different ligand binding domains into the protein. The residue set of the three different sites of the proteins were found. Site 1 annotation USP (1TQ8) contained VAL B 128, GLY B 129, LEU B 130, ALA B 134, GLY B 135, ARG B 136, LEU B 138, GLY B 139, SER B 140, ALA B 143, ASN D 127, VAL D 128, GLY D 129, LEU D 130, GLY D 135, ARG D 136, LEU D 138, GLY D 139, SER D 140, ALA D 143, ASN D 144, ARG D 147. Site B contains VAL A 128, GLY A 129, LEU A 130, ALA A 134, GLY A 135, LEU A 138, GLY A 139, VAL C 128, GLY C 129, LEU C 130, ALA C 134, GLY C 135, LEU C 138, GLY C 139, SER C 140, ALA C 143. Site C contains GLY F 23, THR F 24, ASP F 25, SER F 30, ALA F 50, SER F 51, ALA F 52, ILE F 103, PRO F 107, VAL F 108, LEU F 111, VAL F 125, and GLY F 126. In our present study, Site 1 has been taken for the molecular modelling simulation [Table 2].
Table 2.
Lipinski rule of five values for selected molecule
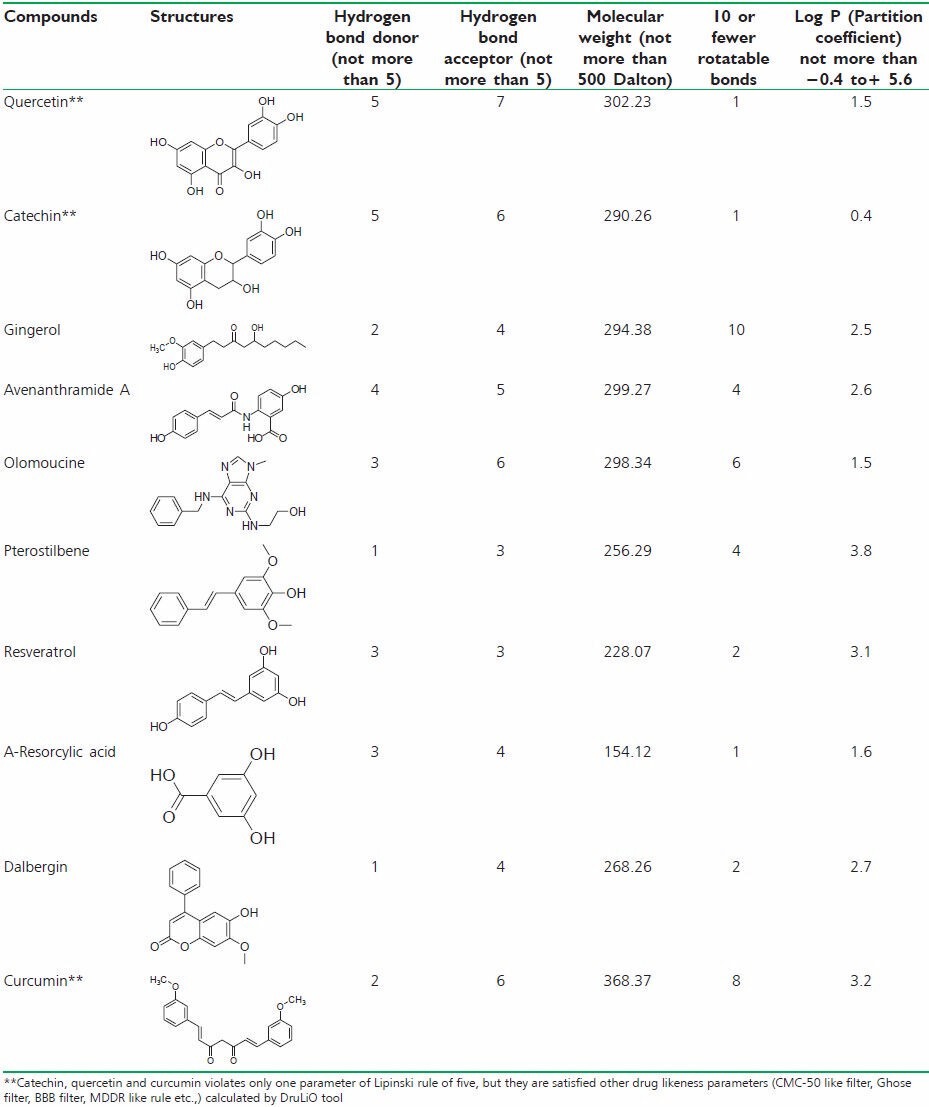
Lipinski rule of five analyses
Lipinski rule of five parameters is important tool for filter the large number of datasets. Hydrogen bond donor, hydrogen bond acceptors, molecular weight, rotatable bonds and partition coefficients are the main parameters to calculate the datasets. The datasets were filtered by Lipinski rule of five tools. Furthermore, the DruLiO tool has been used to calculate the drug-likeness of the selected compounds. The selected compounds have been filtered by CMC-50 such as filter, Ghose filter, BBB filter, MDDR like rule.[16]
Molecular docking analyses
Docking job was undertaken to understand the binding details of the drug in to the target. It is sparingly essential job that is help to understand the binding affinity of the compound toward the target. This will helps to develop competitive inhibitor in future. The docking study was carried out using a described method elsewhere. The binding energy, inhibition constant (Ki), intermolecular energy, Torsional energy, unbound extended energy, reference root mean square and the no. of hydrogen bonds were considered for the analysis. Ten filtered compounds were subjected for the docking study [Table 3]. Here, curcumin showed highest binding energy toward the target. It showed (−9.34 kcal) binding energy with the USP. It also formed one hydrogen bond interaction and two ionic bond interaction with ARG 136, LEU 130 and ASN 144 respectively [Figure 6a]. The study suggested that the hydrogen bond interaction was formed with the carboxyl group of curcumin with the hydrogen of ARG 136 (1.8Å). Ionic bonds have been found with carboxyl group of curcumin with LEU 130 (3.3Å) and ASN 144 (3.4Å) respectively. The carboxyl groups are important for the interaction with the amino acids. Quercetin, Catechin showed − 9.2 kcal and − 8.07 kcal binding energy respectively. Quercetin - USP simulation study showed four hydrogen bonding with ARG 147 (2.1Å), SER 140 (2.8Å), VAL 128 (2.7Å), LEU 138 (3.0Å). Dihydropyran and 3, 4-dihydroxy phenyl ring and 3, 5, 7- trihydroxy group of Quercetin participated hydrogen bonding and ionic interaction with the target. Catechin also has the 3, 4-dihydroxy phenyl ring that participated hydrogen bonding with the target [Figure 6] has been indicated best ligands with their best binding poses. Simulation allows to set a grid field for OA map (oxygen that accepts hydrogen bond), A map (aromatic carbon), HD map (hydrogen that donates hydrogen bond) with 0.5 isovalue and the spacing was set as 0.381Å [Figure 7] expressed the details map field for selected ligand binding site.
Table 3.
Docking results of 10 best polyphenols
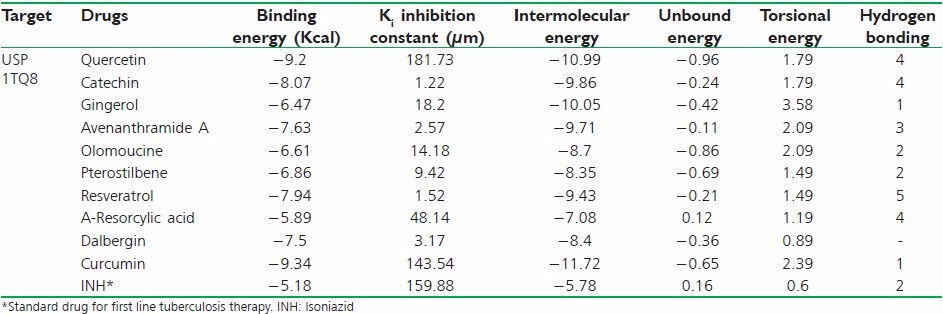
Figure 6.
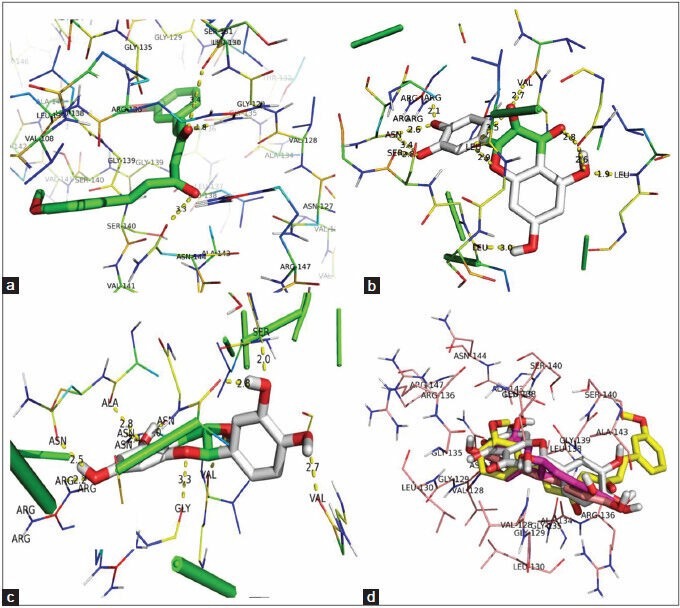
(a) Pictorial representation of curcumin-universal stress protein (USP) simulation. (b) Quercetin-USP simulation representation. (c) Catechin-USP interaction and best binding pose. (d) Superimpose of all best conformation molecules in site 1 binding site annotated by FT site
Figure 7.
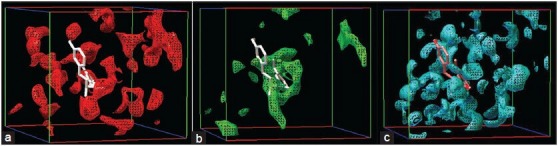
(a) Oxygen that accepts hydrogen (OA) grid area for curcumin (b) Aromatic carbon (a) grid area. (c) Hydrogen bond donor (HD) grid area for curcumin
Recent scenario suggests, drug resistance is one of the major clinical problems for any kind of chemotherapeutic associated treatment. Oxidative stress is also plays an important role in our body, as it is much complicated to establish a hypothesis that, sometimes oxidative stress helps to treat even in cancer, and also it causes several cellular damages in the different organs of our body. Notably, our study focused on the cellular oxidative stress promotes USP up-regulation and therefore it susceptible to bacteria and promotes drug resistance. Under stress, USP is overproduced and through a variety of mechanisms aid the organism in surviving in such uncomfortable conditions.[17] It is also predicted that USPs are helping pathogens, that is, Salmonella, Klebsiella, or Mycobacterium, in invasion of the host organisms;[18,19] which presents potential new opportunities for the pathogenic infection treatment. However, proteomic and transcriptomic analyses have shown that a number of USP genes are significantly upregulated under hypoxic conditions and in response to nitric oxide and carbon monoxide, as well as during M. tuberculosis infection of macrophage cell lines.[21] Hence, there is an urgent need to develop a modern approach, which is less toxic towards the system, which is susceptible to bacterial resistance towards drug. Therefore, our approach toward natural resources may enlighten the future medicine era of treatment. S. M. Hingley-Wilson group reported that M. tuberculosis has 10 USPs, whose function is unknown.[20]
CONCLUSION
To our knowledge, the study depicts the first reported molecular modeling of natural polyphenols as an inhibitor of USP. Our study has been concluded the possible inhibitors those are gifted from nature medicinal source. Expression of USP is distinctly established on Rv1636 strain; therefore the homology modeling helped us to get the desire protein structure in an accurate conformation. After performing the simulation experiments, we can clearly conclude that, Bis-methoxy phenyl Pharmacophore could help to develop the new generation curcumin analogue for future prototypes. In that connection Quercetin, a flavonoid group of compound also showed potent binding efficacy towards the target. Pharmacophoric mapping depicts the future possibilities of natural antioxidants against USP to prevent the drug resistance. The study also indicates some important amino acids like VAL, ARG, SER, ASN, are essential for the binding of drugs in to the target. Furthermore, translational study could help to design a possible combination therapy with antibiotics and natural antioxidants to prevent bacterial antibiotic resistance.
ACKNOWLEDGMENT
The authors are thankful to Vels University (VISTAS) and its management for providing research facilities and encouragement.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Drumm JE, Mi K, Bilder P, Sun M, Lim J, Bielefeldt-Ohmann H, et al. Mycobacterium tuberculosis universal stress protein Rv2623 regulates bacillary growth by ATP-binding: Requirement for establishing chronic persistent infection. PLoS Pathog. 2009;5:e1000460. doi: 10.1371/journal.ppat.1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kvint K, Nachin L, Diez A, Nyström T. The bacterial universal stress protein: Function and regulation. Curr Opin Microbiol. 2003;6:140–5. doi: 10.1016/s1369-5274(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 3.Nachin L, Nannmark U, Nyström T. Differential roles of the universal stress proteins of Escherichia coli in oxidative stress resistance, adhesion, and motility. J Bacteriol. 2005;187:6265–72. doi: 10.1128/JB.187.18.6265-6272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanner MF. Python: A programming language for software integration and development. J Mol Graph Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 5.De Lano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: De Lano Scientific; 2004. [Google Scholar]
- 6.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–8. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ngan CH, Hall DR, Zerbe B, Grove LE, Kozakov D, Vajda S. FTSite: High accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics. 2012;28:286–7. doi: 10.1093/bioinformatics/btr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolton E, Wang Y, Thiessen PA, Bryant SH. Annual Reports in Computational Chemistry. Vol. 4. UK: Elsevier-Oxford; 2008. PubChem: Integrated platform of small molecules and biological activities; pp. 217–40. Ch. 12. [Google Scholar]
- 9.Li Z, Wan H, Shi Y, Ouyang P. Personal experience with four kinds of chemical structure drawing software: Review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J Chem Inf Comput Sci. 2004;44:1886–90. doi: 10.1021/ci049794h. [DOI] [PubMed] [Google Scholar]
- 10.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 11.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 12.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - A program to check the stereochemical quality of protein structures. J App Crystallogr. 1993;26:283–91. [Google Scholar]
- 13.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4:90–8. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forli S, Botta M. Lennard-Jones potential and dummy atom settings to overcome the AUTODOCK limitation in treating flexible ring systems. J Chem Inf Model. 2007;47:1481–92. doi: 10.1021/ci700036j. [DOI] [PubMed] [Google Scholar]
- 15.Aanandhi MV, Bhattacherjee D, Kamalraj R. Synthesis, docking and biological activity of various substituted zolpidem based GABAA inhibitors endowed potent hypnotic and sedative activity. Inventi Rapid Med Chem. 2014;2:1–8. [Google Scholar]
- 16.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 17.Tkaczuk KL, A Shumilin I, Chruszcz M, Evdokimova E, Savchenko A, Minor W. Structural and functional insight into the universal stress protein family. Evol Appl. 2013;6:434–49. doi: 10.1111/eva.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rayan KJ, Ray CG. An Introduction to Infectious Diseases. 4th ed. New York: McGraw Hill; 2004. Sherris Medical Microbiology. [Google Scholar]
- 19.Hensel M. Bacterial Secreted Proteins: Secretory Mechanisms and Role in Pathogenesis. UK, Nottingham: Caister Academic Press; 2009. Secreted Proteins and Virulence in Salmonella enterica. [Google Scholar]
- 20.Hingley-Wilson SM, Lougheed KE, Ferguson K, Leiva S, Williams HD. Individual Mycobacterium tuberculosis universal stress protein homologues are dispensable in vitro. Tuberculosis (Edinb) 2010;90:236–44. doi: 10.1016/j.tube.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mbah AN, Mahmud O, Awofolu OR, Isokpehi RD. Inferences on the biochemical and environmental regulation of universal stress proteins from schistosomiasis parasites. Adv Appl Bioinform Chem. 2013;6:15–27. doi: 10.2147/AABC.S37191. [DOI] [PMC free article] [PubMed] [Google Scholar]