

Abstract
The Polycomb group of epigenetic enzymes represses expression of developmentally regulated genes in higher eukaryotes. This group includes the Polycomb repressive complex 1 (PRC1), which ubiquitylates nucleosomal histone H2A Lys119 using its E3 ubiquitin ligase subunits, Ring1B and Bmi1, together with an E2 ubiquitin-conjugating enzyme, UbcH5c. However, the molecular mechanism of nucleosome substrate recognition by PRC1 or other chromatin enzymes is unclear. Here we present the crystal structure of the Ring1B/Bmi1/UbcH5c E3-E2 complex (the PRC1 ubiquitylation module) bound to its nucleosome core particle substrate. The structure shows how a chromatin enzyme achieves substrate specificity by interacting with multiple nucleosome surfaces spatially distinct from the site of catalysis. Our structure further reveals an unexpected role for the ubiquitin E2 enzyme in substrate recognition, and provides insight into how the related histone H2A E3 ligase, BRCA1, interacts with and ubiquitylates the nucleosome.
The Polycomb group (PcG) proteins are transcriptional repressors that correlate with diverse human cancers and constitute key epigenetic regulators of stem cell self-renewal and lineage development1–5. The PcG proteins assemble into multi-component complexes that remodel chromatin structure both directly and through the establishment and removal of histone post-translational modifications6,7. One of these complexes, the Polycomb repressive complex 1 (PRC1) monoubiquitylates histone H2A lysine 1198,9. Once thought of as a unique complex, it is now clear that human PRC1 exists as a modular subset of complexes with distinct functions and gene targets10,11. All PRC1 complexes share a common core – the RING-type E3 ubiquitin ligase comprised of Ring1B/RNF2 (or Ring1A) and Bmi1/PCGF4 (or one of five other PCGF paralogs) with potential for H2A ubiquitylation. This E3 core combines with accessory subunits to form canonical and variant PRC1s. Variant complexes, especially those containing histone demethylase KDM2B and/or RYBP, appear to be primarily responsible for H2A ubiquitylation10,12–14.
Protein ubiquitylation is accomplished through a cascade of E1, E2, and E3 enzymes15. First an E1 activates and transfers ubiquitin to an E2 conjugating enzyme, forming a thioester between ubiquitin and a cysteine in the E2 active site. Then, in an E3-dependent manner, the C-terminus of ubiquitin is attached to the ε-NH2 of a target protein lysine side chain through an isopeptide bond. RING family E3 ligases, like those found in PRC1, bridge an E2 and a target protein, presenting the target lysine to the E2 active site and constraining the geometry of the ubiquityl-E2 thioester, priming it for catalysis16–18. The E3 is considered the primary specificity determinant of ubiquitylation, reflecting the scarcity of E2s (~40 proteins in human) relative to E3s (more than 600 proteins)19.
While the cognate E2 for PRC1 in vivo is not established, the Ring1B/Bmi1 E3 ligase can pair with one of several E2s, including UbcH5c/UBE2D3, to ubiquitylate nucleosomes in vivo20. The nucleosome core particle is the fundamental unit of the eukaryotic genome formed by 145–147 bp of DNA wrapped around an octameric scaffold of histone proteins (two copies each of H2A, H2B, H3, and H4)21. H2A/H2B dimers and H2A alone are not substrates for Ring1B/Bmi1, suggesting that surfaces formed only in the context of the nucleosome are required for PRC1’s E3 ligase activity22,23. A minimal RING heterodimer of Ring1B and Bmi1 exhibits comparable activity to the full-length complex, implicating the RING domains in both E2 and substrate binding20. However, we lack a fundamental mechanistic understanding of how the Ring1B/Bmi1 E3 ligase interacts with the nucleosome to direct H2A Lys119 ubiquitylation, or more generally, for how chromatin enzymes recognize their nucleosome substrate.
We have crystallized the PRC1 ubiquitylation module, an E2-E3 enzyme complex composed of UbcH5c and the minimal Ring1B/Bmi1 RING heterodimer, bound to its nucleosome core particle (NCP) substrate and solved the structure at 3.3 Å resolution (Supplementary Table 1). To our knowledge this is the first crystal structure of a histone modifying enzyme-nucleosome complex or of a ubiquitin E2-E3-substrate complex. The structure shows that in contrast to most chromatin modification enzymes studied to date, the vast majority of interactions between the PRC1 ubiquitylation module and the nucleosome occur away from the catalytic site. Ring1B and Bmi1 bind to the histone surface of the NCP to orient UbcH5c for H2A ubiquitylation. Surprisingly, UbcH5c also makes direct contacts with the nucleosome core particle that are critical for nucleosome binding and ubiquitylation. This presents a novel role of ubiquitin E2 enzymes in substrate recognition.
Overview of complex
The structure shows one PRC1 ubiquitylation module bound to each disk face of the nucleosome (Fig. 1a, b). The two PRC1 modules more than double the height of the NCP and with the exception of the C-terminal region of UbcH5c, are entirely contained within a cylinder projecting up from the NCP disk surface. Each PRC1 ubiquitylation module forms a crescent with two halves of the crescent contributed by the E2 and E3, respectively. The Ring1B/Bmi1 E3 heterodimer binds to the central histone surface of the NCP. The E2 UbcH5c completes the crescent, following the inner curvature of the DNA from its interface with Ring1B past the DNA end, ultimately terminating above the nucleosomal dyad. In this manner, the Ring1B/Bmi1 E3 ligase positions the active site of UbcH5c over the C-terminal tail of H2A near the target residue Lys119 (Fig. 1c). All three components of the PRC1 ubiquitylation module contact histone and/or DNA components of the nucleosome burying 1470 Å2 of solvent accessible surface.
Figure 1. Crystal structure of PRC1 ubiquitylation module-nucleosome core particle complex.

a, View of the complex looking down on the DNA superhelical axis. b, Orthogonal view of the complex with proximal and distal halves of the structure indicated. c, Zoomed view demonstrating Ring1B/Bmi1 heterodimer positioning UbcH5c active site Cys85 over the H2A C-terminal tail near the H2A Lys119 Cα atom (orange sphere). Crystals contain the minimal RING heterodimer Ring1B(2-116)/Bmi1(2-109).
We selected UbcH5c for our structural studies due to its high activity and thorough biochemical and structural characterization with Ring1B/Bmi1. To overcome the low affinity and salt-sensitive E2-E3 interaction22, we genetically fused the C-terminus of the Ring1B fragment to the N-terminus of UbcH5c, converting the three-polypeptide PRC1 ubiquitylation module into a stable two-polypeptide version. The two fused PRC1 ubiquitylation modules in our structure align well with the previously determined Ring1B/Bmi1/UbcH5c structure22 (rmsd of 1.0 Å over all backbone atoms), suggesting only minor structural changes upon NCP binding and confirming the genetic fusion did not significantly alter the structure of the ubiquitylation module (Extended Data Fig. 1a–c, e). Such validation is especially important given that the fused PRC1 ubiquitylation module is inactive in a ubiquitylation assay due to its inability to undergo E1-mediated transthiolation, presumably because the fused E3 clashes with ubiquitin in the E1 adenylation site24 (Extended Data Fig. 2a, b). The NCP is similarly largely unchanged compared to a published NCP structure using the same Widom 601 positioning sequence25 (rmsd of 0.7 Å over all protein backbone atoms) (Extended Data Fig. 1d, e).
Although the two PRC1 ubiquitylation modules bind their respective nucleosome faces in a similar manner, there are differences particularly at the UbcH5c-nucleosome interfaces. Alignment of the two sides of the structure through the pseudo-two-fold symmetry of the nucleosome reveals near uniformity between the histones on the two sides of the structure (rmsd of 0.7 Å) (Extended Data Fig. 1f–i). In contrast, the PRC1 ubiquitylation modules from the two faces show more variation (rmsd of 1.9 Å and 4.6 Å for the E3 and E2 components, respectively), resulting from a flexing of a hinge at the Ring1B/Bmi1-nucleosome interface. Regions of the PRC1 ubiquitylation module close to this hinge, including the Ring1B/Bmi1 nucleosome binding loops, are very similar on the opposing surfaces of the structure, while regions further from the hinge, notably the UbcH5c-nucleosome interfaces, exhibit larger differences. As a result, UbcH5c approaches the NCP more closely on one side (proximal side) of the structure than the other (distal side).
While we cannot rule out the possibility of plasticity in the UbcH5c-NCP interface, we suspect that the variation in the two halves of the structure result from different environments of each PRC1 module within the crystal lattice. This is not merely a result of the post-crystallization dehydration used to obtain 3.3 Å resolution diffraction as molecular replacement solutions from data collected on less dehydrated crystals with larger unit cells also exhibit similar differences (data not shown). Because the distal side places UbcH5c too far from the nucleosome surface to be consistent with our biochemical studies, we favor the proximal side, which is depicted in the following figures. (Details provided below. The distal side and alignments can be found in Extended Data Fig. 3.) Importantly, the differences between the two sides of the structure influence the extent rather than the presence of interactions at each of the PRC1-NCP interfaces (Extended Data Fig. 1j).
Ring1B/Bmi1-histone interactions
Several basic surfaces of Ring1B and Bmi1 have been shown to be required for nucleosomal ubiquitylation and binding to short duplex DNA22. This informed a structural model docking the Ring1B/Bmi1 heterodimer on DNA near the nucleosome dyad, positioning UbcH5c in proximity to H2A Lys11922. However, recent work implicating the H2A/H2B acidic patch in Ring1B/Bmi1-mediated H2A ubiquitylation in vitro and in vitro26 is inconsistent with this docking model. Our structure reveals that the Ring1B/Bmi1 basic residues anticipated to bind DNA actually interact with histones surfaces, including the H2A/H2B acidic patch. The Ring1B/Bmi1 RING heterodimer, which contacts all four histones, forms a saddle over the N-terminal end of the H2B αC helix anchored on each side by Ring1B- and Bmi1-histone interactions (Extended Data Fig. 3a–c). Ring1B binds to the acidic patch of the H2A/H2B dimer. This interaction, which buries nearly 700 Å2 of solvent accessible surface on the proximal side, is mediated by an intricate network of hydrogen bonds and van der Waals contacts and with few exceptions is limited to protein side chains (Fig. 2a and Extended Data Fig. 3d–g, t, u). Consistent with published mutagenesis experiments22,26, H2A Glu92, Ring1B Lys97 and Arg98, and to a lesser extent Ring1B Lys93 contribute to the Ring1B-nucleosome interface. Ring1B Arg98 inserts into an acidic pocket generated by H2A residues Glu61, Asp90, and Glu92 making charged hydrogen bonds with each of the H2A side chain carboxylates. The depth of this pocket is augmented by a ridge comprised of H2B residues Glu105 and His109, which make van der Waals contacts with the aliphatic region of the Arg98 side chain. This paradigm of nucleosomal recognition mediated by an “arginine-anchor” binding to the H2A/H2B acidic patch is present in all chromatin factor-NCP crystal structures published to date27–30 (Extended Data Fig. 4). Ring1B Lys97 projects into the acidic patch adjacent to Arg98, hydrogen bonding to H2A residues Glu61 and Glu64. Not predicted by previous studies, Ring1B Arg81 also binds to the acidic patch forming both side chain and main chain hydrogen bonds with H2A residues Asp72 and Asn89, respectively. This second arginine-binding site is distinct from sites observed in previous chromatin factor-NCP structures. Further contacts are mediated by Ring1B residues Lys65, Lys85, Lys93, and Val95. With minor exceptions the Ring1B-NCP interface is identical on the proximal and distal faces. (Extended Data Fig. 3f, g) The considerable contribution of charged side chains to the Ring1B-nucleosome interface is consistent with the loss of nucleosomal ubiquitylation by the PRC1 ubiquitylation module at higher ionic strength22.
Figure 2. Ring1B/Bmi1 E3 heterodimer interacts with histone surface.

a, Ring1B-histone acidic patch interactions in cartoon representation with relevant side chains and hydrogen bonds (yellow) highlighted. b, Bmi1-histone interface shown similarly. c, Coomassie blue stained gel of ubiquitylation assay using H2A mutant NCP as indicated. d, Quantitated nucleosome ubiquitylation assay using indicated Ring1B mutants. e, Nucleosome binding curves for fused wild-type and Ring1B mutant E2-E3 complexes. Means and standard deviations are shown in d and e with n = 3 for each data point. Fluorescence is normalized to fit values for unbound and saturated NCP. Concentrations depicted using log scale.
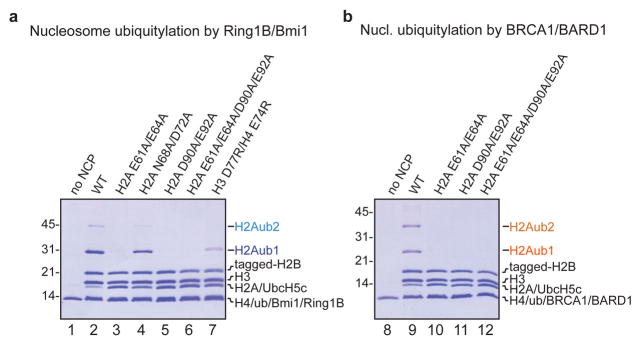
The structural details of the Ring1B-acidic patch interface are validated by our mutagenesis experiments in solution. Tandem alanine mutations of H2A Glu61/Glu64 or Asp90/Glu92 (or the combination thereof) eliminate all detectable nucleosomal ubiquitylation (Fig. 2c and Extended Data Fig. 5a). Nucleosomes harboring the H2A Asn68Ala/Asp72Ala double mutant at the secondary arginine-binding site exhibit roughly half wild-type activity levels. To quantitatively assess the effects of mutations in the PRC1 ubiquitylation module, we established fluorescence-based nucleosome ubiquitylation and binding assays31 (Extended Data Fig. 6). The Ring1B Arg98Ala mutation results in a 50-fold decrease in both nucleosomal ubiquitylation and in the affinity of the fused PRC1 ubiquitylation module for the nucleosome (Fig. 2d, e). Alanine mutations of Ring1B Lys97, Arg81, and Lys93 have similar, yet decreasing effects on nucleosome ubiquitylation and binding, while the Ring1B Lys59Ala mutation away from the binding site has little to no effect on PRC1 function.
In contrast to the large Ring1B-NCP interface, the Bmi1-NCP interface is less extensive, burying only 330 Å2. Bmi1 forms a cap on the C-terminal end of the α1 helix of H3 with a Polycomb-specific RING domain loop insertion (Fig 2b and Extended Data Fig. 3h–k, v, w). This region was previously implicated in PRC1 function given the reduced activity of a Bmi1 Lys62Ala/Arg64Ala mutant22. These residues form charged hydrogen bonds with H4 Glu74 and H3 Asp77, respectively. The intervening Thr63 side chain inserts into a small cavity created by the end of the α1 helix and the neighboring L1 loop of H3, contributing substantial van der Waals contact surface and forming a hydrogen bond with the H3 Gln76 main chain carbonyl oxygen. An additional hydrogen bond is made between Bmi1 Glu33 and H2B Lys108. The distal side Bmi1-NCP interface shares architectural similarity to its proximal counterpart with the exception of insufficient density to model the Lys62 side chain (Extended Data Fig. 3j, k).
Mutagenesis of the Bmi1-NCP interface allows us to assess the importance of this surface relative to the Ring1B-NCP interface. The consequences of the Bmi1 Arg64Ala mutation are comparable to that of Ring1B Lys93Ala, the least deleterious mutant tested at the Ring1B-NCP interface (Extended Data Fig. 7d–f and Supplementary Table 2). Meanwhile alanine mutations of His61 and Lys62 at the Bmi1-NCP interface and Arg45 removed from the interface exhibit near wild-type behavior. A charge reversal mutation of the underlying histone surface (H3 Asp77Arg/H4 Glu74Arg) decreases ubiquitylation by ~80% (Extended Data Fig. 5a). This reduction in activity is smaller than that resulting from alanine mutations at the Ring1B-NCP interface. Paradoxically, a Bmi1 Glu33Ala mutation both drastically decreases nucleosomal ubiquitylation and slightly increases nucleosomal binding affinity. We do not fully understand this phenomenon but it may relate to perturbation of the E3 heterodimer given the location of this side chain at the Ring1B/Bmi1 interface. Overall, our studies suggest a dominant role for Ring1B in PRC1’s nucleosome binding. The 25-fold enhancement of Ring1B activity by Bmi19,20 likely represents the modest role for Bmi1 in NCP binding together with a role in constraining ubiquitin at the Bmi1-ubiquitin interface16–18.
In human PRC1 complexes, Bmi1 can be replaced by five paralogs also know as PCGF (Polycomb Group RING finger) proteins11. The sequence conservation of these paralogs combined with our structural and biochemical data suggest that each PCGF protein RING domain could support H2A ubiquitylation (Extended Data Fig. 8a). Bmi1 Glu33 is absolutely conserved mirroring its importance for nucleosomal ubiquitylation. Moreover, Thr63 and Arg64 show sequence similarity, maintaining both overall side chain size and hydrophilicity. Meanwhile, Lys62, which is dispensable for nucleosomal ubiquitylation, exhibits far less conservation and is replaced by negatively charged side chains in two of the paralogs. The lack of H2A ubiquitylation by canonical PRC1 complexes may reflect masking of the E3 interfaces with either the nucleosome or its E2. Alternatively, the nucleosome surface surrounding H2A Lys119 may become inaccessible, secondary to canonical PRC1-mediated chromatin compaction. Further structural characterization of canonical and variant PRC1 alone and in complex with oligonucleosomes may shed light on this phenomenon.
UbcH5c-nucleosomal DNA interactions
Nucleosomal binding by the Ring1B/Bmi1 E3 ligase minimal RING heterodimer positions the E2 UbcH5c with its active site directly over the H2A C-terminal tail near the target Lys119 (Fig. 1c). However, UbcH5c scarcely contacts the underlying nucleosomal histone surface. The lack of E2 active site-substrate specific interactions may allow UbcH5c to efficiently ubiquitylate multiple substrates by pairing with different E3 ligases, though we cannot rule out nucleosome-specific contacts by other potential E2s. This contrasts the direct recognition of the target lysine and surrounding consensus sequence by the related E2 Ubc9 in the SUMO-RanGAP1-Ubc9-Nup358 E2-E3-product crystal structure32. On the other hand, UbcH5c does interact with the nucleosome using two non-active site surfaces to bind nucleosomal DNA near the DNA end between superhelical location 6 and 7 (SHL 6 and 7) and at the nucleosomal dyad (Fig. 3a, b and Extended Data Fig. 3l–s). These interfaces vary significantly on the proximal and distal sides of the crystal structure. At the DNA end interface, the UbcH5c antiparallel sheet aligns the side chains of His32, His55, and Lys66, directing them toward the DNA backbone (Fig. 3a). This allows Lys66 and His32 to form hydrogen bonds with adjacent DNA phosphate groups on the proximal side of the structure. His55 and Thr58 side chains also approach the neighboring phosphodiester backbone (3.6 and 3.9 Å, respectively). On the distal side of the crystal structure, each of these residues is ~2 Å farther from the nucleosome surface, limiting direct contact to the Lys66 side chain which lies 4.1 Å from the DNA backbone (Extended Data Fig. 3n, o). In addition to these backbone interactions, Asn81 projects into the major groove at the DNA end, although it is unclear in our structure if base specific recognition occurs.
Figure 3. UbcH5c binds to nucleosomal DNA enhancing Ring1B/Bmi1-NCP affinity.
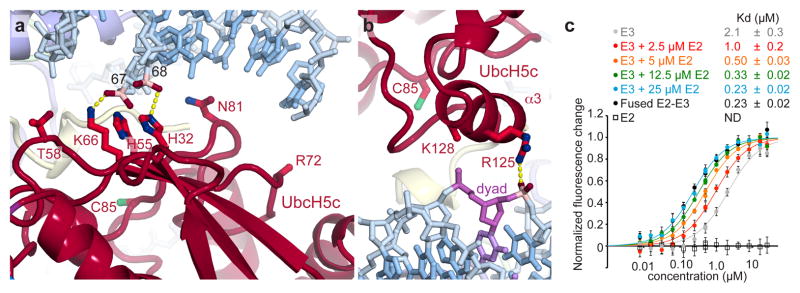
a, UbcH5c antiparallel β-sheet-DNA end interactions. Important phosphates colored pink. The catalytic UbcH5c Cys85 is shown. b, UbcH5c-dyad interactions. The nucleosome dyad nucleotide is colored purple. Arg72 and Lys128 side chains not modeled due to limited electron density. c, Fluorescence quenching NCP binding curves for E3 Ring1B/Bmi1 alone (grey), fused to E2 UbcH5c (black), or with E2 UbcH5c added in trans (colored as shown). E2 UbcH5c alone (open squares) is undetectable under assay conditions as it either fails to bind to the NCP or is undetectable due to the location of the fluorescent probe used to monitor Ring1B/Bmi1 binding. Means and standard deviations are shown with n = 3 for each data point. Fluorescence is normalized to fit values for unbound and saturated NCP. Concentrations depicted using log scale.
UbcH5c also contacts the nucleosomal DNA again as its α3 helix tracks across the nucleosomal dyad (Fig. 3b). On the proximal side, the UbcH5c Arg125 side chain forms a hydrogen bond with the phosphate in position 1. Although the weaker density in this region of the structure prevents modeling of the Lys128 side chain, the Lys128 Cβ is only 3.9 Å from the phosphate backbone at the dyad, allowing for further contacts. A ~3 Å translation of the distal side UbcH5 α3 helix along its axis results in a minor rearrangement of the distal interface (Extended Data Fig. 3r, s). Notably, a hydrogen bond is observed between the Lys128 side chain and the dyad phosphate on the distal side.
Given the novelty of this E2-substrate interaction, we further examined the role of UbcH5c in nucleosomal binding by the PRC1 ubiquitylation module in solution. We initially noted that the fused PRC1 complex binds to the NCP with nearly 10-fold increased affinity as compared to the Ring1B/Bmi1 alone (Fig. 3c and Supplementary Table 2). Titration of UbcH5c in trans incrementally increases the affinity of the Ring1B/Bmi1-NCP interaction, verifying contributions by UbcH5c to nucleosomal binding. At 25 μM UbcH5c (~2.5x the E2-E3 Kd under assay conditions22), the near-saturated, unfused PRC1 ubiquitylation module mimics its fused counterpart both in nucleosome affinity and raw fluorescence change, further validating the use of the fused complex in the structure and to interrogate effects of mutagenesis of all three PRC1 ubiquitylation module components (Fig. 3c and data not shown). Importantly, the Ring1B Asp56Lys mutation that prevents Ring1B-UbcH5c binding22 eliminates affinity changes mediated by UbcH5c in trans (Supplementary Table 2).
Having established a role for UbcH5c in nucleosome binding, we next addressed the contributions from the DNA end and DNA dyad interfaces. The UbcH5c Lys66Glu charge reversal mutation decreased nucleosomal affinity and ubiquitylation approximately 10-fold (Extended Data Fig. 7g–i and Supplementary Table 2). A similar trend was observed with the UbcH5c Arg125Glu/Lys128Glu double mutation, though the nucleosomal affinity of this mutant complex could not be calculated due to aggregation of the complex at high concentrations under assay conditions. The UbcH5c Thr58Glu mutant also exhibited significant though less severe defects in binding and ubiquitylation while the Arg136Glu/Arg139Glu negative control mutant away from the NCP surface exhibited wild-type activity. Altogether, these results implicate both DNA end and DNA dyad interfaces in PRC1 function.
The fused PRC1 ubiquitylation module with the UbcH5c Lys66Glu charge reversal mutant bound less tightly to the nucleosome than the Ring1B/Bmi1 subcomplex alone, suggesting a repulsive effect in addition to loss of any native interactions. Analysis of alanine mutations provides evidence for the role of UbcH5c side chains in nucleosome binding. At the DNA end interface, the UbcH5c Lys66Ala mutation caused a 50% reduction in nucleosome ubiquitylation and a 6-fold decrease in nucleosome affinity (Extended Data Fig. 7j–l and Supplementary Table 2). The neighboring Arg72Ala and His32Ala/His55Ala also showed measurable but more muted effects relative to wild-type proteins. At the dyad interface, the Arg125Ala/Lys128Ala double mutant mimicked the Lys66Ala mutant with a 60% and 6-fold reduction in nucleosome ubiquitylation and binding, respectively. Individual alanine mutations of Arg125 and Lys128 caused nearly identical intermediate effects. Unlike the charge reversal mutations, all fused complexes bearing UbcH5c alanine mutations bound to the nucleosome with affinities weaker than that of the fused wild-type complex but stronger than the Ring1B/Bmi1 subcomplex alone, implicating the side chains in nucleosome binding. All UbcH5c mutants have similar intrinsic activity as measured by lysine aminolysis of ubiquitin charged E2s33 (Extended Data Fig. 2c).
Since the variation between the proximal and distal sides of the structure is greatest at the UbcH5c-DNA end interface, our functional studies present an opportunity to assess the relative authenticity of the two sides. For example, the UbcH5c His32 and His55 side chains are 3.3 and 3.6 Å from the DNA backbone on the proximal side and 5.1 and 7.1 Å on the distal side, respectively. Mutation of both histidines to alanine decreases nucleosomal affinity 1.5-fold while causing a 30% reduction in enzymatic activity. Because the histidine-DNA backbone distances on the proximal side of the structure are more consistent with our biochemical studies, we favor this side as the better representation of the solution state interaction.
Further inspection of the structure suggests potential for additional interactions between UbcH5c and nucleosomal linker DNA (Extended Data Fig. 9a, b). The UbcH5c C-terminal α4 helix rests across the major groove of linear B-form DNA modeled onto the end of the NCP. The DNA-adjacent helical face includes two positively charged and two aromatic side chains. Consistent with this hypothesis, we find that the PRC1 ubiquitylation module binds to nucleosomes centered on 185 bp 601 DNA with twice the affinity of those reconstituted with 147 bp 601 DNA (Extended Data Fig. 9c). The absence of linker DNA required for these interactions in our crystals may explain the apparent flexibility of the α4 helices suggested by weaker electron density and higher B-factors.
In addition to UbcH5c, the Ring1B/Bmi1 E3 ligase can also pair with UbcH5a/UBE2D1, UbcH5b/UBE2D2 and UbcH6/UBE2E120. Meanwhile five other E2s cannot substitute for UbcH5c. These functional classes could result from compatibility differences at the E2-NCP and/or E2-E3 interfaces. Sequence alignment of E2s suggest the possibility that multiple positions including Arg125 combine to determine PRC1 complementarity (Extended Data Fig. 8b).
Nucleosome recognition by BRCA1
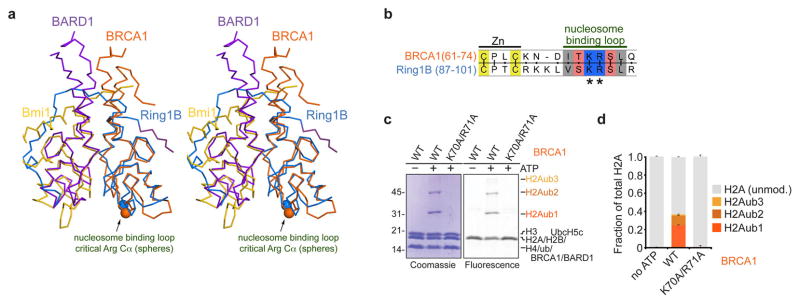
Despite contributions from Bmi1 and UbcH5c, Ring1B is the major contributor to nucleosome recognition as evidenced by buried surface area calculations and the magnitude of effects of Ring1B mutations on nucleosome binding and ubiquitylation. We wondered whether another E3 ligase, BRCA1, recently shown to specifically target nucleosomal H2A Lys127 and Lys 12934, might employ a similar nucleosome recognition pattern. Much like Ring1B, BRCA1 uses its N-terminal RING domain to pair with the BARD1 RING domain to form a heterodimeric RING-type E3 ligase35. Structural and sequence alignment shows a high level of conservation between the nucleosome-binding loop of Ring1B and the corresponding BRCA1 region, including the Lys-Arg motif (Extended Data Fig. 10a, b). We find that disruption of this motif with the Lys70Ala/Arg71Ala double mutation eliminates nucleosomal ubiquitylation by the BRCA1/BARD1 RING heterodimer (Extended Data Fig. 10c, d). A similar loss of activity was observed upon mutation of the H2A/H2B acidic patch (Extended Data Fig. 5b). This suggests the possibility that like Ring1B, BRCA1 uses basic residues in the putative nucleosome binding loop to bind to the nucleosomal H2A/H2B acidic patch. However, since Ring1B and BRCA1 target different H2A residues (Lys119 vs Lys127/Lys129), additional BRCA1/BARD1 contacts may alter its orientation on the nucleosome.
Nucleosomal H2A specificity
The PRC1 ubiquitylation module requires a nucleosome substrate, showing no activity on an H2A/H2B dimer. Our structure indicates that Ring1B, Bmi1 and UbcH5c interact with nucleosomal H2A/H2B, H3/H4 and DNA, respectively, providing a mechanistic basis for nucleosome specificity. We also believe that the nucleosome plays an additional role in presenting the H2A C-terminal tail to the UbcH5c active site. In the context of the H2A/H2B dimer, the H2A C-terminal tail is expected to be unstructured beyond residue 105 assigning an entropic cost to ubiquitylation of Lys11936. However, within the nucleosome, H2A adopts an elongated structure between residues 105 and 117, resting on a platform generated by the underlying H3 surface21. Residues beyond this structured region are more flexible, as suggested by the inability to model beyond Lys118 in many NCP crystal structures25,28–30. As such, the nucleosome both constrains the H2A C-terminal tail trajectory while allowing localized flexibility to reach the UbcH5c active site.
Despite the large number of lysine residues in the nucleosome, the PRC1 ubiquitylation module is specific for H2A Lys119 and to a lesser extent Lys118. In our structure, electron density is weak after Pro117 allowing only main chain atoms to be built to Lys119 and Lys118 on the proximal and distal sides, respectively. Neither main chain trajectory indicates binding to the UbcH5c active site cleft. In order to verify that the UbcH5c positions within our structure are compatible with catalysis, we modeled the tetrahedral ubiquitylation transition state using H2A Lys 118 and 119 on each side of the structure (model for proximal side Lys119 shown in Fig. 4). Aside from the tetrahedral center, ubiquitin makes no contact with the nucleosome surface in these models. This echoes the lack of SUMO-RanGAP1 interactions in the SUMO-RanGAP1-Ubc9-Nup358 E2-E3-product structure32. We find the position of UbcH5c on both sides of the structure is consistent with H2A Lys118 and Lys119 ubiquitylation. Moreover, our structure suggests nucleosome ubiquitylation is limited to these two lysines because UbcH5c is constrained in close proximity to the nucleosome surface, which prevents other lysines from entering the active site. Overall, the multifaceted interaction of the PRC1 ubiquitylation module with the nucleosome and the nucleosomal organization of the H2A C-terminal tail appear to combine to effect H2A ubiquitylation. More generally, our structure illustrates how a chromatin enzyme can use multiple non-active site surfaces for nucleosome recognition and specificity determination. We anticipate this principle to be shared by many chromatin enzymes that like PRC1 function as part of large multi-component complexes.
Figure 4. Model of ubiquitylation transition state complex.
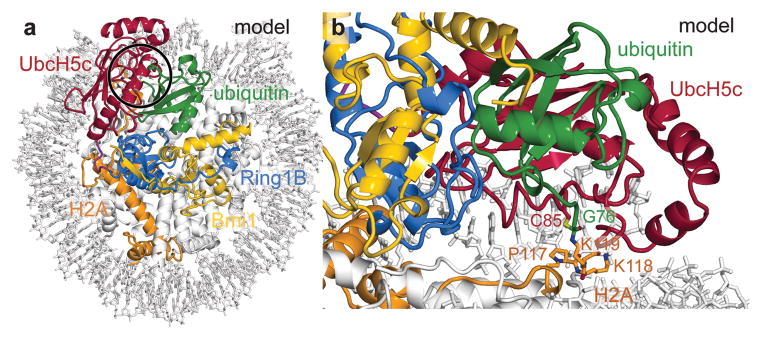
a, View of transition state model looking down on the DNA superhelical axis with region around catalytic site highlighted with black circle. Ubiquitin modeled from the RNF4/UbcH5a/Ub16 (PDB ID 4AP4) structure based on of alignment of its E2 subunit and UbcH5c from the proximal half of the PRC1 ubiquitylation module-nucleosome core particle structure. b, Side view of tetrahedral intermediate model.
Methods
Preparation of proteins, protein complexes, and nucleosome core particles
Complete genes for human Ring1B, Bmi1, UbcH5c, and Uba1 were amplified from HeLa cDNA. Gene fragments of BRCA1 and BARD1 containing the RING domains were amplified from HeLa and U2OS cDNA, respectively. The gene for ubiquitin was synthesized by Invitrogen. UbcH5c(2-147), Uba1(2-1058), and ubiquitin(1-76) were each cloned into pST50Tr vectors with an N-terminal tandem STR-decahistidine (His10) affinity tag. STR-His10-Ring1B(2-116)/Bmi1(2-109) and STR-His10-BRCA1(2-103)/BARD1(27-130) were each cloned into pST44 vectors for polycistronic co-expression37. A similar pST44 vector was created to co-express Bmi1(2-109) with Ring1B(2-116) fused in frame to UbcH5c(2-147) through a linker encoding the protein sequence, GSGSRS. Point mutants were generated by site-directed mutagenesis. The fused human PRC1 ubiquitylation module containing Bmi1(2-109) and STR-His10-Ring1B(2-116)-GSGSRS-UbcH5c(2-157) was expressed in Rosetta(DE3)pLysS Escherichia coli at 18 °C. The complex was enriched by metal-affinity chromatography with Talon resin (Clonetech), the affinity tag removed with tobacco etch virus (TEV) protease and the complex further purified by SourceS cation-exchange chromatography (GE Healthcare). The Ring1B(2-116)/Bmi1(2-109) complex and UbcH5c(2-147) single protein were expressed and purified similarly. STR-His10-Uba1(2-1058) and STR-His10-ubiquitin(1-76) were expressed in CodonPlus(DE3) and BL21(DE3)pLysS Escherichia coli at 18 °C and 37 °C, respectively. Uba1 was purified by metal-affinity chromatography as above, followed by SourceQ anion-exchange chromatography (GE Healthcare) without cleavage of the affinity tag. Ubiquitin was purified similarly with the affinity tag intact using SourceS cation-exchange chromatography. The STR-His10-BRCA1(2-103)/BARD1(27-130) complex was expressed in Rosetta(DE3)pLysS Escherichia coli at 18 °C and purified by metal-affinity chromatography, TEV cleavage, and SourceQ anion-exchange chromatography. All mutants were expressed and purified identically to their wild-type versions including ion exchange chromatography. Histone mutants were generated by site-directed mutagenesis. Recombinant Xenopus histones were expressed, purified and reconstituted with the 147- or 185-bp Widom 601 sequence38 essentially as described previously39 including anion-exchange purification of nucleosome core particles. Fluorescent labeling of histones was performed as described31.
Characterization of proteins and protein complexes
All proteins and complexes were evaluated by dynamic light scattering using a Protein Solutions DynaPro instrument. Scattering was analyzed using the Dynamics v6 software package. All E2 and E3 mutants showed equivalent monodispersity to their wild-type controls (polydispersity < 20%). Due to paradoxical binding and activity results, the secondary structure of Bmi Glu33Ala mutant E3 heterodimer and fused E2-E3 complexes were further analyzed and compared to wild-type controls using a Jasco J-1500 Circular Dichroism Spectrometer. Spectra were obtained at 0.1 mg/ml protein concentration in 2 mM Tris-Cl pH 7.6 containing 150 mM NaF. Data were analyzed using CDPro.
Complex reconstitution and crystallization
The PRC1 ubiquitylation module was reconstituted with nucleosome core particles prepared with 147-bp 601 DNA at a 2.8–3.0:1 module:nucleosome ratio in 20 mM Tris-Cl pH 7.6, 75 mM NaCl, 10 μM ZnSO4, 1 mM DTT. The complex was purified by Superdex 200 size exclusion chromatography (GE Healthcare) in reconstitution buffer supplemented with 0.2 mM PMSF. Pooled fractions were concentrated to ~10 mg/ml by VS500 centrifuge ultrafiltration (Vivascience). Crystallization was performed in modified microbatch40 by combining 1 μl of the concentrated complex with 1 μl 25 mM HEPES pH 7.5 (Hampton), 80 mM NH4NO3 (Hampton), and 3% PEG 2000-MME (Fluka) at 21 °C overlayed by 70 μl Al’s oil (1:1 mixture of silicon oil (Clearco) and mineral oil (Fisher)). Crystals were harvested after 12 to 60 days. Cryoprotection and dehydration was performed by soaking in 25 mM HEPES pH 7.5, 120 mM NH4NO3, 2.5% ethanol, 0.5 mM DTT, and 5% PEG 2000-MME with increasing amounts of PEG 550-MME (Fluka, 0–28% in 4% increments, every 10–15 minutes) at room temperature prior to flash cooling in liquid nitrogen.
Data collection, data processing, model building and refinement
Diffraction data were collected at Advanced Photon Source’s NE-CAT beamline 24-ID-E (0.9792 Å; 100 K). Data were processed using XDS41 and Scala42. The structure was solved by molecular replacement with Phaser43 and polyalanine versions of the PRC1 ubiquitylation module22 (PDB ID 3RPG) and the nucleosome core particle with 145-bp Widom 601 DNA25 (PDB ID 3LZ0) as rigid body search models (Phaser final log likelihood gain 3,700). Crystallographic refinement was performed with iterative automatic refinement using REFMAC544,45, PHENIX46, and CNS47 together with manual model building in COOT48. The structure was refined to 3.3 Å resolution with Rwork/Rfree 19.7%/24.5%. The asymmetric unit contains one copy of the 2:1 PRC1 ubiquitylation module-NCP complex in C2221 space group. The final model includes Bmi1 residues 3–107 for chain K, residues 6–102 for chain M; Ring1B residues 15–116 for chain L, 16–116 for chain N; UbcH5c residues 2–147 for chain L, 2–147 for chain N (residues for UbcH5c are offset by 200 in the PDB file (i.e. 202–347), linker between Ring1B and UbcH5c in final model residues 117–122 for chains L and N); H3 residues 37–134 for chain A, residues 38–134 for chain E; H4 residues 17–102 for chain B, residues 21–102 for chain F; H2A residues 16–119 for chain C, residues 12–118 for chain G; H2B residues 31–125 for chain D, residues 31–125 for chain H; 601 DNA residues −71 to +72 for chain I, residues −72 to +72 for chain J. The histone tails are typically not observed in NCP structures. Above average B-factors are typical for structures containing the NCP especially at resolutions greater than 3 Å30,49–51. Ramachandran statistics were analyzed with PROCHECK52 (88.4% of residues in most favored regions, 11.5% in additional allowed regions, 0.1% in generously allowed regions, 0.1% in disallowed regions – disallowed residue corresponds to Ser122 to in L chain contained in fusion between Ring1B and UbcH5c). Simulated annealing omits maps cumulatively covering the entire structure were generated with CNS. All molecular graphics were prepared with PyMOL53. Buried surface areas and rmsd values for structure alignments were calculated using PyMOL.
Ubiquitylation assays
Ubiquitylation assays were performed by mixing 2 μM NCP, 5 μM STR-His10-ubiquitin, 30 nM STR-His10-Uba1 (E1), 0.375 μM UbcH5c (E2) and 0.375 μM Ring1B(2-116)/Bmi1(2-109) or BRCA1(2-103)/BARD1(27-130) (E3) in 50 mM HEPES pH 7.5, 75 mM NaCl, 1 mM DTT, 10 μM ZnSO4, 2 mM MgCl2, 3 mM ATP. For histone mutants assays, non-fluorescent nucleosomes with a STR-His6 tagged H2B were employed. For PRC1 ubiquitylation module mutants, wild-type recombinant nucleosomes were doped with 5% nucleosomes containing the H2A Thr10Cys mutant labeled with Oregon Green 488 maleimide. Assay samples ranging from 20 to 80 μl were placed in a water bath for 60 min at 30 °C after which assays were quenched with an equivalent volume of 2x SDS-PAGE gel loading dye and boiled for 5 min. Assay samples were separated by SDS-PAGE using 18% gels, scanned with a Typhoon 9410 imager (GE Healthcare; 488 nm excitation; 526 nm emission; PMT 500) and stained with Coomassie brilliant blue. Quantification of assays was performed using Image Quant software by drawing boxes around each band. Each quantified band was background subtracted using an identically sized box in a region of the same lane without signal. As only unmodified H2A, H2Aub1, H2Aub2, and H2Aub3 were observed, the sum of these background-subtracted bands was considered to be the total H2A for each lane. In this manner, fraction of total H2A was calculated for each band. These fractional values could be compared across gels reproducibly (Extended Data Fig. 6). Three technical replicates were performed for each mutant with all mutants of a given type (i.e. all Ring1B mutants) executed simultaneously with both positive and negative controls. In most cases, three gels were run each containing one of the three replicates of each of the mutants in a given dataset. n = 3 was found to be sufficient for reproducible data (Extended Data Fig. 6) with low standard deviations. Assays for E1-mediated transfer of ubiquitin to UbcH5c Cys85Lys mutants were performed as previously described16. Briefly, 100 μM STR-His10-ubiquitin, 1 μM STR-His10-Uba1 (E1), 80 μM UbcH5c or fused PRC1 ubiquitylation module with the Cys85Lys mutant were combined in 50 mM Tris-Cl pH 10, 150 mM NaCl, 8 mM TCEP, 5 mM MgCl2 and incubated at 35 °C for 24 h. Assay samples were separated by SDS-PAGE and stained as described above.
UbcH5c aminolysis assays
To assess intrinsic function of previously uncharacterized UbcH5c mutants, lysine reactivity aminolysis assays were performed basically as previously described33. Briefly, 20 μM UbcH5c (wild-type or mutant) was combined with 20 μM STR-His10-ubiquitin and 0.5 μM STR-His10-Uba1 (E1) in 80 μl of 25 mM sodium phosphate pH 7.0, 150 mM NaCl, 10 mM MgCl2 and 10 mM ATP. Following a 20 min incubation at 37 °C, lysine was added to a final concentration of 50 mM. Aminolysis time points were taken at 0, 10 and 30 min and analyzed by SDS-PAGE. Reduction of the no lysine control was performed with 100 mM DTT.
Nucleosome binding assays
Nucleosome binding assays were performed with nucleosomes reconstituted with a H2B Ser112Cys mutant labeled with Oregon Green 488 maleimide (Life Technologies) with approximately ~50% efficiency and 147- or 185-bp 601 DNA. Fluorescence quenching assays were performed essentially as previously described31. Briefly, chromatin binding proteins (wild-type or mutant fused PRC1 ubiquitylation module or Ring1B/Bmi1 and/or UbcH5c) were diluted in 20 mM Tris-Cl pH 7.6, 100 mM NaCl, 5 mM DTT, 5% glycerol, 0.01% NP-40, 0.01% CHAPS, 0.1 mg/ml BSA to concentrations of 50, 32, 16, 8, 4, 2, 1, 0.5, 0.25, 0.125, 0.062, 0.031, and 0.016 μM. The labeled nucleosome was diluted to 2 nM in identical buffer containing 50 mM NaCl. Twenty microliters of NCP were added to 20 μl of each protein/complex dilution in triplicate in a passivated 384-well plate (Greiner). Following centrifugation, mixing and 10–20 min incubation, plates were scanned with a Typhoon 9410 imager (488 nm excitation; 526 nm emission; PMT 600). Fluorescence was quantitated with ImageQuant software and data was analyzed with ProFit (QuantumSoft). For E2 titration experiments, UbcH5c was added at twice the indicated concentration to each of the Ring1B/Bmi1 dilutions, giving the final indicated concentration following 1:1 mixing with the fluorescent NCP. n = 3 was found to be sufficient for reproducible data (Extended Data Fig. 6) with low standard deviations. One data point of all the data points measured was rejected due to obvious defects in the microplate well. The fused PRC1 ubiquitylation module was most appropriate for measuring affinity constants for mutants of all three components because the higher affinity of the fused complex (relative to E2 or E3 alone) enabled a larger dynamic range for the assay. This allowed the scope of a mutation’s effect to be measured and compared to all other mutants.
BRCA1/BARD1-Ring1B/Bmi1 alignment
The BRCA1/BARD1-Ring1B/Bmi1RING heterodimer alignment was performed using the PyMOL pair_fit command with Cα atoms of BRCA1 residues 22–55 and 60–76 and the corresponding Cα atoms of Ring1B residues 49–79 and 86–102. The alignment gave a rmsd value of 2.69 Å. The BRCA1 loop containing residues 53–59 (corresponding to Ring1B loop residues 80–85) was excluded due to length discrepancy. Divergent regions adjacent to the RING domains were also excluded.
Tetrahedral intermediate model building
To construct the tetrahedral model for the ubiquitylation intermediate, an initial model for ubiquitin was prepared by aligning UbcH5a from the RNF4/UbcH5a/ubiquitin structure16 (PDB ID 4AP4) with UbcH5c in the PRC1 ubiquitylation module-NCP structure. Side chains were built for H2A Lys118 and Lys119 allowing the Lys119 NZ atom to be placed in proximity to the adjacent UbcH5c Cys85 catalytic residue using COOT. A restraint library was constructed using JLigand54 converting ubiquitin Gly76 to ethanolamine and linking its Cβ atom to both the NZ atom of H2A Lys119 and the SG atom of UbcH5c Cys85. The geometry of the model was idealized using REFMAC5. This was repeated for the proximal and distal sides of the structure for H2A Lys 118 and Lys119. Each model resulted in minimal perturbation of the PRC1 ubiquitylation module structure outside of H2A Lys118 and Lys119 residues. Additionally, each model allows addition of H2A residues C-terminal to Lys119 without steric clash.
Extended Data
Extended Data Figure 1. Alignment of PRC1 ubiquitylation module and nucleosome core particle with previously determined sub-structures and comparison of the two halves of the structure.

a, Alignment of PRC1 ubiquitylation model from the proximal (darker colors) and distal (lighter colors) halves of the PRC1 ubiquitylation module-nucleosome core particle structure with the previously determined structure of the PRC1 ubiquitylation module alone22 (grey, PDB ID 3RPG) using all backbone atoms. Similar alignment using b, Ring1B/Bmi1 subcomplex or c, UbcH5c only. d, Alignment of the nucleosome core particle from the PRC1 ubiquitylation module-nucleosome core particle complex (colored) and the previously determined nucleosome core particle complex25 (PDB ID 3LZ0) containing the identical 601 nucleosome positioning sequence. e, Root mean square deviation (rmsd) for each of the alignments calculated over all backbone atoms. f–g, Orthogonal views of an alignment of two copies of the complex following rotation of one copy about the pseudo-two-fold axis of symmetry of the nucleosome. This was accomplished by simultaneously aligning each histone in one copy to its symmetry related counterpart in the other copy. For visualization, the PRC1 ubiquitylation module from one copy of the structure is depicted with lighter colors. h, Zoomed view of the alignment showing PRC1 ubiquitylation module-nucleosome core particle interface. In panel h the proximal and distal PRC1 ubiquitylation modules are shown in dark and light colors, respectively. Hinge indicated by arrows. i, rmsd for each of the components following alignment of the two halves as described above. j, Buried surface area calculations for the indicated interfaces on the proximal and distal sides of the structure.
Extended Data Figure 2. Characterization of fused and unfused UbcH5c.

a, Coomassie stained gel of E1-mediated ubiquitin transfer to UbcH5c Cys85Lys mutant (UbcH5c‡) alone and in the context of the fused PRC1 ubiquitylation module. Ubiquitin is transferred by the E1 Uba1 to the UbcH5c mutant in an ATP dependent manner, black arrow (lanes 1 and 2). No ubiquitin transfer to the fused PRC1 ubiquitylation module is observed (lanes 3 and 4, expected band position for Ring1B-UbcH5c-ubiquitin conjugate indicated by a blue arrow). The Cys85Lys mutant was used to assist in visualization of the UbcH5c-ubiquitin stable isopeptide conjugates. b, Ring1B of the fused PRC1 ubiquitylation module (blue) clashes with ubiquitin in the E1 Uba1 adenylation site (green). Alignment of UbH5c subunit of PRC1 ubiquitylation module22 (PDB ID 3RPG) to Ubc4 in Ubc4/Uba1/ubiquitin structure24 (PDB ID 4II2). Ubc4 and Uba1 are shown pink and grey, respectively. c, Intrisic activity of wild-type and mutant UbcH5c enzymes. Time-course of UbcH5c-ubiquitin thioester aminolysis upon lysine addition. Reactions quenched with non-reducing loading buffer unless 100 mM DTT addition is indicated (†). Asterisk denotes a non-reducible product.
Extended Data Figure 3. Comparison of the proximal and distal Ring1B-, Bmi1-, and UbcH5c-nucleosome interfaces.

Identical views of Ring1B/Bmi1 saddle from a, proximal and b, distal halves of structure and c, overlay. d–g, Ring1B-histone interface views, including d, zoom out with box indicating field of view depicted in zoomed panels from e, proximal and f, distal halves of structure and g, overlay. h–k, Similar views of Bmi1-histone interface. l–s, Simlar views of UbcH5c-DNA interfaces. t–u, 2mFo-DFc difference Fourier transform electron density maps for the Ring1B-nucleosome interface contoured at 1.0 σ on the t, proximal and u, distal sides of the PRC1 ubiquitylation module-nucleosome core particle structure. v–w, Similarly contoured electron density map for the v, proximal and w, distal Bmi1-nucleosome interface. All views are identical to those depicted in Fig. 2 and 3.
Extended Data Figure 4. Chromatin factors use an arginine-anchor to bind the H2A/H2B acidic patch.

Identical views of a, E3 ligase Ring1B (blue), b, viral LANA peptide (purple, PDB ID 1ZLA27), c, chromatin factor RCC1 (purple, PDB ID 3MVD28), d, centromeric protein CENP-C (green, PDB ID 4INM30), and e, silencing protein Sir3 (brown, PDB ID 3TU429) bound the acidic patch. Conserved arginine is shown in space-filling representation. Other arginines involved in binding are shown as sticks. UbcH5c and the Ring1B loop between residues 81 and 98 are not shown for figure clarity.
Extended Data Figure 5. Ring1B/Bmi1- and BRCA1/BARD1-mediated ubiquitylation of nucleosomes containing histone mutations.
Coomassie stained gels of ubiquitylation assays using nucleosomes with the specified histone mutants, E1 Uba1, E2 UbcH5c, STR-His10 tagged ubiquitin and a, Ring1B/Bmi1 or b, BRCA1/BARD1 RING heterodimers. Tagged H2B (STR-His6) was used to prevent electrophoretic comigration with unmodified H2A. Experiment was repeated twice in our laboratory.
Extended Data Figure 6. Fluorescence-based nucleosome ubiquitylation and binding assays.

a, Fluorescence-based nucleosome ubiquitylation assay. Ubiquitylation assays performed with nucleosomes labeled with Oregon Green 488 maleimide on H2A Thr10Cys mutant (H2A T10C-OG488), left. Right, replicates 1, 2 and 3 of wild-type assay run on four different gels and quantified to demonstrate reproducibility across gels. Means and standard deviations shown, n = 4. b, Fluorescence-based nucleosome binding assay. Nucleosomes labeled with Oregon Green 488 on H2B Ser112Cys mutant (H2B S112-OG488), top left. Binding of PRC1 ubiquitylation module leads to partial quenching of the fluorophore allowing affinity measurements to be made, top right. Three technical replicates of fused wild-type PRC1 ubiquitylation module are shown to demonstrate reproducibility, bottom. Means and standard deviations shown for each data point, n = 3.
Extended Data Figure 7. Effects of PRC1 ubiquitylation module mutants on nucleosome ubiquitylation and binding.

a, Representative gel of one replicate of ubiquitylation assay using E1 Uba1, UbcH5c, STR-His10 tagged ubiquitin, nucleosome core particles, and E3 Ring1B/Bmi1 with the indicated Ring1B mutants stained with Coomassie (left) and scanned for fluorescent H2A (right). Nucleosome core particles in the experiment are doped with nucleosome core particles containing the H2A Thr10Cys mutant labeled with Oregon Green 488 maleimide. b, Quantitation of mono- (H2Aub1, dark blue), di- (H2Aub2, blue) and tri-ubiquitylated H2A (H2Aub3, light blue) are shown as a fraction of total H2A. Means and standard deviations from three technical replicates are depicted. Samples from the same experiment were analyzed on different gels, processed in parallel. c, Fluorescence quenching binding curves for fused PRC1 ubiquitylation modules containing the indicated mutations of Ring1B (colored as shown). Means and standard deviations are shown with n = 3 for each data point. Fluorescence is normalized to fit values for unbound and saturated nucleosome core particles. Concentrations depicted using log scale. Experiments as described above for d–f, indicated Bmi1 mutants, g–i, UbcH5c charge reversal mutants and j–l, UbcH5c alanine mutants. Triplicate ubiquitylation assays were repeated at least two times in our laboratory.
Extended Data Figure 8. Alignment of Bmi1 paralogs and E2s.

a, Sequence alignment of segments of Bmi1 with PCGF (Polycomb group RING finger) paralogs also found in PRC1. b, Sequence alignment of UbcH5c and other E2s known to be active (green) or inactive with (red) or of unknown compatibility with Ring1B/Bmi1-mediated nucleosomal ubiquitylation. Key residues discussed in the text are indicated with an asterisk.
Extended Data Figure 9. Linker DNA increases the affinity of the PRC1 ubiquitylation modules for the nucleosome.

a, Linear B-form duplex DNA modeled onto the DNA ends of the PRC1 ubiquitylation module-nucleosome core particle structure. b, The UbcH5c α4 helix occupies the major groove of modeled linker DNA. Several basic and aromatic side chains line the DNA-adjacent face of the α4 helix. c, Linker DNA enhances nucleosomal binding of fused PRC1 ubiquitylation module. Fluorescence quenching binding curves for the wild-type fused PRC1 ubiquitylation module binding to nucleosomes centered on 147 bp (black, grey, and light grey) or 185 bp (blue and light blue) 601 DNA. Technical replicates performed on different days are depicted in different shades of grey and blue. Means and standard deviations are shown with n = 3 for each data point. Fluorescence is normalized to fit values for unbound and saturated nucleosome core particles. Concentrations depicted using log scale.
Extended Data Figure 10. BRCA1 requires similar loop region and H2A/H2B acidic patch for nucleosome ubiquitylation.
a, Alignment of the BRCA1/BARD1 and Ring1B/Bmi1 heterodimers using the RING domains of BRCA1 from the BRCA1/BARD1 NMR structure35 (PDB ID 1JM7) and Ring1B. BRCA1 and BARD1 are shown in orange and purple, respectively; Cα atoms of essential arginine residues are indicated by spheres. b, Sequence alignment of Ring1B and BRCA1 shows conserved nucleosome interacting loop. c, BRCA1 Lys70Ala/Arg71Ala mutation eliminates E3 ligase activity of BRCA1/BARD1 RING heterodimer. Representative gel of one replicate of ubiquitylation assay using E1 Uba1, E2 UbcH5c, STR-His10 ubiquitin, nucleosome core particles, and E3 BRCA1/BARD1 with the indicated mutants stained with Coomassie and scanned for fluorescent H2A. Nucleosome core particles in the experiment are doped with nucleosome core particles containing the H2A Thr10Cys mutant labeled with Oregon Green 488 maleimide. d, Quantitaion of mono- (H2Aub1, dark orange), di- (H2Aub2, orange) and tri-ubiquitylated H2A (H2Aub3, light orange) are shown as a fraction of total H2A. Means and standard deviations from three technical replicates are depicted. Ubiquitylation assay was repeated twice in our laboratory.
Supplementary Material
Acknowledgments
This work was supported by NIGMS grants GM060489-09S1, GM088236 and GM111651 to S.T.. R.K.M. is supported by a Damon Runyon Post-doctoral fellowship (DRG 2107-12).
We would like to thank the staff of APS NE-CAT beamlines 24ID-E and 24ID-C for their assistance during synchrotron data collection; Hemant Yennawar, Neela Yennawar and Julia Fecko at the Penn State Huck Institute X-ray core facility; Allen Minns, Mike Moore and Justin Malloy for reagent preparation; Tavarekere Girish and Jiehuan Huang for assistance with data collection; Shu Wang for providing U2OS cDNA; Ravindra Makde and members of the Tan laboratory and the Penn State Center for Eukaryotic Gene Regulation for helpful discussions.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
R.K.M. designed the study, cloned, purified, and crystallized macromolecules, collected and processed X-ray data, refined and analyzed the structure, performed nucleosome ubiquitylation and binding assays, and wrote the paper. R.C.H. cloned, purified, and crystallized macromolecules and performed nucleosome ubiquitylation assays. S.T. designed the study, cloned and purified macromolecules, collected X-ray data, analyzed the structure and edited the paper. All authors commented on the manuscript.
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data Bank under accession code 4R8P.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature Cell Biology. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 2.Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nature Structural & Molecular Biology. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 3.Laugesen A, Helin K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell. 2014;14:735–751. doi: 10.1016/j.stem.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9:773–784. doi: 10.1038/nrc2736. [DOI] [PubMed] [Google Scholar]
- 5.Crea F, Paolicchi E, Marquez VE, Danesi R. Polycomb genes and cancer: Time for clinical application? Critical Reviews in Oncology / Hematology. 2012;83:184–193. doi: 10.1016/j.critrevonc.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature Reviews Molecular Cell Biology. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz YB, Pirrotta V. A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet. 2013;14:853–864. doi: 10.1038/nrg3603. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 9.Cao R, Tsukada YI, Zhang Y. Role of Bmi-1 and Ring1A in H2A Ubiquitylation and Hox Gene Silencing. Molecular Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Gao Z, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Molecular Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon JA, Kingston RE. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Molecular Cell. 2013;49:808–824. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackledge NP, et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014;157:1445–1459. doi: 10.1016/j.cell.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farcas AM, et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife. 2012;1:e00205–e00205. doi: 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu X, Johansen JV, Helin K. Fbxl10/Kdm2b Recruits Polycomb Repressive Complex 1 to CpG Islands and Regulates H2A Ubiquitylation. Molecular Cell. 2013;49:1134–1146. doi: 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Pickart CM, Pickart CM. Mechanisms underlying ubiquitination. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 16.Plechanovová A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489:115–120. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pruneda JN, et al. Structure of an E3:E2~Ub Complex Reveals an Allosteric Mechanism Shared among RING/U-box Ligases. Molecular Cell. 2012;47:933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nature Structural & Molecular Biology. 2012;19:876–883. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deshaies RJ, Joazeiro CAP. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 20.Buchwald GG, et al. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 2006;25:2465–2474. doi: 10.1038/sj.emboj.7601144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 22.Bentley ML, et al. Recognition of UbcH5c and the nucleosome by the Bmi1/Ring1b ubiquitin ligase complex. EMBO J. 2011;30:3285–3297. doi: 10.1038/emboj.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattiroli F, Uckelmann M, Sahtoe DD, van Dijk WJ, Sixma TK. The nucleosome acidic patch plays a critical role in RNF168-dependent ubiquitination of histone H2A. Nature Communications. 2014;5:3291–3291. doi: 10.1038/ncomms4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen SK, Lima CD. Structure of a ubiquitin E1-E2 complex: insights to E1-E2 thioester transfer. Molecular Cell. 2013;49:884–896. doi: 10.1016/j.molcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasudevan D, Chua EYD, Davey CA. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. Journal of Molecular Biology. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 26.Leung JW, Agarwal P, Canny MD, Gong F, Robison AD. Nucleosome acidic patch promotes RNF168-and RING1B/BMI1-dependent H2AX and H2A ubiquitination and DNA damage signaling. PLoS Genet. 2014 doi: 10.1371/journal.pgen.1004178.s007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbera AJ, et al. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 2006;311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 28.Makde RD, England JR, Yennawar HP, Tan S. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 2010;467:562–566. doi: 10.1038/nature09321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armache KJ, Garlick JD, Canzio D, Narlikar GJ, Kingston RE. Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science. 2011;334:977–982. doi: 10.1126/science.1210915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato H, et al. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science. 2013;340:1110–1113. doi: 10.1126/science.1235532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hieb AR, D’Arcy S, Kramer MA, White AE, Luger K. Fluorescence strategies for high-throughput quantification of protein interactions. Nucleic Acids Research. 2012;40:e33–e33. doi: 10.1093/nar/gkr1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reverter D, Lima CD. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005;435:687–692. doi: 10.1038/nature03588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalb R, Mallery DL, Larkin C, Huang JTJ, Hiom K. BRCA1 Is a Histone-H2A-Specific Ubiquitin Ligase. Cell Reports. 2014 doi: 10.1016/j.celrep.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Z, et al. NMR structure of chaperone Chz1 complexed with histones H2A.Z-H2B. Nature Structural & Molecular Biology. 2008;15:868–869. doi: 10.1038/nsmb.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan S, Kern RC, Selleck W. The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expression and Purification. 2005;40:385–395. doi: 10.1016/j.pep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of Molecular Biology. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 39.Luger KK, Rechsteiner TJT, Richmond TJT. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 40.D’Arcy A, Mac Sweeney A, Haber A. Practical aspects of using the microbatch method in screening conditions for protein crystallization. Methods. 2004;34:323–328. doi: 10.1016/j.ymeth.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnaudo N, et al. The N-terminal acetylation of Sir3 stabilizes its binding to the nucleosome core particle. Nature Structural & Molecular Biology. 2013;20:1119–1121. doi: 10.1038/nsmb.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tachiwana HH, et al. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature. 2011;476:232–235. doi: 10.1038/nature10258. [DOI] [PubMed] [Google Scholar]
- 51.Iwasaki W, et al. Contribution of histone N-terminal tails to the structure and stability of nucleosomes. FEBS Open Bio. 2013;3:363–369. doi: 10.1016/j.fob.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 2010;26:283–291. [Google Scholar]
- 53.The PyMOL Molecular Graphics, Version 1.7.0.5. Shrödinger, LLC; [Google Scholar]
- 54.Lebedev AA, et al. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr. 2012;68:431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.