

Abstract
The conventional route to alkoxyamine hydrochloride derivatives is by reaction of alkyl bromides with N-hydroxyphthalimide or N-hydroxysuccinimide followed by addition of hydrazine and HCl. Transformation of an alkyl bromide to the corresponding alkoxyamine hydrochloride can be accomplished more rapidly in high yield and without using hazardous hydrazine by reaction of (Boc)2NOH (N,N’-di-tert-butoxycarbonylhydroxylamine) and alkyl bromide followed by addition of HCl. Alkoxyamine hydrochlorides are powerful reagents in organic synthesis that can be used to synthesize alkoxyimino derivatives after condensation with a ketone or aldehyde.
Keywords: alkoxyamine, alkyl bromide, O-alkylation
Introduction
Synthesis of alkoxyimino derivatives through condensation of an alkoxyamine hydrochloride with a ketone or aldehyde with is a very powerful tool to introduce a heteroatom, i.e. nitrogen, in organic synthesis.1 There are two current approaches to the synthesis of R-ONH2 (alkoxyamino derivatives). One approach involves conversion of an alcohol to R-ONH2 using: a) by displacement of an alcohol using N-hydroxyphthalimide under Mitsunobu conditions and subsequent treatment with hydrazine2 or b) by direct amination through reaction of an alcohol and a substituted oxaziridine.3 Another approach consists of substitution of R-Br/I with N-hydroxyphthalimide (Gabriel synthesis), N-hydroxysuccinimide or another N-protected hydroxylamine.4 It appears that new methods for the above conversion are needed, and here we have developed a method through which R-ONH2 can be synthesized rapidly and in high yield from R-Br by reaction with (Boc)2NOH.
Results
N,N’-di-tert-Butoxycarbonylhydroxylamine ((Boc)2NOH) 1 was synthesized from BzONH2.HCl as a white solid in high yield.5 Reaction of (Boc)2NOH and R-Br with Hünig’s base (DIPEA) or DBU in DMF at room temperature is slow and normally requires 12-24 h for reaction completion. The reaction rate can be accelerated by heating at 50 °C in DMF to achieve completion typically in 1 to 2 h. Unlike the synthesis of R-ONH2 using R-Br and N-hydroxyphthalimide or N-hydroxysuccinimide, by this method hydrazine is not needed to convert the acylated N-hydroxy adduct to R-ONH2, and the Boc protecting groups can be removed easily in acidic conditions. Normally, the intermediate R-ON(Boc)2 is dissolved in CH2Cl2 and treated with 4 M HCl (16 eq) in dioxane at room temperature, and the mixture stirred for 6-12 h. The resulting R-ONH2.HCl can be isolated as a precipitate by filtration.
Thus, we have demonstrated a practical and efficient synthetic route to R-ONH2.HCl from R-Br in two steps. The product R-NH2.HCl can be easily isolated in high yield mainly by precipitation. The starting (Boc)2NOH is very stable for several months at room temperature and for more than 1 year at 4 °C. Although several methods for the synthesis of R-NH2.HCl from R-Br are available, improved methods to obtain R-ONH2 are needed to overcome some of the existing drawbacks, most notably use of toxic and hard to remove hydrazine in the Gabriel synthesis. One important biological application of R-ONH2.HCl (e.g. 5b) is in the synthesis of N4-alkoxy modified cytidine derivatives (Scheme 2), which after phosphorylation have proven to be potent and selective ligands of P2Y nucleotide receptors.1d,1e The target O-substituted hydroxylamine compounds are also useful for the orthogonal labeling of proteins and surfaces of cells and biomaterials.1a,1b,5
Scheme 2.

Intended biological application of alkoxyamine hydrochloride 5b and related derivatives for the study of P2Y nucleotide receptor agonists, such as 9 (MRS4062).
Chemical Synthesis
N,N’-di-tert-Butoxycarbonylhydroxylamine (1)
Compound 1 was obtained by a modification of two literature procedures (Supporting information).6,7 The product 1 was isolated as a homogeneous, white crystalline solid. 1H NMR (400 MHz, CDCl): 1.50. 13C NMR (100 MHz, CDCl3): 150.9, 84.5, 28.0. Melting point (0C): 87.4 ± 0.5. HRMS EI m/z (M – H); found: 232.1186 (M – H+)−; calc for C10H18O5N: 232.1190. The alkoxyamine hydrochloride derivatives (2b – 6b) were prepared using the synthetic routes shown in Scheme 1. A typical reaction procedure to obtain R-ONH2 is as follows: To a magnetically stirred mixture of 6a (0.36 mmol, 83 mg, 1 eq) and 1 (0.34 mmol, 79 mg, 0.95 eq) in 0.5 mL of DMF was added DBU (0.42 mmol, 60 μL, 1.2 eq) at room temperature under N2. Then, the mixture was heated to 50 °C and stirred for 2 h. Reaction was monitored using TLC. After completion, solvent was removed and the mixture was dissolved in EtOAc (50 mL) and washed with water and brine. The organic phase was dried (Na2SO4) and re-dissolved in CH2Cl2 (1 mL) in a round bottom flask, treated with 4M HCl in dioxane (5.7 mmol, 1.4 mL, 16 eq) and stirred overnight. The resulting white precipitate was filtered and washed with 1 mL of CH2Cl2 and dried to obtain 6b (0.30 mmol, 65 mg, 87%).
Scheme 1.
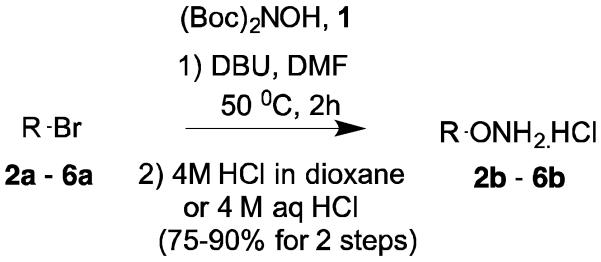
General reaction condition for O-alkylation of hydroxylamine in two steps.
O-(3-(4-Methoxyphenyl)propyl)hydroxylamine hydrochloride (6b)
1H NMR (400 MHz, MeOD): 7.06 (d, J1 = 8.52 Hz, 2H), 6.80 (d, J1 = 8.50 Hz, 2H), 3.97 (t, J1 = 6.44 Hz, 2H), 3.72 (s, 3H), 2.62 (t, J1 = 7.44 Hz, 2H), 1.94 (m, 2H). 13C NMR (100 MHz, MeOD): 158.2, 132.5, 128.9, 113.5, 74.1, 54.2, 30.2, 29.2. HRMS ESI m/z (M+H) found: 182.1183; calc for C10H16NO2: 182.1181.
Table 1.
Results of reaction of R-Br and (Boc)2NOH, varying group R.
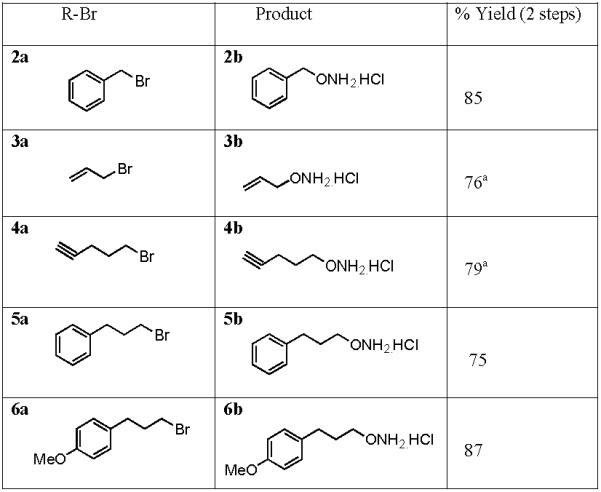
|
Product was isolated from 4 M aqueous HCl.
Acknowledgements
Support from the Intramural Research Program of NIDDK, National Institutes of Health is acknowledged.
Footnotes
Supporting Information: Supplemental data for this article can be accessed on the publisher’s website.
Contributor Information
P. Suresh Jayasekara, Molecular Recognition Section, Laboratory of Bioorganic Chemistry.
Kenneth A. Jacobson, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892
References
- 1.(a) Lemieux GA, Yarema KJ, Jacobs CL, Bertozzi CR. J. Am. Chem. Soc. 1999;121:4278–4279. [Google Scholar]; (b) Perouzel E, Jorgensen MR, Keller M, Miller AD. Bioconjug. Chem. 2003;14:884–898. doi: 10.1021/bc034068q. [DOI] [PubMed] [Google Scholar]; (c) Sadamoto R, Niikura K, Ueda T, Monde K, Fukuhara N, Nishimura SI. J. Am. Chem. Soc. 2004;126:3755–3761. doi: 10.1021/ja039391i. [DOI] [PubMed] [Google Scholar]; (d) Maruoka H, et al. J. Med. Chem. 2011;23(12):4018–33. doi: 10.1021/jm101591j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jayasekara PS, et al. Med. Chem. Commun. 2013;4(8):1156–1165. [Google Scholar]
- 2.Palandoken H, Bocian CM, McCombs MR, Nantz MH. Tetrahedron Letters. 2005;46:39, 6667–6669. [Google Scholar]
- 3.Choong IC, Ellman JC. J. Org. Chem. 1999;64:6528–6529. doi: 10.1021/jo990490h. [DOI] [PubMed] [Google Scholar]
- 4.Rougny Annie, Daudon Marc. Bulletin de la Societe Chimique de France. 1976;5-6(Pt. 2):833–838. [Google Scholar]
- 5.Fleissner MR, Brustad EM, Kálai T, Altenbach C, Cascio D, Peters FB, Hideg K, Peuker S, Schultz PG, Hubbell WL. Proc. Natl. Acad. Sci. USA. 2009;106:21637–21642. doi: 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gouliaev AH, Brown WD, Wätjen F. 2004 Mar 04; PCT Int. Appl., 2004018466.
- 7.Chilkoti A, Gao W. 2010 Aug 26; PCT Int. Appl., 2010096422.