

Abstract
It is now a well-accepted view that cation-driven Cl− transporters in neurons are involved in determining the intracellular Cl− concentration. In the present review, we propose that additional factors, which are often overlooked, contribute substantially to the Cl− gradient across neuronal membranes. After briefly discussing the data supporting and opposing the role of cation–chloride cotransporters in regulating Cl−, we examine the participation of the following factors in the formation of the transmembrane Cl− gradient: (i) fixed ‘Donnan’ charges inside and outside the cell; (ii) the properties of water (free vs. bound); and (iii) water transport through the cotransporters. We demonstrate a steep relationship between intracellular Cl− and the concentration of fixed negative charges on macromolecules. We show that in the absence of water transport through the K+–Cl− cotransporter, a large osmotic gradient builds at concentrations below or above a set value of ‘Donnan’ charges, and show that at any value of these fixed charges, the reversal potential for Cl− equates that of K+. When the movement of water across the membrane is a source of free energy, it is sufficient to modify the movement of Cl− through the cotransporter. In this scenario, the reversal potential for Cl− does not closely follow that of K+. Furthermore, our simulations demonstrate that small differences in the availability of freely diffusible water between inside and outside the cell greatly affect the Cl− reversal potential, particularly when osmolar transmembrane gradients are minimized, for example by idiogenic osmoles. We also establish that the presence of extracellular charges has little effect on the chloride reversal potential, but greatly affects the effective inhibitory conductance for Cl−. In conclusion, our theoretical analysis of the presence of fixed anionic charges and water bound on macromolecules inside and outside the cell greatly impacts both Cl− gradient and Cl− conductance across neuronal membranes.
The old dogma: Cl− passively distributed
For many decades, physiologists have lived with the knowledge that intracellular Na+ and K+ are actively transported and with the dogma that Cl− is passively distributed across the plasma membrane (Coombs et al. 1955). Certainly, since the mid-1950s, it has been well established that most cells (with a few exceptions, e.g. dog and sheep red blood cells) maintain a high intracellular K+ concentration ([K+]i) while extruding Na+ through the use of an ATP-consuming enzyme that exchanges three Na+ ions for two K+ ions. Discovery of the Na+–K+ pump or ATPase was made by Robert Post and Jens Christian Skou in 1957 (Post & Jolly, 1957; Skou, 1957). Thus, it is now well recognized that intracellular Na+ and K+ are kept in pseudo-equilibrium, far away from electrochemical potential equilibrium, by the action of a pump and leak system (Tosteson & Hoffman, 1960). Intracellular Cl−, in contrast, has long been regarded to be passively distributed across the plasma membrane or distributed according to its electrochemical potential equilibrium (Boyle & Conway, 1941; Hodgkin & Horowicz, 1949). Chloride will be at thermodynamic equilibrium when the combined chemical and electrical force of the ion is equal on both sides of the membrane. The Cl− potential is the electrical force generated by the diffusion of chloride ions across the membrane, which is driven by the chemical concentration gradient existing across the membrane:
| (1) |
where R is the gas constant (8.314 J K−1 mol−1), Z the valence of the ion (−1 for Cl−) and F the Faraday constant (9.65 × 104 C mol−1).
The membrane potential of a cell (Vm) is defined by the diffusion of positive and negative charges across its plasma membrane; Vm is mathematically represented by the Goldman equation, which accounts for individual ion permeability (Pion) and concentration on each side of the membrane, as follows:
| (2) |
As the permeability of K+ is far greater than that of Na+ and Cl−, the Goldman equation simplifies to the Nernst equation, i.e. the equilibrium potential for K+:
| (3) |
At rest, the membrane potential varies from cell type to cell type from −40 to −80 mV. Chloride is at equilibrium if its reversal potential, shown in eqn (4), is equal to the membrane potential.
| (4) |
Figure 1 plots [Cl−]i as a function of [Cl−]o at different membrane potentials. It can be seen that for a neuron with a membrane potential of −75 mV and a [Cl−]o of 140 mm [typical Ringer solution or artificial cerebrospinal fluid (CSF)], [Cl−]i at rest is slightly below 8 mm; and if [Cl−]o is lowered to 110 mm (as in vivo), [Cl−]i at rest is closer to 6 mm. This concentration is in the range of what has been measured in mature neurons (6–13 mm; Misgeld et al. 1986; Thompson et al. 1988b; Owens et al. 1996; Ehrlich et al. 1999). As we will discuss in the “Distinct roles for [A−]i and [A−]o” section, the extracellular chloride concentration ([Cl−]o) is more complex than what we have introduced here, because fixed extracellular anions locally alter [Cl−]o at the neuronal membrane.
Figure 1. Relationship between intra- and extracellular chloride concentrations based on the Nernst equation.
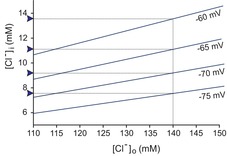
The linear relationship was plotted at membrane potentials ranging from −60 to −75 mV. Values of [Cl−]i for an artificial CSF chloride concentration of 140 mm are indicated by blue arrowheads. Values of [Cl−]i for more a physiological [Cl−]o of 110 mm are indicated by the intercepts on the y-axis. Note that for physiological membrane potentials of −70 to −75 mV, the neuronal intracellular Cl− concentration is low (<10 mm).
Chloride is in fact not passively distributed and the birth of a new dogma: the SLC12A transporters
Inhibitory neurotransmission is mediated by the activation of ligand-gated Cl− channels, such as the GABA receptor type-A (GABAA receptor) and the glycine receptor. The function of these two receptor types counteracts the action of glutamate receptors, which provide the nervous system excitatory drive. Based on intracellular recordings of inhibitory postsynaptic potentials, it became clear in the late 1970s to early 1980s that Cl− must not be passively distributed across the membrane, else GABAA receptor activation would not change the resting membrane potential. In fact it was then rapidly realized, against the old dogma, that peripheral neurons and immature CNS neurons accumulate Cl− well above the thermodynamic equilibrium potential, while mature CNS neurons have Cl− concentrations lower than expected based on the thermodynamic equilibrium potential (reviewed by Delpire, 2000; Ben-Ari, 2002; Blaesse et al. 2009).
The idea that mature neurons need an ‘active’ Cl− extrusion mechanism to set the concentration to very low values arose from the observation of hyperpolarizing responses to the activation of anion-selective ligand-gated channels opened by the action of the neurotransmitters glycine and GABA (Misgeld et al. 1986; Thompson et al. 1988a), reflecting influx of anions along an electrochemical gradient. However, a variety of artifacts affect the electrophysiological measurement of ECl, from dialysis of the cytoplasm by the electrode solution (Coombs et al. 1955) to increases in cell volume and chloride induced by the trauma of slice preparation (Dzhala et al. 2012). Techniques that reduce dialysis artifacts, including perforated-patch recordings using the cation-selective ionophore gramicidin (Ebihara et al. 1995) and dual cell-attached measurements of the driving force for Cl− (Tyzio et al. 2008), indicate higher concentrations of cytoplasmic Cl−, as do Cl−-sensitive fluorophores (Kuner & Augustine, 2000). This last technique indicates that non-traumatized mature neurons have [Cl−]i values that are normally distributed, with medians ranging from 6 to 14 mm (Berglund et al. 2006; Glykys et al. 2014).
The molecular identification (cloning) of electroneutral cation–chloride cotransporters (Delpire et al. 1994; Payne et al. 1996) and the demonstration of their expression in neurons in the 1990s (Payne et al. 1996; Plotkin et al. 1997a,b1997b; Lu et al. 1999) provided mechanisms suitable to explain why Cl− sits away from the thermodynamic equilibrium potential. Indeed, these transporters can be viewed as ‘pumps’ that are able to drive one ion uphill (against its driving force) by using the gradient energy of other cotransported ionic species (concept of ‘secondary active’ transporters), and all of this in an electroneutral fashion. Thus, an Na+–K+–2Cl− cotransporter, such as NKCC1 (SLC12A2), would be able to drive Cl− inside neurons by using the energy of the Na+ gradient generated by the Na+/K+ pump, whereas a K+–Cl− cotransporter, e.g. KCC2 (SLC12A5), would be able to drive Cl− out of neurons by using the energy of the K+ gradient (Delpire, 2000; Ben-Ari, 2002; Blaesse et al. 2009).
Whether Na+–K+–2Cl− cotransporter function truly results in Cl− accumulation is a matter of argument. The debate seems to be settled for sensory afferent neurons, which demonstrate [Cl−]i in the range of 30–40 mm. The pioneering work of Javier Alvarez-Leefmans in amphibian dorsal root ganglion neurons first identified the Na+–K+–2Cl− cotransporter as the main mechanism for inward transport of Cl− (Alvarez-Leefmans et al. 1988). Using ion-selective microelectrodes, he demonstrated an inward Cl− movement that was directly dependent upon external Cl−, Na+ and K+ ions. Interestingly for the purpose of this review, the frog dorsal root ganglion neuron study reported the effects of only furosemide and bumetanide (drugs known in clinical medicine as Lasix and Bumex, used to inhibit Na+ and water reabsorption in the thick ascending loop of Henle, and thus referred to as loop diuretics) on the recovery of [Cl−]i following incubation of the cells in a Na+-free solution and their return to a regular saline. As anticipated, the loop diuretics greatly affected the recovery of [Cl−]i. What the study did not do is to test whether application of the Na+–K+–2Cl− cotransporter inhibitors in control conditions resulted in a progressive decrease of [Cl−]i towards equilibrium, nor did it test whether the recovery of [Cl−]i after the Na+-free conditions in the presence of the Na+–K+–2Cl− cotransporter inhibitors occurred over a much longer period of time. To summarize, the study clearly demonstrated that the Na+–K+–2Cl− cotransporter constitutes a pathway for Cl− movement in these cells, but did not settle the involvement of the cotransporter in setting [Cl−]i to 30–40 mm.
This was later addressed in mouse dorsal root ganglion neurons, which were shown to express NKCC1, one of the two isoforms of the Na+–K+–2Cl− cotransporter (Plotkin et al. 1997a). Dorsal root ganglion neurons isolated from NKCC1 knockout mice demonstrated an EGABA that was 30 mV more hyperpolarized than the EGABA of control dorsal root ganglion neurons (EGABA is the potential at which GABA reverses its effect; as a first approximation, it generally equals ECl). In fact, in knockout neurons, EGABA was extremely close to the resting membrane potential (Sung et al. 2000). From this study, it was clear that the lack of NKCC1 expression resulted in the absence of Cl− accumulation above electrochemical potential equilibrium. Note that for these experiments, dorsal root ganglion neurons were isolated from adult mice which, in contrast to central neurons, undergo minimal decrease in [Cl−]i during development (Mao et al. 2012).
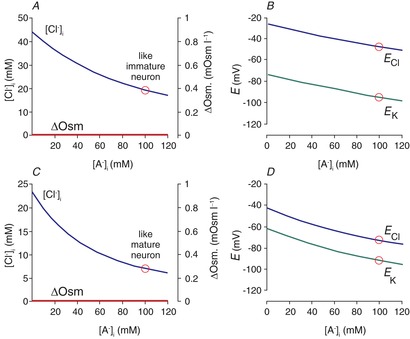
It was best demonstrated in cortical (Owens et al. 1996) and auditory brainstem neurons (Ehrlich et al. 1999) that CNS neurons undergo a developmental decrease in [Cl−]i (Fig. 2A and B). This decrease coincides with the decreased expression of NKCC1 and increased expression of KCC2 (Plotkin et al. 1997b; Clayton et al. 1998; Lu et al. 1999; Dzhala et al. 2005). Complete absence of KCC2 in the mouse results in respiratory failure at birth, causing lethality (Hübner et al. 2001), whereas disruption of the major isoform (KCC2b) results in seizure activity in pups that also leads to postnatal lethality (Woo et al. 2002). Thus, KCC2 function is unquestionably critical to proper neuronal function. Whether the phenotypes were related to a deficiency in fast regulation of Cl− upon neuronal activity or related to an intraneuronal chloride concentration that is in equilibrium with the membrane potential has not yet been resolved.
Figure 2. Developmental decrease of [Cl−]i measured in neurons.
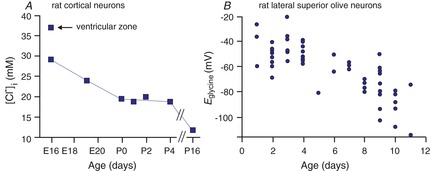
A, [Cl−]i in rat cortical neurons is plotted as a function of age from late embryonic (E) stages to postnatal (P) day 16. The data are replotted from Owens et al. (1996). B, the reversal potential for glycine (Eglycine) measured in individual rat lateral superior olive neurons as a function of postnatal age, P1–P11. Data are redrawn from Ehrlich et al. (1999).
Evidence questioning the role of SLC12A transporters
The Na+–K+–2Cl− cotransporter NKCC1 is also expressed in central neurons (Plotkin et al. 1997a), where it is developmentally regulated (Plotkin et al. 1997b; Yamada et al. 2004; Dzhala et al. 2005). While the downregulation coincides in many brain regions with the developmental decrease in [Cl−]i, is the transporter truly involved in Cl− accumulation? In a study published in the Journal of Neuroscience in 2009 (Sipilä et al. 2009), gramicidin perforated-patch recordings demonstrated that EGABA in young (postnatal day 3–4) CA3 pyramidal neurons from NKCC1 knockout mice was 16 mV more negative than in age-matched control mice, indicating that the cotransporter does participate in Cl− accumulation, but a similar study also using NKCC1-deficient mice (although of a different background strain) showed a much smaller shift in EGABA (Pfeffer et al. 2009).
In knockout experiments, it is not easy to differentiate the substantial developmental effects of NKCC1 knockout (Wang & Kriegstein, 2008, 2011) from transport effects. Thus, many studies have used acute pharmacological blockade of NKCC1 with low concentrations of the loop diuretic bumetanide. A 12 mV shift, corresponding to a 10 mm calculated change in [Cl−]i, was measured in postnatal day 2 rat neocortical neurons exposed to 20 μm bumetanide (Yamada et al. 2004), whereas only a 3 mV shift in EGABA, corresponding to a 3 mm decrease in [Cl−]i, was observed in postnatal day 4–6 rat CA1 pyramidal neurons exposed to bumetanide (Dzhala et al. 2005).
In auditory brainstem neurons, which demonstrate the nice developmental decrease in [Cl−]i (evidenced by the change in the reversal potential for glycine (Eglycine) shown in Fig. 2B), expression of NKCC1 is minimal at early age, and therefore, the cotransporter cannot account for the Cl− accumulation (Balakrishnan et al. 2003). The situation is similar in developing retinal neurons, which exhibit high [Cl−]i early in development to the same extent in wild-type and NKCC1 knockout cells (Zhang et al. 2007). However, GABA-gated calcium transients in neurons in olfactory brain slices are reduced by bumetanide, suggesting that GABAergic depolarization requires the action of NKCC1 in these cells (Sun et al. 2012). Thus, a series of experiments using different preparations and techniques have produced conflicting data regarding the role of NKCC1 in chloride accumulation.
Some of these differences in results may be explained by the finding of Dzhala et al. (2012) that the trauma involved in slice preparation results in a superficial layer of 40–100 μm in the slice in which neurons are swollen, caspases are activated and [Cl−]i is elevated. The elevated [Cl−]i in these injured neurons was reduced by bumetanide. This raises the possibility that in some experimental series, the strong dependence of [Cl−]i on bumetanide might be a function of traumatic injury during slice preparation.
Another potential explanation for these disparate results is the wide range of [Cl−]i observed in healthy neurons (Ebihara et al. 1995; Kuner & Augustine, 2000; Dzhala et al. 2005; Berglund et al. 2006; Glykys et al. 2009). Electrophysiologists (including ourselves) cannot afford to record from hundreds of neurons, and in addition, develop preferences as to what sorts of cell properties are most likely to result in a stable recording. In light of what we will discuss in the next section, regarding water transport by KCC2, it is not surprising that an electrophysiologist who is visualizing neurons prior to patching might obtain different results regarding the effects of transport inhibitors on ECl, depending on his/her preferences regarding the degree of turgor in neurons selected for patching. How ‘crinkly’ the membrane of the neuron appears when patching under visual guidance, and the amplitude of the initial voltage deflection when patching blindly, will select for neurons with a different turgor, i.e. osmotic pressure, and thus, different EGABA.
A third explanation would be the osmolarity of the extracellular fluid used in the experiment. Indeed, NKCC1 was shown to be an obligate transporter of water, along with salt in roughly isotonic proportions (Hamann et al. 2010). Thus, NKCC1-mediated Cl− transport will be influenced by the free energy of water transport. This could produce substantial experimental variances if the osmolarity of the perfusate differed from the osmolarity to which the neurons had most recently been equilibrated (in vivo for acute slices or in the culture media for organotypic and dissociated cell cultures). This possibility will be discussed in more detail in the next sections.
Diffusion equilibrium and Donnan effect
To introduce the concept of diffusion equilibrium, before we consider the more complex case of a cell, let us first consider the equilibrium that develops between a small and rigid compartment separated from a second compartment of infinite volume by a membrane that is equally permeable to both Na+ and Cl−. Wall rigidity allows us to ignore any osmotic effect (water movement) that occurs during ion disequilibrium. In this example (Fig. 3), if we place 1 mm NaCl in the first compartment and 10 mm NaCl in the other compartment, we can immediately see that to reach equilibrium, both Na+ and Cl− diffuse so that 10 mm of each species is on either side, or 9 mm NaCl has to be added to the first compartment. After ions redistribute and a new equilibrium is reached, the energy distribution for each ion must be equal (otherwise more net diffusion will take place) and therefore:
Figure 3. Model of a two-compartment system starting with different NaCl concentrations separated by a permeable membrane.
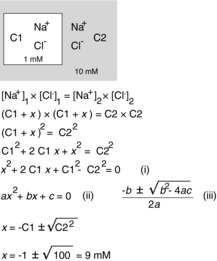
We derive the equation for the new equilibrium that results from salt redistribution. The derivation leads to an equation (i), which has the general form of a quadratic equation (ii), and the solution to which is given by the traditional formula (iii). The ‘x’ is the amount (or concentration) of NaCl that moves from compartment C2 to compartment C1. Using equation (iii), x is calculated to be 9 mm. Note that this model has the following two important assumptions: there is no water movement, because the membrane between the compartments is postulated to be rigid; and the volume of C2 is infinite.
or:
Note that the ion ratio is inversed for cation and anion, due to Z = +1 for Na+ and Z = −1 for Cl−, or:
or:
or:
| (5) |
Second, to minimize charge separation:
| (6) |
Solving this system of equations (eqns (5) and (6) is straightforward in this simplistic example, because it gives 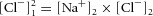 or, for the example in which ions are added to the first compartment until equilibrium with the second compartment is reached, [Cl−]1 = √100 = 10. Note that there is a quadratic relationship between work and [Cl−]1, which is made more obvious when solving for the change in [Cl−]1 (see Fig. 3).
or, for the example in which ions are added to the first compartment until equilibrium with the second compartment is reached, [Cl−]1 = √100 = 10. Note that there is a quadratic relationship between work and [Cl−]1, which is made more obvious when solving for the change in [Cl−]1 (see Fig. 3).
Living cells are much more complex systems. For one thing, HCO3− is present on both sides of the membrane, and via hydration/dehydration reactions, is freely diffusible across the membrane as CO2. For another thing, cells contain many osmotically active substances besides permeable cations and anions. In fact, in the majority of cells chloride and HCO3− are minor constituents of the cytoplasmic anionic milieu. Most anions are moieties of macromolecules: phosphates, sulfates and amino acids that are negatively charged at physiological intracellular pH. Donnan addressed the effect of an intracellular impermeable anion (A) on the distribution of permeable cations and Cl− across the membrane (Donnan, 1911). The conditions are all the same as for Fig. 3, apart from intracellular charge distribution, where:
| (7) |
The extracellular Na+, K+ and Cl− concentrations are set to 140, 4 and 110 mm, respectively. Note that these extracellular concentrations result in what physiological textbooks refer to as an anion gap, which we eliminate by adding 24 mm HCO3− and 10 mm impermeable [large molecules] anions ([A−]o). Diffusion equilibrium necessitates that the product of diffusible ions inside and outside is also equal. In the case of membranes expressing the K+– Cl− cotransporter KCC2, the permeability of K+ and Cl− is greatly increased via facilitated codiffusion, and the permeability of other ions can be neglected, so that from eqn (5):
| (8) |
Equations (7) and (8) are also the necessary conditions for a Donnan equilibrium, in which the distribution of permeable ions across a membrane is altered by the presence of the impermeable anion, [A−]i. Combining eqns (7) and (8) provides the following quadratic equation:
| (9) |
whose solution is eqn (10):
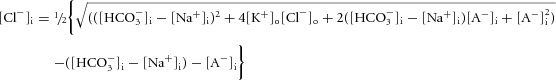 |
(10) |
Figure 4A demonstrates the solutions to eqn (10) for different values of [A−]i, assuming that [Na+]i = 10, [K+]o = 4 and [HCO3−]i = 24. In this set of solutions, [Cl−]i varies with [A−]i. These paired values of [A−]i and [Cl−]i all satisfy eqns (7) and (8). The range of [Cl−]i in solutions is restricted to 3–15 mm; higher values of [Cl−]i satisfy eqn (7), but not eqn (8). This is because of the limitations imposed by the extracellular side of eqn (8); [K+]o is very small and fixed, so that the product [K+]o × [Cl−]o is also modest. The intracellular product, [K+]i × [Cl−]i, exceeds [K+]o × [Cl−]o when [Cl−]i > [K+]o. At values of [Cl−]i higher than 15 mm, these low values of [K+]i (∼20 mm) can no longer be reconciled with eqn (7) (for physiological values of [Na+]i).
Figure 4. Modelling the effect of intracellular impermeant anions.
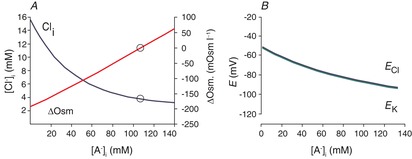
A, effect of the intracellular concentration of impermeant anions ([A−]i) on [Cl−]i and the transmembrane osmotic gradient using the solution to eqn (10) with [Na+]i = 10, [K+]o = 4, [Cl−]o = 110, [HCO3−]o = 24 and [HCO3−]i = 24. Open circles indicate the condition for [Cl−]i = [K+]o. B, values of ECl and EK correspond to the conditions and solutions of eqn (10) illustrated in A. Note that for all values of [A−]i, EK ≈ ECl, as predicted from rearrangement of eqn (8) ([K+]i /[K+]o = [Cl−]o/[Cl−]i; these are the variables in the Nernst equations for EK and ECl).
This solution brings up the following three issues.
Equation (8) constrains [Cl−]i ≈ [K+]o, but ECl is rarely measured to be as negative as predicted by this (ECl ≈ EK), and ECl and [Cl−]i vary in a way that cannot be reconciled with this constraint. These problems have been validated by measurements made with relatively non-invasive techniques, including perforated-patch recordings (Ebihara et al. 1995; Akaike, 1996; Pathak et al. 2007), as well as fluorimetric measurements of [Cl−]i (Berglund et al. 2006; Glykys et al. 2014). These studies report [Cl−]i to be normally distributed around means of 6–14 mm, with substantial variability in [Cl−]i from cell to cell. Dual cell-attached patch recordings (Tyzio et al. 2008) are also non-invasive but report only the difference between ECl and the resting membrane potential (RMP). These studies show a similar variability and an ECl that is more positive than RMP, which in turn is more positive than EK. These issues are summarized in Fig. 5, which illustrates a variety of concentrations of intra- and extracellular immobile anions, and the chaos this imposes on solutions to eqns (7) and (8).
The value of EK is too positive for most values of [A−]i. Over the corresponding range of values for [Cl−]i, ECl and EK vary from −55 to −98 mV (Fig. 4B). While this is within the range of measured ECl, EK is generally never less negative than −90 mV.
The osmotic gradient across the membrane (ΔOsm, the difference between intracellular and extracellular osmolarity) is illustrated for different solutions in Fig. 4A. For solutions other than [Cl−]i = [K+]o, the values of ΔOsm are too large to be sustainable by neuronal membranes. This suggests that to avoid a catastrophic osmotic gradient, neurons must maintain a [Cl−]i that is essentially equal to [K+]o, but this does not agree with the experimental findings cited in the preceding paragraphs.
Figure 5. Modelling the effect of intracellular impermeant anions when the transporter responsible for the ion permeability also carries water.
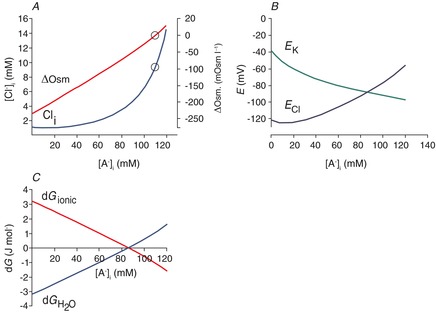
A, effect of [A−]i on [Cl−]i and the transmembrane osmotic gradient; solutions to eqns (7) and (15), with [K+]o = 3.5, [Na+]i = 10, [HCO3−]o = 24 and [HCO3−]i = 24. The concentration of free water was set at 27.8 mol l−1 (this is half the value of pure water) on the extracellular side, and 27.74 mol l−1 on the intracellular side, so that there was 0.22% less free water in the intracellular vs. extracellular space, favouring inward water flux. The coupling ratio, the number of water molecules transported per KCl pair, was set at 500. B, EK and ECl were calculated from eqn (9); EK is appropriately negative for physiological values of [A−]i and [Cl−]i that are solutions to eqn (11) for the conditions illustrated in A. In the physiological range, ECl varies from well above to well below action potential threshold. C, the free energy associated with water transport, 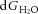 , and the free energy associated with KCl transport, dGionic, add to 0 for the solutions to eqn (22) illustrated in A.
, and the free energy associated with KCl transport, dGionic, add to 0 for the solutions to eqn (22) illustrated in A.
To reconcile these difficulties, ECl must be free to take on values other than EK. This requires some other source of free energy that can be added to loosen the strict linkage of the potassium and chloride transmembrane gradients discussed in relationship to eqns (7) and (8). One way to reconcile these difficulties is to propose stoichiometries other than one K+: one Cl−, as was proposed for NKCC1 (Brumback & Staley, 2008). However, in contrast to NKCC1, there is only one electroneutral stoichiometry for KCC2; the number of potassium ions transported must equal the number of chloride ions. Another source of free energy that may contribute to eqn (8) is the movement of water through the transporter. Most central neurons do not contain aquaporins, that is, they do not have water-permeable channels in their membranes (Papadopoulos & Verkman, 2013). Thus, neurons might be able to sustain a difference in water concentration relative to the extracellular space. Additionally, cation–chloride cotransporters, such as KCC2 and NKCC1, transport water along with cations and chloride in roughly isosmotic ratios (MacAulay et al. 2004; Hamann et al. 2005). The transporters can be considered as isotonic salt water transporters (Zeuthen, 1994). In this case, the free energy of transport is not only a sum of the electrochemical work of ion transport [eqn (8)], but also the work of water transport against its concentration gradient (assuming negligible hydrostatic pressure (Eijkel & van den Berg, 2005; Mollajew et al. 2010); then eqns (5) and (8) become:
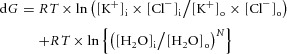 |
(11) |
where dG is the change in Gibbs free energy per transport cycle, N the number of H2O molecules moved per KCl pair, R the gas constant (8.314 J K−1 mol−1), T the temperature in degrees kelvin; and [H2O], the free water concentration, represents the water available for diffusion across the membrane, as follows:
 |
(12) |
The volume displaced by macromolecules (Schellman, 2003) is an important factor in measurements of water per unit volume, although it does not affect the free energy of transmembrane water movement and will not be considered further here. Restricted water is water that is constrained to the hydration layers of macromolecules (Timasheff, 2002). Restricted water would be the term representing the oncotic pressure if we were using a Starling approach to water flux across the membrane (Starling, 1896). Restricted water does not participate in solute-mediated phenomena, such as osmotic pressure and freezing point alteration (Fullerton & Cameron, 2007). Although the layers of restricted water are only a few molecules wide (Ebbinghaus et al. 2007), the density of macromolecules in the cytoplasm and possibly the extracellular matrix is high (Ellis, 2001; Ellis & Minton, 2003). Thus, the amount of restricted water is thought to be of the order of 50% of all water in the cytoplasm (Luby-Phelps, 2000; Fullerton & Cameron, 2007).
Although we are not aware of similar analyses for the extracellular space, the diffusion of small molecules through the extracellular space of the brain has been studied extensively, and the effective diffusion coefficients are characterized by the term ‘tortuosity’ (Syková & Nicholson, 2008). Although many factors may contribute to tortuosity, a key factor is the fraction of water in which solutes can move, because the extracellular space is a matrix or gel comprised of negatively charged glycosaminoglycans (Murakami et al. 1993; Brandtlow & Zimmerman, 2000). Diffusion coefficients in the extracellular space are 0.3–0.5 of the expected value using a variety of measurement techniques (Syková & Nicholson, 2008). For our analyses, we assumed that 50% of bulk water in the intra- and extracellular space is restricted, and 50% is available for diffusion across the membrane.
At equilibrium, dG = 0, and any difference in the gradients in K+ and Cl− must be matched by a corresponding difference in the water potential, as follows
| (13) |
At equilibrium, the logarithms must add to 0. This occurs when the products of the logarithmic elements = 1, as follows:
| (14) |
or:
| (15) |
Equation (15) is analogous to eqns (5) and (8), with an additional term describing the water diffusion potential; N, the number of water molecules transported per KCl pair, is ∼500 (MacAulay et al. 2004). The size of this exponent makes eqns (22)–(15) extremely sensitive to intracellular vs. extracellular differences in the amount of diffusible water that is lost to hydration layers of macromolecules, as well as to differences in the osmolarity.
Owing to the difficulty in solving eqns (7) and (22) formally, we obtained numerical solutions to the equations and created Fig. 5A. The figure shows the relationship between [A−]i, [Cl−]i and the osmotic gradient. There are several differences that can be appreciated by considering the three issues discussed for eqn (8), as follows.
The ECl no longer needs to be equal to EK. The ECl and EK can now diverge at different values of [A−]i (Fig. 5B), because there is another free energy contribution to eqn (8) (the free energy of water transport in eqns (22)–(15)). The balance between the net free energy changes associated with KCl cotransport vs. water transport for different [A−]i is shown in Fig. 5C.
The EK has an appropriately negative value for the range of physiological values of [A−]i and [Cl−]i (Fig. 5B).
Regarding the osmotic gradient, the [H2O]i and [H2O]o are inversely proportional to the osmolarity, so that the higher the omolarity, the lower the [H2O]i and [H2O]o (eqn (12). When the concentrations of intracellular and extracellular diffusible water are equal, then the driving force for water diffusion arises from the transmembrane osmotic gradient. For values of [A−]i and [Cl−]i that generate transmembrane osmotic pressure gradients favouring inward water flux, there is an oppositely directed diffusive force favouring KCl exit and vice versa. For example, when the cytoplasm is hypertonic relative to the extracellular space, the product of [K+]i × [Cl−]i is much greater than the product of [K+]o × [Cl−]o, and these two forces balance (Fig. 5C). Thus, there is no net KCl efflux and no water influx despite the osmotic pressure gradient across the neuronal membrane.
Osmotic pressure is not the only way to balance the KCl transmembrane gradient. Another means by which the water potential might balance KCl transport is by a gradient of the concentration of diffusible water, or conversely, differences in the amount of water that is restricted to hydration shells in the intra- and extracellular space. This can also be considered as an oncotic pressure gradient that influences the movement of water (Starling, 1896). Owing to the high ratio of water transport to ion transport, the transmembrane differences in restricted water can be quite small. Figure 5A was calculated assuming a transmembrane difference of 0.11% in the amount of water that is available for diffusion (i.e. 50% of the pure water concentration, or 27.8 mol l−1 in the extracellular space, and 49.89%, or 27.74 mol l−1, in the intracellular space). The interaction between the free energy for water transport and the free energy of ion transport is illustrated in Fig. 6A–C. Figure 6D demonstrates that there are many water potentials for which the free energy of water transport exceeds the ion transport energies for all physiological values of K+ and Cl−, so that eqn (15) is never satisfied and no equilibrium is available.
Figure 6. Free energies for water and KCl transport by KCC2 at different values of [Cl−]i.
A, conditions are as described for Fig. 5. At the value of [A−]i = 109 mm, the osmotic gradient is zero in Fig. 5A. The free energy of KCl cotransport, the free energy of water movement across the membrane and the sum of these free energies [eqns (12) and (15)] are shown for different values of [Cl−]i. The sum of the ionic and water-movement free energies is zero for [Cl−]i = 9.3 mm; this is the equilibrium point for the cotransport of potassium, chloride and water via KCC2. B, the same relationships are shown for a different value of [A−]i, 87 mm, to illustrate that the free energy for ionic transport is zero at a different value of [Cl−]i from the free energy minimum for water transport or the minimum of the sum of the free energies of ion and water transport. In this example, the equilibrium value of [Cl−]i is 4.3 mm. C, a second free energy minimum exists for the parameters shown in B, at a [Cl−]i of 116 mm. The high-[Cl−]i solutions are not illustrated in Fig. 5 because they are not observed experimentally and because the corresponding transmembrane osmotic gradient is so high (in this case, 167 mosmol l−1). D, for many values of intra- and extracellular diffusible water, eqn (15) cannot be satisfied. In this case, the sum of ionic and free water transport is never zero because the gradient for water diffusion is too large. The difference in intra- vs. extracellular diffusible water was set to 0.36% in this example; other parameters are the same as for Fig. 5.
The solutions illustrated in Fig. 5 look very different from those illustrated in Fig. 4. Most notably, [Cl−]i decreases instead of increases with decreasing [A−]i, and EK becomes more positive than ECl at low [A−]i. These changes occur because at low [A−]i the osmolarity decreases; that is, the free water concentration increases according to eqn (12). This reverses the free water gradient, now favouring water efflux. The only way to balance the total free energy of transport in these conditions is for the K+ and Cl− gradients to favour KCl influx into the cell, and this happens for [Cl−]i × [K+]i < [Cl−]o × [K+]o. Therefore, low values of [A−]i are associated with low values of [Cl−]i, such that the gradient favouring water efflux is balanced by the KCl gradient favouring KCl influx. Intracellular K+ is also low in these circumstances because to maintain electroneutrality, [K+]i varies with the sum of [A−]i + [Cl−]i. Consequently, EK becomes relatively positive at low [A−]i in Fig. 5B.
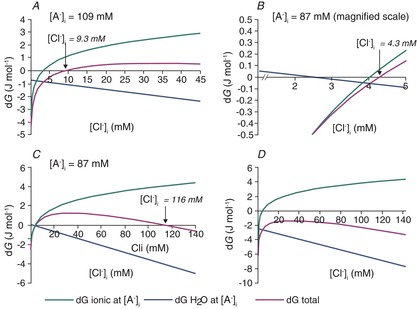
If idiogenic osmoles correct the osmotic gradient so that the intra- and extracellular osmolarity are always equal, then KCC2-mediated ion transport and the differences in intra- and extracellular restricted water are the only contributors to the total free energy of KCC2-mediated transport. In this case, the KCC2 equilibrium solutions to eqns (7) and (15) shown in Fig. 7A have the same shape as the solutions to the Donnan eqns (7) and (8) in Fig. 4. However, Fig. 7 differs somewhat from Fig. 4 because the free water concentration gradient influences the [Cl−]i at which KCC2 transport comes to equilibrium. The free water concentration gradient is not altered by [A−]i when osmotic differences are corrected by idiogenic osmoles, so EK and ECl are offset by a fixed potential for all values of [A−]i (Fig. 7B). The transmembrane free water concentration gradient therefore determines the range of [Cl−]i and [A−]i that can be maintained at equilibrium, including relatively high concentrations of [Cl−]i that would be typical of immature neurons (Fig. 7A) or lower values typical of more mature neurons (Fig. 7C). In each case, EK and ECl are offset by a potential that is proportional to the size of the water potential (Fig. 7B and D). Thus, when idiogenic osmoles correct any osmotic gradient that develops, variations in [A−]i and the transmembrane free water gradient support a wide range of equilibrium values of [Cl−]i.
Figure 7. Effects of correction of osmotic gradients with idiogenic osmoles on [Cl−]i, ECl and EK.
This figure was calculated using the same parameters as for Figs 5 and 6A, except that the transmembrane osmotic gradient was set to zero for all tested values of [A−]i and [Cl−]i, the intracellular water was set at 27.7 mol l−1, while the extracellular free water remained at 27.8 mol l−1, corresponding to a 0.36% difference in water concentration. A, The osmotic gradient is zero for all values of [A−]i and [Cl−]i, because differences are made up by intracellular idiogenic osmoles. In these conditions, solutions to eqns (7) and (15) support quite high [Cl−]i at equilibrium, even though Cl− cotransport is via KCC2, not NKCC1. B, EK and ECl calculated for the solutions of eqns (7) and (15) shown in A. The fixed difference between EK and ECl at all values of [A−]i represents the free energy in the 0.36% free water transmembrane concentration gradient. C, lower values of [Cl−]i, more typical of mature neurons, are solutions to eqns (7) and (15) for different values of [A−]i when the intracellular free water concentration is 27.76 mol l−1, or 0.14% less than the extracellular space. D, EK and ECl for the solutions illustrated in C. These values are typical of the range reported in mature neurons.
Distinct roles for [A−]i and [A−]o
Figure 5B illustrates how changes in [A−]i alter ECl. As discussed above (eqn 4), the equilibrium potential for Cl− is calculated from the Nernst equation, which varies as the natural logarithm of the ratio of [Cl−]o to [Cl−]i. This ratio is very sensitive to small changes in the denominator, because [Cl−]i is fairly low to begin with. For the same reasons, ECl is less sensitive to changes in [Cl−]o, because this value is much higher than [Cl−]i. For example, a 4 mm change in [Cl−]i from 2 to 6 mm changes the transmembrane ratio by a factor of three and changes ECl from −90 to −70 mV. However, a 4 mm change in [Cl−]o from 110 to 114 mm changes the transmembrane Cl− ratio by only 4%, and consequently, changes ECl by <1 mV. Therefore, it is to be expected that changes in [A−]o, which will directly affect [Cl−]o, have more modest effects on ECl. The effects of [A−]o on ECl are further blunted by the changes in [Cl−]i induced by changes in [A−]o. As [A−]o increases, [Cl−]o decreases, the [K+]o × [Cl−]o product decreases, and thus, a proportional decrease in [K+]i × [Cl−]i will be induced by KCC2 (assuming a stable transmembrane osmotic gradient). This has the effect of offsetting any changes in ECl induced by changes in [A−]o and [Cl−]o. The remarkably modest effect of [A−]o on ECl is illustrated in Fig. 8A; for a given value of [A−]i, ECl changes depending on the water diffusion gradient, but this relationship is unchanged by a 50 mm increase or decrease in [A−]o. Glykys et al. (2014) provided experimental support for the induction of compensatory changes [Cl−]i to re-establish equilibrium after changes in [Cl−]o. The [A−]o was altered by removing sulfates from the extracellular matrix with chondroitinase ABC. Replacement of the lost sulfates with extracellular chloride increased [Cl−]o and induced large increases in [Cl−]i to rebalance the free energy of ion and water transport by KCC2.
Figure 8. Extracellular anion concentration ([A−]o) vs. ECl and GABAA current.
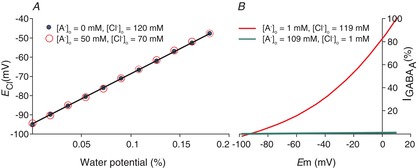
A, ECl is the same for two very different values of [A−]o, 0 and 50 mm, over a range of water potentials represented by the fractional difference in restricted water in the intra- vs. extracellular space. This occurs because KCC2 transports KCl until a new equilibrium is reached. Given that changes in [A−]o do not change the transmembrane K+ and H2O potentials, ECl cannot change either. Plotted values are solutions to eqn 15. B, the Goldman–Hodgkin–Katz current equation is plotted at different membrane potentials for two extreme values of [A−]o, 109 and 1 mm, with corresponding values of [Cl−]o of 1 and 109 mm. At 1 mm [Cl−]o, [Cl−]i is 0.03 mm. At these low values of [Cl−]i and [Cl−]o, there are very few Cl− ions available to carry current through the channel; thus, the maximal slope of the I–V curve is 100 times more shallow for the high [A−]o values (in which [Cl−]o = 1 mm) than for the low [A−]o values (in which [Cl−]o = 109 mm).
It is important to note that the anatomical distribution of [A−]o will determine the extent to which [Cl−]o is altered. For example, if [A−]o extended only a few micrometres from the neuronal membrane, [Cl−]o in the bulk CSF would probably be the most important extracellular determinant of the free energy of transmembrane Cl− flux. In this case, modelling of three compartments (such as depicted for two compartments in Fig. 3) indicates that fast exchange or movement of ions between different extracellular compartments, at steady state, will result in no effect of [A−]o on [Cl−]o. However, if [A−]o is significant throughout the extracellular space, and bulk CSF is separated from this extracellular matrix by the ependymal lining, then the most important extracellular parameter determining the free energy of neuronal transmembrane Cl− flux will be [A−]o. Thus, fixed extracellular charges will impact transmembrane gradients only if the extracellular matrix is uniform and substantially isolated from the large reservoirs of ventricular CSF. While there is evidence that the first assumption is true (Glykys et al. 2014), the second assumption has still to be addressed experimentally.
The [A−]o and the amplitude of anion fluxes
In contrast to the modest effects of [A−]o on ECl, [A−]o has a much more robust effect on the effective conductance of electrogenic transmembrane permeabilities, such as that gated by the GABAA receptor. For example, increases in [A−]o decrease [Cl−]o and induce compensatory decreases in [Cl−]i (described in the preceding paragraph). The net effect is a reduction in charge carriers available to carry inward anionic flux (outward, inhibitory transmembrane currents). The Goldman–Hodgkin–Katz current equation (Hodgkin & Katz, 1949) is linear with respect to [Cl−]i and [Cl−]o, as follows:
| (16) |
Figure 8B illustrates the effects on the GABAA conductance for the same conditions plotted in Fig. 6A. In this situation, [A−]o is a key determinant of the amplitude of inward chloride fluxes for a given transmembrane permeability, while [A−]i determines the reversal potential (Fig. 5B). Modelling studies have demonstrated that for dendritic signal processing, the conductance values can provide multiplication and division functions, while the reversal potential can provide addition and subtraction functionality (Sejnowski et al. 1998).
We have made the case that in theory, a change in the Cl− concentration in cells can be explained solely by a change either in the amount of large molecules (proteins) that carry negative charges inside the cell or in the gradient for water diffusion across the membrane (Fig. 9). The concentration of these immobile intracellular charges is sufficient to explain a change in [Cl−]i without the need to involve a primary (ATPase) or a non-equilibrative secondary active transport mechanism. Thus, the developmental decrease in [Cl−]i that is observed in neurons (Fig. 2) might be accounted for by an increase in the number of nucleic acids bearing negatively charged phosphate groups and proteins comprised of amino acids that are negatively charged at physiological pH.
Figure 9. Complexity of transmembrane and intracellular chloride gradients in the presence of charged immobile anions.
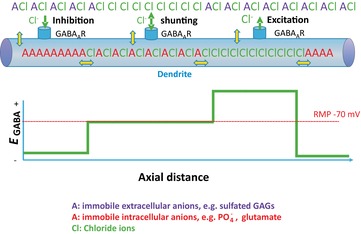
Yellow double-headed arrows represent boundaries at which KCl and water movement must be at equilibrium, assuming that neurons do not expend continuous energy transporting KCl in order to maintain static chloride gradients. In the presence of varying local concentrations of immobile anions, a variety of Donnan equilibria could exist, so that EGABA could vary between GABAergic synapses along a single dendrite, as long as at each yellow double-headed arrow eqn (15) was satisfied.Abbreviation: RMP, resting membrane potential.
This hypothesis was addressed experimentally by measuring [Cl−]i (using the genetically encoded chlomeleon as a chloride sensor) and [A−]i (using SYT064 binding of nucleic acids). A negative correlation was measured over a very large number of neurons, consistent with [A−]i and [Cl−]i varying inversely ( Figs 4 and 5A; Glykys et al. 2014). More importantly, the levels of [A−]i were experimentally manipulated in brain slices by incubating the slices with gluconate, pyruvate and d-lactate. This manipulation resulted in a corresponding reduction in [Cl−]i. Likewise, a negative correlation was observed between [Cl−]o and [A−]o (constituted mostly of sulfated proteoglycans of the extracellular matrix), and reducing the levels of [A−]o by treating brain slices with chondroitinase ABC resulted in a significant increase in local [Cl−]o. Thus, this study provides the first experimental evidence for a major role of impermeant charges and challenges the dogma that expression and activity of specific secondary active transporters exclusively ‘sets’ the [Cl−]i to low or high levels. Additional support for this idea comes from synaptic physiologists who for decades have exploited the inverse relationship between [A−]i and [Cl−]i to change [Cl−]i and EGABA by altering the amount of impermeant anion in their electrode solutions (e.g. Fig. 9A of Staley & Proctor, 1999).
These considerations also cast light on another recent problem in neurobiology, the subcellular differences in the reversal potential for GABAA anionic currents. Electrophysiological recordings have suggested that these currents, primarily carried by Cl− but with a minor contribution from HCO3−, have reversal potentials that are several millivolts more positive in the axon initial segment compared with the soma in mature neurons (Szabadics et al. 2006). Differences of a similar size have been observed between dendrites and somata (Duebel et al. 2006). Given that the impermeant anions in the cytoplasm are associated with very large macromolecules comprised of repeating nucleic acids and amino acids, not only do these charges not cross the membrane, they are also unlikely to move in the cytoplasm. For example, the polyglutamate chains attached to microtubules could provide substantially fixed local anionic charges in the dendritic cytoplasm (Liu et al. 2013). Donnan equilibria have been carefully described for a variety of such immobile ions, from protein gels to osmotic media in slow-release pharmaceutical preparations (Ricka & Tanaka, 1984). Thus, Donnan equilibria can exist not only between the intra- and extracellular fluids, but also between different subcellular compartments containing different [A−]i, as long as the mobility of these intracellular anions is very low. These equilibrium points are illustrated by double-headed yellow arrows in Fig. 9.
The relationships of [A−]i and [Cl−]i between different subcellular regions and the corresponding osmotic gradients can therefore be obtained by solving eqns (7) and (15), where [K+]o and [Cl−]o would be replaced by the K+ and Cl− concentrations in the intracellular compartment that is being compared with the intracellular compartment, in which Cl− = [Cl−]i. This approach is attractive because it is an equilibrium solution; no energy is expended to maintain subcellular differences in [Cl−]i. In contrast, if regional, oppositely directed Cl− transport were responsible for maintaining these subcellular gradients, then the free diffusion of Cl− through the cytoplasm (Kuner & Augustine, 2000) would result in very costly short-circuiting Cl− currents between these subcellular regions, and the chloride gradient would be very sensitive to transport inhibition, which has not been observed (Zhang et al. 2007; Glykys et al. 2014).
If [A−]i and [A−]o rather than KCC2 determine [Cl−]i and [Cl−]o, what is the role of KCC2? The major role of KCC2 (the major neuronal K+–Cl− cotransporter) is to maintain the complex balance between the potentials for water and ion flux across the membrane. For example, KCC2 provides a pathway for the rapid, electrically silent recovery of [Cl−]i after perturbations due to electrogenic Cl− fluxes. This view is supported by data showing that manipulations that tend to raise [Cl−]i in mature neurons, i.e. long GABA applications or GABA application coupled to depolarization steps, result in fast [Cl−]i recovery (Staley & Proctor, 1999; Wagner et al. 2001; Jin et al. 2005; Pathak et al. 2007). This recovery is delayed in immature neurons, where KCC2 expression is minimal or absent (Yamada et al. 2004), or in neurons lacking KCC2 expression (Zhu et al. 2005). Thus, KCC2 maintains [Cl−]i and ECl. However, KCC2 does not set, i.e. determine, [Cl−]i; this is done by the concentrations of impermeant anions in the intra- and extracellular space, as well as by the macromolecules and osmoles that determine the water potential.
Equation (11) permits higher values of [Cl−]i than eqn (8), but the range of feasible [Cl−]i is still limited (Fig. 5A) unless idiogenic osmoles are allowed to reduce transmembrane osmotic gradients. Young cells may have very low [A−]i because they have not had time to manufacture the macromolecules that comprise [A−]i in adult neurons. This may lead to values of transmembrane osmotic gradients that are not safe or desirable due to water leakage through other channels and transporters. This problem can be alleviated by expression of NKCC1 in place of KCC2. It is important to re-emphasize that when isolating [Cl−]i in eqn (11) (free energy), not only is the gradient energy of diffusible water (second term) a significant component, but the K+ gradient across the plasma membrane (part of the first term) is also critical. This gradient is maintained by the consumption of ATP by the Na+–K+-ATPase. In the absence of Na+–K+ pump activity, the K+ gradient collapses, and consequently, the energy for the generation of a Cl− gradient dissipates. Therefore, while our model is adding Donnan charges and water transport as key factors in determining [Cl−]i, its dependence on the action of the Na+–K+ pump is still essential.
Outlook and conclusions
Many of the issues discussed here have direct bearing on pathophysiologies into which we have little insight so far. For example, if many neurons maintain balanced gradients for ionic and water diffusion across their cytoplasmic membranes, then damage to the membrane that increases the ionic or water permeability could lead to deranged Cl− homeostasis and GABA signalling, which may contribute to early seizures after brain injury. Protein hydration is very sensitive to pH and other denaturants, which could induce large changes in the gradient for water diffusion and corresponding changes in [Cl−]i.
In conclusion, caution needs to be exercised when addressing the reasons for which intracellular Cl− sits at a particular concentration. One does not want to replace the dogma that Cl− is passively distributed in cells with a new dogma that cation–chloride cotransporter expression and function defines the level of intracellular Cl−. While there is no doubt that the function of the cation–chloride cotransporter is critical in providing a pathway for rapid recovery of Cl− upon challenge, it is not at all clear that cotransporter activity itself sets the resting concentration of the anion. Despite a relatively high CSF Cl− concentration (above 100 mm), and therefore, a seemingly significant driving force for Cl− invasion, the presence of large immobile molecules carrying negative charges inside the cell can explain exclusion of the anion and a resulting low concentration. We demonstrated here that adding other sources of free energy, e.g. gradients for water diffusion, in equilibrium equations allows for larger ranges in resting intracellular Cl− concentrations. Some of the concepts advanced here have consequences that can now be tested experimentally.
Additional information
Competing interests
None declared.
Funding
Support for the authors’ research comes from the National Institutes of Health grants GM074771, DK093501 to E.D. and NS040109, NS074772 to K.J.S.
References
- Akaike N. Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Prog Biophys Mol Biol. 1996;65:251–264. doi: 10.1016/s0079-6107(96)00013-2. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Gamiño SM, Giraldez F, Nogueron I. Intracellular chloride regulation in amphibian dorsal root ganglion neurons studied with ion-selective microelectrodes. J Physiol. 1988;406:225–246. doi: 10.1113/jphysiol.1988.sp017378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan V, Becker M, Löhrke S, Nothwang HG, Güresir E, Friauf E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci. 2003;23:4134–4145. doi: 10.1523/JNEUROSCI.23-10-04134.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Krieger P, Loo LS, Wang D, Cant NB, Feng G, Augustine GJ, Kuner T. Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 2006;35:207–228. doi: 10.1007/s11068-008-9019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Boyle PJ, Conway EJ. Potassium accumulation in muscle and associated changes. J Physiol. 1941;100:1–63. doi: 10.1113/jphysiol.1941.sp003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandtlow CE, Zimmerman DR. Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev. 2000;80:1267–1290. doi: 10.1152/physrev.2000.80.4.1267. [DOI] [PubMed] [Google Scholar]
- Brumback AC, Staley KJ. Thermodynamic regulation of NKCC1-mediated Cl− cotransport underlies plasticity of GABAA signaling in neonatal neurons. J Neurosci. 2008;28:1301–1312. doi: 10.1523/JNEUROSCI.3378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolf JS, Smith RL. Ontogeny of cation–Cl− cotransporter expression in rat neocortex. Brain Res Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Coombs JS, Eccles JC, Fatt P. The specific ionic conductances and the ionic movements across the motoneuronal membrane that produce the inhibitory post-synaptic potential. J Physiol. 1955;130:326–374. doi: 10.1113/jphysiol.1955.sp005412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E. Cation-chloride cotransporters in neuronal communication. News Physiol Sci. 2000;15:309–312. doi: 10.1152/physiologyonline.2000.15.6.309. [DOI] [PubMed] [Google Scholar]
- Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl− cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994;269:25677–25683. [PubMed] [Google Scholar]
- Donnan FG. Theorie der Membrangleichgewichte und Membranpotentiale bei Vorhandensein von nicht dialysierenden Elektrolyten. Ein Beitrag zur physikalisch-chemischen Physiologie [The theory of membrane equilibrium and membrane potential in the presence of a non-dialyzable electrolyte. A contribution to physical-chemical physiology] Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 1911;17:572–581. [Google Scholar]
- Duebel J, Haverkamp S, Schleich W, Feng G, Augustine GJ, Kuner T, Euler T. Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron. 2006;49:81–94. doi: 10.1016/j.neuron.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Dzhala V, Valeeva G, Glykys J, Khazipov R, Staley K. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J Neurosci. 2012;32:4017–4031. doi: 10.1523/JNEUROSCI.5139-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus S, Kim SJ, Heyden M, Yu X, Heugen U, Gruebele M, Leitner DM, Havenith M. An extended dynamical hydration shell around proteins. Proc Natl Acad Sci U S A. 2007;104:20749–20752. doi: 10.1073/pnas.0709207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara S, Shirato K, Harata N, Akaike N. Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. J Physiol. 1995;484:77–86. doi: 10.1113/jphysiol.1995.sp020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Lohrke S, Friauf E. Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurones is due to age-dependent Cl− regulation. J Physiol. 1999;520:121–137. doi: 10.1111/j.1469-7793.1999.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkel JC, van den Berg A. Water in micro- and nanofluidics systems described using the water potential. Lab Chip. 2005;5:1202–1209. doi: 10.1039/b509819j. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;20:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Minton AP. Join the crowd. Nature. 2003;425:27–28. doi: 10.1038/425027a. [DOI] [PubMed] [Google Scholar]
- Fullerton GD, Cameron IL. Water compartments in cells. Methods Enzymol. 2007;428:1–28. doi: 10.1016/S0076-6879(07)28001-2. [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala V, Egawa K, Balena T, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ. Local impermeant anions set the GABAA reversal potential. Science. 2014;343:670–675. doi: 10.1126/science.1245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann S, Herrera-Perez JJ, Bundgaard M, Alvarez-Leefmans FJ, Zeuthen T. Water permeability of Na+–K+–2Cl− cotransporters in mammalian epithelial cells. J Physiol. 2005;568:123–135. doi: 10.1113/jphysiol.2005.093526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann S, Herrera-Perez JJ, Zeuthen T, Alvarez-Leefmans FJ. Cotransport of water by the Na+–K+–2Cl− cotransporter NKCC1 in mammalian epithelial cells. J Physiol. 2010;588:4089–4101. doi: 10.1113/jphysiol.2010.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol. 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Liu Y, Garnham CP, Roll-Mecak A, Tanner ME. Phosphinic acid-based inhibitors of tubulin polyglutamylases. Bioorg Med Chem Lett. 2013;23:4408–4412. doi: 10.1016/j.bmcl.2013.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–568. [PubMed] [Google Scholar]
- Luby-Phelps K. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int Rev Cytol. 2000;192:189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- MacAulay N, Hamann S, Zeuthen T. Water transport in the brain: role of cotransporters. Neuroscience. 2004;129:1031–1044. doi: 10.1016/j.neuroscience.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Mao S, Garzon-Muvdi T, Difulvio M, Chen Y, Delpire E, Alvarez FJ, Alvarez-Leefmans FJ. Molecular and functional expression of cation-chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J Neurophysiol. 2012;108:834–852. doi: 10.1152/jn.00970.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science. 1986;232:1413–1415. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- Mollajew R, Zocher F, Horner A, Wiesner B, Klussmann E, Pohl P. Routes of epithelial water flow: aquaporins versus cotransporters. Biophys J. 2010;99:3647–3656. doi: 10.1016/j.bpj.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Taguchi T, Ohtsuka A, Kkuta A. Neurons with strongly negative-charged surface-coats in adult rat brain as detected by staining with cationic iron colloid. Arch Histol Cytol. 1993;56:13–21. doi: 10.1679/aohc.56.13. [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MBE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Stein V, Keating DJ, Maier H, Rinke I, Rudhard Y, Hentschke M, Rune GM, Jentsch TJ, Hübner CA. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E. Expression of the Na+-K+-2Cl− cotransporter BSC2 in the nervous system. Am J Physiol Cell Physiol. 1997a;272:C173–C183. doi: 10.1152/ajpcell.1997.272.1.C173. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 1997b;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Post RL, Jolly PC. The linkage of sodium, potassium, and ammonium active transport across the human erythrocyte membrane. Biochim Biophys Acta. 1957;25:118–128. doi: 10.1016/0006-3002(57)90426-2. [DOI] [PubMed] [Google Scholar]
- Ricka J, Tanaka T. Swelling of ionic gels: quantitative performance of the Donnan theory. Macromolecules. 1984;17:2916–2921. [Google Scholar]
- Schellman JA. Protein stability in mixed solvents: a balance of contact interaction and excluded volume. Biophys J. 2003;85:108–125. doi: 10.1016/S0006-3495(03)74459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejnowski TJ, Koch C, Churchland PS. Computational neuroscience. Science. 1998;241:1299–1306. doi: 10.1126/science.3045969. [DOI] [PubMed] [Google Scholar]
- Sipilä ST, Huttu K, Yamada J, Afzalov R, Voipio J, Blaesse P, Kaila K. Compensatory enhancement of intrinsic spiking upon NKCC1 disruption in neonatal hippocampus. J Neurosci. 2009;29:6982–6988. doi: 10.1523/JNEUROSCI.0443-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta. 1957;23:494–401. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. J Physiol. 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling E. On the absorption of fluids from the connective tissue spaces. J Physiol. 1896;19:175–185. doi: 10.1113/jphysiol.1896.sp000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Yu Z, Wang W, Liu X. Both NKCC1 and anion exchangers contribute to Cl− accumulation in postnatal forebrain neuronal progenitors. Eur J Neurosci. 2012;35:661–672. doi: 10.1111/j.1460-9568.2012.08007.x. [DOI] [PubMed] [Google Scholar]
- Sung K-W, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na–K–2Cl cotransporter null mice. J Neurosci. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syková E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett. 1988a;89:49–54. doi: 10.1016/0304-3940(88)90479-x. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Relative contributions of passive equilibrium and active transport to the distribution of chloride in mammalian cortical neurons. J Neurophysiol. 1988b;60:195–124. doi: 10.1152/jn.1988.60.1.105. [DOI] [PubMed] [Google Scholar]
- Timasheff SN. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry. 2002;41:13473–13482. doi: 10.1021/bi020316e. [DOI] [PubMed] [Google Scholar]
- Tosteson DC, Hoffman JF. Regulation of cell volume by active cation transport in high and low potassium sheep red cells. J Gen Physiol. 1960;44:169–194. doi: 10.1085/jgp.44.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Minlebaev M, Rheims S, Ivanov A, Jorquera I, Holmes GL, Zilberter Y, Ben-Ari Y, Khazipov R. Postnatal changes in somatic γ-aminobutyric acid signalling in the rat hippocampus. Eur J Neurosci. 2008;27:2515–2528. doi: 10.1111/j.1460-9568.2008.06234.x. [DOI] [PubMed] [Google Scholar]
- Wagner S, Sagiv N, Yarom Y. GABA-induced current and circadian regulation of chloride in neurones of the rat suprachiasmatic nucleus. J Physiol. 2001;537:853–869. doi: 10.1111/j.1469-7793.2001.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci. 2008;28:5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex. 2011;21:574–587. doi: 10.1093/cercor/bhq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo N-S, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyper-excitability and epilepsy associated with disruption of the mouse neuronal-specific K–Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuthen T. Cotransport of K+, Cl− and H2O by membrane proteins from choroid plexus epithelium of Necturus maculosus. J Physiol. 1994;478:203–219. doi: 10.1113/jphysiol.1994.sp020243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LL, Delpire E, Vardi N. NKCC1 does not accumulate chloride in developing retinal neurons. J Neurophysiol. 2007;98:266–277. doi: 10.1152/jn.00288.2007. [DOI] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]