

Abstract
Neuronal networks of the thalamus are the target of extensive cholinergic projections from the basal forebrain and the brainstem. Activation of these afferents can regulate neuronal excitability, transmitter release, and firing patterns in thalamic networks, thereby altering the flow of sensory information during distinct behavioural states. However, cholinergic regulation in the thalamus has been primarily examined by using receptor agonist and antagonist, which has precluded a detailed understanding of the spatiotemporal dynamics that govern cholinergic signalling under physiological conditions. This review summarizes recent studies on cholinergic synaptic transmission in the thalamic reticular nucleus (TRN), a brain structure intimately involved in the control of sensory processing and the generation of rhythmic activity in the thalamocortical system. This work has shown that acetylcholine (ACh) released from individual axons can rapidly and reliably activate both pre- and postsynaptic cholinergic receptors, thereby controlling TRN neuronal activity with high spatiotemporal precision.
Michael Beierlein received his PhD at Brown University, studying the properties of chemical and electrical synapses in neocortical networks in the laboratory of Dr Barry Connors. He pursued postdoctoral studies with Dr Rafael Yuste at Columbia University and with Dr Wade Regehr at Harvard Medical School. He is now an Assistant Professor at the University of Texas Medical School at Houston. His laboratory employs a combination of electrophysiology, imaging, and optogenetics to explore the functional properties of distinct types of synapses and local networks in the thalamocortical system.
 |
Introduction
GABAergic neurons of the thalamic reticular nucleus (TRN) are intimately involved in the bidirectional information transfer between thalamus and neocortex, as well as between different thalamic nuclei (Jones, 2007). Thalamic output generated by thalamocortical (TC) relay neurons leads to the activation of neocortical networks via TC synapses in layers 4 and 5/6 of neocortex. TC neurons are the target of extensive cortical feedback, generated by corticothalamic neurons in layer 6. Both of these long-range connections between thalamus and neocortex form axon collaterals onto TRN neurons, which in turn project onto TC neurons (Pinault, 2004). Thus, sensory processing in TC neurons is controlled by TRN-mediated feedforward inhibition triggered by corticoreticular inputs as well as by TRN-mediated feedback or lateral inhibition evoked by thalamoreticular inputs (Fig. 1C).
Figure 1. Cholinergic signalling modes in the thalamic reticular nucleus.
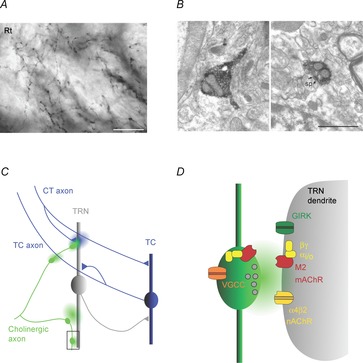
A, axonal arborizations with small diameter varicosities formed by cholinergic afferents in the rat TRN (Rt). Axons are immunostained for choline acetyltransferase (ChAT), the enzyme responsible for ACh synthesis. Scale bar, 20 μm. B, micrographs show two examples of axonal varicosities (labelled dark) formed by cholinergic axons in the TRN. Left image depicts an unmyelinated axon containing vesicles leading into a varicosity, without an obvious synaptic junction. Right image shows synaptic junction (outlined by arrows), formed onto a postsynaptic spine (sp). Scale bar, 1 μm. Images reproduced with permission from Parent & Descarries (2008). C, schematic diagram illustrates thalamic circuitry as well as possible targets of ACh signalling in the TRN. Cholinergic afferents (green) contact distal dendrites of TRN neurons and generate E–I cholinergic responses, probably mediated by ultrastructurally defined synapses (area outlined by square and shown in more detail in D). In addition, ACh released from en passant varicosities could act over larger distances and activate extrasynaptic receptors expressed on TRN dendrites, nAChRs expressed by thalamocortical (TC) axons or receptors expressed at presynaptic corticothalamic (CT) terminals. In turn, glutamate release from CT axons could modulate cholinergic signalling by acting on pre- or postsynaptic receptors at cholinergic synapses. D, schematic diagram illustrating the cellular mechanisms underlying cholinergic synaptic signalling in the TRN, based on the findings in Sun et al. (2013). ACh release activates postsynaptic ionotropic nAChRs (probably α4β2) and metabotropic Gi/o-coupled M2 mAChRs, which are expressed both pre- and postsynaptically. M2 mAChR activation in the postsynaptic membrane triggers the liberation of the βγ subunit complex, in turn leading to the opening of G protein-coupled inwardly rectifying K+ (GIRK) channels. Presynaptic M2 mAChRs are responsible for autoinhibition of ACh release, probably generated by βγ subunit complex-mediated inhibition of presynaptic Ca2+ channels.
Extensive research over the last several decades has provided ample evidence that activity in thalamic circuits formed by TRN and TC neurons is constantly regulated by a number of afferent systems, whose activation depends on the behavioural state of the animal (Lee & Dan, 2012). Perhaps the best understood of these systems are cholinergic afferent projections which arise from two distinct areas. Cholinergic neurons in the pedunculopontine and laterodorsal tegmental nuclei of the brainstem innervate virtually all thalamic nuclei, including TRN. In vivo, these neurons show persistent increases in activity rates during wakefulness and arousal (Boucetta et al. 2014). TRN is the target of a separate projection formed by basal forebrain (BF) cholinergic neurons (Hallanger et al. 1987). Transient increases in BF neuronal activity are implicated in regulating a number of processes on more rapid time scales, such as sleep–wake transitions, learning, sensory processing and attention (Parikh et al. 2007; Letzkus et al. 2011; Pinto et al. 2013; Han et al. 2014). As reviewed elsewhere, BF cholinergic control of the majority of these processes is assumed to be mediated by the rapid activation of distinct neuronal circuits in neocortex (Arroyo et al. 2014; Muñoz & Rudy, 2014), while the involvement of TRN remains largely unexplored.
The cellular actions of acetylcholine (ACh) on thalamic neurons are well documented, but we know little about the spatiotemporal dynamics governing endogenous ACh signalling in thalamic circuits. TC neurons express M1 and M3 muscarinic acetylcholine receptors (mAChRs), whose activation leads to the closure of potassium conductances mediated by TASK and TREK channels (Bista et al. 2012) and depolarization of the resting membrane potential. This allows TC neurons to undergo a switch in their response mode from burst firing to tonic firing, in large part mediated by the inactivation of low-threshold T-type Ca2+ channels (McCormick & Bal, 1997). Membrane depolarization can similarly be generated by the activation of nicotinic acetylcholine receptors (nAChRs) (Zhu & Uhlrich, 1997). However, how nAChRs and mAChRs are recruited under physiological conditions is not well understood. In vivo studies have documented long-lasting membrane depolarizations in TC neurons evoked by only brief extracellular stimulus trains applied to the brainstem cholinergic region (Hu et al. 1989a; Curro Dossi et al. 1991; Steriade, 2004). These data have led to the idea that brainstem cholinergic afferents globally regulate TC cell firing mode by influencing populations of neurons over long time scales (Descarries et al. 1997).
TRN neurons also express both nAChRs and mAChRs, but their respective activation appears to generate opposing effects on resting membrane potential. Activation of cholinergic afferents in the brainstem leads to a biphasic response, with a fast nAChR-mediated depolarization followed by long-lasting mAChR-mediated hyperpolarization (Hu et al. 1989b). Such biphasic responses have also been observed in vitro, following brief applications of ACh (Lee & McCormick, 1995). Other work has emphasized that activation of cholinergic afferents primarily acts to inhibit TRN neuronal firing (Pinault & Deschênes, 1992), an effect that can be mimicked by the application of the cholinergic agonist carbachol in TRN (Hirata et al. 2006). This has led to the hypothesis that cortical desynchronization observed during attentional tasks is at least in part mediated by BF-evoked inhibition of TRN neurons, resulting in disinhibition of TC neurons and a switch into tonic firing mode, allowing them to more faithfully respond to rapid changes in sensory afferent activity (Harris & Thiele, 2011).
To help resolve these conflicting data requires a better understanding of the cellular mechanisms underlying cholinergic signalling. Anatomical studies have provided clues for the existence of several distinct signalling modes. Cholinergic afferents into TRN form a dense network of unmyelinated axonal arborizations (Fig. 1A) which form numerous, more or less equally spaced synaptic varicosities, with an average diameter of ∼0.6 μm. While about 40% of these terminals show ultrastructural specializations indicative of conventional synapses, a majority of potential release sites do not appear to be associated with the postsynaptic membrane (Fig. 1B), suggesting that fast synaptic transmission and certain forms of volume transmission might coexist. To help distinguish between these possibilities requires insights into how ACh released by physiologically realistic activity patterns activates nAChRs and mAChRs expressed in the somatodendritic membrane. However, such information has been lacking until recently, in part due to the absence of an in vitro model of cholinergic transmission.
Biphasic cholinergic synaptic signalling in TRN neurons
In a recent study carried out using brain slices of the somatosensory thalamus we employed conventional extracellular stimulation using patch pipettes and isolated putative cholinergic inputs using receptor antagonists for fast glutamatergic and GABAergic synaptic transmission (Sun et al. 2013). Single stimuli applied locally in the TRN generated highly reliable biphasic excitatory–inhibitory (E–I) responses in all postsynaptic TRN neurons examined (Fig. 2A), with an initial short-latency nAChR-mediated EPSC (nEPSC) followed by a long-lasting mAChR-mediated IPSC (mIPSC). Fast-latency nEPSCs had slow kinetics as compared to AMPA receptor (AMPAR)-mediated EPSCs (decay time constants of 100–300 ms, Fig. 2B) and were mediated by non-α7 nAChRs (most likely α4β2), whereas long-latency mIPSCs (decay time constants of 500–700 ms) were triggered by the activation of M2 mAChRs, thereby leading to the opening of a G protein-coupled inwardly rectifying potassium (GIRK) conductance (Fig. 1D). These data indicate that in the TRN, both nAChRs and mAChRs are expressed near sites of ACh release and can be recruited following low-frequency afferent activity. While ACh-mediated E–I synaptic signalling may seem highly unusual, similar responses have been described in a number of other systems (Blankenship et al. 1971; Yarosh et al. 1988; Glowatzki & Fuchs, 2000), though the mechanisms generating biphasic responses vary between preparations. Studies employing brief agonist applications have revealed additional examples. In layer 5 neocortical neurons I–E responses are generated by activation of only M1 mAChRs, with sustained excitation preceded by transient inhibition mediated by phospholipase C-dependent Ca2+ increases from internal stores and the activation of Ca2+-dependent SK conductances (Gulledge & Stuart, 2005; Gulledge et al. 2009). GABAergic neurogliaform cells in layer 1 can generate E–I responses mediated by α4β2 nAChRs and M1 mAChRs whose activation similarly results in the opening of SK conductances (Brombas et al. 2014). It remains to be determined if biphasic responses in neocortical neurons can also be generated by endogenously released ACh.
Figure 2. Properties of biphasic cholinergic synaptic signalling.
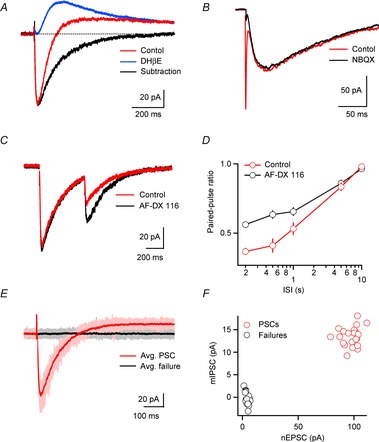
TRN neurons were recorded with a K+-based internal solution, except for the recordings shown in B–D, which were obtained with a Cs+-based internal solution. Cholinergic synaptic transmission was isolated by including antagonists of glutamatergic and GABAergic synaptic transmission, except for data shown in B. A, E–I postsynaptic response (red trace) evoked by single stimuli applied locally. The EPSC was blocked by the nAChR antagonist DHβE, while the remaining IPSC (blue trace) was eliminated by the mAChR antagonist atropine (not shown). Subtraction of the IPSC from the biphasic response yields time course of nicotinic EPSC (nEPSC, black trace). B, comparison of the time course of the postsynaptic responses evoked by cholinergic and glutamatergic afferents to TRN. Single stimuli evoked EPSCs with a fast and a slow component (red trace) in a TRN neuron. The fast component generated by activation of corticoreticular and thalamoreticular inputs was blocked by the AMPAR antagonist NBQX and the slow component (black trace) was blocked by the nAChR antagonist DHβE (not shown). C, nEPSCs evoked by paired stimuli (inter-stimulus interval (ISI) 500 ms) prior to (red) and following application of the M2 mAChR antagonist AF-DX 116 (10 μm, black) in a representative recording. Blocking M2 mAChRs leads to an increase in nEPSC amplitude evoked by the second stimulus, showing that ACh release is controlled by autoinhibition. Note that the nEPSC amplitude evoked by the first stimulus remains unchanged, indicating the ACh release is not controlled by tonic activation of presynaptic mAChRs. D, summary (n = 5) showing nEPSC2/nEPSC1 (paired-pulse ratio) for different ISIs in control (red circles) and AF-DX 116 (black circles). E, activation of individual presynaptic cholinergic axons elicits E–I postsynaptic responses. Overlay of individual trials evoked at a constant stimulus intensity, showing both PSCs (light red) and response failures (grey), with averages for the two groups shown in red and black, respectively. F, graph plots mIPSC amplitude against nEPSC amplitude of individual trials (n = 38), for data shown in E. Note that nEPSCs and mIPSCs occur in common trials. Data adapted from Sun et al. (2013).
In addition to acting on nAChRs and mAChRs expressed in TRN dendrites, synaptically released ACh also activated M2 mAChRs expressed presynaptically, thereby mediating autoinhibition (Fig. 2C). Cholinergic responses evoked by paired-pulses displayed considerable synaptic depression over a range of interstimulus intervals, mediated by a combination of presynaptic (e.g. vesicle depletion) and postsynaptic mechanisms (e.g. receptor desensitization). Paired-pulse depression was partially relieved following block of presynaptic M2 mAChRs, showing that even single stimuli lead to the activation of autoreceptors expressed on presynaptic terminals (Fig. 2D). The mechanisms for M2 mAChR-mediated reduction in transmitter release have not been further investigated, but probably involve the inhibition of presynaptic Ca2+ channels (Vogt & Regehr, 2001, Fig. 1D).
To examine the possibility that nAChRs and mAChRs are recruited by different cholinergic afferents or that the synchronous ACh release from many presynaptic fibres is required to activate either receptor system, we recorded from TRN neurons and activated individual presynaptic inputs at threshold, using minimal stimulation. Even under these conditions, E–I signalling was observed for all neurons examined (Fig. 2E and F). Individual trials either resulted in response failures (due to failure of axonal stimulation) or in biphasic responses with surprisingly low amplitude variability, indicating that ACh release occurs at multiple release sites, with a high initial release probability (but see below for alternative explanation). Moreover, the large size of single-axon-mediated nEPSCs (∼50 pA) indicates that synchronous activity of only a small number of cholinergic inputs can strongly influence TRN neuronal activity.
Are biphasic cholinergic synaptic responses mediated by conventional synapses? While studies examining the ultrastructure of cholinergic terminals have described small terminals that contact distal dendrites of TRN neurons (Hallanger & Wainer, 1988; Parent & Descarries, 2008), many varicosities do not appear to be associated with postsynaptic membranes, suggesting the existence of multiple signalling modes (Fig. 1B). Studies performed in neocortex have shown that layer 1 interneurons express both α7 as well as non-α7 nAChRs and that BF cholinergic afferents activate these two populations of nAChRs via distinct signalling modes (Arroyo et al. 2012; Bennett et al. 2012). Fast nEPSCs mediated by α7 nAChRs were found to be highly variable in amplitude from trial to trial and were insensitive to perturbations of ACh breakdown, suggesting signalling via conventional synapses. In contrast, slower nEPSCs mediated by non-α7 nAChRs displayed smaller trial-to-trial fluctuations and were highly sensitive to perturbations of ACh breakdown, implying that they are mediated by some form of volume transmission (Arroyo et al. 2014). Similarly, nEPSCs in the TRN also displayed very low trial-to-trial variability (∼0.1 for single-axon responses, Fig. 2E and F) and showed dramatic prolongation following block of ACh breakdown. In principle, this would be consistent with the activation of dendritic nAChRs and mAChRs by ACh released from multiple sites and with the diffusion of ACh over considerable distances. However, under our conditions, nEPSCs were generated at latencies comparable to AMPAR-mediated EPSCs (∼3.5 ms). Furthermore, activation of even single presynaptic axons was sufficient to generate postsynaptic responses in TRN, showing that pooling of ACh released from multiple afferents is not required for the activation of postsynaptic receptors. It is possible that postsynaptic responses are generated by the synaptic activation of high-affinity nAChRs with slow kinetics, as has been proposed for slow nAChR-mediated components at the cholinergic motoneuron–Renshaw cell synapse (d’Incamps & Ascher, 2014). Understanding the cellular mechanisms as well as the functional consequences of the cholinergic signalling remains an important area for future investigations.
Control of TRN neuronal activity by cholinergic inputs
The location of cholinergic synapses on distal dendrites of TRN neurons (Hallanger & Wainer, 1988) and their slow kinetics as compared to glutamatergic synapses would suggest that ACh release merely modulates TRN neuronal activity. In marked contrast with this assumption, current clamp recordings showed that activation of even individual cholinergic inputs could reliably trigger action potentials (Fig. 3B). Interestingly, ACh-evoked action potential generation required the activation of postsynaptic low-threshold T-type Ca2+ channels (Fig. 3C). In TRN neurons, the most abundant subtype expressed is CaV3.3 with a smaller contribution of CaV3.2 (Astori et al. 2011) and it is well established that their activation is intimately linked to the generation of state-dependent burst firing, a property that enables networks of TRN and TC neurons to engage in oscillatory activity (Lüthi, 2013). More recent imaging studies have shown that these conductances are expressed non-uniformly in the somatodendritic membrane, with higher densities at increasing distance from the soma (Fig. 3A; Crandall et al. 2010). This suggests an important role of T-type Ca2+ channels in the amplification of synaptic inputs that contact the distal dendrites, including not only cholinergic afferents (Sun et al. 2013) but also glutamatergic feedback projections from neocortex (Crandall et al. 2010). For cholinergic inputs, it is possible that action potential initiation from resting membrane potentials is mediated by the generation of a dendritic Ca2+ spike which then propagates towards the initiation zone for Na+ spikes in the axon. The fact that action potentials can also be evoked in tonic mode suggests that T-type conductances expressed in dendrites are not fully inactivated by somatic depolarization (Errington & Connelly, 2011; Deleuze et al. 2012).
Figure 3. Cholinergic synaptic inputs triggers TRN neuronal firing via activation of T-type Ca2+ channels.
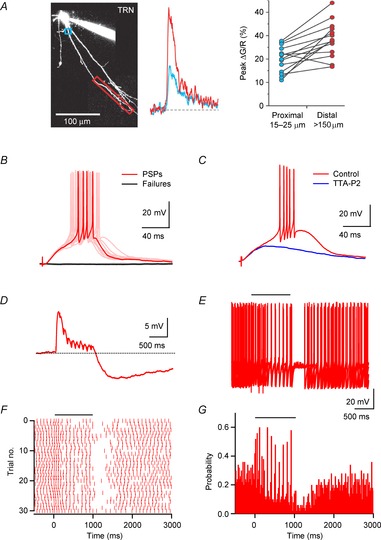
A, somatic burst discharges produce robust Ca2+ responses in distal dendrites of TRN neurons, mediated by the activation of T-type Ca2+ channels. Left, image of TRN neuron filled with the green Ca2+-sensitive dye Fluo-4 and the red Ca2+-insensitive dye Alexa 594, obtained using 2-photon laser scanning microscopy. Boxes in blue and red outline regions from which Ca2+ measurements were obtained. Middle, Ca2+ responses in the proximal and distal dendrite of a different cell, evoked by burst firing at the soma. Fluorescence changes are expressed as ΔG/R, calculated as the change in Fluo-4 fluorescence, normalized to the average fluorescence of Alexa 594. Right, summary data (n = 14 dendrites) showing larger burst-evoked Ca2+ transients in distal dendrites (red) as compared to transients at more proximal dendrites (blue). Data reproduced with permission from Crandall et al. (2010). B, activation of cholinergic inputs triggers burst firing in TRN neuron. A representative experiment shows individual trials of either PSPs generating action potentials (red) or response failures (black), evoked at a constant stimulus intensity, indicating activation of a single presynaptic axon. C, activation of postsynaptic T-type Ca2+ channels is required for ACh-evoked neuronal firing. Representative recording shows cholinergic input-evoked burst firing prior to and following application of the specific T-type Ca2+ channel antagonist TTA-P2. D–G, cholinergic inputs entrain ongoing neuronal activity. Data shown are from the same neuron. D, postsynaptic E–I response evoked by a brief stimulus train (10 stimuli, 10 Hz), for a TRN neuron held at −60 mV. E, the same stimulus train (indicated by horizontal bar) was applied during ongoing action potential activity evoked by depolarizing current steps (6 s, 120 pA). Shown are 5 consecutive trials. F, raster plot showing the timing of spikes in consecutive trials during and following stimulus train (onset of synaptic stimulation at t = 0 ms). G, poststimulus time histogram (bin size 20 ms), compiled for 58 consecutive trials. Data adapted from Sun et al. (2013).
Studies in vivo have reported that cholinergic neurons in the brainstem and the BF can fire at rates of ∼5–15 Hz (Hassani et al. 2009; Boucetta et al. 2014), with BF neurons often generating rhythmic bursts of action potentials during active waking (Lee et al. 2005). This raises the question of how cholinergic synaptic inputs triggered by stimulus trains can influence postsynaptic firing. As described above, nAChR-mediated excitation is tightly controlled by both autoinhibition and postsynaptic hyperpolarization, mediated by pre- and postsynaptic M2 mAChRs, respectively. As a consequence, nAChR-mediated EPSPs are curtailed by mIPSPs during stimulus trains, followed by a long-lasting mIPSP at the end of stimulation (Fig. 3D). However, this does not appear to limit the influence of cholinergic afferent activity to the onset of cholinergic stimulation. In TRN neurons that generate sustained action potential activity (mimicked by depolarizing pulses) cholinergic responses evoked by stimulus trains can entrain postsynaptic firing with high temporal precision (Fig. 3E–G). It remains to be determined how E–I synaptic signalling and intrinsic conductances expressed in TRN dendrites can dynamically interact to control neuronal firing under more realistic conditions.
Remaining questions
Our findings have shown that cholinergic afferents to TRN can generate highly reliable biphasic responses in all TRN neurons, thereby tightly regulating TRN neuronal output. Nevertheless, a number of important questions remain unanswered. Many cholinergic release sites in TRN are not associated with postsynaptic dendrites and it is possible that ACh release from these terminals is responsible for slow and sustained activation of either extrasynaptic receptors in TRN dendrites or presynaptic receptors expressed by nearby glutamatergic or GABAergic synapses, thereby mediating heterosynaptic plasticity (Fig. 1C). Furthermore, slow changes in ACh levels might be involved in the activation of nAChRs expressed in TC axons (Kawai et al. 2007). In turn, it will be important to understand if and how fast cholinergic synaptic strength can be modulated on different time scales, for example by local increases in glutamate, or by the release of endocannabinoids from TRN neurons (Sun et al. 2011).
A more fundamental issue is the origin of synaptic afferents giving rise to fast E–I signalling. TRN is unique in that it receives cholinergic afferents from both BF and the brainstem. Given the prevalence of fast cholinergic responses mediated by BF cholinergic afferents in neocortex (Letzkus et al. 2011; Arroyo et al. 2012), hippocampus (Bell et al. 2011; Gu & Yakel, 2011), and the olfactory bulb (Ma & Luo, 2012), it would be tempting to conclude that E–I signalling is similarly mediated by BF neuronal activity but this awaits direct anatomical and physiological confirmation. If BF neurons are in fact responsible for the recruitment of TRN neurons, it raises the interesting possibility of a rapid and spatiotemporally precise cholinergic control of sensory processing and perception on two distinct levels, in the thalamus as well as in neocortex.
Glossary
- AMPAR
AMPA receptor
- BF
basal forebrain
- E–I
excitatory–inhibitory
- mAChR
muscarinic acetylcholine receptor
- mIPSC
mAChR-mediated IPSC
- nAChR
nicotinic acetylcholine receptor
- nEPSC
nAChR-mediated EPSC
- TC
thalamocortical
- TRN
thalamic reticular nucleus
Additional information
Competing interests
None declared.
Funding
Our research is supported by funds from the NIH (Grant NS077989).
References
- Arroyo S, Bennett C, Aziz D, Brown SP, Hestrin S. Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J Neurosci. 2012;32:3859–3864. doi: 10.1523/JNEUROSCI.0115-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Hestrin S. Nicotinic modulation of cortical circuits. Front Neural Circuits. 2014;8:30. doi: 10.3389/fncir.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Luthi A. The CaV3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A. 2011;108:13823–13828. doi: 10.1073/pnas.1105115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KA, Shim H, Chen CK, McQuiston AR. Nicotinic excitatory postsynaptic potentials in hippocampal CA1 interneurons are predominantly mediated by nicotinic receptors that contain α4 and β2 subunits. Neuropharmacology. 2011;61:1379–1388. doi: 10.1016/j.neuropharm.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C, Arroyo S, Berns D, Hestrin S. Mechanisms generating dual-component nicotinic EPSCs in cortical interneurons. J Neurosci. 2012;32:17287–17296. doi: 10.1523/JNEUROSCI.3565-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista P, Meuth SG, Kanyshkova T, Cerina M, Pawlowski M, Ehling P, Landgraf P, Borsotto M, Heurteaux C, Pape HC, Baukrowitz T, Budde T. Identification of the muscarinic pathway underlying cessation of sleep-related burst activity in rat thalamocortical relay neurons. Pflugers Arch. 2012;463:89–102. doi: 10.1007/s00424-011-1056-9. [DOI] [PubMed] [Google Scholar]
- Blankenship JE, Wachtel H, Kandel ER. Ionic mechanisms of excitatory, inhibitory, and dual synaptic actions mediated by an identified interneuron in abdominal ganglion of Aplysia. J Neurophysiol. 1971;34:76–92. doi: 10.1152/jn.1971.34.1.76. [DOI] [PubMed] [Google Scholar]
- Boucetta S, Cisse Y, Mainville L, Morales M, Jones BE. Discharge profiles across the sleep-waking cycle of identified cholinergic, GABAergic, and glutamatergic neurons in the pontomesencephalic tegmentum of the rat. J Neurosci. 2014;34:4708–4727. doi: 10.1523/JNEUROSCI.2617-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brombas A, Fletcher LN, Williams SR. Activity-dependent modulation of layer 1 inhibitory neocortical circuits by acetylcholine. J Neurosci. 2014;34:1932–1941. doi: 10.1523/JNEUROSCI.4470-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signalling in thalamic reticular neurons. J Neurosci. 2010;30:15419–15429. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curro Dossi R, Pare D, Steriade M. Short-lasting nicotinic and long-lasting muscarinic depolarizing responses of thalamocortical neurons to stimulation of mesopontine cholinergic nuclei. J Neurophysiol. 1991;65:393–406. doi: 10.1152/jn.1991.65.3.393. [DOI] [PubMed] [Google Scholar]
- Deleuze C, David F, Béhuret S, Sadoc G, Shin HS, Uebele VN, Renger JJ, Lambert RC, Leresche N, Bal T. T-type calcium channels consolidate tonic action potential output of thalamic neurons to neocortex. J Neurosci. 2012;32:12228–12236. doi: 10.1523/JNEUROSCI.1362-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Gisiger V, Steriade M. Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol. 1997;53:603–625. doi: 10.1016/s0301-0082(97)00050-6. [DOI] [PubMed] [Google Scholar]
- d'Incamps BL, Ascher P. High affinity and low affinity heteromeric nicotinic acetylcholine receptors at central synapses. J Physiol. 2014 doi: 10.1113/jphysiol.2014.273128. (in press; DOI: 10.1113/jphysiol.2014.273128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington AC, Connelly WM. Dendritic T-type Ca2+ channels: giving a boost to thalamic reticular neurons. J Neurosci. 2011;31:5551–5553. doi: 10.1523/JNEUROSCI.0067-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science. 2000;288:2366–2368. doi: 10.1126/science.288.5475.2366. [DOI] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallanger AE, Levey AI, Lee HJ, Rye DB, Wainer BH. The origins of cholinergic and other subcortical afferents to the thalamus in the rat. J Comp Neurol. 1987;262:105–124. doi: 10.1002/cne.902620109. [DOI] [PubMed] [Google Scholar]
- Hallanger AE, Wainer BH. Ultrastructure of ChAT-immunoreactive synaptic terminals in the thalamic reticular nucleus of the rat. J Comp Neurol. 1988;278:486–497. doi: 10.1002/cne.902780403. [DOI] [PubMed] [Google Scholar]
- Han Y, Shi YF, Xi W, Zhou R, Tan ZB, Wang H, Li XM, Chen Z, Feng G, Luo M, Huang ZL, Duan S, Yu YQ. Selective activation of cholinergic basal forebrain neurons induces immediate sleep-wake transitions. Curr Biol. 2014;24:693–698. doi: 10.1016/j.cub.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Harris KD, Thiele A. Cortical state and attention. Nat Rev Neurosci. 2011;12:509–523. doi: 10.1038/nrn3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Henny P, Jones BE. Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep-wake cycle. J Neurosci. 2009;29:11828–11840. doi: 10.1523/JNEUROSCI.1259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata A, Aguilar J, Castro-Alamancos MA. Noradrenergic activation amplifies bottom-up and top-down signal-to-noise ratios in sensory thalamus. J Neurosci. 2006;26:4426–4436. doi: 10.1523/JNEUROSCI.5298-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Steriade M, Deschenes M. The effects of brainstem peribrachial stimulation on neurons of the lateral geniculate nucleus. Neuroscience. 1989a;31:13–24. doi: 10.1016/0306-4522(89)90027-4. [DOI] [PubMed] [Google Scholar]
- Hu B, Steriade M, Deschenes M. The effects of brainstem peribrachial stimulation on perigeniculate neurons: the blockage of spindle waves. Neuroscience. 1989b;31:1–12. doi: 10.1016/0306-4522(89)90026-2. [DOI] [PubMed] [Google Scholar]
- Jones EG. The Thalamus. Cambridge, New York: Cambridge University Press; 2007. [Google Scholar]
- Kawai H, Lazar R, Metherate R. Nicotinic control of axon excitability regulates thalamocortical transmission. Nat Neurosci. 2007;10:1168–1175. doi: 10.1038/nn1956. [DOI] [PubMed] [Google Scholar]
- Lee KH, McCormick DA. Acetylcholine excites GABAergic neurons of the ferret perigeniculate nucleus through nicotinic receptors. J Neurophysiol. 1995;73:2123–2128. doi: 10.1152/jn.1995.73.5.2123. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE. Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci. 2005;25:4365–4369. doi: 10.1523/JNEUROSCI.0178-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Dan Y. Neuromodulation of brain states. Neuron. 2012;76:209–222. doi: 10.1016/j.neuron.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Luthi A. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature. 2011;480:331–335. doi: 10.1038/nature10674. [DOI] [PubMed] [Google Scholar]
- Lüthi A. Sleep spindles: where they come from, what they do. Neuroscientist. 2013;20:243–256. doi: 10.1177/1073858413500854. [DOI] [PubMed] [Google Scholar]
- Ma M, Luo M. Optogenetic activation of basal forebrain cholinergic neurons modulates neuronal excitability and sensory responses in the main olfactory bulb. J Neurosci. 2012;32:10105–10116. doi: 10.1523/JNEUROSCI.0058-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- Muñoz W, Rudy B. Spatiotemporal specificity in cholinergic control of neocortical function. Curr Opin Neurobiol. 2014;26C:149–160. doi: 10.1016/j.conb.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent M, Descarries L. Acetylcholine innervation of the adult rat thalamus: distribution and ultrastructural features in dorsolateral geniculate, parafascicular, and reticular thalamic nuclei. J Comp Neurol. 2008;511:678–691. doi: 10.1002/cne.21868. [DOI] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–154. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Pinault D, Deschênes M. Muscarinic inhibition of reticular thalamic cells by basal forebrain neurones. Neuroreport. 1992;3:1101–1104. doi: 10.1097/00001756-199212000-00017. [DOI] [PubMed] [Google Scholar]
- Pinto L, Goard MJ, Estandian D, Xu M, Kwan AC, Lee SH, Harrison TC, Feng G, Dan Y. Fast modulation of visual perception by basal forebrain cholinergic neurons. Nat Neurosci. 2013;16:1857–1863. doi: 10.1038/nn.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Acetylcholine systems and rhythmic activities during the waking–sleep cycle. Prog Brain Res. 2004;145:179–196. doi: 10.1016/S0079-6123(03)45013-9. [DOI] [PubMed] [Google Scholar]
- Sun YG, Pita-Almenar JD, Wu CS, Renger JJ, Uebele VN, Lu HC, Beierlein M. Biphasic cholinergic synaptic transmission controls action potential activity in thalamic reticular nucleus neurons. J Neurosci. 2013;33:2048–2059. doi: 10.1523/JNEUROSCI.3177-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YG, Wu CS, Lu HC, Beierlein M. Target-dependent control of synaptic inhibition by endocannabinoids in the thalamus. J Neurosci. 2011;31:9222–9230. doi: 10.1523/JNEUROSCI.0531-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh CA, Acosta CG, Ashe JH. Modification of nicotinic ganglionic transmission by muscarinic slow postsynaptic potentials in the in vitro rabbit superior cervical ganglion. Synapse. 1988;2:174–182. doi: 10.1002/syn.890020209. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Uhlrich DJ. Nicotinic receptor-mediated responses in relay cells and interneurons in the rat lateral geniculate nucleus. Neuroscience. 1997;80:191–202. doi: 10.1016/s0306-4522(97)00095-x. [DOI] [PubMed] [Google Scholar]