

Abstract
The magnocellular neurosecretory cells of the hypothalamus (MNCs) synthesize and secrete vasopressin or oxytocin. A stretch-inactivated cation current mediated by TRPV1 channels rapidly transduces increases in external osmolality into a depolarization of the MNCs leading to an increase in action potential firing and thus hormone release. Prolonged increases in external osmolality, however, trigger a reversible structural and functional adaptation that may enable the MNCs to sustain high levels of hormone release. One poorly understood aspect of this adaptation is somatic hypertrophy. We demonstrate that hypertrophy can be evoked in acutely isolated rat MNCs by exposure to hypertonic solutions lasting tens of minutes. Osmotically evoked hypertrophy requires activation of the stretch-inactivated cation channel, action potential firing, and the influx of Ca2+. Hypertrophy is prevented by pretreatment with a cell-permeant inhibitor of exocytotic fusion and is associated with an increase in total membrane capacitance. Recovery is disrupted by an inhibitor of dynamin function, suggesting that it requires endocytosis. We also demonstrate that hypertonic solutions cause a decrease in phosphatidylinositol 4,5-bisphosphate in the plasma membranes of MNCs that is prevented by an inhibitor of phospholipase C (PLC). Inhibitors of PLC or protein kinase C (PKC) prevent osmotically evoked hypertrophy, and treatment with a PKC-activating phorbol ester can elicit hypertrophy in the absence of changes in osmolality. These studies suggest that increases in osmolality cause fusion of internal membranes with the plasma membrane of the MNCs and that this process is mediated by activity-dependent activation of PLC and PKC.
Key points
Acutely isolated rat magnocellular neurosecretory cells (MNCs) display reversible hypertrophy when exposed to hypertonic saline (325 mosmol kg−1) for tens of minutes.
Osmotically evoked hypertrophy is prevented by agents that block Na+ channels, TRPV1 channels (which mediate an osmotically evoked depolarization in these cells), L-type Ca2+ channels, and exocytotic fusion and is associated with an increase in whole-cell capacitance.
Exposure to hypertonic saline activates phospholipase C leading to a decrease in plasma membrane phosphatidylinositol 4,5-bisphosphate.
Inhibition of phospholipase C or protein kinase C prevents osmotically evoked hypertrophy and activation of protein kinase C evokes hypertrophy in the absence of an increase in osmolality.
These results suggest that osmotically evoked hypertrophy is triggered by an increase in MNC action potential firing, leading to Ca2+ influx, the activation of phospholipase C, an increase in diacylglycerol, and the activation of protein kinase C.
Introduction
The magnocellular neurosecretory cells (MNCs) of the supraoptic and paraventricular nuclei of the hypothalamus are osmosensitive (Bourque, 2008) in that their electrical excitability is altered by changes in external osmolality. These cells regulate body fluid balance by releasing more vasopressin (VP) and oxytocin (OT) as extracellular osmolality increases (Bourque, 2008). MNCs show a decrease in volume and thus plasma membrane tension in response to acute increases in osmolality and lack the acute compensatory mechanisms (Zhang & Bourque, 2003) that limit osmotically evoked volume changes in most cell types (Hoffmann et al. 2009). This enables MNCs to faithfully transduce increases in osmolality into a depolarizing current through activation of a stretch-inactivated cation channel (SIC; Oliet & Bourque, 1993) mediated by a variant of the transient receptor potential cation channel vanilloid subfamily member 1 (TRPV1) channel (Sharif Naeini et al. 2006). Increases in osmolality lasting hours, however, also activate a broad and dramatic functional and structural transformation of the MNCs that is thought to enable sustained high levels of hormone release. Similar changes occur in OT-releasing MNCs during lactation and parturition, when the release of OT is elevated (Theodosis et al. 2008). Osmotically evoked adaptations include structural changes such as retraction of the glial processes surrounding the MNCs (Theodosis et al. 2008) and increased synaptic innervation (Tasker et al. 2002), and also functional changes such as up-regulation of many genes (Ghorbel et al. 2003), and increases in the cell surface expression of many channels and receptors (Shuster et al. 1999; Tanaka et al. 1999; Hurbin et al. 2002; Zhang et al. 2007). One aspect of this adaptation is a marked and reversible hypertrophy of the MNC somata (Miyata & Hatton, 2002).
The sequence of events that triggers and mediates MNC hypertrophy is unknown and difficult to elucidate using in vivo models. We therefore sought to test whether exposure to hypertonic saline could evoke hypertrophy in MNCs acutely isolated from adult rats. We observed that MNCs undergo hypertrophy when exposed to increases in external osmolality lasting tens of minutes and that this occurs in response to increases in external osmolality that would be expected to occur physiologically. We present evidence that the initiation and maintenance of osmotically induced hypertrophy is activity dependent and occurs through soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE)-dependent exocytotic fusion of internal membranes with the MNC plasma membrane. Furthermore, we show that exposure of isolated MNCs to hypertonic solutions causes a rapid increase in the activity of the enzyme phospholipase C and that this activation appears to be central to the initiation of osmotically evoked hypertrophy. Our results demonstrate a mechanism that is likely to underlie at least part of the osmotically induced hypertrophy that has been observed in mammalian MNCs in situ and suggest that MNC somata may undergo dynamic structural regulation in vivo in response to changes in external osmolality within the physiological range.
Methods
Ethical approval
This work was approved by the University of Saskatchewan's Animal Research Ethics Board, and adhered to the Canadian Council on Animal Care guidelines for humane animal use.
Animals and cell preparation
MNCs were isolated using a protocol described previously (Liu et al. 2005) and identified using the criterion established by Oliet & Bourque (1992), i.e. a maximal cross-sectional area (CSA) greater than 160 μm2. In brief, male Long–Evans rats (200–300 g) were anaesthetized with halothane and killed by decapitation. The brain was removed and blocks of tissue containing most of the two supraoptic nuclei were excised. The tissue blocks were incubated with an oxygenated (100% O2) Pipes solution (pH 7.1) composed of (in mm): NaCl, 110; KCl, 5; MgCl2, 1; CaCl2, 1; Pipes, 20; glucose, 25; and containing trypsin (Type XI, 0.6 mg ml−1) for 90 min at 34°C. After incubation, the tissues were then transferred into oxygenated Pipes solution without trypsin for 30 min at room temperature. Finally, the tissues were gently triturated with fire-polished pipettes to disperse the cells, which were plated onto glass-bottomed culture dishes and kept at room temperature for use the same day. Hippocampal neurons were isolated from hippocampal tissue blocks obtained from adult rats using a similar protocol. The osmolalities of the external solutions were adjusted before each experiment to 295 ± 3 mosmol kg−1, or as noted in the text, using a VAPRO pressure osmometer (WESCOR; Logan, UT, USA) by adding mannitol as required.
Hypertrophy experiments
In some experiments, the MNCs were perfused with oxygenated isotonic Pipes saline, switched to hypertonic saline at the indicated osmolality, and then returned to isotonic saline. In other experiments, MNCs were exposed to stationary bath solutions of defined osmolality, with or without the addition of chemicals, as indicated in the text. Healthy-looking MNCs (typically 2–3 per dish) were photographed at the indicated times with a cooled CCD camera attached to a Zeiss (Jena, Germany) Axiovert 200 inverted fluorescence microscope using a ×40 objective. The maximal circumference of the cell soma was traced and the CSA determined using ImageJ (NIH). MNCs that failed to shrink in response to application of hypertonic solution or to recover toward baseline when they were returned to isotonic solution were considered unhealthy and were excluded from further analysis. Following rapid shrinkage, most MNCs showed a slow hypertrophy to at least their baseline CSA in both the perfusion studies shown in Fig. 1B (12 out of 15 MNCs treated with 325 mosmol kg−1 and 10 out of 12 MNCs treated with 305 mosmol kg−1), Fig. 1C (in the presence of bumetanide; 10 out of 12), and Fig. 2D (10 out of 13), and for the stationary bath experiments shown in Fig. 1D (17 out of 21 MNCs), Fig. 2B (21 out of 24), and Fig. 2C (in the presence of the scrambled version of the TAT-NSF700scr peptide; 19 out of 19). We do not know if the MNCs that do not hypertrophy are a distinct subset of MNCs or have incurred some form of damage during the isolation procedure that prevents them from being activated by hypertonic saline or from undergoing hypertrophy. We did not include data on MNCs that did not hypertrophy in the plots shown to give a better indication of the hypertrophic response. Inclusion of the MNCs that did not undergo hypertrophy in response to hypertonic treatment does not change the level of significance of any of the statistical comparisons shown in the Results. Data were normalized by dividing each measurement by the mean CSA of that cell during the control period and are expressed as mean ± SEM. For the fluorescent images shown in Fig. 1A, MNCs were incubated with the membrane dye CellMask Orange (Invitrogen; Carlsbad, CA, USA; 5 μg ml−1) for 5 min and then rinsed with isotonic saline three times. Fluorescence imaging was performed as described below.
Figure 1. Increases in osmolality evoke reversible hypertrophy in osmosensitive supraoptic neurons but not hippocampal neurons.
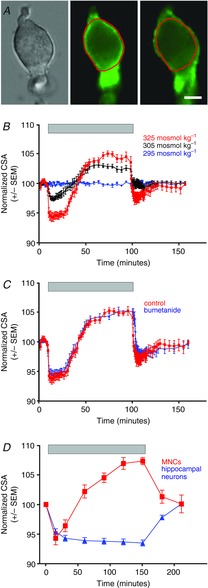
A, images of an acutely isolated MNC showing osmotically evoked cell shrinkage and hypertrophy. The image on the left shows a DIC image of an isolated MNC in isotonic saline. The two images to the right show the fluorescence of a plasma membrane dye (CellMask Orange; see Methods) in the same cell 5 and 80 min after administration of hypertonic saline. The red line shows the perimeter of the cell under isotonic conditions for comparison. Note that the cell in the centre image shows shrinkage relative to the red line and the right image shows enlargement relative to the red line. The scale bar indicates 10 μm. B, perfusion of oxygenated hypertonic saline (325 and 305 mosmol kg−1) causes isolated MNCs to shrink and then hypertrophy over tens of minutes (n = 12 and 10, respectively) whereas perfusion with isotonic saline (295 mosmol kg−1) has no effect. The period of perfusion of hypertonic saline is indicated by the bar at the top of the plot. Return to isotonic saline causes the cells to return to their original size. C, the response to perfusion of hypertonic saline (325 mosmol kg−1) was not affected by the presence of bumetanide (10 μm; n = 10), which is an inhibitor of the Na+–K+–Cl− co-transporter NKCC1. The response of the MNCs to perfusion of hypertonic saline (325 mosmol kg−1) is shown for comparison (and is labelled ‘control’). D, similar results were seen with MNCs that were maintained in a stationary bath that was switched to a hypertonic saline (325 mosmol kg−1) and then back to isotonic saline (n = 17). Isolated hippocampal neurons respond with shrinkage, but not hypertrophy, to this treatment (n = 20).
Figure 2. The initiation and maintenance of osmotically evoked hypertrophy depends upon cell firing and Ca2+ influx and involves exocytotic fusion.
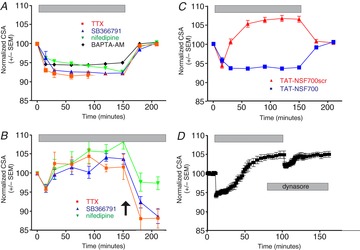
A, hypertrophy is prevented by treatment with tetrodotoxin (“TTX”; 0.2 μm; n = 24), SB336791 (1.5 μm; n = 26), nifedipine (10 μm; n = 27), or BAPTA-AM (10 μm; n = 20). B, hypertrophy is reversed in hypertonic saline by application (at arrow) of TTX (0.2 μm; n = 6), SB355791 (1.5 μm; n = 7), or nifedipine (10 μm; n = 7). C, hypertrophy is prevented by administration of the cell-permeant peptide TAT-NSF700 (n = 57), which blocks SNARE-mediated exocytotic fusion, but not the scrambled version of the peptide (TAT-NSF700scr; n = 19). D, the administration of dynasore (80 μm), an inhibitor of dynamin-mediated endocytosis, inhibits recovery from osmotically evoked hypertrophy (n = 10).
Electrophysiological methods
The plasma membrane capacitances of acutely isolated rat MNCs were determined using whole-cell patch clamp at room temperature. The values for MNCs exposed to hypertonic (325 mosmol kg−1) saline for 90 min or more were compared to those of MNCs maintained in isotonic (295 mosmol kg−1) saline. Borosilicate glass capillaries (1.2 mm o.d., 0.68 mm i.d.; A-M Systems; Carlsborg, WA, USA) were used to pull patch pipettes on a P-97 horizontal pipette puller (Sutter Instrument Company; Novato, CA, USA) and fire-polished using a microforge (Narashige; Tokyo, Japan). They were filled with an internal solution containing (in mm): 140 KCl, 10 Hepes, 1 MgCl2, 1 EGTA, and 1 Mg-ATP (pH 7.2) and had a resistance of 2–4 MΩ. The whole-cell membrane capacitances of MNCs were estimated using an EPC-9 amplifier (HEKA Elektronik; Lambrecht/Pfalz, Germany) controlled with PULSE software (HEKA), using the Auto-CSlow function of PULSE. Data are expressed as mean ± SEM.
Immunocytochemistry
Acutely isolated rat MNCs were incubated in Pipes saline with or without the PLC inhibitor U73122 (Enzo Life Sciences; Farmingdale, NY, USA) for 20 min and then stimulated with either hypertonic Pipes saline (325 mosmol kg−1) or isotonic saline containing 10 μm oxotremorine (Sigma) for 5 min. The control cells were left untreated. The cells were then subjected to phosphatidylinositol 4,5-bisphosphate (PIP2) immunostaining in rat MNCs using a modification of a published protocol (Hammond et al. 2006). Briefly, the cells were fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde and 0.1% glutaraldehyde for 20–25 min at room temperature. Following three washes with PBS, the cells were blocked with solution containing 10% donkey serum and 0.5% saponin for 1 h. The cells were then incubated with a mouse monoclonal PIP2 antibody (Enzo Life Sciences; 1:1000) overnight at 4°C. The dishes were washed with PBS three times and incubated for another 1 h with donkey anti-mouse secondary antibody (Invitrogen Alexa Fluor 488, 1:1000). After three washes with PBS, Citifluor mounting solution (Citifluor Ltd; Gore, QC, Canada) was added to the dishes and cells were then viewed using a Zeiss inverted Axiovert 200 microscope with appropriate filter sets and a ×40 objective and images were captured using a cooled CCD camera. The images were analysed using ImageJ software (NIH) by tracing the perimeter of each soma by following the line of greatest fluorescence (disregarding processes) and determining the mean fluorescence of pixels on that line. The mean intensities of staining for all MNCs in each treatment group were normalized to the mean fluorescence of all of the control cells on each experimental day. Data are expressed as normalized mean ± SEM.
Chemicals
All chemicals, unless stated otherwise, were from Sigma-Aldrich Corporation (St Louis, MO, USA). The TAT-NSF700 peptide and its scrambled version were purchased from AnaSpec, Inc. (Fremont, CA, USA) and were used at a concentration of 1.2 μm.
Results
We sought to determine if an increase in osmolality can trigger hypertrophy in MNCs acutely isolated from adult rats and, if so, to elucidate the underlying mechanisms. We used the maximal cross-sectional area (CSA) of the MNCs to monitor changes in volume, as has been used previously (Zhang & Bourque, 2003), and observed that treatment with hypertonic saline caused rapid cell shrinkage followed by slower cell enlargement. This is illustrated in Fig. 1A, which shows an acutely isolated MNC and the shrinkage and enlargement of that cell following treatment with hypertonic saline. Note that the fluorescent membrane dye used to obtain these images was for demonstration purposes only; in all of the other experiments we measured the cell perimeter using differential interference contrast (DIC) images of the MNCs. To determine the time course of these changes, MNCs (n = 12) were perfused with an oxygenated saline solution with an osmolality close to the normal set point in the rat (i.e. “isotonic” or 295 mosmol kg−1) and then switched to a hypertonic saline (325 mosmol kg−1). MNCs rapidly shrunk to approximately 94% of control (a reduction of mean CSA from 363 ± 36 μm2 to 343 ± 36 μm2; Fig. 1B), but after a delay of about 20 min started to hypertrophy and achieved a peak size of approximately 105% of control (381 ± 38 μm2) after about 1 h (Fig. 1B). The mean CSA during the shrunken and enlarged states (measured 5 and 75 min after the beginning of perfusion of hypertonic saline, respectively) were both significantly different than the mean baseline CSA (using a one-way repeated measures analysis of variance test; P < 0.01 in both cases). Smaller amounts of shrinkage and hypertrophy were observed (Fig. 1B) when MNCs were perfused with 305 mosmol kg−1 saline (98% and 103%; n = 10), but these differences were also significant (using a one-way repeated measures analysis of variance test; P < 0.01 in both cases). MNCs rapidly recovered to their control size when returned to isotonic saline and no changes in size were observed in MNCs maintained for similar time periods in isotonic saline. The mean CSA during the shrunken and enlarged states following perfusion with 325 mosmol kg−1 or 305 mosmol kg−1 saline were also significantly different from the mean CSA of MNCs perfused with isotonic saline for similar periods (using a two-way analysis of variance; P < 0.01 in all cases). The hypertrophic response did not appear to be altered by inhibition of the Na+–K+–Cl− cotransporter NKCC1, which is commonly involved in cell volume regulation, by the antagonist bumetanide (10 μm; Fig. 1C). Experiments that were conducted using a stationary bath showed a similar pattern of hypertrophy in response to hypertonic saline (Fig. 1D), but acutely isolated hippocampal neurons did not display osmotically evoked hypertrophy (Fig. 1D), suggesting that the response is specific to the MNCs.
Preincubation with the Na+ channel blocker tetrodotoxin (TTX; 0.2 μm) prevented hypertrophy (Fig. 2A), demonstrating that the response is dependent upon the activation of action potentials. Hypertrophy was also prevented by SB366791 (1.5 μm), which blocks TRPV1 channels (and more specifically the SIC; Sharif-Naeini et al. 2008), suggesting that activation of the SIC is necessary for hypertrophy, by the cell-permeant Ca2+ chelator BAPTA-AM (10 μm), suggesting that an increase in intracellular Ca2+ is required, and by the L-type Ca2+ channel blocker nifedipine (10 μm), suggesting that the effect depends on Ca2+ influx through L-type Ca2+ channels (Fig. 2A). These data suggest that increases in external osmolality cause MNC shrinkage, leading to the activation of the SIC, an increase in the firing of action potentials, and an increase in Ca2+ influx through L-type Ca2+ channels, and that the resultant increase in intracellular Ca2+ somehow activates hypertrophy. The addition of TTX, SB366791, or nifedipine to MNCs in hypertonic solutions following a hypertrophic response caused its reversal (Fig. 2B), suggesting that the maintenance of hypertrophy is dependent on continued electrical activity and Ca2+ influx and that the cessation of Ca2+ influx leads to the reversal of the process. These data also suggest that MNCs continue to fire action potentials even when their surface area has been significantly enlarged and that hypertrophy does not therefore decrease activity of the SIC.
We attempted to block the hypertrophic response using TAT-NSF700 (Matsushita et al. 2005), a peptide that prevents SNARE-mediated exocytotic fusion by blocking the function of N-ethylmaleimide-sensitive factor (NSF). Although the presence of a scrambled version of the peptide had no apparent effect on the response of the MNCs to increased osmolality, hypertrophy was virtually eliminated by preincubation with TAT-NSF700 (n = 57; Fig. 2C), suggesting that hypertrophy depends on SNARE-mediated exocytotic fusion. The mean CSA of hypertrophied MNCs incubated with 325 mosmol kg−1 saline in the presence of the scrambled peptide was significantly larger than the mean CSA of MNCs incubated with 325 mosmol kg−1 saline in the presence of TAT-NSF700 (using a two-way analysis of variance; P < 0.01). Dynasore (80 μm), an inhibitor of dynamin-dependent endocytosis, was applied to MNCs in hypertonic saline (325 mosmol kg−1) to test whether the rapid recovery of MNC cell size following hypertrophy requires membrane internalization. Dynasore prevented the recovery of MNCs to their original size when they were returned to isotonic saline, suggesting that the recovery process involves endocytotic retrieval of membrane from the MNC plasma membrane (Fig. 2D).
We tested whether osmotically evoked hypertrophy was associated with an increase in plasma membrane area by measuring the cell capacitance of isolated MNCs using whole-cell patch clamp techniques. We found (Fig. 3) that the whole-cell capacitance was larger in MNCs that had been exposed to hypertonic (325 mosmol kg−1) solutions for at least 90 min (16.7 ± 0.4 pF; n = 71) compared to that of MNCs maintained in isotonic (295 mosmol kg−1) solution (15.6 ± 0.3 pF; n = 66; P < 0.05). These data support the hypothesis that the hypertrophic response involves the fusion of internal membranes with the MNC plasma membrane.
Figure 3. Exposure to hypertonic saline causes an increase in the total plasma membrane capacitance of isolated MNCs.
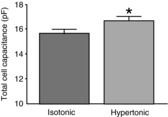
The bars indicate the mean capacitance measured using whole-cell patch clamp in isolated cells maintained in isotonic (295 mosmol kg−1) or hypertonic (325 mosmol kg−1) saline for at least 90 min. MNCs exposed to hypertonic saline had a greater total membrane capacitance (16.7 ± 0.4 pF; n = 71) than MNCs maintained in isotonic saline (15.6 ± 0.3 pF; n = 66). Data are expressed as mean ± SEM (*P < 0.05).
Activation of PKC by diacylglycerol (DAG has been implicated in translocation of Ca2+ channels to the cell surface in molluscan neuroendocrine cells (Strong et al. 1987) and of transient receptor potential channels in neurons (Morenilla-Palao et al. 2004) and we therefore sought to determine whether such a mechanism could be involved in osmotically evoked fusion of internal membranes with the MNC plasma membrane. DAG is produced by the cleavage of PIP2 by the enzyme PLC and we therefore tested whether exposure to high osmolality causes a decrease in PIP2 immunoreactivity in isolated MNCs. We found robust PIP2 immunoreactivity in the plasma membrane of acutely isolated MNCs and that this immunoreactivity was reduced by exposure to hypertonic saline (Fig. 4A and B). When the data from all cells are normalized to the mean intensity of staining in control cells we found that the level of staining in MNCs treated for 5 min with hypertonic saline (72.5 ± 3.4; n = 254 cells in 7 experiments) was decreased compared to that in control cells (100 ± 3.8; n = 276 cells in 7 experiments; P < 0.01 using a paired t test), and that this difference was prevented by pretreatment with the PLC inhibitor U73122 (104.7 ± 2.8; n = 303 cells in 7 experiments). These data suggest that exposure to hypertonic saline causes a decrease in membrane PIP2 levels through the activation of PLC. Treatment of MNCs with the muscarinic receptor agonist oxotremorine also causes a decrease in PIP2 immunoreactivity (68 ± 4.3; n = 155 cells in 4 experiments; P < 0.05 using a paired t test) that is prevented by U73122 (97.7 ± 3.9; n = 127 cells in 4 experiments).
Figure 4. Exposure to hypertonic saline causes a decrease in immunoreactivity to PIP2 in the plasma membrane of isolated MNCs.
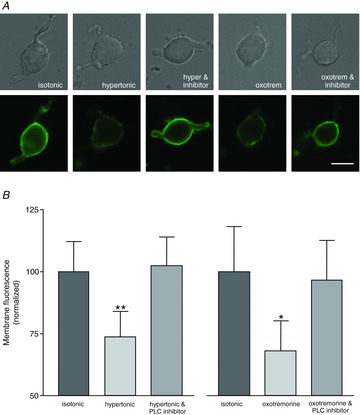
A, images of isolated MNCs using either differential interference contrast images (upper panels) or fluorescence images showing immunoreactivity for PIP2 (lower panels). MNCs were maintained in isotonic saline (control), or exposed to hypertonic saline (hypertonic), hypertonic saline with the PLC inhibitor U73122 (‘hyper & inhibitor’), the muscarinic agonist oxotremorine (‘oxotrem’), or oxotremorine and U73122 (‘oxotrem & inhibitor’). B, the bar graph to the left shows the normalized immunoreactivity to PIP2 in MNCs maintained in isotonic saline (control; 100.0 ± 12.0; n = 276 cells in 7 experiments) exposed to hypertonic saline (73.7 ± 10.5; n = 254 cells in 7 experiments), and hypertonic saline with the PLC inhibitor U73122 (102.4 ± 11.6; n = 303 cells in 7 experiments). The bar graph on the right shows the normalized immunoreactivity to PIP2 in MNCs maintained in isotonic saline (control; 100.0 ± 18.2; n = 139 cells in 4 experiments), exposed to the muscarinic agonist oxotremorine (68.1 ± 12.1; n = 155 cells in 4 experiments), and exposed to oxotremorine and U73122 (96.6 ± 16.0; n = 127 cells in 4 experiments). Data are expressed as mean normalized fluorescence intensity ± SEM (*P < 0.05; **P < 0.01).
We then exposed MNCs to hypertonic solutions in the presence of inhibitors of PLC and PKC to test whether the activation of PLC is required for osmotically evoked hypertrophy. MNCs exposed to hypertonic saline (325 mosmol kg−1) in the presence of either a PLC inhibitor (U73122; 1 μm) or a PKC inhibitor (bisindolylmaleimide I; 1 μm) displayed osmotically induced cell shrinkage but not hypertrophy (Fig. 5A). The mean CSA of MNCs at the end of the incubation with 325 mosmol kg−1 saline in the presence of either of the two inhibitors was significantly smaller than the mean CSA of MNCs incubated in their absence (using a two-way analysis of variance; P < 0.01 in both cases). Furthermore, application of the PKC activator phorbol 12-myristate 13-acetate (0.1 μm) induced hypertrophy in the absence of an increase in osmolality in 7 out of 10 cells tested. The mean response of the cells that showed enlargement is shown in Fig. 5A. The inactive phorbol ester 4α-phorbol 12-myristate 13-acetate (0.1 μm) caused no change in cell size (not shown). The mean CSA of MNCs treated with the PKC activator was significantly larger than the mean CSA of MNCs treated with the inactive phorbol analogue (using a two-way analysis of variance; P < 0.01). Hypertrophy was also evoked by addition of the Ca2+ ionophore A23187 (10 μm) in isotonic solution or by exposure to isotonic saline with an elevated (25 mm) concentration of K+ (Fig. 5B), which would be expected to depolarize the resting membrane potential of the MNCs to about −40 mV. This depolarization could result in Ca2+ influx by triggering the firing of action potentials or it could cause influx of Ca2+ through the low-voltage-activated L-type Ca2+ channels that are expressed in MNCs (Fisher & Bourque, 1995). Hypertrophy evoked by high K+ concentrations was also prevented by the presence of U73122 (1 μm; Fig. 5B). The mean CSA of MNCs incubated with high K+ saline was significantly larger than the mean CSA of MNCs incubated with high K+ saline in the presence of the PLC inhibitor (using a two-way analysis of variance; P < 0.01). These results are consistent with the hypothesis that osmotically evoked hypertrophy depends upon activity-dependent Ca2+ influx leading to the activation of PLC and, through an increase in the concentration of DAG, activation of PKC.
Figure 5. Osmotically evoked hypertrophy is prevented by inhibitors of PLC or PKC and hypertrophy may be activated by a Ca2+ ionophore or by exposure to high K+ saline.
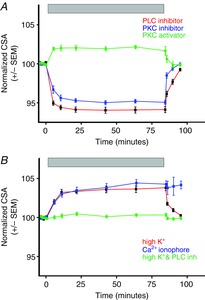
A, exposure (indicated by grey bar) to hypertonic (325 mosmol kg−1) saline causes MNC shrinkage, but not hypertrophy, in the presence of the PLC inhibitor U73122 (1 μm) or the PKC inhibitor bisindolylmaleimide I (1 μm). Exposure to the PKC activator phorbol 12-myristate 13-acetate (0.1 μm; green symbols and line) induced hypertrophy in the absence of an increase in osmolality. B, exposure (indicated by grey bar) to either high K+ saline (25 mm) or to the Ca2+ ionophore A23187 (10 μm) causes MNCs to hypertrophy. The hypertrophic effect of high K+ saline is blocked by the PKC inhibitor bisindolylmaleimide I (1 μm).
Discussion
The MNCs and the astrocytes that surround them undergo a remarkable structural and functional transformation in response to sustained increases in external osmolality. The astrocytes in both the hypothalamus and the neurohypophysis retract their processes from around the MNCs (Theodosis et al. 2008) and the latter astrocytes also show an up-regulation of L-type Ca2+ channels (Wang et al. 2009). In addition to the hypertrophy of the MNC somata, MNCs show increased synaptic innervation (Tasker et al. 2002), up-regulation of the genes coding for VP (Zingg et al. 1986) and other peptides (Ghorbel et al. 2003; Hindmarch et al. 2006), an increase in L-type Ca2+ current in MNC somata (Zhang et al. 2007), an increase in the expression of the V1a vasopressin receptor on the MNC membrane (Hurbin et al. 2002), translocation of the dynorphin receptor to the MNC terminal plasma membrane (Shuster et al. 1999), and an increase in the amplitude of transient and persistent Na+ currents (Tanaka et al. 1999). The somatic hypertrophy that forms the focus of this work was first identified using electron microscopy to observe the increase in CSA of MNC somata in rats that had been dehydrated. MNCs have been reported to actually double in size (Armstrong et al. 1977). This increase appears to involve an increase in total membrane area as MNCs isolated from dehydrated rats have been shown to have membrane capacitance values that are 20–33% larger than those from normally hydrated rats (Tanaka et al. 1999; Di & Tasker, 2004; Zhang et al. 2007). The time course of hypertrophy may be quite rapid; changes in cell size have been detected in rat MNCs following water deprivation of as little as 2 h (Hatton & Walters, 1973) and injection of hypertonic saline can cause some structural changes in 30 min (Tweedle et al. 1993). The mechanisms underlying hypertrophy and its physiological consequences are poorly understood, in part because hypertrophy is difficult to study using in vivo models.
Our data suggest that some portion of the hypertrophic response can be observed in acutely isolated MNCs and that no other cells are required to initiate this process. The fact that these changes can be evoked by changes in the concentration of mannitol in the bathing solution suggests that an increase in external osmolality is a sufficient trigger. Osmotically evoked hypertrophy is dependent on activation of TRPV1 channels, on the firing of action potentials, on Ca2+ influx through L-type Ca2+ channels, and on SNARE-dependent exocytotic fusion. We do not know the source of the internal membranes that are responsible for hypertrophy, but it is unlikely to be due primarily to the fusion of neuropeptide-containing granules because osmotically evoked release of VP from MNC somata is slow (Leng & Ludwig, 2008) and because there are not likely to be enough neuropeptide-containing granules to induce such an increase in total membrane area. It therefore appears likely that hypertrophy involves transfer of membrane from a large internal source such as the endoplasmic reticulum, but it could also involve the fusion of specialized membrane vesicles or granules to mediate the translocation of specific membrane proteins to the plasma membrane. We have shown that an osmotically evoked increase in the activity of PLC is required for the initiation of hypertrophy and that activation of PKC is necessary and sufficient to cause MNC enlargement. It will be interesting to determine the mechanism by which PKC activation triggers membrane transfer to the MNC plasma membrane.
Acute osmotically evoked changes in MNC size are not associated with changes in membrane capacitance (Zhang & Bourque, 2003) and thus our observations suggest a novel mechanism for MNC hypertrophy. Although we observed an increase in the mean CSA of MNCs from the shrunken state to the hypertrophied state of about 11% (i.e. from 343 to 381 μm2), the increase in cell membrane capacitance was only about 7%. The smaller increase in cell capacitance probably reflects the fact that the capacitance measurement includes membrane that is not on the somatic cell surface, such as that in the MNC processes and in the large membrane reserve that MNCs possess (Zhang & Bourque, 2003). Increasing the volume of the MNC soma by a given amount would therefore be expected to cause a somewhat lower increase in the total membrane area (and the measured membrane capacitance). Both the measurement of CSA changes and the change in capacitance, however, are markedly lower than the changes evoked by water deprivation or salt loading (see above). The extent of the increase under our conditions may be limited by the time of exposure, by the absence of most of the MNC dendritic tree, or by the absence of signalling molecules that are derived from a cell type that is present in vivo but absent from our preparation (e.g. the surrounding astrocytes).
Osmotically evoked hypertrophy is of particular interest in the MNCs because their osmosensitivity is thought to depend on a stretch-inactivated cation current (Oliet & Bourque, 1993) mediated by TRPV1 channels (Sharif Naeini et al. 2006) that are activated by the decrease in membrane tension caused by cell shrinkage (Zhang & Bourque, 2003). The MNCs have been shown to respond to hypertonic saline by shrinking and remaining shrunk for up to 6 min, suggesting that they do not display acute cell volume regulation in response to osmotically evoked cell shrinkage (Zhang & Bourque, 2003). Our results are consistent with this report because hypertrophy occurs only after a significant delay (see Fig. 1) and depends on mechanisms distinct from those underlying the acute cell volume regulatory mechanisms observed in many other neuron types. It is important to note, however, that water molecules will always tend to flow in or out of the cell to equalize the internal and external osmolality and therefore the increases in cell volume observed in vivo or by us in vitro must be accompanied by mechanisms to increase the ionic content of the MNC cytoplasm. The lack of effect of bumetanide suggests that the activity of the Na+–K+–Cl− co-transporter NKCC1 is not required, but the mechanism underlying the increase in cytoplasmic volume in MNCs remains to be determined.
The increase in MNC membrane during osmotically evoked hypertrophy has implications on the mechanisms by which TRPV1 channels mediate MNC osmosensitivity. We observed that hypertrophy rapidly reverses when Ca2+ influx into the MNCs is suppressed by the block of the TRPV1 channels, Na+ channels, or L-type Ca2+ channels (see Fig. 2B). The maintenance of hypertrophy therefore depends on the continuation of action potential firing. This suggests either that the addition of new membrane to the MNC plasma membrane does not alter the membrane tension, thereby allowing the TRPV1 channels to continue to be in an active state, or that a different mechanism is involved in maintaining the activity of the TRPV1 channels in MNCs following hypertrophy. It is possible, for example, that TRPV1 channels are regulated both by membrane tension and by one or more signalling molecules (which could include PIP2) or that hypertrophy leads to an increase in TRPV1 activity by causing translocation of the channels to the MNC plasma membrane.
Although the physiological significance of MNC hypertrophy remains unclear, it is possible that the fusion of internal membranes mediates the translocation of specific channels, receptors, or other membrane proteins to the MNC plasma membrane. This process could be involved, for example, in the dehydration-induced increase in the cell surface expression of V1a vasopressin receptors (Hurbin et al. 2002), Na+ currents (Tanaka et al. 1999), dynorphin receptors (Shuster et al. 1999), and L-type Ca2+ channels (Zhang et al. 2007), and the Ca2+-dependent translocation of N-type Ca2+ channels (Tobin et al. 2011). The activation of PKC by DAG has been implicated in analogous forms of translocation, including that of Ca2+ channels in molluscan neuroendocrine cells (Strong et al. 1987) and of TRPV1 in an oocyte expression system (Morenilla-Palao et al. 2004), and we therefore tested whether PKC could play a role in triggering MNC hypertrophy. Our data suggest that hypertrophy is dependent upon activation of both PLC and PKC. The activation of PKC is sufficient to activate at least part of the response, although the small size of the response to PKC activator alone may suggest that other triggers, for example intracellular Ca2+, may contribute to the full response. Evidence of whether the hypertrophic response does involve the translocation of channels and receptors awaits further study. PKC-mediated translocation of Ca2+ channels or TRPV1 channels could play an important role in MNC osmosensitivity. Ca2+ channels have been observed on intracellular granules in MNCs (Fisher et al. 2000) and this could represent an internal pool that is available for translocation to the MNC membrane.
The osmotically evoked increase in PLC activity could also be important in mediating osmosensitivity by regulating MNC activity in other ways. PIP2 has been shown to regulate the activity of a large number of ion channels, and in particular both TRP channels and M-type K+ currents (Suh & Hille, 2005). The latter is important because we identified an M-type K+ current in the MNCs (Liu et al. 2005; Zhang et al. 2009). We also showed that this current is suppressed by muscarinic activation (Zhang et al. 2009) and our current data are consistent with the hypothesis that this occurs by the G-protein-mediated activation of PLC, as occurs in other neurons (Suh & Hille, 2005). M-currents are low threshold, slow K+ currents and their modulation has important effects on the excitability of many central neurons (Brown & Passmore, 2009) and it is possible that they are important in MNC physiology as well. We showed that when MNCs are subjected to whole-cell patch clamp and then exposed to an increase in external osmolality, there is an increase in this M-type current (Zhang et al. 2009). Our current data show that osmotic activation of PLC decreases PIP2 and would therefore be expected to decrease the amplitude of the M-type currents. It is possible that the activity of PLC and/or the regulation of PIP2 levels is altered during whole-cell patch clamp and that our earlier results do not therefore reflect the physiological mechanism of osmotic regulation of M-type current. It is also possible that the M-current is regulated in some way other than by changes in PIP2. We are currently working to resolve this contradiction.
Our data suggest that osmotically evoked, activity- and Ca2+-dependent exocytotic fusion may underlie part or all of the hypertrophy observed in MNCs following water deprivation or salt loading. Hypertrophy occurred in response to modest changes in osmolality suggesting that the size of MNCs may be regulated in vivo in a dynamic fashion as the electrical activity of the MNCs responds to changes in external osmolality. The full significance of this phenomenon is not clear, but it could represent a mechanism for osmotically induced translocation of channels and receptors to the MNC plasma membrane and could contribute to the adaptive response of MNCs to sustained high osmolality. Our data suggest that this process is mediated by an activity-dependent increase in PLC activity, leading to an increase in PKC activity. The PLC-mediated decrease in PIP2 and increase in DAG and inositol 1,4,5-trisphosphate (IP3) could also play a number of other important roles in regulating ion channel function in MNCs. Our data therefore have important implications for acute and longer-term osmosensitivity of the MNCs.
Acknowledgments
The authors would like to thank Xuan Thanh Vo for his excellent technical assistance.
Glossary
- CSA
cross-sectional area
- DAG
diacylglycerol
- MNC
magnocellular neurosecretory cell
- NSF
N-ethylmaleimide-sensitive factor
- OT
oxytocin
- PKC
protein kinase C
- PLC
phospholipase C
- PIP2
phosphatidylinositol 4,5-bisphosphate
- SIC
stretch-inactivated cation channel
- SNARE
soluble NSF attachment protein receptor
- TRPV1
transient receptor potential cation channel vanilloid subfamily member 1
- VP
vasopressin
Additional information
Competing interests
The authors have no competing interests.
Author contributions
L.S., V.B., P.L.R., N.M. and A.T. collected, analysed and interpreted data. T.E.F. conceived and designed the experiments and drafted the paper; L.S. and V.B. revised it critically. All experiments were done in the Department of Physiology at the University of Saskatchewan. All authors approved of the final version of the manuscript.
Funding
This work was funded by an operating grant from the Canadian Institutes of Health Research Regional Partnership Program in association with the Saskatchewan Health Research Foundation (ROP 94985).
References
- Armstrong WE, Gregory WA, Hatton GI. Nucleolar proliferation and cell size changes in rat supraoptic neurons following osmotic and volemic challenges. Brain Res Bull. 1977;2:7–14. doi: 10.1016/0361-9230(77)90019-3. [DOI] [PubMed] [Google Scholar]
- Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519–531. doi: 10.1038/nrn2400. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Tasker JG. Dehydration-induced synaptic plasticity in magnocellular neurons of the hypothalamic supraoptic nucleus. Endocrinology. 2004;145:5141–5149. doi: 10.1210/en.2004-0702. [DOI] [PubMed] [Google Scholar]
- Fisher TE, Bourque CW. Voltage-gated calcium currents in the magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol. 1995;486:571–580. doi: 10.1113/jphysiol.1995.sp020835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher TE, Carrion-Vazquez M, Fernandez JM. Intracellular Ca2+ channel immunoreactivity in neuroendocrine axon terminals. FEBS Lett. 2000;482:131–138. doi: 10.1016/s0014-5793(00)02043-3. [DOI] [PubMed] [Google Scholar]
- Ghorbel MT, Sharman G, Leroux M, Barrett T, Donovan DM, Becker KG, Murphy D. Microarray analysis reveals interleukin-6 as a novel secretory product of the hypothalamo-neurohypophyseal system. J Biol Chem. 2003;278:19280–19285. doi: 10.1074/jbc.M209902200. [DOI] [PubMed] [Google Scholar]
- Hammond GR, Dove SK, Nicol A, Pinxteren JA, Zicha D, Schiavo G. Elimination of plasma membrane phosphatidylinositol (4,5)-bisphosphate is required for exocytosis from mast cells. J Cell Sci. 2006;119:2084–2094. doi: 10.1242/jcs.02912. [DOI] [PubMed] [Google Scholar]
- Hatton GI, Walters JK. Induced multiple nucleoli, nucleolar margination, and cell size changes in supraoptic neurons during dehydration and rehydration in the rat. Brain Res. 1973;59:137–154. doi: 10.1016/0006-8993(73)90256-4. [DOI] [PubMed] [Google Scholar]
- Hindmarch C, Yao S, Beighton G, Paton J, Murphy D. A comprehensive description of the transcriptome of the hypothalamoneurohypophyseal system in euhydrated and dehydrated rats. Proc Natl Acad Sci U S A. 2006;103:1609–1614. doi: 10.1073/pnas.0507450103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Hurbin A, Orcel H, Alonso G, Moos F, Rabie A. The vasopressin receptors colocalize with vasopressin in the magnocellular neurons of the rat supraoptic nucleus and are modulated by water balance. Endocrinology. 2002;143:456–466. doi: 10.1210/endo.143.2.8643. [DOI] [PubMed] [Google Scholar]
- Leng G, Ludwig M. Neurotransmitters and peptides: whispered secrets and public announcements. J Physiol. 2008;586:5625–5632. doi: 10.1113/jphysiol.2008.159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Zhang W, Fisher TE. A novel osmosensitive voltage gated cation current in rat supraoptic neurones. J Physiol. 2005;568:61–68. doi: 10.1113/jphysiol.2005.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Morrell CN, Lowenstein CJ. A novel class of fusion polypeptides inhibits exocytosis. Mol Pharmacol. 2005;67:1137–1144. doi: 10.1124/mol.104.004275. [DOI] [PubMed] [Google Scholar]
- Miyata S, Hatton GI. Activity-related, dynamic neuron-glial interactions in the hypothalamo-neurohypophysial system. Microsc Res Tech. 2002;56:143–157. doi: 10.1002/jemt.10012. [DOI] [PubMed] [Google Scholar]
- Morenilla-Palao C, Planells-Cases R, García-Sanz N, Ferrer-Montiel A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem. 2004;279:25665–25672. doi: 10.1074/jbc.M311515200. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Bourque CW. Properties of supraoptic magnocellular neurones isolated from the adult rat. J Physiol. 1992;455:291–306. doi: 10.1113/jphysiol.1992.sp019302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Bourque CW. Mechanosensitive channels transduce osmosensitivity in supraoptic neurons. Nature. 1993;364:341–343. doi: 10.1038/364341a0. [DOI] [PubMed] [Google Scholar]
- Sharif-Naeini R, Ciura S, Bourque CW. TRPV1 gene required for thermosensory transduction and anticipatory secretion from vasopressin neurons during hyperthermia. Neuron. 2008;58:179–185. doi: 10.1016/j.neuron.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Sharif Naeini R, Witty MF, Seguela P, Bourque CW. An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci. 2006;9:93–98. doi: 10.1038/nn1614. [DOI] [PubMed] [Google Scholar]
- Shuster SJ, Riedl M, Li X, Vulchanova L, Elde R. Stimulus-dependent translocation of κ opioid receptors to the plasma membrane. J Neurosci. 1999;19:2658–2664. doi: 10.1523/JNEUROSCI.19-07-02658.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong JA, Fox AP, Tsien RW, Kaczmarek LK. Stimulation of protein kinase C recruits covert calcium channels in Aplysia bag cell neurons. Nature. 1987;325:714–717. doi: 10.1038/325714a0. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Cummins TR, Ishikawa K, Black JA, Ibata Y, Waxman SG. Molecular and functional remodeling of electrogenic membrane of hypothalamic neurons in response to changes in their input. Proc Natl Acad Sci U S A. 1999;96:1088–1093. doi: 10.1073/pnas.96.3.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Di S, Boudaba C. Functional synaptic plasticity in hypothalamic magnocellular neurons. Prog Brain Res. 2002;139:113–119. doi: 10.1016/s0079-6123(02)39011-3. [DOI] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- Tobin VA, Douglas AJ, Leng G, Ludwig M. The involvement of voltage-operated calcium channels in somato-dendritic oxytocin release. PloS One. 2011;6:e25366. doi: 10.1371/journal.pone.0025366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedle CD, Smithson KG, Hatton GI. Rapid synaptic changes and bundling in the supraoptic dendritic zone of the perfused rat brain. Exp Neurol. 1993;124:200–207. doi: 10.1006/exnr.1993.1190. [DOI] [PubMed] [Google Scholar]
- Wang D, Yan B, Rajapaksha WR, Fisher TE. The expression of voltage-gated Ca2+ channels in pituicytes and the up-regulation of L-type Ca2+ channels during water deprivation. J Neuroendocrinol. 2009;21:858–866. doi: 10.1111/j.1365-2826.2009.01906.x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Star B, Rajapaksha WR, Fisher TE. Dehydration increases L-type Ca2+ current in rat supraoptic neurons. J Physiol. 2007;580:181–193. doi: 10.1113/jphysiol.2006.126680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang D, Liu XH, Kosala WR, Rajapaksha JS, Fisher TE. An osmosensitive voltage-gated K+ current in rat supraoptic neurons. Eur J Neurosci. 2009;29:2335–2346. doi: 10.1111/j.1460-9568.2009.06772.x. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Bourque CW. Osmometry in osmosensory neurons. Nat Neurosci. 2003;6:1021–1022. doi: 10.1038/nn1124. [DOI] [PubMed] [Google Scholar]
- Zingg HH, Lefebvre D, Almazan G. Regulation of vasopressin gene expression in rat hypothalamic neurons. Response to osmotic stimulation. J Biol Chem. 1986;261:12956–12959. [PubMed] [Google Scholar]