

Abstract
It has recently been shown that dynorphin A (Dyn A), an endogenous agonist of the κ-opioid receptor (KOR), directly inhibits proopiomelanocortin (POMC) neurons in the hypothalamus through activation of G-protein-coupled inwardly rectifying K+ channels (GIRKs). This effect has been proposed to be mediated by the putative κ2-opioid receptor (KOR-2), and has been suggested as a possible mechanism for the orexigenic actions of KOR agonists. Using whole-cell voltage clamp recordings in brain slice preparations, the present study demonstrates that Dyn A (1 or 5 μm) induces an outward current in POMC neurons that is reversed by the highly selective μ-opioid receptor (MOR) antagonist CTAP and is absent in mice lacking MORs. Additionally, the KOR-2-selective agonist GR89696 binds MORs on POMC neurons but fails to induce an outward current. Similar to Dyn A, the KOR-selective antagonist nor-binaltorphimine (nor-BNI) lacked specificity when used at sufficiently high concentrations. Maximal concentrations of the MOR-selective agonist DAMGO induced outward currents in POMC neurons that were completely reversed by a relatively high concentration of nor-BNI. Experiments using a half-maximal concentration of DAMGO demonstrate that nor-BNI must be used at concentrations <100 nm to avoid non-specific actions of the antagonist at MORs located on POMC neurons. These data suggest that direct inhibition of POMC neurons by Dyn A is mediated through the MOR, not the KOR-2, which is consistent with previous studies demonstrating that Dyn A can act at the μ-opioid receptor (MOR) when present in high concentrations.
Key points
Dynorphin A inhibits anorexigenic hypothalamic proopiomelanocortin neurons by activating a potassium conductance.
Although dynorphin A is considered selective for κ-opioid receptors, the present data show that the dynorphin A-induced potassium conductance is reversed by a μ-opioid receptor-selective antagonist and is absent in mice lacking functional μ-opioid receptors.
Thus, μ-opioid receptors mediate the inhibition of proopiomelanocortin neurons caused by dynorphin A, consistent with other studies showing that κ-opioid receptor-selective agonists and antagonists can act at the μ-opioid receptor when used at sufficiently high concentrations.
The results indicate that if dynorphin A inhibits proopiomelanocortin neurons to increase food intake, it does so either by activating μ-opioid receptors on these neurons or by inhibiting κ-opioid receptors located in the presynaptic compartment of proopiomelanocortin neurons.
Introduction
κ-Opioid receptor (KOR) agonists, including the endogenous peptide dynorphin A (Dyn A), potently stimulate food intake (Morley & Levine, 1983). It was recently demonstrated that Dyn A is capable of direct inhibition of anorexigenic proopiomelanocortin (POMC) neurons of the arcuate nucleus of the hypothalamus via activation of G-protein-coupled inwardly rectifying K+ channels (GIRKs; Zhang & van den Pol, 2013). This led to the hypothesis that Dyn A-induced inhibition of POMC neurons may be an important component in the stimulation of food intake by KOR agonists (Zhang & van den Pol, 2013). It was also proposed that Dyn A-induced inhibition of POMC neurons was mediated by the putative κ2 subtype of the κ-opioid receptor (KOR-2), the presence of which had been overlooked in previous studies characterizing opioid regulation of POMC neurons (Pennock & Hentges, 2011) due to the unique pharmacological properties of the KOR-2.
Pharmacological and receptor binding studies led to the hypothesis that two KOR subtypes are expressed in brain tissue (reviewed in Wollemann et al. 1993). Electrophysiological studies have also observed pharmacological differences between pre- and postsynaptic KORs; the synthetic KOR-selective agonist U69593 acts as a full agonist at presynaptic KORs but as a partial agonist at postsynaptic KORs in the ventral tegmental area (VTA; Ford et al. 2007). However, the gene that codes for the KOR-2 has yet to be identified, and there is evidence that putative KOR-2 binding that occurs in the brain is actually non-selective binding to other opioid receptors (Simonin et al. 2001).
If two pharmacologically distinct KOR subtypes exist on the somato-dendritic region of POMC neurons and POMC terminals, the postsynaptic population of receptors may be selectively targeted. Varying distribution of KOR subtypes could also allow for differential regulation of pre- and postsynaptic receptors, particularly as Dyn A concentrations change in response to physiological conditions. This would make the KOR-2 an intriguing target for those wishing to develop therapies that increase food intake through the inhibition of POMC neurons, as other Gαi/o-coupled receptors that directly inhibit POMC neurons through GIRK activation also disinhibit POMC neurons through the inhibition of GABA release.
Dyn A is a selective agonist of the KOR, activating the receptor at nanomolar concentrations (Goldstein & James, 1984; Zhang et al. 1998). However, Dyn A has also been shown to bind and act as an agonist at the μ-opioid receptor (MOR), albeit with an EC50 value 50–100 times greater than that observed at the KOR (Goldstein & James, 1984; James & Goldstein, 1984; Chavkin et al. 1985; Goldstein & Naidu, 1989; Mulder et al. 1989; Emmerson et al. 1994; Raynor et al. 1994). Dyn A-induced inhibition of POMC neurons occurs with an EC50 in the micromolar range (Zhang & van den Pol, 2013), well above the concentration range that is selective for the KOR (Mulder et al. 1989; Grudt & Williams, 1993). Thus, it is possible that Dyn A-induced inhibition of POMC neurons is not mediated through either KOR subtype, but is instead due to activation of the MOR.
The present studies use pharmacological and receptor knockout approaches to show that Dyn A-induced inhibition of POMC neurons occurs at high concentrations as a consequence of MOR activation. Thus, endogenous KOR ligands most probably exert their effects via KORs located on presynaptic inputs to POMC neurons and POMC neuron terminals.
Methods
Ethical approval
All animal use procedures were approved by the Colorado State University Institutional Animal Care and Use Committee and met United States Public Health guidelines and the policies and regulations for animal experimentation described by The Journal of Physiology.
Animals
Mice expressing discosoma red (dsRed) or enhanced green fluorescent protein (eGFP) driven by the POMC promoter (Cowley et al. 1999; Hentges et al. 2009) were backcrossed onto a C57BL/6 background for >12 generations. Mice expressing a mutant μ-opioid receptor allele (MOR–; Matthes et al. 1996) were obtained from The Jackson Laboratory (B6.129S2-Oprm1tm1Kff/J) and were backcrossed onto the C57BL/6 background for >12 generations prior to purchase. MOR−-expressing mice were crossed with POMC–eGFP animals to produce μ-opioid receptor knockout mice (MOR−/−) expressing eGFP in POMC neurons (POMC–eGFP–MOR−/−). All mice received tap water and standard rodent chow ad libitum. Animals were housed at a controlled temperature (22–24°C) with a 12 h light–dark cycle. Transgenic mice were identified using standard PCR genotyping.
Brain slice preparation
Fifty-eight mice were used over the course of this study. Brain slices were prepared from 6- to 12-week-old male and female POMC–dsRed, POMC–eGFP and POMC–eGFP–MOR−/− mice. Before being killed, mice were deeply anaesthetized using isoflurane. Mice were quickly decapitated and brains were rapidly removed and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21 NaHCO3 and 11 glucose. ACSF solutions were adjusted to pH 7.5 and saturated with 95% O2–5% CO2 mixture. Sagittal brain slices were prepared at a thickness of 240 μm using a VT 1200S vibratome (Leica). Slices containing the arcuate nucleus of the hypothalamus were transferred into warm ACSF (37°C) containing 15 μm MK-801 immediately after collection. At least 45 min passed between collection and transfer of slices to the recording chamber.
Electrophysiological recording
After being transferred to the recording chamber, slices were continuously perfused (∼2 ml min−1) with warm (37°C) ACSF saturated with 95% O2–5% CO2. Recordings were made using pipettes with a resistance of 1.5–2.5 MΩ after being filled with an internal solution containing (in mm): 57.5 potassium methyl sulfate, 57.5 KCl, 20 NaCl, 1.5 MgCl2, 5 Hepes (K+ salt), 0.1 EGTA, 2 Mg-ATP, 0.5 Na-GTP, 10 phosphocreatine, pH 7.3. mIPSCs were recorded with an internal solution that replaced potassium methyl sulfate and KCl with caesium methane sulfonate and CsCl, respectively. POMC neurons were identified by the presence of dsRed or eGFP fluorescence. After obtaining a seal with >1 GΩ resistance, negative pressure was used to rupture the cellular membrane and enter whole-cell mode. Recordings were made at a holding potential of −60 mV with no series resistance compensation. Postsynaptic currents were obtained in 1 s sweeps taken every 11 s using Axograph X software. Each sweep contained a 50 ms −10 mV voltage step to monitor access and input resistance over the course of the recording. I–V curves were constructed using nine −10 mV voltage steps starting at a holding potential of −50 mV. Each step was 100 ms in length with 200 ms between steps. This protocol was repeated 200 ms after the end of the first set of voltage steps. The two data sweeps for each holding potential were then averaged. The currents induced by the voltage step protocol were not removed from any current traces (Fig. 1A, C, E and F; Fig. 2A–F; Fig. 3A–C). DAMGO-, dynorphin A- and baclofen-induced postsynaptic currents were recorded in the presence of 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μm), bicuculline methiodide (BMI; 10 μm) and tetrodotoxin (TTX; 300 nm). Miniature IPSCs were collected at 10 kHz and digitally filtered at 1 kHz. Events were collected during 15 s sweeps that were repeated every 15 s and detected using Axograph X software based on rise time kinetics. Events with a rise time <100 μs were rejected. mIPSCs were recorded in the presence of DNQX (10 μm) and TTX (300 nm).
Figure 1. Dynorphin A-induced outward currents measured from POMC neurons are reversed by the μ-opioid receptor-selective antagonist CTAP.
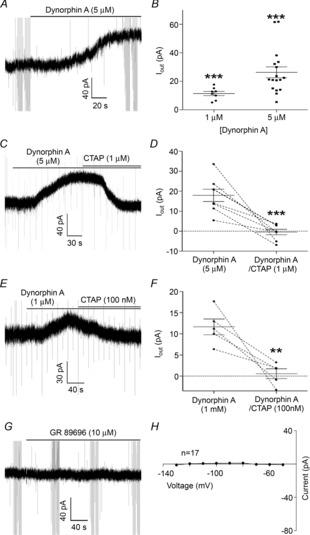
A, a sample trace of an outward current induced by Dyn A (5 μm). B, compiled data for the magnitude of the outward current induced by Dyn A (1 or 5 μm). C, a sample trace demonstrating the reversal of an outward current induced by Dyn A (5 μm) by the MOR-selective antagonist CTAP (1 μm). D, compiled data showing the amplitude and the Dyn A-induced current before and after addition of CTAP (1 μm). E, a sample trace demonstrating the reversal of an outward current induced by Dyn A (1 μm) by CTAP (100 nm). F, compiled data showing the amplitude of the outward current induced by Dyn A before and after the addition of CTAP (1 μm). G, a sample trace from a recording showing that the putative KOR-2 agonist GR 89696 induces no outward current in POMC neurons. H, a subtracted I–V plot showing no difference between the I–V relationship of POMC neurons before and after GR 89696 application. P values are represented by asterisks: **P < 0.01, ***P < 0.001.
Figure 2. GR89696 binds to μ-opioid receptor.

A, a sample trace demonstrating that the outward current induced by DAMGO (10 μm) is reversed by GR89696 (10 μm). B, a sample trace demonstrating the occlusion of a DAMGO-induced outward current by pre-application of GR89696. C, a sample trace demonstrating that GR89696 pre-application does not occlude the outward current induced by baclofen (30 μm). D, compiled data showing the amplitude of the outward current induced by DAMGO (10 μm) before and after application of GR89696 (10 μm). E, subtracted I–V plots showing the currents induced by both DAMGO and baclofen in the presence of GR89696 at voltages ranging from −50 to −130 mV in −10 mV steps. P values are represented by asterisks: **P < 0.01.
Figure 3. Dynorphin A-induced outward currents are absent in POMC neurons lacking the μ-opioid receptor.
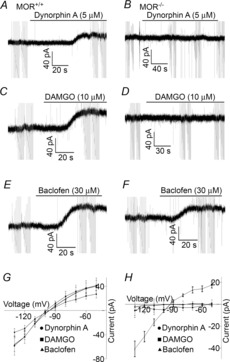
A and B, sample traces demonstrating the effects of Dyn A in wild-type and MOR knockout mice. Dyn A induces an outward current in MOR+/+ but not MOR−/− mice. C and D, similar traces are shown for DAMGO. E and F, sample traces demonstrating a baclofen-induced outward current in both MOR+/+ and MOR−/− mice. G and H, subtracted I–V relationships demonstrating the currents induced by Dyn A, DAMGO and baclofen at holding potentials ranging from −50 to −130 mV in −10 mV steps in MOR+/+ and MOR−/− mice.
Drugs
[d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin acetate (DA-MGO; Sigma, St. Louis, Missouri, USA), dynorphin A (1–13) (Dyn A; Phoenix Pharmaceuticals, Burlingame, California, USA), R(+)-baclofen hydrochloride (Sigma), (–)-bicuculline methiodide (BMI; Tocris, Bristol, UK), GR 89696 fumurate (Tocris), and tetrodotoxin citrate (TTX; Tocris) were prepared in distilled water (at least 1000:1 of the final concentration). 6,7-Dinitroquinoxaline-2,3-dione (DNQX; Sigma) and (+)-MK-801 (Sigma) were prepared in DMSO (at least 10,000:1 of the final concentration). Drugs were diluted in ACSF to achieve final concentrations.
Statistics
Data were analysed using a one-sample or paired Student's t test. Data sets containing multiple repeated measures were analysed using a repeated-measures ANOVA and Tukey's multiple comparison post hoc test. Induced outward currents <5 pA, were not included in analyses as clear reversal was difficult to assess. Using this criterion, the number of cells excluded for each drug and concentration used was as follows: 10 μm DAMGO (2 of 17 cells), 500 nm DAMGO (4 of 11 cells), 5 μm Dyn A (2 of 21 cells), 1 μm Dyn A (10 of 18), 30 μm baclofen (1 of 14 cells). All data points are represented as the mean ± SEM. Differences were considered significant if P < 0.05.
Results
Dynorphin A (Dyn A)-induced outward currents are reversed by CTAP
As reported previously (Zhang & van den Pol, 2013), Dyn A (1 μm or 5 μm) was found to induce a clear outward current in POMC neurons (11.4 ± 1.5 pA and 26.2 ± 3.8 pA, respectively; 8 out of 18 cells responded to 1 μm, 19 out of 21 cells responded to 5 μm, only responding cells are included in statistics; P < 0.001, one-sample t test; Fig. 1A, B and E). To determine if this outward current is mediated by activation of the putative KOR-2 (Zhang & van den Pol, 2013), or rather by actions at the MOR (Chavkin et al. 1985), the MOR-selective antagonist CTAP was applied immediately after a peak Dyn A current was observed. CTAP (1 μm) caused in a rapid reversal of the outward current induced by Dyn A (5 μm) in all cells examined (Dyn A = 17.9 ± 3.1 pA, Dyn A–CTAP = −0.42 ± 1.4 pA; n = 8 cells from 4 mice; P < 0.001, paired t test; Fig. 1C and D). To further examine the possibility that the outward current induced by Dyn A was due exclusively to activation of the MOR, the experiment was repeated with a lower concentration of both Dyn A (1 μm) and CTAP (100 nm). The outward current induced by Dyn A was reversed by CTAP in all recordings (Dyn A = 11.7 ± 1.9 pA, Dyn A–CTAP = 0.6 ± 1.2 pA; n = 5 cells from 4 mice; P = 0.007, paired t test; Fig. 1E and F). Finally, the putative KOR-2 agonist GR 89696 (10 μm) was applied to determine whether any KOR-2-mediated outward currents could be detected in POMC neurons. No outward current was detected at a holding potential of −60 mV (−0.6 ± 0.7 pA; n = 17 cells from 8 mice; P = 0.41, one-sample t test; Fig. 1G and H), and the subtracted I–V relationship showed no detectable currents at any of the holding potentials examined (−130 to −50 mV; n = 17 cells from 8 mice; P = 0.13, repeated measures ANOVA; Fig. 1H). Together, these data suggest that Dyn A-induced outward currents on POMC neurons result from activation of the MOR, not the KOR-2.
The putative KOR-2-selective agonist GR89696 binds the μ-opioid receptor
It has been previously reported that putative KOR-2 binding in the brain can be accounted for by non-selective binding of KOR-2 agonists to other opioid receptors (Simonin et al. 2001). Although GR89696 did not induce changes in the electrical properties of POMC neurons in the present study (Fig. 1G and H), GR89696 does appear to bind to MORs on POMC neurons. Outward currents induced by DAMGO (10 μm) were reversed by GR89696 (10 μm) in all cells examined (DAMGO = 24.2 ± 4.6 pA, DAMGO–GR89696 = −1.2 ± 0.5 pA; n = 5 cells from 3 mice; P = 0.003, paired t test; Fig. 2A and D). Similarly, pre-application of 10 μm GR89696 occluded DAMGO-induced outward currents in POMC neurons (−0.06 ± 0.9 pA at −60 mV, n = 9 cells from 5 mice; P = 0.94, one-sample t test; Fig. 2B and E), but not outward currents induced by the GABAB receptor agonist baclofen (32.8 ± 12.2 pA at −60 mV, n = 4 cells from 2 mice; P = 0.07, one-sample t test; Fig. 2C and E). These data are consistent with previously reported findings (Simonin et al. 2001) that putative KOR-2 ligands can bind non-selectively to other opioid receptors.
Dynorphin A-induced outward currents are absent in μ-opioid receptor knockout mice
To verify that Dyn A-induced outward currents are mediated by the MOR, current–voltage relationships for both the MOR-selective agonist DAMGO and Dyn A were constructed in POMC–eGFP mice and POMC–eGFP mice that were crossed with mice containing a mutant μ-opioid receptor allele that results in a loss of MOR function (POMC–eGFP–MOR−/−). If Dyn A-induced inhibition of POMC neurons can be mediated by something other than the MOR, the absence of functional MORs should have no consequence on Dyn A actions. Consistent with the above studies, both DAMGO (10 μm) and Dyn A (5 μm) induced inwardly rectifying currents with reversal potentials near the K+ equilibrium potential in POMC neurons expressing the MOR (−98.4 ± 1.2 mV for DAMGO, n = 5 cells from 4 mice; −96.5 ± 4.2 mV for Dyn A, n = 9 cells from 5 mice; P < 0.0001, repeated measures ANOVA; Fig. 3A, C and G). As expected, DAMGO did not induce a current in POMC neurons lacking MORs when held at −60 mV (−1.4 ± 1.2 pA; P = 0.29, one-sample t test) or at any other holding potential tested (−50 to −130 mV, P = 0.74, repeated measures ANOVA, n = 4 cells from 2 mice; Fig. 3D and H). Similarly, Dyn A did not produce a detectable current at any of the holding potentials examined (0.5 ± 0.7 pA at −60 mV, P = 0.48 by one-sample t test; P = 0.99 using repeated measures ANOVA on data from −50 to −130 mV holding potentials; n = 6 cells from 3 mice; Fig. 3B and H). To ensure that Gαi/o-coupled GPCRs were still generally functional in POMC–eGFP–MOR−/− animals, the effect of the GABAB receptor agonist baclofen on whole cell currents were recorded in POMC neurons with and without MORs. In both populations, baclofen induced an inwardly rectifying current with a reversal potential near the equilibrium potential of K+ (MOR+/+ = −92 ± 7.4 mV, n = 3 cells from 2 mice; MOR−/− = −91.6 ± 1.6 mV, 5 cells from 4 mice; Fig. 3E–H).
To ensure that the κ-opioid system was unaffected by knockout of the MOR, the inhibition of presynaptic GABA release by Dyn A was examined in MOR−/− and MOR+/+ mice. In MOR−/− mice, Dyn A (100 nm) induced robust inhibition of mIPSC frequency measured from POMC neurons (baseline = 7.5 ± 1.9 Hz, Dyn A = 3.4 ± 0.7 Hz) and this was reversed by nor-BNI (50 nm, to 8.8 ± 1.7; P = 0.003; repeated measures ANOVA; n = 5 cells from 4 mice; Fig. 4A and B). While DAMGO (10 μm) strongly inhibits mIPSC frequency in MOR+/+ mice (∼80% reduction; Pennock & Hentges, 2011), DAMGO-induced inhibition of mIPSC frequency was essentially absent in MOR−/− mice (∼3.8% reduction as opposed to the ∼80% reduction observed in wild-type mice, 7.9 ± 1.6 Hz baseline vs. 7.6 ± 1.5 Hz in 10 μm DAMGO; n = 8 cells from 4 mice; P = 0.02, paired t test; Fig. 4C and D). The lack of Dyn A-induced postsynaptic currents in the MOR knockout provides strong evidence that Dyn A inhibits POMC neurons via actions at the MOR.
Figure 4. Presynaptic κ-opioid receptors are still present and functional in the μ-opioid receptor knockout mouse.
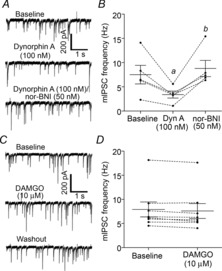
A, sample traces demonstrating the inhibition of mIPSCs measured from POMC neurons by Dyn A (100 nm), as well as the reversal of Dyn A-induced inhibition of mIPSCs by the KOR-selective antagonist nor-BNI (50 nm), in MOR knockout mice. B, compiled data for the inhibition of mIPSCs by Dyn A and the reversal of Dyn A-induced inhibition by nor-BNI. Using Tukey's post hoc test, mIPSC frequency was found to be significantly different than the baseline mIPSC frequency (a) and the mIPSC frequency in the presence of nor-BNI (50 nm, b). The baseline mIPSC frequency and mIPSC frequency in the presence of nor-BNI (50 nm) were not significantly different. C, sample traces demonstrating a lack of DAMGO-induced inhibition of mIPSCs measured from POMC neurons in MOR knockout mice. D, compiled data for the effects of DAMGO on mIPSC frequency in MOR knockout mice.
Nor-BNI is non-selective for the KOR at sufficiently high concentrations
Dyn A-induced currents measured from POMC neurons have been attributed to actions at the KOR-2 in part because the current is blocked by the KOR-selective antagonist nor-BNI (Zhang & van den Pol, 2013). However, nor-BNI can also act as an antagonist of the MOR when used at sufficiently high concentrations (Emmerson et al. 1994; Raynor et al. 1994). To determine if the concentration of nor-BNI used in previous experiments was sufficient to act on the MOR, nor-BNI (1 μm) was used to reverse outward currents induced by a maximal concentration of DAMGO (10 μm). Similar to previous studies, nor-BNI (1 μm) was sufficient to reverse the outward current induced by Dyn A (5 μm Dyn A = 36.4 ± 10.4 pA, with nor-BNI = 1.9 ± 1.7 pA; P = 0.01, paired t test; n = 5 cells from 3 mice; Fig. 5A and D). However, nor-BNI (1 μm) was also sufficient to reverse the outward current induced by a relatively high concentration of DAMGO (10 μm) in all cells examined (DAMGO = 31.7 ± 9.0 pA, nor-BNI = −0.7 ± 1.3 pA; P = 0.009, paired t test; n = 5 cells from 3 mice; Fig. 5B and E). To determine the concentration of nor-BNI needed to avoid antagonism of the MOR, outward currents were induced with an ∼EC50 concentration of DAMGO (500 nm; Pennock & Hentges, 2011), then cells were exposed to increasing concentrations of nor-BNI. DAMGO (500 nm) induced an outward current that was partially reversed by a low concentration of nor-BNI (DAMGO = 12.8 ± 2.7 pA, 100 nm nor-BNI = 6.5 ± 1.5 pA; P = 0.003 DAMGO vs. baseline, one-sample t test; n = 7 cells from 5 mice; Fig. 5C and F) and almost completely reversed by a higher concentration of nor-BNI (500 nm nor-BNI = 2.1 ± 0.9 pA; P < 0.001, repeated measures ANOVA; n = 7 cells from 5 mice; Fig. 5C and F). Thus, it appears that nor-BNI must be used at concentrations <100 nm to avoid actions at MORs on POMC neurons.
Figure 5. MORs on POMC neurons are antagonized by nor-BNI at sufficiently high concentrations.
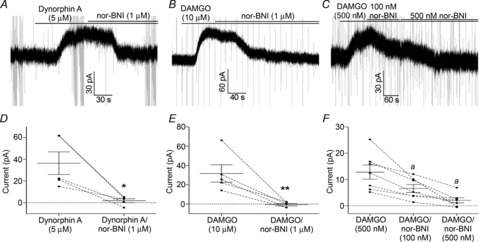
A, a sample trace demonstrating the reversal of an outward current induced by Dyn A (5 μm) by nor-BNI (1 μm). B, the same concentration of nor-BNI (1 μm) is also sufficient to reverse the outward current induced by higher concentration of DAMGO (10 μm). D and E, compiled data showing the reversal of outward currents induced by Dyn A and DAMGO by nor-BNI (1 μm). C, dose-dependent inhibition of an outward current induced by a near EC50 concentration of DAMGO (500 nm) by 100 nm and 500 nm nor-BNI. F, compiled data demonstrating the reversal of the outward current induced by DAMGO (500 nm) by 100 nm and 500 nm nor-BNI. The amplitude of the outward current induced by DAMGO (500 nm) in the presence of nor-BNI (500 nm) is significantly different than that of DAMGO (500 nm) alone using Tukey's post hoc test (a). P values are represented by asterisks: *P < 0.05, **P < 0.01.
Discussion
The present data demonstrate that Dyn A can activate an inwardly rectifying K+ current on the somato-dendritic region of POMC neurons and that this is mediated by the MOR, and not the putative KOR-2 receptor. Dyn A must be present in micromolar concentrations to induce currents in POMC neurons, which is sufficient to activate the MOR. Further, the presence of functional MORs is required for Dyn A to induce currents in POMC neurons. These results are consistent with previous studies demonstrating a lack of KOR expression on the somato-dendritic region of POMC neurons (Pennock & Hentges, 2011; Dicken et al. 2012).
Selectivity of dynorphin A and nor-BNI for KORs on POMC neurons
As shown in a previous study (Zhang & van den Pol, 2013), Dyn A induced a robust outward current in POMC neurons when applied at micromolar concentrations that was reversed with nor-BNI. However, the present study demonstrates that the concentrations of Dyn A needed to induce outward currents in POMC neurons are sufficient to produce non-specific effects at the MOR. This was demonstrated by the complete reversal of Dyn A-induced outward currents by the MOR-selective antagonist CTAP, and by the complete reversal of DAMGO-induced currents by nor-BNI at the concentration (1 μm) used in the previous study to reverse the putative κ2 effect (Zhang & van den Pol, 2013). The mismatched binding to receptors by classical MOR or KOR ligands is consistent with previous studies demonstrating that the selectivity of Dyn A and nor-BNI diminishes when used at relatively high concentrations (Goldstein & James, 1984; James & Goldstein, 1984; Chavkin et al. 1985; Goldstein & Naidu, 1989; Mulder et al. 1989; Emmerson et al. 1994; Raynor et al. 1994). In contrast to the high concentrations of Dyn A needed to cause a postsynaptic outward current in POMC neurons, Dyn A caused robust inhibition of GABA release onto POMC neurons when present in a much lower concentration (100 nm) and this effect was reversed by a concentration of nor-BNI (50 nm) that is selective for the KOR. These concentrations are consistent with previous studies using Dyn A and nor-BNI to selectively target KORs (Grudt & Williams, 1993; Simmons & Chavkin, 1996; Margolis et al. 2003; Ford et al. 2007).
Dyn A-induced outward currents are absent in MOR−/− mice
If Dyn A-induced currents were mediated by the KOR-2, the current should still be present in mice lacking the MOR. Although presynaptic KORs were still functional in MOR−/− animals, no Dyn A-induced postsynaptic currents were detected in POMC neurons lacking the MOR. There was also no outward current detected using the putative KOR-2 agonist GR89696 in wild-type mice. However, it was determined that GR89696 binds, but does not activate, MORs on POMC neurons. These findings agree with previous work demonstrating that all putative KOR-2 binding can be accounted for by non-selective binding at other opioid receptors (Simonin et al. 2001).
Whereas the present work did not find a GR89696-mediated current in POMC neurons, a previous study found this putative KOR-2 agonist to hyperpolarize POMC neurons (Zhang & van den Pol, 2013). The reasons for this difference between the two studies are not completely clear. One possibility is that GR89696 acts on upstream cells to alter the activity of POMC neurons, although Zhang & van den Pol (2013) did not find GR89696 to change spontaneous IPSCs or EPSCs in POMC neurons. We cannot rule out a potential age dependence to the GR89696 effect. The mean age of mice used in the present study was likely to be older than that in the previous study since the range used here was 6–12 weeks compared to 2–7.3 weeks in Zhang & van den Pol (2013). An additional difference between the current and previous study is the use of voltage clamp versus current clamp. However, since the GR89696-induced hyperpolarization of POMC neurons observed by Zhang & van den Pol (2013) was reported to occur through activation of a GIRK conductance, it is unlikely that a corresponding outward current would not be detectable in experiments performed in voltage clamp. Additional assays to detect the presence and function of KOR-2 on POMC neurons and further studies with GR89696 may help fully discern the reasons for the apparent discrepancy between these two studies.
Possible roles for endogenous Dyn A in the regulation of POMC neurons
Although the present study suggests that no form of KOR is present on the somato-dendritic region of POMC neurons, endogenous Dyn A may still play a role in the regulation of POMC neurons in vivo through presynaptic actions. KORs are present on the presynaptic terminals of POMC neurons and potently inhibit neurotransmitter release (Dicken et al. 2012). Inhibition of transmitter release directly at the level of POMC terminals would be expected to increase food intake since POMC neuron-derived transmitters are largely anorexigenic (reviewed in Cone, 2005 and Mercer et al. 2013). It is also possible that Dyn A directly hyperpolarizes the somato-dendritic region of POMC neurons, albeit through the MOR. As shown in the present study, Dyn A activates somato-dendritic MORs on POMC neurons when used at a sufficient concentration. However, it is unknown whether Dyn A concentrations in vivo reach concentrations sufficient to induce activation of the MOR. The selectivity profile of Dyn A may also change depending on how the peptide is processed. For example, Dyn A (1–8) shows higher selectivity for the MOR than KOR (Goldstein & Naidu, 1989). If Dyn A-expressing neurons forming synapses with POMC neurons release a shorter fragment of Dyn A such as Dyn A (1–8) this may produce activation of the MOR.
Conclusions
Although dynorphin A is capable of inducing direct inhibition of the somato-dendritic region of POMC neurons, the present study found such inhibition is mediated by activation of the MOR. There is significant evidence that the KOR system is an important endogenous regulator of energy balance, and KOR agonists have known orexigenic actions. The present results suggest that if KOR agonists act at POMC neurons to increase food intake and body weight, they do so either via inhibition of presynaptic release or by activating MORs to reduce the activity of POMC neurons.
Glossary
- BMI
(–)-bicuculline methiodide
- CTAP
d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2
- DAMGO
[d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin acetate
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- dsRed
discosoma red
- Dyn A
dynorphin A (1–13)
- eGFP
enhanced green fluorescent protein
- GIRK
G-protein-coupled inwardly rectifying K+ channel
- GPCR
G-protein-coupled receptor
- KOR
κ-opioid receptor
- KOR-2
κ2-opioid receptor
- MOR
μ-opioid receptor
- nor-BNI
nor-binaltorphimine
- POMC
proopiomelanocortin
- TTX
tetrodotoxin
Additional information
Competing interests
The authors have no competing financial interests.
Author contributions
R.L.P. and S.T.H. conceived and designed the experiments. R.L.P. collected and analysed the data, R.L.P. and S.T.H. interpreted the data and wrote and revised the manuscript. Both authors approve of the final version of the manuscript.
Funding
This work was supported by NIH R01DK078749 (S.T.H.), NIH 1 F31DA035586 (R.L.P.) and the John H. Venable Memorial Scholarship (R.L.P.).
References
- Chavkin C, Henriksen SJ, Siggins GR, Bloom FE. Selective inactivation of opioid receptors in rat hippocampus demonstrates that dynorphin-A and -B may act on mu-receptors in the CA1 region. Brain Res. 1985;331:366–370. doi: 10.1016/0006-8993(85)91565-3. [DOI] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Dicken MS, Tooker RE, Hentges ST. Regulation of GABA and glutamate release from proopiomelanocortin neuron terminals in intact hypothalamic networks. J Neurosci. 2012;32:4042–4048. doi: 10.1523/JNEUROSCI.6032-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson PJ, Liu MR, Woods JH, Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther. 1994;271:1630–1637. [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, James IF. Site-directed alkylation of multiple opioid receptors. II. Pharmacological selectivity. Mol Pharmacol. 1984;25:343–348. [PubMed] [Google Scholar]
- Goldstein A, Naidu A. Multiple opioid receptors: ligand selectivity profiles and binding site signatures. Mol Pharmacol. 1989;36:265–272. [PubMed] [Google Scholar]
- Grudt TJ, Williams JT. kappa-Opioid receptors also increase potassium conductance. Proc Natl Acad Sci U S A. 1993;90:11429–11432. doi: 10.1073/pnas.90.23.11429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci. 2009;29:13684–13690. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James IF, Goldstein A. Site-directed alkylation of multiple opioid receptors. I. Binding selectivity. Mol Pharmacol. 1984;25:337–342. [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Mercer AJ, Hentges ST, Meshul CK, Low MJ. Unraveling the central proopiomelanocortin neural circuits. Front Neurosci. 2013;7:19. doi: 10.3389/fnins.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley, Levine Involvement of dynorphin and the kappa opioid receptor in feeding. Peptides. 1983;4:797–800. doi: 10.1016/0196-9781(83)90069-4. [DOI] [PubMed] [Google Scholar]
- Mulder AH, Wardeh G, Hogenboom F, Frankhuyzen AL. Selectivity of various opioid peptides towards delta-, kappa; and mu-opioid receptors mediating presynaptic inhibition of neurotransmitter release in the brain. Neuropeptides. 1989;14:99–104. doi: 10.1016/0143-4179(89)90065-6. [DOI] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci. 2011;31:281–288. doi: 10.1523/JNEUROSCI.4654-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- Simmons ML, Chavkin C. k-Opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fibre neurotransmitter release. Mol Pharmacol. 1996;50:80–85. [PubMed] [Google Scholar]
- Simonin F, Slowe S, Becker JA, Matthes HW, Filliol D, Chluba J, Kitchen I, Kieffer BL. Analysis of [3H]bremazocine binding in single and combinatorial opioid receptor knockout mice. Eur J Pharmacol. 2001;414:189–195. doi: 10.1016/s0014-2999(01)00822-6. [DOI] [PubMed] [Google Scholar]
- Wollemann M, Benyhe S, Simon J. The kappa-opioid receptor: evidence for the different subtypes. Life Sci. 1993;52:599–611. doi: 10.1016/0024-3205(93)90451-8. [DOI] [PubMed] [Google Scholar]
- Zhang S, Tong Y, Tian M, Dehaven RN, Cortesburgos L, Mansson E, Simonin F, Kieffer B, Yu L. Dynorphin A as a potential endogenous ligand for four members of the opioid receptor gene family. J Pharmacol Exp Ther. 1998;286:136–141. [PubMed] [Google Scholar]
- Zhang X, van den Pol AN. Direct inhibition of arcuate proopiomelanocortin neurons: a potential mechanism for the orexigenic actions of dynorphin. J Physiol. 2013;591:1731–1747. doi: 10.1113/jphysiol.2012.248385. [DOI] [PMC free article] [PubMed] [Google Scholar]