

Abstract
Diurnal release of the orexin neuropeptides orexin-A (Ox-A, hypocretin-1) and orexin-B (Ox-B, hypocretin-2) stabilises arousal, regulates energy homeostasis and contributes to cognition and learning. However, whether cellular correlates of brain plasticity are regulated through orexins, and whether they do so in a time-of-day-dependent manner, has never been assessed. Immunohistochemically we found sparse but widespread innervation of hippocampal subfields through Ox-A- and Ox-B-containing fibres in young adult rats. The actions of Ox-A were studied on NMDA receptor (NMDAR)-mediated excitatory synaptic transmission in acute hippocampal slices prepared around the trough (Zeitgeber time (ZT) 4–8, corresponding to 4–8 h into the resting phase) and peak (ZT 23) of intracerebroventricular orexin levels. At ZT 4–8, exogenous Ox-A (100 nm in bath) inhibited NMDA receptor-mediated excitatory postsynaptic currents (NMDA-EPSCs) at mossy fibre (MF)–CA3 (to 55.6 ± 6.8% of control, P = 0.0003) and at Schaffer collateral–CA1 synapses (70.8 ± 6.3%, P = 0.013), whereas it remained ineffective at non-MF excitatory synapses in CA3. Ox-A actions were mediated postsynaptically and blocked by the orexin-2 receptor (OX2R) antagonist JNJ10397049 (1 μm), but not by orexin-1 receptor inhibition (SB334867, 1 μm) or by adrenergic and cholinergic antagonists. At ZT 23, inhibitory effects of exogenous Ox-A were absent (97.6 ± 2.9%, P = 0.42), but reinstated (87.2 ± 3.3%, P = 0.002) when endogenous orexin signalling was attenuated for 5 h through i.p. injections of almorexant (100 mg kg−1), a dual orexin receptor antagonist. In conclusion, endogenous orexins modulate hippocampal NMDAR function in a time-of-day-dependent manner, suggesting that they may influence cellular plasticity and consequent variations in memory performance across the sleep–wake cycle.
Key points
Orexins are well described for their excitatory actions on feeding- and arousal-promoting brain centres; we describe here an inhibitory action of orexin-A on synaptic NMDA receptors in hippocampus, a key area for synaptic plasticity and memory formation.
Orexin-A inhibited NMDA receptor responses at mossy fibre–CA3 connections through postsynaptically expressed orexin-2 receptors, whereas a minor inhibition was observed at Schaffer collateral–CA1 connections, and no effect occurred at non-mossy-fibre excitatory synapses in CA3.
Exogenously applied orexin-A inhibited NMDA receptors in slices prepared during the rats’ resting phase, when endogenous orexin levels are low, but not in slices prepared in the active phase, when endogenous orexins peak.
Through intraperitoneal administration of an orexin receptor antagonist during the active period, exogenous orexin-A-mediated inhibition was restored in the slice.
Endogenous orexins suppress hippocampal synaptic NMDA receptor function in a diurnally cyclic manner, probably restraining synaptic plasticity and learning during certain periods of waking.
Introduction
From rodent to human, a few thousand to ten-thousand neurones in the lateral hypothalamus form a brain-wide axonal projection system that regulates many physiological and behavioural processes via releasing two orexin peptides, orexin-A (Ox-A, hypocretin-1) and orexin-B (Ox-B, hypocretin-2). The densest projections formed by these orexinergic neurones are found in wake-promoting and feeding control centres (Peyron et al. 1998), in the mesolimbic dopaminergic reward system (Fadel & Deutch, 2002), and in the spinal cord autonomic tracts (van den Pol, 1999). Today, a complex picture of orexinergic function is emerging, in which energy homeostasis and arousal stability are coordinated with adaptation of the organism to environmental challenge (Li et al. 2013). In line with orexin neurones promoting waking and energy expenditure, orexinergic concentration in the cerebral spinal fluid (Zhang et al. 2004) and in the lateral hypothalamic area (Yoshida et al. 2001) is high (hundreds of nanomolar) during the active state, whereas it declines during the resting phase. Via the G-protein-coupled orexin-1 receptors (OX1Rs) and orexin-2 receptors (OX2Rs), orexin peptides mediate excitatory effects through postsynaptic depolarisation or facilitation of repetitive action potential discharge at subcortical and cortical levels of the wake-promoting system (Horvath et al. 1999; Brown et al. 2001; Eriksson et al. 2001; Bayer et al. 2002; Burlet et al. 2002; Zhang et al. 2010), through presynaptic facilitation of neurotransmitter release (van den Pol et al. 1998; Burlet et al. 2002; Li et al. 2002; Lambe & Aghajanian, 2003), or through promoting insertion of NMDA receptor (NMDAR) subunits at synaptic membranes (Borgland et al. 2006).
More recent studies implicate orexins in cognitive processes, involving attention and memory formation (Jaeger et al. 2002; Lambe et al. 2005; Deadwyler et al. 2007; Boschen et al. 2009; Sears et al. 2013). Activation of arousal systems, such as the locus coeruleus and the basal forebrain (Boschen et al. 2009; Sears et al. 2013; Soya et al. 2013), and stimulation of thalamocortical circuits (Lambe et al. 2005; Deadwyler et al. 2007; Mair & Hembrook, 2008) are some of the brain mechanisms by which orexins promote cognitive performance. In contrast, few and partially contradictory pieces of evidence support a hippocampal involvement in orexinergic actions on cognitive function. Hippocampus-dependent spatial memory, as assessed with rats in the Morris Water Maze, reportedly is compromised by hippocampal or intracerebroventricular infusion of either agonists or antagonists of orexin receptors (Aou et al. 2003; Akbari et al. 2006, 2007). Ox-A also attenuates NMDAR-dependent long-term potentiation (LTP) and NMDAR-mediated field potentials at Schaffer collateral (SC)–CA1 synapses in vitro (Aou et al. 2003; Selbach et al. 2004; Doreulee et al. 2009), whereas it indirectly upregulates glutamatergic synaptic strength at different hippocampal synapses (Selbach et al. 2004; Walling et al. 2004) and boosts LTP in dentate gyrus (DG) in vivo (Wayner et al. 2004). Immunohistochemistry consistently reveals the presence of orexinergic fibres throughout hippocampal areas, but fibre density is low (Peyron et al. 1998; Selbach et al. 2004; Morales et al. 2006). OX1Rs and OX2Rs are expressed in all hippocampal areas at the mRNA and protein levels (Hervieu et al. 2001; Marcus et al. 2001; Cluderay et al. 2002; Ito et al. 2008). Taken together, these studies suggest that orexin neurones innervate the hippocampal formation to, directly and/or indirectly, regulate synaptic function. However, whether endogenous orexins indeed act on hippocampal circuits and what their primary synaptic targets are has not been investigated.
Using ex vivo slice recordings and pharmacological interference with orexin signalling in the rat, this study defines the role of orexins at hippocampal excitatory synapses by focusing on the following questions. Do orexins directly control hippocampal excitatory synaptic transmission? If yes, what is the role played by synaptic NMDARs? Do endogenous orexins act on these targets in a manner consistent with their time-of-day-dependent release?
We found that exogenously applied Ox-A inhibited NMDAR function at two major hippocampal synapses, with a stronger effect at mossy fibre (MF)–CA3 compared to SC–CA1 synapses. Our data also indicate that MF-NMDAR function is suppressed by orexins released during periods of waking, while they recover during rest, suggesting a diurnal regulation of NMDAR properties at a key synapse of the hippocampal trisynaptic circuit.
Methods
Animals
Pregnant (E14) Sprague–Dawley dams were obtained from Janvier Labs (Le Genest – St Isle, France) and kept in a 12 h/12 h light/dark (LD) cycle (lights on at 07.00 h), with food and water provided ad libitum, in a room controlled for temperature and humidity (∼24°C, 30–40%) in standard Makrolon cages (22 cm × 39 cm). For some experiments, mothers with their pups (postnatal day (P)7) were shifted to a LD cycle from 14.00 h to 02.00 h (lights on at 14.00 h), through increasing time steps of 1–2 h per day. The rats were kept in these conditions for at least one additional week before use. Experiments were performed on P19–P30 offspring. All experimental procedures conformed to the policies of the Veterinary Office of the Canton de Vaud.
Immunohistochemistry
Several 3-week-old rats were kept under deep anaesthesia using 4–5% isofluorane and were transcardially perfused with 5–10 ml of 0.9% NaCl followed by ∼500 ml of an ice-cold fixative solution (4% paraformaldehyde in 0.1 mm phosphate buffer, pH 7.4). The brains were extracted, post-fixed in the same fixative for 2 h at 4°C and cryopreserved in increasing sucrose gradients (10–20–30% in PBS) at 4°C. Coronal sections (45 μm) were cut with a freezing microtome and collected in PBS. Free-floating sections were incubated with 2% methanol and 0.3% H2O2 in PBS for 30 min at room temperature (RT) to quench the endogenous peroxidases. Sections were washed in PBS, kept for 45 min in a blocking solution containing 2% normal horse serum and 0.3% Triton X-100 in PBS, and then incubated overnight at 4°C with an Ox-A or Ox-B primary antibody (goat polyclonal antibodies raised against the C-terminus of Ox-A or Ox-B of human origin; 1:1000 diluted in blocking solution, Santa Cruz Biotechnology, Heidelberg, Germany). After several rinses with PBS, sections were incubated with the Biotin-SP-conjugated secondary antibody (1:500 diluted in PBS, Biotin-SP-conjugated AffiPure Donkey Anti-Goat IgG, Jackson ImmunoResearch, Suffolk, UK) for 2 h at RT. Subsequently, sections were washed with a Tris-buffered saline (TBS, 0.1 m Tris and 0.9% NaCl, pH 8) and incubated in the avidin–biotin–peroxidase complex (Vectastain Elite ABC-peroxidase kit, Vector Labs Inc., Peterborough, UK) diluted 1:100 in the same Tris-buffered solution. After being rinsed in a different TBS (0.05 m Tris and 0.9% NaCl, pH 7.6), sections were incubated with 3,3′-diaminobenzidine (DAB, 0.04%) and 0.015% H2O2 diluted in TBS (0.05 m Tris and 0.9% NaCl, pH 7.6), to allow the DAB oxidation and precipitation. As a negative control, the immunostaining procedure was performed in the absence of the primary antibodies and resulted in a lack of labelling. The sections were mounted on glass slides (SuperFrostPlus, Menzel-Gläser, Braunschweig, Germany), dehydrated in an ascending series of ethanol, cleared in xylol and coverslipped with Eukitt mounting medium (Reactolab, Servion, Switzerland). Illustrations for Fig. 1 were taken from the sections of one rat.
Figure 1. Ox-A reduces NMDA-EPSCs at MFs and at SCs.
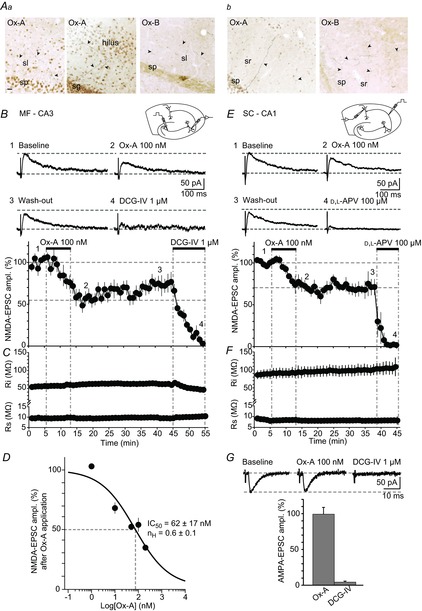
Aa and b, images depicting immunostainings for Ox-A or Ox-B proteins in areas CA3/DG (a) and CA1 (b). Ox-A- and Ox-B-containing fibres (some indicated with filled arrowheads), sparse and of different size, are found throughout CA3, DG and CA1. sp, s. pyramidale; sl, s. lucidum; sr, s. radiatum; sg, s. granulosum. Scale bar for both a and b: 20 μm. B, top, schematic diagram shows hippocampal slice with positioning of recording and stimulation electrodes. Middle, representative NMDA-EPSCs (mean of two successive sweeps), recorded from a CA3 pyramidal neurone in a slice prepared at ZT 4, are shown for baseline, during maximal Ox-A suppression, after wash-out and following DCG-IV application to confirm MF origin. Numbers correspond to the time points in the graph shown below. Horizontal dashed lines refer to baseline holding current and peak of the NMDA-EPSC before Ox-A application. Bottom, mean NMDA-EPSC amplitudes, expressed as percentage of baseline, are plotted against time (n = 10). Horizontal dashed lines indicate mean EPSC levels before and after 10 min of Ox-A application. C, time course of Ri and Rs corresponding to B. Vertical lines indicate time period of Ox-A and DCG-IV application in B and C. D, concentration–response curve for Ox-A-induced inhibition of MF-NMDA-EPSCs, with 1, 10, 50, 100 and 200 nm tested (n = 2–10 recordings from different cells per concentration). Data were fitted with a Hill equation, yielding IC50 = 62 ± 17 nm (value indicated by dotted lines) and a Hill coefficient nH = 0.6 ± 0.1. E, top, schematic diagram as in B. Middle, representative SC-NMDA-EPSCs recorded from a CA1 pyramidal neurone in a slice prepared at ZT 4, are shown for baseline, in Ox-A, after wash-out and in d,l-APV. Numbers and horizontal dashed lines are as in B. Bottom, graph of time course of Ox-A actions on SC-NMDA-EPSCs (n = 4), in percentage of baseline. F, corresponding time course of Ri and Rs, vertical lines as in C. G, top, representative MF-AMPA-EPSCs (mean of 2 successive sweeps) in a CA3 pyramidal neurone recorded in a slice prepared at ZT 4, during baseline, in Ox-A and after DCG-IV application. Horizontal dashed lines are used as in B and E. Bottom, bar graph representing mean AMPA-EPSC amplitudes expressed as percentage of baseline (n = 3). Ox-A effect was measured 10 min after application onset; DCG-IV blocked the response. Data are presented as mean ± SEM.
Electrophysiological recordings
Rats were deeply anaesthetised with 4–5% isofluorane and immediately decapitated. Hippocampal slices were prepared according to two previously described procedures, depending on the type of NMDAR-mediated response recorded. The standard time of slice preparation was at Zeitgeber time (ZT) 4 (corresponding to 4 h into the resting phase). For some experiments, slices were prepared at ZT 8 or ZT 23, as specified in the text. For most of the recordings of NMDA-EPSCs at MF–CA3 synapses (MF-NMDA-EPSCs), hippocampi were dissected and cut as described (Kwon & Castillo, 2008a) to optimally preserve the integrity of MF projections. For some of the initial recordings of MF-NMDA-EPSCs, for SC–CA1 NMDA-EPSCs and for iontophoretic NMDAR-mediated currents (NMDA-currents), transverse slices from whole brains were prepared (Bischofberger et al. 2006). For slice preparation from dissected hippocampi, the brain was quickly immersed in an ice-cold oxygenated (95%–5%, O2–CO2) sucrose solution containing (in mm): 213.3 sucrose, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3, 7 MgCl2, 0.5 CaCl2, 25 glucose, 1.7 l(+)-ascorbic acid. Entire hippocampi were gently removed and cut perpendicular to their longitudinal axis while being fixed in an agar cube glued to the stage of the vibratome (HM650V, Microm-Zeiss AG, Feldbach, Switzerland). Slices (400 μm) were incubated in a recovery chamber filled with a solution made of sucrose solution and ACSF, mixed at 1:1, for 30 min at RT, which was subsequently replaced by standard ACSF. The ACSF contained (in mm): 117.7 NaCl, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3, 1.2 MgCl2, 2 CaCl2, 25 glucose, 1.7 l(+)-ascorbic acid. For parasagittal hippocampal slices cut from entire brain, the brain was quickly immersed in an ice-cold oxygenated solution containing (in mm): 58.8 NaCl, 105 sucrose, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3, 7 MgCl2, 0.5 CaCl2, 25 glucose, 1.7 l(+)-ascorbic acid and 2 kynurenic acid. Slices (400 μm) were cut with the vibratome and incubated at 35°C for 30 min in a storage chamber filled with ACSF, followed by cooling to RT.
For whole-cell voltage-clamp recordings, patch pipettes (3–3.5 MΩ) were pulled from borosilicate glass (TW150F-A, WPI, Berlin, Germany) and filled with the following intracellular solution (in mm): 120 caesium methanesulphonate, 10 CsCl, 10 Hepes, 8 NaCl, 0.5 EGTA, 4 Mg-ATP, 10 phosphocreatine, adjusted to pH 7.3–7.4 with CsOH and to 300–305 mosmol l–1. Na-GTP (0.2 mm) and N-(2,6-dimethylphenylcarbamoylmethyl) TEA chloride (QX-314 chloride, 2 mm) were freshly added and the solution was filtered and kept on ice.
Slices were perfused with oxygenated ACSF in the recording chamber at a rate of ∼3–3.5 ml min−1 and recordings were performed at RT. Pyramidal neurone layers were visualised using near-infrared differential interference contrast microscopy (BX51WI, Olympus, Volketswil, Switzerland), but cells were recorded ‘semi-blind’ or ‘blind’ at depths >100 μm below the surface to target cells with largely preserved neurites and maximal vitality. Voltage-clamp recordings were obtained using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), filtered at 2 kHz and sampled at 10 kHz, using Clampex 10.2 (Molecular Devices). Series resistance (Rs) was monitored during the experiments and changes >15% were not accepted. A liquid junction potential of ∼−7 mV, measured as previously described (Neher, 1992), was not compensated for. NMDA-EPSCs were pharmacologically isolated through continuous bath application of oxygenated ACSF containing (in μm): 40 6,7-dinitroquinoxaline-2,3-dione (DNQX), 100 picrotoxin, 2(2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2hydroxypropyl] (phenylmethyl) phosphinic acid hydrochloride (CGP55845) to block AMPA, GABAA and GABAB receptors, respectively, and recorded at +30 mV. The NMDAR co-agonist glycine (1 μm) was also included in the bath. AMPA-EPSCs of MF-CA3 synapses (MF-AMPA-EPSCs) were recorded at −60 mV in ACSF supplemented with (in μm): 100 picrotoxin, 2 CGP55845, 100 d,l-2-amino-5-phosphonopentanoic acid (d,l-APV). In this case, TTX was included at 30 nm to avoid polysynaptic contamination (Kwon & Castillo, 2008b).
Synaptic currents were elicited by brief current pulses (100 μs, 50–150 μA) through monopolar (saline-filled patch pipette) or bipolar tungsten electrodes (FHC Inc., Bowdoin, ME, USA). Mossy fibres were stimulated by placing the stimulation electrode in the granule cell layer of the DG, whereas associational/commissural synapses and entorhinal inputs, referred to as non-MF synapses, were activated in the hilus/stratum (s.) lucidum of CA3, and SCs were recruited in s. radiatum of the CA1. To ascertain the origin of MF-induced responses, the group II metabotropic glutamate receptor (mGluR2) agonist (2′S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV), 1 μm, was always applied at the end of the experiments (Kamiya et al. 1996). When the EPSC amplitude was reduced by >80%, EPSCs were included as MF-mediated responses. When the reduction was ≤60%, recordings were considered of predominantly non-MF origin. SC-NMDA-EPSCs were routinely blocked by d,l-APV (100 μm) at the end of the experiments. Five experiments yielding intermediate DCG-IV sensitivity (60–80%) were included only in the linear regression analysis between DCG-IV and Ox-A sensitivity (Fig. 2D).
Figure 2. Ox-A does not modify NMDA-EPSCs at non-MF–CA3 synapses.
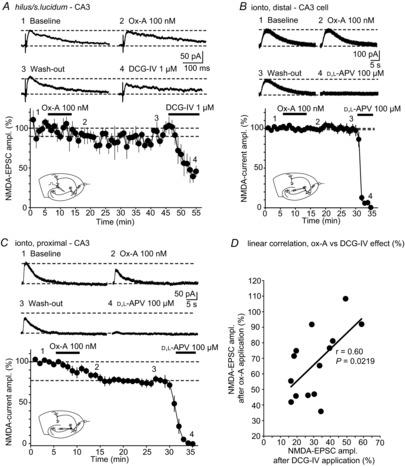
A, top, representative NMDA-EPSCs (mean of 2 successive sweeps), elicited through hilum/s. lucidum stimulation and recorded in a slice prepared at ZT 4, are shown for baseline, during maximal Ox-A action, after wash-out and after DCG-IV application. Numbers correspond to the time points in the graph shown below. Horizontal dashed lines refer to baseline holding current and peak of NMDA-EPSC before Ox-A application. Bottom, time course of mean NMDA-EPSC amplitudes as a percentage, with horizontal lines indicating 100% and percentage suppression of Ox-A, measured 10 min after application onset (n = 4). DCG-IV induced a partial block of NMDA-EPSCs, indicating that a mixture of MFs and non-MFs was recruited. Inset, schematic diagram of recording configuration, indicating in particular the positioning of the stimulation pipette in the hilus/s. lucidum. B, similar experiment to that in A, except that iontophoretic (ionto) application of NMDA was used to elicit NMDA-currents at distal dendritic sites (see inset). The pipette was positioned ∼250 μm from the cell body to activate NMDARs preferentially at non-MF synapses. Corresponding representative traces and time course are shown (n = 8). Horizontal lines are used as in A. Data are presented as mean ± SEM. C, same experiment as B, but for iontophoretic NMDA application on proximal dendritic sites. In B and C, d,l-APV fully blocked the iontophoretic current responses. D, least-squares linear regression analysis of DCG-IV sensitivity and Ox-A sensitivity for 14 recordings with low to intermediate suppression by DCG-IV (to 15–60% of control amplitude), which includes the four cells presented in A. The slope of the regression curve is 1.02, with r = 0.60 and P = 0.0219.
Iontophoretic NMDA-currents were elicited through a patch pipette filled with 10 mm NMDA (in ACSF), placed either at the proximal apical dendrite (within 100 μm from the soma in s. lucidum) or at the distal apical dendrite (> ∼250 μm from the soma in s. radiatum) to preferentially activate MF or non-MF inputs, respectively. NMDA was ejected once per minute by applying currents of 200–500 nA and 1 s duration. Cellular currents were recorded at +30 mV in ACSF containing (in μm): 1 glycine, 40 DNQX, 100 picrotoxin, 2 CGP55845 and 0.5 TTX. Retention currents of 10–20 nA were used in between ejections. The ejection area was visualised by fluorescence microscopy after including Alexa 594 (1 μm) in the pipette, which covered 94 ± 20 μm of proximal dendritic length (n = 3 measurements). d,l-APV (100 μm) was applied at the end of the recording to block the NMDA-currents.
Ox-A was applied through the bath and dissolved at the desired concentration immediately before application. Time points of Ox-A application were between 114 and 405 min after slice preparation for both MF stimulation and iontophoretic NMDA application. In none of these experimental series was there a significant correlation between the time of recording and the amplitude of the Ox-A effect, as assessed by a least-squares linear fit (Pearson's correlation coefficient, r = 0.04 and 0.15 for MF stimulation (n = 10) and NMDA iontophoresis (ZT 4, n = 12), respectively; P > 0.05 for both).
Analysis of electrophysiological recordings
Current amplitudes were measured in a 4 ms (EPSCs) or 200 ms (iontophoresis) time window around the peak of the event. Inhibitory effects of Ox-A were calculated when steady-state was reached, typically 10 min after onset of bath application. DCG-IV effects were measured after 10 min and d,l-APV after 5 min of wash-in. Currents were normalised to the corresponding mean baseline amplitudes. The decay time of currents was calculated by dividing the charge transfer by the mean amplitude of the NMDA-EPSCs or NMDA-currents (Longordo et al. 2009). Charge transfer was calculated between the peak time and the time of return to baseline (for EPSCs ∼1 s after peak, for iontophoretic currents ∼30 s after the peak). The IC50 and the Hill coefficient for Ox-A-suppression of MF-NMDA-EPSCs were calculated by fitting the data with the Hill equation. Least-squares linear fits were performed in JMP 9.0.0 (SAS Campus Drive, Cary, NC, USA).
Behavioural monitoring and almorexant injections
For these experiments, animals were shifted to a LD cycle of 14.00 h to 02.00 h. At P20–21, each rat was transferred singly into a larger cage (53 cm × 31.5 cm) with 34 cm-high transparent walls and with a dark bedding made of various tree barks (Zoosano AG, Basadingen, Switzerland) or Cellu-Dri Soft bedding (Shepherd Specialty Papers Tecnilab-BMI, Someren, The Netherlands). As enrichment, pieces of black cardboard were put in the cage. Animals were monitored for their sleep–wake behaviour over 48 h (24 h baseline trial (Bsl), 24 h injection trial (Inj)) through tracking the spontaneous locomotor activity (distance travelled per unit of recording time), using an infrared-sensitive camera (Ikegami ICD47E) together with Ethovision XT8 software (Noldus Information Technology, Wageningen, The Netherlands). The dual orexin receptor antagonist (2R)-2-{(1S)-6,7-dimethoxy-1-[2-(4-trifluoromethylphenyl)-ethyl]-3,4-dihydro-1H-isoquinolin-2-yl}-N-methyl-2-phenyl-acetamide (almorexant, Alm) was suspended in 200 μl of 0.25% methyl cellulose (Sigma-Aldrich, Buchs, Switzerland) immediately before injections and injected i.p. at 100 mg kg−1 under red light illumination. In a preliminary trial test, we observed that the action of Alm (100 mg kg−1) on the rats’ resting time lasted for up to ∼3 h after the injection. We administered two injections at ZT 18 and 21 of the Inj trial to maintain the resting state for the entire second half of the dark, active phase. In parallel to the rat group injected with Alm (Alm group), the control group (Veh group) was injected at the same ZTs with equal volumes of 0.25% methyl cellulose, the vehicle (Veh). After the two injections, rats were removed from the cage at ZT 23 and anaesthetised within 30 s for slice preparation.
Analysis of behavioural data
The resting time was calculated as an hourly percentage of time during which the rat's centre of gravity moved at <1 cm s−1. This threshold was set during a visually identified resting episode. For the 24 h Bsl trial, mean resting times were calculated for the 12 h light and 12 h dark periods. For the 24 h Inj trial, means of 5 h resting time (ZT 18–23) during the injections were taken for comparison between animals injected with Alm or Veh.
Drugs and chemicals
Picrotoxin, d,l-APV, DNQX, CGP55845 hydrochloride and kynurenic acid were purchased from Abcam (Cambridge, UK); TTX was from Latoxan (Valence, France); NMDA, glycine, d,l-propranolol and methyl cellulose were from Sigma-Aldrich; Ox-A, DCG-IV, N-(2-methyl-6-benzoxazolyl)-N′-1,5-naphthyridin-4-yl urea (SB334857), scopolamine hydrobromide and prazosin hydrochloride were from Tocris-R&D systems; 1-(2,4-dibromophenyl)-3-[(4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-yl] urea (JNJ10397049) was a generous gift from Dr Mauro Corsi and Dr Corrado Corti (Aptuit, Verona, Italy). Alm was kindly provided by Professor Mehdi Tafti and Dr Anne Vassalli (Center for Integrative Genomics, University of Lausanne (CIG-UNIL)).
Statistics
All data are given as means ± SEM. Statistics were done using Student's t tests as follows. Electrophysiological data were assessed with two-tailed paired Student's t tests comparing raw datasets for baseline and test conditions in Excel. Two-tailed paired Student's t tests were also used to compare resting times of rats across the light–dark cycle and between the Bsl and the Inj trials. Unpaired Student's t tests were used for comparison between conditions using normalised data both in electrophysiological and behavioural analyses. Correlation coefficients of linear regressions were obtained in JMP 9.0.0. The level of significance was set at P < 0.05.
Results
Orexin-A suppresses NMDA-EPSCs at MF synapses through a postsynaptic mechanism
We first tested whether 3-week-old Sprague–Dawley rats expressed immunoreactivity for orexins in hippocampus. With antibodies recognising the C-terminus of Ox-A and Ox-B, previously used for hippocampal immunostaining of orexins (Morales et al. 2006), we identified heavily stained cell bodies and fibres in the lateral hypothalamic area (not shown), consistent with the described location of orexin neurones (Peyron et al. 1998). Individual immunoreactive fibres were clearly present within the CA3 and CA1 areas, as well as in the DG (Fig. 1Aa and b). Ox-A- or Ox-B-expressing fibres, ranging from ∼20 to ∼270 μm in length within the 45 μm-thick coronal section, were sparse and located throughout all layers, crossing s. pyramidale in CA3 and CA1 to enter s. lucidum and s. radiatum, respectively. In the DG, Ox-A- and Ox-B-positive short fibres appeared mostly in close proximity to the granule cell layer.
To evaluate the physiological effects of orexin on hippocampal NMDARs, we prepared acute hippocampal slices at ZT 4 (see Methods), corresponding to 11.00 h in the light phase (lights on at 07.00 h). Pharmacologically isolated NMDA-EPSCs were evoked at 0.033 Hz in CA3 or CA1 pyramidal neurones held at +30 mV. Stimulation was achieved via monopolar or bipolar extracellular electrodes positioned in the DG for MF stimulation and in s. radiatum for stimulation of SCs (see Methods and insets in Fig. 1B and E). Mean baseline NMDA-EPSCs had an amplitude of 57 ± 5 pA (n = 10) in CA3 neurones, and 87 ± 8 pA in CA1 neurones (n = 4). Orexinergic actions were examined through bath application of Ox-A, which shows comparable affinity for both orexin receptor types (Sakurai et al. 1998). At 100 nm, Ox-A decreased the amplitude of NMDA-EPSCs to 55.6 ± 6.8% (38 ± 4 pA, n = 10, P = 0.0003) of baseline at MF–CA3 (Fig. 1B), and to 70.8 ± 6.3% (61 ± 9 pA, n = 4, P = 0.013) at SC–CA1 synapses (Fig. 1E). The effect reached a maximum after 7–10 min of application and recovered partially in the case of MF-NMDA-EPSCs upon wash-out of Ox-A. Holding current remained unaltered after Ox-A application in these cells (651 ± 50 pA vs. 653 ± 59 pA in Ox-A, P = 0.9 for MF–CA3 recordings, 344 ± 52 pA vs. 341 ± 52 pA, P = 0.5 for SC–CA1 recordings). The input resistance (Ri) weakly increased 10 min after Ox-A application (for CA3 pyramidal neurones: 53 ± 4 MΩ vs. 59 ± 5 MΩ in Ox-A, P = 0.009, Fig. 1C; for CA1 pyramidal neurones: 89 ± 12 MΩ vs. 95 ± 14 MΩ, P = 0.059, Fig. 1F). Continuous monitoring of cellular Rs indicated no changes during the recordings (<10% change in DCG-IV vs. baseline, Fig. 1C and ∼10% change in d,l-APV vs. baseline, Fig. 1F). Ox-A slowed the decay time course of the synaptically elicited MF-NMDA-EPSCs in 9/10 cells (145 ± 14 ms vs. 211 ± 24 ms after Ox-A application, P = 0.0008) but not of SC-NMDA-EPSCs (208 ± 8 ms vs. 237 ± 17 ms, P = 0.2, see Methods). The mGluR2 agonist DCG-IV (1 μm) largely abolished NMDA-EPSCs at MFs (7.6 ± 1.4 pA, n = 10, P < 0.0001, see Methods, Fig. 1B). The NMDA-EPSCs recorded at SC–CA1 synapses were fully blocked by the NMDAR antagonist d,l-APV (100 μm, n = 4, Fig. 1E).
The inhibitory effect of Ox-A on NMDA-EPSCs at MF–CA3 synapses was concentration -dependent (Fig. 1D): NMDA-EPSCs were not affected by 1 nm Ox-A, but reduced to a minimum of 33.5 ± 2.3% of baseline amplitude by 200 nm Ox-A (n = 2–10 per concentration tested). This concentration range corresponds to that found in the cerebral spinal fluid (Yoshida et al. 2001) and overlaps with several concentration–functional response relations of heterologously expressed mouse OX2Rs (Chen & Randeva, 2004; Wang et al. 2014). Fitting a sigmoidal Hill function yielded an IC50 of 62 ± 17 nm and a Hill coefficient of 0.6 ± 0.1 (Fig. 1D). In all subsequent experiments, Ox-A was applied at 100 nm to induce a close-to-maximal suppression.
In contrast to NMDA-EPSCs, no suppressant actions of Ox-A were found at MF-AMPA-EPSCs. Amplitudes remained unaltered after 10 min in the continuous presence of Ox-A (99.3 ± 9.5% of control, n = 3, P = 0.78), while they were fully blocked by DCG-IV (1 μm, Fig. 1G).
To assess the synaptic specificity of Ox-A actions, we recorded NMDA-EPSCs formed by associational/commissural and entorhinal connections, referred to as non-MF connections. Stimulation of the s. lucidum/hilar regions elicited NMDA-EPSCs showing an intermediate to weak sensitivity to DCG-IV (40–60% reduction), indicating that a mixture of fibres of both MF and non-MF origin was recruited. The EPSCs were not affected by bath application of Ox-A (89.5 ± 7.1%, n = 4, P = 0.31, Fig. 2A) and no significant changes occurred in holding currents after Ox-A application (656 ± 64 pA vs. 558 ± 63 pA in Ox-A, P = 0.2). Values for Ri were 44 ± 6 MΩ vs. 57 ± 5 MΩ in Ox-A (P = 0.053). We further assessed Ox-A-mediated modulation of NMDARs on distal sites of CA3 cells by placing an iontophoresis pipette filled with NMDA (10 mm) at the distal apical dendrite of CA3 pyramidal neurones (s. radiatum, ∼250 μm from the soma, see inset in Fig. 2B). Local pressure application of NMDA on CA3 cells has been previously used to study G-protein-mediated regulation of NMDARs (Benquet et al. 2002). NMDA-currents had a mean amplitude of 85 ± 12 pA and a mean decay time of 10.2 ± 1.0 s (n = 8, Fig. 2B), comparable to previous observations (Benquet et al. 2002). Ox-A (100 nm) applied in the bath neither affected amplitude (98.7 ± 3.6%, 84 ± 12 pA, n = 8, P = 0.73) nor decay time (9.6 ± 1.0 s, P = 0.12) of these currents that were fully blocked by d,l-APV (100 μm) at the end of the experiments (Fig. 2B). In contrast, NMDA-currents elicited iontophoretically on the proximal apical dendrite (within 100 μm from the soma, see inset in Fig. 2C), the site of MF synapses (Claiborne et al. 1986), were reduced by Ox-A to 77.8 ± 3.9% (n = 9, P = 0.016, Fig. 2C). Baseline NMDA-currents had a mean amplitude of 95 ± 13 pA and a mean decay time of 8.2 ± 1.6 s, whereas, in the presence of Ox-A, amplitudes were 78 ± 11 pA and decay time 7.5 ± 1.4 s. No recovery was observed up to 20 min after wash-out. Currents were fully blocked by d,l-APV (100 μm) at the end of the experiment (Fig. 2C). In these experiments, the Ox-A effects were smaller, probably because NMDARs at both MF and non-MF synapses were recruited. Nevertheless, NMDA-current measurements offered a rapid and reliable assay for further characterisation of Ox-A actions on postsynaptic NMDARs.
Taken together, we identified a specific inhibitory effect of Ox-A on NMDA-EPSCs at two sets of hippocampal synapses, with a major suppression at MF–CA3 and a moderate effect at SC–CA1 synapses. Moreover, within the CA3 area, Ox-A effects were confined to MF–CA3 synapses. This conclusion was further supported by the fact that the Ox-A sensitivity of NMDA-EPSCs depended linearly on the DCG-IV sensitivity, which is an indicator of the relative presence of MFs in the stimulated fibre bundle (n = 14, correlation coefficient r = 0.60, P = 0.0219; Fig. 2D). For the further analysis of Ox-A actions, the remainder of the study was focused on the MF–CA3 pathway.
Based on the clear Ox-A effects on iontophoretic NMDA-currents, and on the lack of such on AMPA-EPSCs, it is likely that Ox-A acted primarily post- rather than presynaptically. To further corroborate this, we examined whether Ox-A affected low-frequency facilitation (LFF), a well-characterised presynaptic form of short-term plasticity at MFs that sensitively reports on presynaptic receptor-induced changes in release characteristics (for review, see Evstratova & Tóth, 2014). Baseline NMDA-EPSCs elicited at 0.033 Hz were followed by a train of 20 pulses at 1 Hz during which NMDA-EPSC amplitude increased by 2- to 3-fold (297 ± 77% of baseline, n = 5, Fig. 3A and B). Subsequent application of Ox-A (100 nm) decreased the amplitude of the NMDA-EPSCs, but LFF developed to a comparable extent during 1 Hz stimulation (328 ± 97%, n = 5, P = 0.69 compared to LFF in control, Fig. 3A–C), thus excluding a primarily presynaptic mechanism for Ox-A-inhibition of NMDAR-mediated synaptic responses.
Figure 3. Ox-A leaves low-frequency facilitation of MF synapses unaltered.
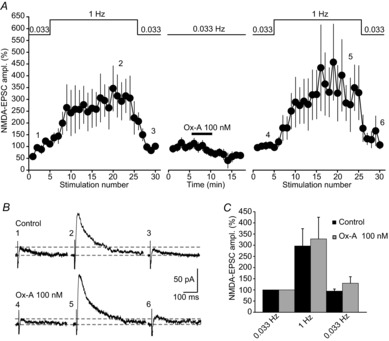
A, mean time course of LFF of MF-NMDA-EPSCs (n = 5), evoked by increasing the stimulation frequency from 0.033 Hz to 1 Hz (indicated on top of the graph). Note that, although EPSCs show a gradual run-up during the initial 0.033 Hz stimulation, a strong, several-fold increase in EPSC amplitude occurred during the 20 pulses at 1 Hz that recovered after 0.033 Hz stimulation was resumed. After Ox-A (100 nm) application, NMDA-EPSC amplitude decreased, but LFF persisted. EPSC amplitudes before and in Ox-A were normalised to their respective baseline values at 0.033 Hz and set to 100%. B, raw traces (mean of 2 sweeps) corresponding to an experiment shown in A. Note that baseline NMDA-EPSCs were reduced in Ox-A, but LFF was fully developed. Numbers correspond to the time points in the graph shown in A. Horizontal lines refer to baseline holding current and peak NMDA-EPSC at 0.033 Hz. C, bar graphs summarising mean relative values of LFF in control (black bars) and in Ox-A (grey bars) (n = 5). The amplitude of LFF was obtained by dividing the mean EPSC amplitudes during the last 5 sweeps in the 1 Hz train by the mean of the 5 baseline sweeps. Relative values of LFF were not altered in Ox-A. Steady-state EPSC amplitudes at 0.033 Hz after the 1 Hz train are also indicated. Data are presented as mean ± SEM.
Orexin-A suppresses NMDA-currents through OX2Rs
In situ hybridisation studies suggest that the OX2R is predominantly expressed in the CA3 area (Marcus et al. 2001). We examined the actions of potent and selective orexin receptor antagonists to test the functional involvement of OX1Rs and OX2Rs in Ox-A actions on NMDA-currents. SB334867 selectively blocks OX1Rs in acute brain slices (Borgland et al. 2006; Li et al. 2010), whereas JNJ10397049 abrogates OX2R actions in vitro (Borgland et al. 2008; Gozzi et al. 2011). When used at these previously established concentrations in the bathing solution, Ox-A-mediated actions on NMDA-currents elicited on proximal dendrites persisted in SB334867 (1 μm) (82.6 ± 1.7%, n = 7, P = 0.00003, Fig. 4A and Da), whereas they were entirely blocked by JNJ10397049 (1 μm) (97.3 ± 3.7%, n = 8, P = 0.34, Fig. 4B and Db). The extent of current reduction in SB334867 was comparable to that obtained when Ox-A was applied alone (P = 0.49), whereas it was different in JNJ10397049 (P = 0.003). In addition, Ox-A actions were largely maintained when applied in the continuous presence of a cocktail of cholinergic (scopolamine hydrobromide, 10 μm) and adrenergic receptor antagonists (prazosin hydrochloride, 1 μm and d,l-propranolol, 1 μm) shown previously to interfere with Ox-A actions on hippocampal excitatory transmission (Selbach et al. 2004; Doreulee et al. 2009) (81.4 ± 2.7%, n = 5, P = 0.69 compared to control, Fig. 4C and Dc). Therefore, Ox-A acts primarily through postsynaptic OX2Rs to directly suppress NMDAR-mediated transmission at MF synapses.
Figure 4. Ox-A actions on MF-NMDARs are mediated by OX2Rs.
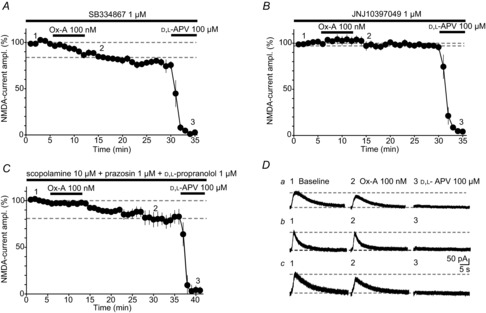
A and B, pharmacological dissection of Ox-A actions through continuous bath application of the OX1R antagonist SB334867 (A), or the OX2R antagonist JNJ10397049 (B). Time courses for both pharmacological tests, performed on iontophoretic NMDA-currents in slices prepared at ZT 4, indicate that Ox-A-induced suppression of NMDA-currents was maintained in SB334867 (1 μm, n = 7, A), but abolished in JNJ10397049 (1 μm, n = 8, B). C, Ox-A actions were assessed in the presence of a cocktail of scopolamine (non-selective muscarinic antagonist, 10 μm), prazosin and d,l-propranolol (α- and β-adrenergic antagonists, respectively, 1 μm each) (n = 5). Ox-A inhibition developed fully, but more slowly than in the absence of these antagonists. D, representative NMDA-currents for experiments in A, B and C. Lowercase letters correspond to the experiments in A, B and C, respectively, and numbers to the points in the respective time courses. Horizontal lines indicate baseline holding and peak NMDA-current values before Ox-A application. Data are presented as mean ± SEM.
Ox-A-mediated suppression of NMDA-currents varies with time of day
Is NMDAR function modified by endogenous orexins released within the hippocampus? The recent history of the animal's sleep–wake behaviour prior to being killed can be traced in several synaptic characteristics of acute brain slices (Kopp et al. 2006; Kurotani et al. 2008; Lanté et al. 2011). Endogenous orexins fluctuate across the day, notably also in discrete brain areas innervated by orexinergic fibres (Yoshida et al. 2001; Zhang et al. 2004). Therefore, we examined whether exogenous Ox-A effects on NMDA-EPSCs varied depending on the time of day of slice preparation.
Slices were cut at ZT 23, corresponding to the last hour of the dark phase, during which Ox-A reaches a peak, and compared to recordings from slices at ZT 4 or at ZT 8, at which Ox-A concentrations are their lowest (Zhang et al. 2004), and at which the majority of our recordings were performed (Figs 4). Application of exogenous Ox-A (100 nm) did not affect NMDA-current amplitudes in brain slices prepared at ZT 23 (97.6 ± 2.9% baseline, n = 12, P = 0.42, Fig. 5A–C). On the contrary, in slices prepared in the period of low endogenous orexin levels (ZT 4 and ZT 8), bath application of Ox-A invariably decreased the amplitude of NMDA-currents (77.2 ± 3.5% for ZT 4, n = 12, P = 0.006 and 80.4 ± 2.3% for ZT 8, n = 4, P = 0.022, Fig. 5A–C). Was the time-of-day dependence of Ox-A actions due to elevated amounts of endogenous orexins present in slices at ZT 23, but not at ZT 4 or ZT 8? High ambient neurotransmitter concentrations can be maintained in slices, as described for adenosine in the MF–CA3 synaptic environment (Moore et al. 2003). If endogenous orexin concentration remained high in our case, the actions of exogenous Ox-A would be attenuated. However, the OX2R antagonist JNJ10397049 (1 μm), when applied for a period of at least 25 min, did not affect NMDA-currents (100.3 ± 3.7% of amplitude before drug application, n = 6, P = 0.87, Fig. 5D), arguing against a tonic suppressive action of ambient orexins in the slice.
Figure 5. Exogenous Ox-A effects are dependent on the time-of-day of slice preparation.
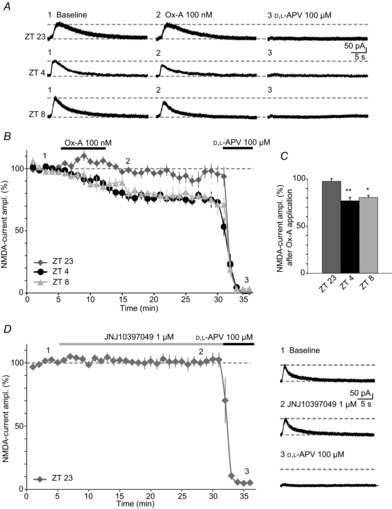
A, representative NMDA-currents evoked iontophoretically on proximal CA3 dendrites, in slices prepared during different ZTs, as indicated to the left of the traces. Traces in the presence of Ox-A and d,l-APV are also shown. Time points chosen for slice preparation correspond to the peak of Ox-A cerebral spinal fluid levels (ZT 23), and to the minimum (ZT 4, ZT 8) (Zhang et al. 2004). Note that Ox-A inhibitory effects are minor at ZT 23, but fully developed at ZT 4 and ZT 8. B, time course of Ox-A actions in slices prepared at these three different times of day (ZT 23, n = 12; ZT 4, n = 12; ZT 8, n = 4), with mean NMDA-EPSC amplitudes, expressed as percentage of baseline. At ZT 4, 9 datapoints presented in Fig. 2C were included. Horizontal line indicates 100%. C, bar graphs representing mean Ox-A inhibition at these different ZT values. Asterisks represent significance relative to baseline NMDA-currents. *P < 0.05; **P < 0.01. D, left, time course of NMDA-currents in the continuous presence of JNJ10397049 (1 μm). NMDA-currents remained unaltered over >25 min (n = 6), arguing against a tonic suppression by endogenous orexins present in the slice. Responses were fully blocked by d,l-APV. Dotted line indicates 100%. Right, representative traces corresponding to the graph on the left. Dotted lines denote baseline holding and peak NMDA-current amplitude before drug wash-in. Data are presented as mean ± SEM.
Even without the continued presence of the ligand, orexinergic modulation induced during waking could persist in the slice and attenuate or occlude the actions of exogenously added Ox-A. Such modifications could have taken place at the level of the OX2Rs or the NMDARs, occluding further inhibitory effects of Ox-A. In this case, NMDA-EPSCs recorded in slices prepared at ZT 23 should be smaller compared to those at ZT 4 or ZT 8. Such comparison, however, is experimentally difficult due to variable recruitment of MFs that, even in the dissected hippocampal preparation (Kwon & Castillo, 2008a), are left intact in the slice to only a very limited extent and at very restricted sites. To further define the role of endogenous orexins in the observed time-of-day dependence, we chose to pharmacologically interfere with orexinergic signalling prior to slice preparation. Almorexant (Alm) is a potent dual orexin receptor antagonist (Brisbare-Roch et al. 2007; Dugovic et al. 2009; Kang et al. 2009; Morairty et al. 2012) that crosses the blood–brain barrier (Brisbare-Roch et al. 2007) and that acts as a preferential OX2R antagonist in vivo (Dugovic et al. 2009; Mang et al. 2012). Video monitoring was used to follow the activity during the LD cycle in two groups of rats, one injected with Alm (Alm group, n = 8, 100 mg kg−1 i.p. in 0.25% methyl cellulose suspension), the other one with vehicle (Veh) only (Veh group, n = 6, 0.25% methyl cellulose). For each group, a 24 h undisturbed baseline activity (Bsl Alm, Bsl Veh) was followed by a 24 h period when injections of Alm (Inj Alm) or Veh (Inj Veh) in the dark, active phase were applied. Activity monitoring during the Bsl trials indicated that both Alm and Veh groups were well entrained to the LD cycle, showing consistently higher resting time during the light (ZT 1–12) than during the dark phase (ZT 13–24) (Bsl Alm: mean 12 h resting time, ZT 1–12: 63 ± 2.3%; ZT 13–24: 45.1 ± 1.8%, n = 8, P = 0.018; Bsl Veh: mean 12 h resting time, ZT 1–12: 67 ± 3.8%; ZT 13–24: 44.9 ± 3.4%, n = 6, P = 0.077, Fig. 6Aa and c). Moreover, both rat groups showed a delayed and transient increase in resting time during ZT 16–18, described previously as a secondary recovery phase after an initial peak of activity in the dark (Franken et al. 1991; Brisbare-Roch et al. 2007; Lanté et al. 2011). In the Inj Alm trials, two injections of Alm at ZT 18 and ZT 21 were applied to ensure OXR block during the entire second half of the dark phase (see Methods). Veh injections occurred at equal ZTs in the Inj Veh trial. After Alm injections, rats rested significantly more than during the corresponding times in the Bsl Alm trial (mean 5 h resting time, ZT 18–23: Inj Alm, 66.5 ± 1.8%; Bsl Alm, 38.8 ± 1.5%, P = 0.0006, Fig. 6Ab and d). On the contrary, in the Veh group, resting behaviour was not different in the Bsl Veh and Inj Veh trials (mean 5 h resting time, ZT 18–23: Inj Veh, 33.2 ± 2.9%, Bsl Veh: 39.8 ± 3.7%, P = 0.28, Fig. 6Ab and d), confirming the sedative action induced by Alm but not Veh injections. When recording from CA3 cells in slices prepared at ZT 23 from Alm-injected animals, we found that bath-applied Ox-A (100 nm) suppressed NMDA-currents to 87.2 ± 3.3% of baseline (n = 11, P = 0.002, Fig. 6Ba). Conversely, exogenous Ox-A (100 nm) did not affect the amplitude of NMDA-currents in the Veh-injected group (97.4 ± 3.1%, n = 8, P = 0.53, Fig. 6Bb), in agreement with the results from undisturbed animals killed at the same ZT.
Figure 6. Almorexant administration restores the actions of exogenous Ox-A on NMDARs.
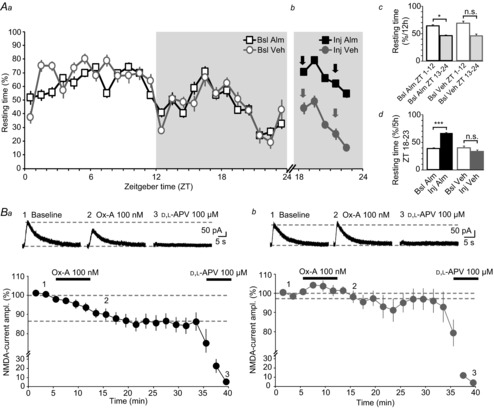
Aa, distribution of resting time of the Veh and Alm groups of rats across the LD cycle, during the Bsl Alm (squares) and Bsl Veh (circles) trials. Resting time was quantified as %time h−1 below a visually set threshold of locomotor activity (see Methods). Rats rested more during the light phase (ZT 1–12, unshaded portion of the graph) compared to the dark phase (ZT 13–24, shaded) (Alm group, n = 8; Veh group, n = 6). Ab, mean resting time of the Alm and Veh groups over ZT 18–23 of the Inj trials, during which two i.p. injections of Alm or Veh were administered at ZT 18 and ZT 21 (arrows). The Alm group rested more than in the corresponding 5 h period in the preceding Bsl Alm trial, while resting time of the Veh group was unaltered. Ac and d, summarising bar graphs of mean resting times for the 12 h light and 12 h dark periods (Ac) and for the 5 h dark periods after Alm or Veh injection, respectively (Ad). In the Veh group, all but one animal showed a decreased resting time in the dark compared to the light phase of the Bsl trial, rendering P non-significant (0.077) for the right column pair in Ac. Exclusion of this animal would decrease P to 0.008. The animal was kept for further analysis as it showed regular behaviour in the subsequent Inj trial. Ba and b, Ox-A actions on iontophoretic NMDA-currents in slices prepared at ZT 23 from the Alm (Ba) and Veh (Bb) groups. The actions of bath-applied Ox-A were recovered in the Alm group (n = 11), but not in the Veh group (n = 8). Horizontal lines represent 100% and mean Ox-A-induced suppression. Data are presented as mean ± SEM. n.s., P > 0.05; *P < 0.05; ***P < 0.001.
Therefore, by inhibiting the orexinergic system through the injection of Alm, we prevented the build-up of endogenous orexin's suppressant action on NMDARs during this waking period, leaving NMDARs available for modulation by exogenous orexin.
Discussion
We have identified the NMDARs as a target for orexin at two separate hippocampal excitatory synapses. This finding adds a novel facet to the repertoire of orexin's actions: in addition to its widely demonstrated predominant excitatory effects (Leonard & Kukkonen, 2014), the peptide is inhibitory for synaptic NMDAR-mediated transmission in a brain area central for mnemonic functions. Orexin-mediated inhibition is most strongly expressed and direct at MFs, moderate at SCs, and is absent at non-MF synapses in the CA3 area, indicating a synaptic specificity of Ox-A actions not only amongst pyramidal cell types, but also within subsets of synapses established on the dendritic arbor of the same neurone. We further found that endogenous orexins regulate NMDAR-mediated responses according to their daily fluctuations, perhaps contributing to a diurnal variation of NMDAR function in the hippocampus. Therefore, our study has implications for the consequences of waking on hippocampal excitatory communication, and, consequently, for diurnal variability in memory performance.
The identification of a hippocampally targeted action of orexin on excitatory synapses complements expression studies describing structural and molecular substrates for orexinergic signalling in the hippocampus. Orexinergic fibres enter the hippocampus via a dorsal ascending pathway (Peyron et al. 1998) and innervate both cellular and dendritic strata across hippocampal subfields, including the hilus, s. lucidum in CA3 (Morales et al. 2006) and s. radiatum in CA1 (Selbach et al. 2004). Using Ox-A- and Ox-B-specific antibodies, we confirmed a hippocampus-wide invasion by orexinergic fibres, in particular in dendritic synapse-rich regions in both CA3 and CA1 of young adult rat. Moreover, mRNA for OX1Rs locates to the CA1 area, whereas OX2R mRNA is predominantly found in the CA3 area (Marcus et al. 2001), consistent with our identification of OX2Rs as mediators of Ox-A actions. In addition to pyramidal cells (Hervieu et al. 2001; Cluderay et al. 2002), OXRs are also present on hilar and s. lucidum interneurones (Hervieu et al. 2001; Ito et al. 2008), possibly representing additional, yet unexplored, sites of action for the endogenous peptides within hippocampal circuits.
Aside from several studies suggesting a role of orexins in hippocampus-dependent memory formation (Jaeger et al. 2002; Aou et al. 2003; Yang et al. 2013), evidence for direct orexinergic actions on hippocampal circuitry is scarce (Wayner et al. 2004; Akbari et al. 2006, 2007). Several studies indicate that cholinergic and monoaminergic nuclei, both heavily innervated by orexinergic fibres, regulate hippocampal activity in response to orexinergic excitation (Walling et al. 2004; Doreulee et al. 2009; Stanley & Fadel, 2012). The present study used an isolated hippocampal slice preparation from young adult animals and identified a suppression of NMDAR-mediated synaptic transmission that developed over minutes and recovered only partially, or not at all, even after prolonged wash-out. The effect of Ox-A could be long-lasting (see Wang et al. 2014), but a poor wash-out of the peptide once it had deeply penetrated into the slice and reached the recorded neurone could also prevent the return to baseline responses. Additionally, OX1Rs bound to Ox-A can maintain G-protein-mediated signalling despite being internalised (Milasta et al. 2005; Xu et al. 2012). The suppression was largely preserved in the presence of a cocktail of neurotransmitters, found in previous reports to cooperate with Ox-A in modulating hippocampal excitatory synapses (Selbach et al. 2004; Doreulee et al. 2009). Also, our experiments were carried out in the continuous presence of GABAA and GABAB receptor blockers to exclude an influence of GABA release from OXR-expressing interneurones on MF–CA3 circuitry. NMDA-EPSC inhibition was thus mediated, to a major extent, by a direct action of Ox-A on hippocampal NMDARs.
The effect of Ox-A was observed only on the NMDA- and not on the AMPA-component of the MF-EPSCs, arguing in favour of a postsynaptically delimited Ox-A action. Consistent with this finding, LFF, a sensitive measure of presynaptic function that reports on cumulative increases in presynaptic residual Ca2+ levels (Salin et al. 1996), was unaffected in the presence of Ox-A. Finally, Ox-A suppressed NMDA-currents elicited through direct NMDA application specifically on dendritic sites where MF terminals arrive. Altogether, although a possible expression of OX2Rs at presynaptic sites still needs to be definitely excluded, our combined results indicate that Ox-A-induced suppression of MF-NMDA-EPSCs is a predominantly postsynaptic process. In the in vivo situation, however, the possibility remains that orexinergic fibres act presynaptically on MF synapses through dynorphin and glutamate, well-described co-transmitters at orexinergic terminals (Chou et al. 2001; Schöne et al. 2012), both of which modulate glutamate release from MF terminals (Weisskopf et al. 1993; Nicoll & Schmitz, 2005).
Inhibitory effects of Ox-A were found at MF synapses, but not at non-MF synapses. Placing the stimulation electrode in the granule cell layer allowed us to record almost pure MF responses in the CA3 pyramidal neurone, as indicated by their near-total block by DCG-IV. While MFs form giant terminals at predominantly proximal dendritic portions in s. lucidum, non-MF inputs impinge on more distal portions of CA3 apical dendrites, in s. radiatum (Claiborne et al. 1986). These findings thus suggest that Ox-A acts in a dendritically compartmentalised manner, modulating NMDARs at the proximally located MF synapses, while sparing those at non-MF synapses. Similar spatially delimited effects were observed upon iontophoretic NMDA application to 100 μm-portions of the dendrite: only proximally, but not distally positioned NMDA pulses produced NMDA-currents susceptible to Ox-A. The MF-delimited action could be explained by a selective localisation of OX2Rs to s. lucidum but not to s. radiatum, a possibility to be further tested with immunohistochemistry. Alternatively, molecular characteristics of NMDARs or localised signalling cascades could play a role. For example, lamination of expression patterns has been described for NMDAR subunits, with GluN2A dominating in s. lucidum and GluN2B in s. radiatum (Watanabe et al. 1998). A functional compartmentalisation to s. lucidum exists for protein kinase C (PKC)-mediated stimulatory actions on NMDARs (Kwon & Castillo, 2008a) and for synaptic mGluR-mediated activation of a slow cationic conductance (Heuss et al. 1999).
MF-NMDARs were long considered of minor relevance for normal MF-mediated synaptic transmission, in part due to their low expression level compared to AMPARs (Fritschy et al. 1998; Watanabe et al. 1998), and due to the presence of marked NMDAR-independent forms of long-term plasticity (Nicoll & Schmitz, 2005). Overcoming this traditional view, several recent studies now indicate that MF-NMDARs contribute importantly to CA3 pyramidal cell excitability and plasticity (Tsukamoto et al. 2003; Kwon & Castillo, 2008a; Rebola et al. 2008, 2011; Astori et al. 2010; Hunt et al. 2013). Electron microscopy revealed that CA3 spines receiving MF boutons express the NMDAR-modulating adenosinergic A2A and mGluR group I receptors in close proximity to the synapse (Rebola et al. 2008; Hunt et al. 2013). The OX2R-mediated downregulation of NMDARs thus suggests a dedicated arrangement of G-protein-coupled receptors on CA3 spines to up- or downregulate an NMDAR pool with important roles in basal and activity-dependent modification of MF function.
GluN2A-containing but not GluN2B-containing NMDARs were detected at MF synapses (Fritschy et al. 1998; Watanabe et al. 1998) and are therefore the likely final targets of OX2R activation. Consistent with a possible removal of GluN2A from the synapse are the observed slow-down of the NMDA-EPSC decay time course in the presence of Ox-A, the slow time course of Ox-A actions, and the poor recovery. In the ventral tegmental area, however, OX2Rs up- rather than downregulate GluN2A-containing NMDARs through a mechanism involving PKC (Borgland et al. 2006). Why OX2Rs do not couple to PKC, known to be present at MFs and to be recruited during LTP of NMDA-EPSCs (Kwon & Castillo, 2008a), is unclear. Interesting in this context is that mice have two OX2R variants that, upon heterodimerisation, produce receptors with novel signalling properties (Wang et al. 2014). Therefore, additional signalling pathways, such as OX2R-mediated activation of extracellular mitogen-activating protein kinases, extracellular signal-regulated kinases, coupling to Gi/o-proteins and subsequent receptor modification need to be considered (Zhu et al. 2003; Selbach et al. 2010; Guo & Feng, 2012; Wang et al. 2014). The considerable number of possible pathways involved, together with the comparatively small size of the synaptic response in Ox-A and the consequent difficulty in reliably assessing time courses, as well as the reduced effect of Ox-A on iontophoretic currents, leave an assessment of the mechanistic bases of Ox-A-mediated modulation open for future investigation.
Surprisingly, Ox-A actions were not observed in slices prepared from rats at the end of their active period. Instead, several hours of rest were required to observe a suppression of NMDA-EPSCs by the peptide. Furthermore, treating animals with the dual orexin receptor antagonist Alm augmented their resting time during the late dark phase and partially restored the effect for exogenous Ox-A. This supports the idea that the endogenous orexin level in hippocampus shows diurnal fluctuations (Yoshida et al. 2001) and is involved in a time-of-day dependence of Ox-A-induced NMDAR regulation. The persistence of endogenous orexinergic actions in the slice is not due to the continued presence of a high orexinergic tone in the slice, but probaby to maintained effects at the OX2R or the NMDAR level. Thus, high orexinergic levels during waking could downregulate OX2Rs, rendering slices prepared then unresponsive to exogenous Ox-A. Additionally, NMDAR function could already be downregulated, occluding further Ox-A actions. Our work is the first to demonstrate that acute slices preserve some of the functional consequences of endogenous orexinergic levels present at the moment of brain dissection. However, alternative possibilities remain. For example, a circadian fluctuation in the molecular constitution of NMDARs (Ishida et al. 1994) could explain diurnal insensitivity to Ox-A. Furthermore, although unlikely based on our in vitro results, potential secondary contributions from afferent modulatory systems regulated by orexin cannot be excluded. Thus, the Alm Inj group showed an almost doubled resting time compared to the Veh Inj group in the same 5 h of the dark phase, probably accompanied by an overall decrease in the activity of orexin-driven wake-promoting systems that could, in turn, modulate responsiveness to Ox-A. Hippocampus-selective manipulation of orexinergic activity is needed to distinguish amongst these possibilities.
Time-of-day-dependent modification of properties at excitatory synapses has been previously reported for the amplitude of the evoked field potential in rodent DG (Barnes et al. 1977), for transcallosal synaptic strength in rat cortex (Vyazovskiy et al. 2008), for the constitution of AMPARs at synapses of layer 5 somatosensory cortex pyramidal neurones (Lanté et al. 2011) and for the expression of synaptic adhesion molecules (El Helou et al. 2013). The present work is the first to suggest that the wake-promoting compound Ox-A, accumulating during the active phase, could be directly involved in such modulation. Interestingly, diurnal fluctuations of Ox-A in hippocampus drive a rhythmic increase in amyloid-β peptide in hippocampus, a pathogenic factor of Alzheimer's disease (Kang et al. 2009). Together with our results, this suggests that prolonged wakefulness and associated elevation of orexin deteriorates hippocampal function and imposes a risk for pathology in the long term. This is not only in line with the fact that circadian time of day affects cognitive performance (Goel et al. 2013), but also that sleep deprivation compromises hippocampal synaptic function (Kopp et al. 2006) and can trigger neurodegeneration (Cirelli, 2006). In addition, hippocampal neuronal degeneration due to sleep deprivation-induced epileptical seizures is amplified by the action of orexins (Ni et al. 2014).
To conclude, our study provides mechanistic insights into how the waking state modifies synapses, bringing to attention the consequences of elevated orexin levels on molecular processes important for hippocampal learning. The wake-promoting neurochemical environment could eventually limit the susceptibility of hippocampal circuits to undergo plastic strengthening. We propose that such insights should be considered in further discussions of general concepts on how waking and sleeping drive changes in synaptic function (Tononi & Cirelli, 2014).
Acknowledgments
We thank Drs Mehdi Tafti and A. Vassalli for providing us with almorexant, and Drs Corrado Corti and Mauro Corsi for making the JNJ10397049 available to us. M.P. is indebted to Drs P. Castillo and P. Haeger for teaching her the hippocampal slice preparation for mossy fibre recordings in rat, and to Dr C. Mulle for introducing her to the mouse slice preparation. Dr Castillo additionally provided invaluable advice towards the final version of the manuscript. We thank Drs U. Gerber and C. Peyron for helpful discussions in the course of the study, and Drs S. Astori, L. Fernandez, S. Lecci and C. Pellegrini for critical comments on the manuscript.
Glossary
- Alm
almorexant
- d,l-APV
d,l-2-amino-5-phosphonopentanoic acid
- DG
dentate gyrus
- LD
light/dark
- LFF
low-frequency facilitation
- LTP
long-term potentiation
- MF
mossy fibre
- mGluR
metabotropic glutamate receptor
- NMDAR
NMDA receptor
- PKC
protein kinase-C
- Ox-A
orexin-A
- Ox-B
orexin-B
- OX1R
orexin-1 receptor
- OX2R
orexin-2 receptor
- Ri
input resistance
- Rs
series resistance
- RT
room temperature
- s.
stratum
- SC
Schaffer collateral
- TBS
Tris-buffered saline
- Veh
vehicle
- ZT
Zeitgeber time
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
M.P. carried out all experiments and analyses and contributed to writing the paper; F.L. contributed to the first experiments; C.M. and E.W. helped with immunohistochemistry and writing; A.L. helped with analysis, supervised the project and wrote the paper. All authors read and approved the final version of the manuscript.
Funding
This work was funded by the Swiss National Science Foundation (Grant Nos 129810 and 146244) and by Etat de Vaud.
Author's present address
F. Longordo: Eccles Institute of Neuroscience, John Curtin School of Medical Research, Australian National University, GPO Box 334, Canberra City, ACT 2600, Australia.
References
- Akbari E, Naghdi N, Motamedi F. Functional inactivation of orexin 1 receptors in CA1 region impairs acquisition, consolidation and retrieval in Morris water maze task. Behav Brain Res. 2006;173:47–52. doi: 10.1016/j.bbr.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Akbari E, Naghdi N, Motamedi F. The selective orexin 1 receptor antagonist SB-334867-A impairs acquisition and consolidation but not retrieval of spatial memory in Morris water maze. Peptides. 2007;28:650–656. doi: 10.1016/j.peptides.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Aou S, Li X-L, Li A-J, Oomura Y, Shiraishi T, Sasaki K, Imamura T, Wayner MJ. Orexin-A (hypocretin-1) impairs Morris water maze performance and CA1-Schaffer collateral long-term potentiation in rats. Neuroscience. 2003;119:1221–1228. doi: 10.1016/s0306-4522(02)00745-5. [DOI] [PubMed] [Google Scholar]
- Astori S, Pawlak V, Köhr G. Spike-timing-dependent plasticity in hippocampal CA3 neurons. J Physiol. 2010;588:4475–4488. doi: 10.1113/jphysiol.2010.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA, McNaughton BL, Goddard GV, Douglas RM, Adamec R. Circadian rhythm of synaptic excitability in rat and monkey central nervous system. Science. 1977;197:91–92. doi: 10.1126/science.194313. [DOI] [PubMed] [Google Scholar]
- Bayer L, Eggermann E, Saint-Mleux B, Machard D, Jones BE, Mühlethaler M, Serafin M. Selective action of orexin (hypocretin) on nonspecific thalamocortical projection neurons. J Neurosci. 2002;22:7835–7839. doi: 10.1523/JNEUROSCI.22-18-07835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benquet P, Gee CE, Gerber U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J Neurosci. 2002;22:9679–9686. doi: 10.1523/JNEUROSCI.22-22-09679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc. 2006;1:2075–2081. doi: 10.1038/nprot.2006.312. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Storm E, Bonci A. Orexin B/hypocretin 2 increases glutamatergic transmission to ventral tegmental area neurons. Eur J Neurosci. 2008;28:1545–1556. doi: 10.1111/j.1460-9568.2008.06397.x. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Boschen KE, Fadel JR, Burk JA. Systemic and intrabasalis administration of the orexin-1 receptor antagonist, SB-334867, disrupts attentional performance in rats. Psychopharmacology (Berl) 2009;206:205–213. doi: 10.1007/s00213-009-1596-2. [DOI] [PubMed] [Google Scholar]
- Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S, Mueller C, Nayler O, van Gerven J, de Haas SL, Hess P, Qiu C, Buchmann S, Scherz M, Weller T, Fischli W, Clozel M, Jenck F. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13:150–155. doi: 10.1038/nm1544. [DOI] [PubMed] [Google Scholar]
- Brown RE, Sergeeva O, Eriksson KS, Haas HL. Orexin A excites serotonergic neurons in the dorsal raphe nucleus of the rat. Neuropharmacology. 2001;40:457–459. doi: 10.1016/s0028-3908(00)00178-7. [DOI] [PubMed] [Google Scholar]
- Burlet S, Tyler CJ, Leonard CS. Direct and indirect excitation of laterodorsal tegmental neurons by hypocretin/orexin peptides: implications for wakefulness and narcolepsy. J Neurosci. 2002;22:2862–2872. doi: 10.1523/JNEUROSCI.22-07-02862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Randeva HS. Genomic organization of mouse orexin receptors: characterization of two novel tissue-specific splice variants. Mol Endocrinol. 2004;18:2790–2804. doi: 10.1210/me.2004-0167. [DOI] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C. Cellular consequences of sleep deprivation in the brain. Sleep Med Rev. 2006;10:307–321. doi: 10.1016/j.smrv.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Claiborne BJ, Amaral DG, Cowan WM. A light and electron microscopic analysis of the mossy fibers of the rat dentate gyrus. J Comp Neurol. 1986;246:435–458. doi: 10.1002/cne.902460403. [DOI] [PubMed] [Google Scholar]
- Cluderay JE, Harrison DC, Hervieu GJ. Protein distribution of the orexin-2 receptor in the rat central nervous system. Regul Pept. 2002;104:131–144. doi: 10.1016/s0167-0115(01)00357-3. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Porrino L, Siegel JM, Hampson RE. Systemic and nasal delivery of orexin-A (hypocretin-1) reduces the effects of sleep deprivation on cognitive performance in nonhuman primates. J Neurosci. 2007;27:14239–14247. doi: 10.1523/JNEUROSCI.3878-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreulee N, Alania M, Chikovani M, Chkhartishvili B, Skhirtladze C. Orexin-A induces long-term depression of nmda responses in CA-1 field of hippocampal slices. Georgian Med News. 2009:65–70. [PubMed] [Google Scholar]
- Dugovic C, Shelton JE, Aluisio LE, Fraser IC, Jiang X, Sutton SW, Bonaventure P, Yun S, Li X, Lord B, Dvorak CA, Carruthers NI, Lovenberg TW. Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J Pharmacol Exp Ther. 2009;330:142–151. doi: 10.1124/jpet.109.152009. [DOI] [PubMed] [Google Scholar]
- El Helou J, Bélanger-Nelson E, Freyburger M, Dorsaz S, Curie T, La Spada F, Gaudreault P-O, Beaumont É, Pouliot P, Lesage F, Frank MG, Franken P, Mongrain V. Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci U S A. 2013;110:9974–9979. doi: 10.1073/pnas.1221381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9279. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evstratova A, Tóth K. Information processing and synaptic plasticity at hippocampal mossy fiber terminals. Front Cell Neurosci. 2014;8:28. doi: 10.3389/fncel.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel J, Deutch AY. Anatomical substrates of orexin-dopamine interactions: lateral hypothalamic projections to the ventral tegmental area. Neuroscience. 2002;111:379–387. doi: 10.1016/s0306-4522(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Franken P, Tobler I, Borbély AA. Sleep homeostasis in the rat: simulation of the time course of EEG slow-wave activity. Neurosci Lett. 1991;130:141–144. doi: 10.1016/0304-3940(91)90382-4. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Weinmann O, Wenzel A, Benke D. Synapse-specific localization of NMDA and GABAA receptor subunits revealed by antigen-retrieval immunohistochemistry. J Comp Neurol. 1998;390:194–210. [PubMed] [Google Scholar]
- Goel N, Basner M, Rao H, Dinges DF. Circadian rhythms, sleep deprivation, and human performance. Prog Mol Biol Transl Sci. 2013;119:155–190. doi: 10.1016/B978-0-12-396971-2.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzi A, Turrini G, Piccoli L, Massagrande M, Amantini D, Antolini M, Martinelli P, Cesari N, Montanari D, Tessari M, Corsi M, Bifone A. Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PLoS One. 2011;6:e16406. doi: 10.1371/journal.pone.0016406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Feng P. OX2R activation induces PKC-mediated ERK and CREB phosphorylation. Exp Cell Res. 2012;318:2004–2013. doi: 10.1016/j.yexcr.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervieu GJ, Cluderay JE, Harrison DC, Roberts JC, Leslie RA. Gene expression and protein distribution of the orexin-1 receptor in the rat brain and spinal cord. Neuroscience. 2001;103:777–797. doi: 10.1016/s0306-4522(01)00033-1. [DOI] [PubMed] [Google Scholar]
- Heuss C, Scanziani M, Gähwiler BH, Gerber U. G-protein-independent signaling mediated by metabotropic glutamate receptors. Nat Neurosci. 1999;2:1070–1077. doi: 10.1038/15996. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Peyron C, Diano S, Ivanov A, Aston-Jones G, Kilduff TS, Van den Pol AN. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comp Neurol. 1999;415:145–159. [PubMed] [Google Scholar]
- Hunt DL, Puente N, Grandes P, Castillo PE. Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat Neurosci. 2013;16:1049–1059. doi: 10.1038/nn.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N, Matsui M, Mitsui Y, Mishina M. Circadian expression of NMDA receptor mRNAs, ε3 and ζ1, in the suprachiasmatic nucleus of rat brain. Neurosci Lett. 1994;166:211–215. doi: 10.1016/0304-3940(94)90488-x. [DOI] [PubMed] [Google Scholar]
- Ito N, Yabe T, Gamo Y, Nagai T, Oikawa T, Yamada H, Hanawa T. I.c.v. administration of orexin-A induces an antidepressive-like effect through hippocampal cell proliferation. Neuroscience. 2008;157:720–732. doi: 10.1016/j.neuroscience.2008.09.042. [DOI] [PubMed] [Google Scholar]
- Jaeger LB, Farr SA, Banks WA, Morley JE. Effects of orexin-A on memory processing. Peptides. 2002;23:1683–1688. doi: 10.1016/s0196-9781(02)00110-9. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp C, Longordo F, Nicholson JR, Lüthi A. Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci. 2006;26:12456–12465. doi: 10.1523/JNEUROSCI.2702-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotani T, Yamada K, Yoshimura Y, Crair MC, Komatsu Y. State-dependent bidirectional modification of somatic inhibition in neocortical pyramidal cells. Neuron. 2008;57:905–916. doi: 10.1016/j.neuron.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008a;57:108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Role of glutamate autoreceptors at hippocampal mossy fiber synapses. Neuron. 2008b;60:1082–1094. doi: 10.1016/j.neuron.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe EK, Aghajanian GK. Hypocretin (orexin) induces calcium transients in single spines postsynaptic to identified thalamocortical boutons in prefrontal slice. Neuron. 2003;40:139–150. doi: 10.1016/s0896-6273(03)00598-1. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. J Neurosci. 2005;25:5225–5229. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanté F, Toledo-Salas J-C, Ondrejcak T, Rowan MJ, Ulrich D. Removal of synaptic Ca2+-permeable AMPA receptors during sleep. J Neurosci. 2011;31:3953–3961. doi: 10.1523/JNEUROSCI.3210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CS, Kukkonen JP. Orexin/hypocretin receptor signalling: a functional perspective. Br J Pharmacol. 2014;171:294–313. doi: 10.1111/bph.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chen F, Ye J, Chen X, Yan J, Li Y, Xiong Y, Zhou Z, Xia J, Hu Z. The modulation of orexin-A on HCN currents of pyramidal neurons in mouse prelimbic cortex. Cereb Cortex. 2010;20:1756–1767. doi: 10.1093/cercor/bhp241. [DOI] [PubMed] [Google Scholar]
- Li J, Hu Z, de Lecea L. The hypocretins/orexins: integrators of multiple physiological functions. Br J Pharmacol. 2013;171:332–350. doi: 10.1111/bph.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao X-B, Sakurai T, van den Pol AN. Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron – a potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci. 2002;22:9453–9464. doi: 10.1523/JNEUROSCI.22-21-09453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longordo F, Kopp C, Mishina M, Luján R, Lüthi A. NR2A at CA1 synapses is obligatory for the susceptibility of hippocampal plasticity to sleep loss. J Neurosci. 2009;29:9026–9041. doi: 10.1523/JNEUROSCI.1215-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair RG, Hembrook JR. Memory enhancement with event-related stimulation of the rostral intralaminar thalamic nuclei. J Neurosci. 2008;28:14293–14300. doi: 10.1523/JNEUROSCI.3301-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang GM, Dürst T, Bürki H, Imobersteg S, Abramowski D, Schuepbach E, Hoyer D, Fendt M, Gee CE. The dual orexin receptor antagonist almorexant induces sleep and decreases orexin-induced locomotion by blocking orexin 2 receptors. Sleep. 2012;35:1625–1635. doi: 10.5665/sleep.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- Milasta S, Evans NA, Ormiston L, Wilson S, Lefkowitz RJ, Milligan G. The sustainability of interactions between the orexin-1 receptor and β-arrestin-2 is defined by a single C-terminal cluster of hydroxy amino acids and modulates the kinetics of ERK MAPK regulation. Biochem J. 2005;387:573–584. doi: 10.1042/BJ20041745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Nicoll RA, Schmitz D. Adenosine gates synaptic plasticity at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2003;100:14397–14402. doi: 10.1073/pnas.1835831100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morairty SR, Revel FG, Malherbe P, Moreau JL, Valladao D, Wettstein JG, Kilduff TS, Borroni E. Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS One. 2012;7:e39131. doi: 10.1371/journal.pone.0039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales A, Bonnet C, Bourgoin N, Touvier T, Nadam J, Laglaine A, Navarro F, Moulin C, Georges B, Pequignot JM, Bezin L. Unexpected expression of orexin-B in basal conditions and increased levels in the adult rat hippocampus during pilocarpine-induced epileptogenesis. Brain Res. 2006;1109:164–175. doi: 10.1016/j.brainres.2006.06.075. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Ni LY, Zhu MJ, Song Y, Liu XM, Tang JY. Pentylenetetrazol-induced seizures are exacerbated by sleep deprivation through orexin receptor-mediated hippocampal cell proliferation. Neurol Sci. 2014;35:245–252. doi: 10.1007/s10072-013-1495-5. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Carta M, Lanore F, Blanchet C, Mulle C. NMDA receptor-dependent metaplasticity at hippocampal mossy fiber synapses. Nat Neurosci. 2011;14:691–693. doi: 10.1038/nn.2809. [DOI] [PubMed] [Google Scholar]
- Rebola N, Luján R, Cunha RA, Mulle C. Adenosine A2a receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu W-S, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöne C, Cao ZF, Apergis-Schoute J, Adamantidis A, Sakurai T, Burdakov D. Optogenetic probing of fast glutamatergic transmission from hypocretin/orexin to histamine neurons in situ. J Neurosci. 2012;32:12437–12443. doi: 10.1523/JNEUROSCI.0706-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears RM, Fink AE, Wigestrand MB, Farb CR, de Lecea L, Ledoux JE. Orexin/hypocretin system modulates amygdala-dependent threat learning through the locus coeruleus. Proc Natl Acad Sci U S A. 2013;110:20260–20265. doi: 10.1073/pnas.1320325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach O, Bohla C, Barbara A, Doreulee N, Eriksson KS, Sergeeva OA, Haas HL. Orexins/hypocretins control bistability of hippocampal long-term synaptic plasticity through co-activation of multiple kinases. Acta Physiol (Oxf) 2010;198:277–285. doi: 10.1111/j.1748-1716.2009.02021.x. [DOI] [PubMed] [Google Scholar]
- Selbach O, Doreulee N, Bohla C, Eriksson KS, Sergeeva OA, Poelchen W, Brown RE, Haas HL. Orexins/hypocretins cause sharp wave- and θ-related synaptic plasticity in the hippocampus via glutamatergic, gabaergic, noradrenergic, and cholinergic signaling. Neuroscience. 2004;127:519–528. doi: 10.1016/j.neuroscience.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Soya S, Shoji H, Hasegawa E, Hondo M, Miyakawa T, Yanagisawa M, Mieda M, Sakurai T. Orexin receptor-1 in the locus coeruleus plays an important role in cue-dependent fear memory consolidation. J Neurosci. 2013;33:14549–14557. doi: 10.1523/JNEUROSCI.1130-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EM, Fadel J. Aging-related deficits in orexin/hypocretin modulation of the septo-hippocampal cholinergic system. Synapse. 2012;66:445–452. doi: 10.1002/syn.21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto M, Yasui T, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Mossy fibre synaptic NMDA receptors trigger non-Hebbian long-term potentiation at entorhino–CA3 synapses in the rat. J Physiol. 2003;546:665–675. doi: 10.1113/jphysiol.2002.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. J Neurosci. 1999;19:3171–3182. doi: 10.1523/JNEUROSCI.19-08-03171.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB. Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J Neurosci. 1998;18:7962–7971. doi: 10.1523/JNEUROSCI.18-19-07962.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Walling SG, Nutt DJ, Lalies MD, Harley CW. Orexin-A infusion in the locus ceruleus triggers norepinephrine (NE) release and NE-induced long-term potentiation in the dentate gyrus. J Neurosci. 2004;24:7421–7426. doi: 10.1523/JNEUROSCI.1587-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Pan Y, Zhang R, Bai B, Chen J, Randeva HS. Heterodimerization of mouse orexin type 2 receptor variants and the effects on signal transduction. Biochim Biophys Acta. 2014;1843:652–663. doi: 10.1016/j.bbamcr.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y. Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci. 1998;10:478–487. doi: 10.1046/j.1460-9568.1998.00063.x. [DOI] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Phelix CF, Oomura Y. Orexin-A (hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides. 2004;25:991–996. doi: 10.1016/j.peptides.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Zalutsky RA, Nicoll RA. The opioid peptide dynorphin mediates heterosynaptic depression of hippocampal mossy fibre synapses and modulates long-term potentiation. Nature. 1993;362:423–427. doi: 10.1038/362423a0. [DOI] [PubMed] [Google Scholar]
- Xu TR, Ward RJ, Pediani JD, Milligan G. Intramolecular fluorescence resonance energy transfer (FRET) sensors of the orexin OX1 and OX2 receptors identify slow kinetics of agonist activation. J Biol Chem. 2012;287:14937–14949. doi: 10.1074/jbc.M111.334300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zou B, Xiong X, Pascual C, Xie J, Malik A, Sakurai T, Xie XS. Hypocretin/orexin neurons contribute to hippocampus-dependent social memory and synaptic plasticity in mice. J Neurosci. 2013;33:5275–5284. doi: 10.1523/JNEUROSCI.3200-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, Mignot E, Nishino S. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–1081. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kolaj M, Renaud LP. Ca2+-dependent and Na+-dependent K+ conductances contribute to a slow AHP in thalamic paraventricular nucleus neurons: a novel target for orexin receptors. J Neurophysiol. 2010;104:2052–2062. doi: 10.1152/jn.00320.2010. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zeitzer JM, Yoshida Y, Wisor JP, Nishino S, Edgar DM, Mignot E. Lesions of the suprachiasmatic nucleus eliminate the daily rhythm of hypocretin-1 release. Sleep. 2004;27:619–627. doi: 10.1093/sleep/27.4.619. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Miwa Y, Yamanaka A, Yada T, Shibahara M, Abe Y, Sakurai T, Goto K. Orexin receptor type-1 couples exclusively to pertussis toxin-insensitive G-proteins, while orexin receptor type-2 couples to both pertussis toxin-sensitive and -insensitive G-proteins. J Pharmacol Sci. 2003;92:259–266. doi: 10.1254/jphs.92.259. [DOI] [PubMed] [Google Scholar]