

Abstract
Sudden unexpected death in epilepsy (SUDEP) is the leading cause of death in patients with refractory epilepsy. Defects in central control of breathing are important contributors to the pathophysiology of SUDEP, and serotonin (5-HT) system dysfunction may be involved. Here we examined the effect of 5-HT neurone elimination or 5-HT reduction on seizure risk and seizure-induced mortality. Adult Lmx1bf/f/p mice, which lack >99% of 5-HT neurones in the CNS, and littermate controls (Lmx1bf/f) were subjected to acute seizure induction by maximal electroshock (MES) or pilocarpine, variably including electroencephalography, electrocardiography, plethysmography, mechanical ventilation or pharmacological therapy. Lmx1bf/f/p mice had a lower seizure threshold and increased seizure-induced mortality. Breathing ceased during most seizures without recovery, whereas cardiac activity persisted for up to 9 min before terminal arrest. The mortality rate of mice of both genotypes was reduced by mechanical ventilation during the seizure or 5-HT2A receptor agonist pretreatment. The selective serotonin reuptake inhibitor citalopram reduced mortality of Lmx1bf/f but not of Lmx1bf/f/p mice. In C57BL/6N mice, reduction of 5-HT synthesis with para-chlorophenylalanine increased MES-induced seizure severity but not mortality. We conclude that 5-HT neurones raise seizure threshold and decrease seizure-related mortality. Death ensued from respiratory failure, followed by terminal asystole. Given that SUDEP often occurs in association with generalised seizures, some mechanisms causing death in our model might be shared with those leading to SUDEP. This model may help determine the relationship between seizures, 5-HT system dysfunction, breathing and death, which may lead to novel ways to prevent SUDEP.
Key points
Sudden unexpected death in epilepsy is the leading cause of death in patients with refractory epilepsy.
Respiratory and cardiac impairment induced by a seizure have been identified as possible causes of seizure-related death, but which is more important has been the subject of debate.
Serotonin has been linked to seizure control, but whether it is primarily anti-convulsant or proconvulsant remains controversial.
In this study we induced seizures in mice with a genetic deletion of serotonin neurones and their phenotypically normal littermates while recording EEG, EMG, ECG and breathing, and assessed the effects of seizures on breathing, cardiac activity and survival
Serotonin and serotonin neurones are involved in setting the seizure threshold, regulating seizure severity and preventing mortality, and death in at least one seizure model is due to respiratory arrest, which can be prevented with selective serotonin reuptake inhibitor treatment or 5-HT2A receptor activation.
Introduction
Epilepsy affects millions of individuals world-wide (Banerjee 2009). Patients, families and many clinicians have recently begun to appreciate that patients with epilepsy have a greater chance of sudden death. Sudden unexpected death in epilepsy (SUDEP) is the leading cause of death for patients with refractory epilepsy, with estimates of cumulative life-time risk ranging from 10 to 50% (Lhatoo & Sander, 2005; Tomson et al. 2005). Understanding how seizures can result in death is therefore of critical importance.
Respiratory, cardiac and autonomic dysfunction, as well as primary electrocerebral shutdown, have been proposed as causes of SUDEP (Tupal & Faingold, 2006; Bateman et al. 2008; Goldman et al. 2009; Lhatoo et al. 2010; Sowers et al. 2013), but evidence is lacking to support any conclusions about whether any of these is the primary cause of death in the majority of SUDEP cases. Recently, there have been suggestions that SUDEP might be caused by dysfunction of the serotonin (5-HT) system. Evidence for this is purely circumstantial, and includes the following: (1) 5-HT plays an important role in breathing and arousal (Buchanan et al. 2008), two brainstem functions involved in SUDEP; (2) Htr2c knockout mice have a high incidence of SUDEP-like deaths (Brennan et al. 1997), and selective serotonin reuptake inhibitors (SSRIs) prevent SUDEP-like deaths in DBA/1 and DBA/2 mice (Tupal & Faingold, 2006; Faingold et al. 2011); (3) SSRIs decrease the desaturation that occurs after seizures in many patients, and which may be a biomarker for increased risk of SUDEP (Bateman et al. 2010a) and (4) there are many similarities between SUDEP and sudden infant death syndrome (SIDS; Richerson & Buchanan, 2011; Sowers et al. 2013), and SIDS is linked to a defect in the 5-HT system (Kinney et al. 2009). Many lines of evidence indicate that 5-HT can decrease the frequency and severity of seizures in animal models of epilepsy and in epilepsy patients (Bagdy et al. 2007). In addition, several commonly used anti-epileptic drugs increase extracellular 5-HT concentration, which may contribute to the mechanism of action of these agents (Okada et al. 1992; Dailey et al. 1997; Ahmad et al. 2005; Bagdy et al. 2007).
For a person who dies suddenly from a seizure to be classified as SUDEP, they must have epilepsy. However, there is no evidence that the pathophysiological events that cause death are different during a seizure in a person with epilepsy than they are in a person with a normal brain. Thus, understanding how seizures cause respiratory arrest and death in a non-epileptic brain may lend insight into how to combat SUDEP. Here we used two paradigms of acute seizure induction in Lmx1bf/f/p mice (Zhao et al. 2006) to better understand the relationship between seizures, 5-HT, breathing and death. We also examined the effect of subacute depletion of 5-HT with para-chlorophenylalanine (PCPA). In the absence of 5-HT neurones, seizure susceptibility and seizure-related mortality were both increased. Reduction of 5-HT with PCPA increased seizure susceptibility, but not mortality. The primary cause of death was respiratory failure, and providing respiratory support, blocking 5-HT transporters or activating 5-HT2A receptors prevented mortality.
Methods
Ethical approval
All procedures and protocols performed on Lmx1bf/f/p and WT mice were approved by the Institutional Animal Care and Use Committee at Yale University. PCPA depletion studies were performed at the University of Iowa in C57BL/6N mice obtained from Charles River (Wilmington, MA, USA). These studies were approved by the Institutional Animal Care and Use Committee at the University of Iowa. Animals that did not succumb during the experimental paradigm were killed with an overdose of sodium pentobarbital (150 mg kg−1, i.p.).
Experimental animals
Adult (26–40 g) Lmx1bf/f mice (which carry two floxed Lmx1b alleles, but are phenotypically normal; this allowed us to use littermates as controls) and mice homozygous for floxed Lmx1b and hemizygous for ePet1-Cre (Lmx1bf/f; e-Pet1-Cre/+ or Lmx1bf/f/p) were housed in standard cages in a 12 h light/12 h dark regimen with food and water available ad libitum. Generation, breeding and genotyping of Lmx1bf/f/p mice have been previously described (Zhao et al. 2006; Hodges et al. 2009). Briefly, Lmx1bf/f females were mated with Lmx1bf/f/p males to produce progeny of these two genotypes in a slightly less than 1:1 ratio (Hodges et al. 2009).
The phenotype of Lmx1bf/f/p mice has been described previously. These mice have decreased growth rate and prolonged apnoeas until postnatal day 12 (Hodges et al. 2009), and as adults have reduced baseline ventilation (Hodges et al. 2008), impaired hypercapnic ventilatory response (Hodges et al. 2008), impaired thermoregulation (Hodges et al. 2008), impaired arousal response to CO2 (Buchanan & Richerson, 2010), normal locomotor activity (Zhao et al. 2006), decreased sensitivity to mechanical stimuli (Zhao et al. 2007a), enhanced inflammatory pain response (Zhao et al. 2007a), impaired analgesic response to anti-depressant drugs (Zhao et al. 2007a), reduced opioid-mediated analgesia at the supraspinal level (Zhao et al. 2007b) and lack of sexual mating preference (Liu et al. 2011; Zhang et al. 2013). They are known to not display compensatory up-regulation in dopamine or noradrenaline within the CNS (Zhao et al. 2006) and they have normal blood, liver and intestinal 5-HT levels (Zhao et al. 2006) presumably due to normal peripheral 5-HT synthesis by the non-neuronal isoform of TPH1 (Walther et al. 2003).
Experimental protocols
Mice were subjected to one of the following experimental protocols.
Protocol 1
To determine the effect of 5-HT neurones on acute chemical seizure induction, 32 mice (8 Lmx1bf/f and 8 Lmx1bf/f/p mice of each sex) were implanted with EEG headmounts and subjected to seizure induction with graded doses of pilocarpine.
Protocol 2
To determine the effect of 5-HT neurones on the threshold for maximal electroshock (MES)-induced seizures, 24 non-instrumented mice (6 Lmx1bf/f and 6 Lmx1bf/f/p mice of each sex) were subjected to stimulation with increasing current intensities from 0 to 10 mA to determine the minimum current required to induce a seizure.
Protocol 3
To determine the effect of 5-HT neurones on MES-induced seizure severity and mortality, three separate sets of 24 non-instrumented mice (6 Lmx1bf/f and 6 Lmx1bf/f/p mice of each sex) were subjected to seizure induction at 10, 30 or 50 mA (a total of 72 mice).
Protocol 4
To determine the effect of acute depletion of 5-HT in C57BL/6N mice (with 5-HT neurones otherwise normal) on MES-induced seizure severity and mortality, C57BL/6N mice were treated with vehicle (n = 11) or PCPA methyl ester (n = 6) at a dose of 800 mg kg−1 i.p. once a day for 5 days prior to seizure induction with MES (50 mA). 5-HT levels in the blood, medulla and forebrain of C57BL/6N (n = 7), PCPA-treated (n = 6) and Lmx1bf/f/p (n = 7) mice were measured following MES and brain removal after the animals had been killed.
Protocol 5
To determine the physiological mechanism of death resulting from MES-induced seizures, 11 male Lmx1bf/f mice were implanted with EEG headmounts and ECG electrodes, and allowed to recover for 7–10 days. They were then subjected to MES-induced seizures within a plethysmography chamber. This allowed simultaneous recording of EEG, ECG and breathing.
Protocol 6
To obtain information about which 5-HT receptors are involved, separate sets of seven non-instrumented male Lmx1bf/f and Lmx1bf/f/p mice were subjected to MES-induced seizures 3–5 s prior to initiation of mechanical ventilation or following pretreatment with vehicle, 2,5-dimethoxy-4-iodoamphetamine (DOI, 0.3 or 1 mg kg−1; 5-HT2 agonist), citalopram (20 mg kg−1; SSRI), TCB-2 (10 mg kg−1; 5-HT2A agonist) or MK-212 (10 mg kg−1; 5-HT2C agonist), or combinations of ketanserin (10 mg kg−1; 5-HT2 antagonist) plus citalopram (20 mg kg−1), ketanserin (10 mg kg−1) plus DOI (1 mg kg−1), MDL 11,939 (10 mg kg−1; 5-HT2A antagonist) plus DOI (1 mg kg−1), or RS102221(10 mg kg−1; 5-HT2C antagonist) plus DOI (1 mg kg−1) (Table 1).
Table 1.
List of pharmacological treatments
| Drug | Dose (mg kg−1) |
|---|---|
| DOI | 0.3 |
| 1* | |
| Citalopram | 20 |
| Ketanserin + Citalopram | 10 |
| 20 | |
| TCB-2 | 10 |
| MK-212 | 10 |
| DOI + Ketanserin | 1 |
| 10 | |
| DOI + MDL11,939 | 1 |
| 10 |
Lmx1bf/f/p mice only.
EEG data acquisition and analysis
EEG headmounts (8201; Pinnacle Technology Inc., Lawrence, KS, USA) were implanted as previously described (Hodges et al. 2008; Buchanan & Richerson, 2010). Briefly, under either ketamine (100 mg kg−1 i.p.) and xylazine (10 mg kg−1 i.p.), or isoflurane (0.5–2% inhaled) anaesthesia the skull was exposed, and the headmount was attached to the skull with two 0.1 inch (anterior) and two 0.125 inch (posterior) stainless steel machine screws (000-120; Pinnacle Technology). The base of the headmount and screw heads were coated with dental acrylic (Jet Acrylic; Lang Dental, Wheeling, IL, USA) and the skin sutured was closed leaving only the headmount socket exposed. Animals received pre- and post-operative analgesia with meloxicam (0.3 mg kg−1 i.p. pre-op; 0.05 mg kg−1 day−1 post-op in drinking water for 7 days) and recovered for at least 10 days before being studied. A subset of animals, as described above, was also implanted with ECG electrodes (MS303-76; Plastics One, Inc., Roanoke, VA, USA) at the same time as EEG headmount implantation. The suture pad of one lead was sutured to muscle in the left chest wall and the other to muscle in the right axilla. The wires were then routed under the skin over the nuchal muscles with the electrode pedestal protruding through the scalp posterior to the EEG headmount.
Animals were fitted with a preamplifier (8202-SL; Pinnacle Technology) inserted into the sockets of the implanted headmount, introduced to the recording chamber and allowed to acclimate as described below. EEG leads were then passed through a commutator (#8204; Pinnacle Technology) and into an analog conditioning amplifier (Model 440 Instrumentation Amplifier; Brownlee Precision Co., San Jose, CA, USA). Data were digitised with an analog-to-digital converter (PCI-6221; National Instruments, Austin, TX, USA) in a desktop computer and acquired using software custom written using MATLAB (Mathworks, Natick, MA, USA). EEG signals were amplified (×50,000), band-pass filtered (0.3–200 Hz) and digitised (1000 samples s–1).
Chemical seizure induction
After an acclimation period in the recording chamber of at least 1 h, animals received scopolamine methyl nitrate (1 mg kg−1 i.p.; Sigma-Aldrich, St Louis, MO, USA) to reduce the peripheral effects of pilocarpine (Turski et al. 1984). After 15 min, animals received the first dose of pilocarpine (50 mg kg−1 i.p.; Sigma-Aldrich). Subsequent pilocarpine injections (50 mg kg−1 i.p.) were given every 20 min until status epilepticus was achieved. This graded dosing scheme was modelled after published paradigms (Groticke et al. 2007; Winawer et al. 2007; Trindade-Filho et al. 2008; Muller et al. 2009). Animals were briefly removed from the recording chamber to receive each injection. To control for potential circadian effects on seizure susceptibility and sensitivity to cholinomimetics all experiments commenced between 08.30 and 09.30 h. Seizures were scored behaviourally on a modified Racine scale (Racine, 1972) as follows: 0, behavioural arrest; 1 and 2 (grouped together), facial automatisms, tremor, tail stiffening, head bobbing, body jerks; 3, single limb myoclonus; 4, bilateral myoclonus, rearing, non-sustained tonic–clonic activity; 5, recurrent (<2 min apart) and/or sustained tonic–clonic seizures (status epilepticus); 6, death.
The graded pilocarpine dosing regimen was used here to induce acute seizures and assess differences in seizure susceptibility and severity between the sexes and genotypes. The more commonly used protocol for pilocarpine is to administer a large dose to induce status epilepticus, which in turn leads to epileptogenesis and eventually spontaneous, recurrent seizures over many days to weeks. While at sufficiently high cumulative pilocarpine doses our mice entered status epilepticus and most ultimately succumbed to it, we used this as a model to characterise seizure susceptibility in these mice. We did not recover animals following status epilepticus and monitor for development of spontaneous seizures. We report the dose to reach status epilepticus and the mortality rate, but because it is impossible to separate mortality from status epilepticus in this chemical-induction model we do not include these results in our discussion of seizure-related mortality. This protocol was also not a model of SUDEP, as seizures were not spontaneous, but instead was a model of acute seizure-induced death. However, as discussed above, there are no data available to indicate that the mechanisms of death are different in an epileptic mouse compared to a mouse without epilepsy. Collection of data here are the first step in determining whether this is true or not.
Electrical seizure induction
A separate cohort of mice was used to study acute seizures induced by MES. After an acclimation period of 15 min, mice received single electroshock stimulations (10, 30 or 50 mA; 0.2 s; 60 Hz) via ear electrodes (modified, toothless, stainless steel alligator clips). Many mice succumbed to the seizure. Those that survived were killed with an overdose of isoflurane (30% in propylene glycol). Seizure intensity was assessed by calculation of the extension to flexion ratio (E/F ratio) – the length of time the hind limbs were extended beyond 90° divided by the length of time the hind limbs were flexed (≤90°). Higher E/F ratios correlate with widespread propagation of epileptiform activity (Anderson et al. 1986). E/F ratio determinations were made off-line by post hoc video review. In a separate set of animals, the minimum current required to induce seizures was determined by stimulating animals with increasing current (1, 3, 5, 10 mA; 5 min between stimulations) until seizures occurred.
5-HT depletion with PCPA
PCPA methyl ester was delivered at 800 mg kg−1 i.p. (in 0.3 ml of 0.9% NaCl) between 07.00 and 10.00 h for four consecutive days. After each injection the animals experienced transient lethargy and mild hypothermia. On the fifth day an injection was given at 05.30 h to allow the lethargy and hypothermia to subside prior to MES delivery. Volume-matched saline injections were given to control mice.
Measurement of brain and serum 5-HT levels
Animals received a dose of flunixin meglumine (Flunixamide; 2.5 mg kg−1 s.c.) 2 h prior to whole blood collection and brain removal for pre-procedural analgesia. Under 1.5–3.0% isoflurane via precision vaporizer (Summit Isoflurane), a thoracotomy was performed and whole blood samples were collected via cardiac puncture into syringe with EDTA for a final concentration of 2 mg ml−1. Samples were mixed in a 1:1 ratio with the extraction solvent (0.8 m perchloric acid, 0.1 m ascorbic acid and 10 mm EDTA), vortexed for 15 s, spun in a microfuge at 10,000 g for 10 min, and the supernatant was injected into a Phenomenex Nucleosil C18 HPLC column, eluted with a mobile phase [89.5% 0.1 m trichloroacetic acid, 10−2 m sodium acetate, 10−4 m EDTA and 10.5% methanol (pH 3.8)] at 0.7 ml min−1 using a Waters 515 HPLC pump for measurement on an Antec Decade detector.
HPLC (HTEC-500; EiCOM USA, San Diego, CA, USA) was used to verify that PCPA caused a reduction in 5-HT content in the forebrain and medulla. Following flunixamide (2.5 mg kg−1 s.c.) analgesia and under 1.5–3.0% isoflurane anaesthesia, animals were perfused with chilled PBS (1 m). This flushed the blood out of the vascular system to avoid contamination of measurements of brain 5-HT levels by the high levels of 5-HT in platelets. A transverse cut was made rostral to the superior colliculus to separate the lower brainstem from the forebrain. The tissues were flash frozen on dry ice, and homogenised with a sonicator (Sonic Dismembrator 60; Fisher Scientific, Houston, TX, USA) at 1 W for 45 s in 100 μl mobile phase solution (80% 0.1 m citrate-acetate buffer, pH 3.5, 20% methanol, with 220 mg l−1 sodium octane sulfonate and 5 mg ml−1 EDTA). Homogenates and plasma were spiked with 40 ng 2,3-dihydroxybenzoic acid (2,3-DHBA; internal standard) and extracted by centrifugation at 20,000 g at 4°C for 20 min. The supernatant was then filtered with aluminium oxide and a 0.45 μm syringe filter, and 10 μl was transferred to a 96 well plate within a 4°C auto sampler. The supernatant was then automatically injected into a CA-ODS pre-column and SC-30DS column and eluted with the mobile phase at a flow rate of 200 μl min−1. 5-HT and 5-hydroxyindoleacetic acid (5-HIAA) standards were used to generate daily calibration curves, using 2,3-DHBA as the internal standard.
Plethysmography
For quantification of ventilation during seizures induced by MES, a plethysmography chamber was fitted with an ultra-low pressure/high sensitivity pressure transducer (DC002NDR5; Honeywell International, Minneapolis, MN, USA). The analog output from the pressure transducer was digitised by an A-D converter (PCI-6221; National Instruments), displayed on a computer monitor in real time using an acquisition program custom written in MATLAB and saved on a computer hard drive. Prior to seizure induction the signal was calibrated by delivering metered breaths (300 μl; 150 breaths min−1) via a mechanical ventilator (Mini-Vent; Harvard Apparatus) to the recording chamber. Breathing parameters including respiratory rate (RR), tidal volume (VT) and minute ventilation (VE) were assessed with software custom written in MATLAB using previously described methods (Hodges et al. 2008).
Ventilatory support during seizures
In a subset of experiments mice were provided with ventilatory support during seizures to determine if they could be resuscitated. Three to five seconds after induction of seizures with MES, artificial breaths (150 breaths min−1; 300 μl stroke volume) were delivered to animals via a length of tygon tubing (3/16 inch inner diameter) connected to a mechanical ventilator (Model 683; Harvard Apparatus). Ventilator-delivered breaths were continued until the end of the tonic phase of the seizure.
Drug delivery prior to MES
Citalopram hydrobromide, MDL 11,939, MK-212, RS 102221 hydrochloride and TCB-2 were obtained from Tocris Bioscience (Ellisville, MO, USA). DOI hydrochloride and ketanserin tartrate were obtained from Sigma-Aldrich. Citalopram hydrobromide, DOI hydrochloride, TCB-2 and MK-212 were delivered via i.p. injections 30 min prior to seizure induction with MES. For antagonist–agonist combination studies ketanserin, MDL 11,939 or RS102221 were delivered via i.p. injection 45 min prior to MES, and agonists or citalopram were delivered 30 min prior to MES. All drugs have been shown to have CNS activity when given systemically at the same concentrations.
Statistical analyses
Interactions between genotype, pilocarpine dosage and latency to each Racine grade seizure, as well as interactions between genotype, sex, electrical stimulus intensity and E/F ratio were analysed using two-way ANOVA or paired t test as appropriate. Survival analyses were conducted using Kaplan–Meier analysis with log rank test or logistic regression as appropriate. The effects of mechanical ventilation or pretreatment with citalopram, DOI, TCB-2, MK-212, ketanserin plus citalopram, ketanserin plus DOI, MDL 11,939 plus DOI, and RS 102221 plus DOI on seizure survival were analysed with Wilcoxon signed ranks test. The significance threshold was P < 0.05 for all analyses. Statistical analyses were performed with Systat 11.0 and Microsoft Excel by an investigator blind to the genotype. Data expressed as x ± y are mean ± SEM unless stated otherwise. All error bars are SEM.
Results
Lmx1bf/f/p mice were more susceptible to the acute convulsant effect of pilocarpine
Seizures of all Racine grades were recorded in both Lmx1bf/f and Lmx1bf/f/p mice following pilocarpine treatments (Fig. 1). Scopolamine alone followed by subsequent vehicle injections did not result in seizures in either genotype. All Lmx1bf/f/p mice achieved Racine grade 1 or 2 seizures following the first injection of pilocarpine (Fig. 1A), as did all of the male Lmx1bf/f mice. No female mice achieved grade 1 or 2 seizures after the first injection, but all reached grades 1 or 2 after the second injection. The majority of Lmx1bf/f/p mice further progressed to grade 3 seizures following the first pilocarpine injection. No Lmx1bf/f mice experienced grade 3 seizures following the first pilocarpine injection (Fig. 1A).
Figure 1. Increased susceptibility to and mortality from pilocarpine-induced seizures in Lmx1bf/f/p mice.

A, mean maximum Racine grade achieved after each cumulative pilocarpine dose in male (squares) and female (circles) Lmx1bf/f (solid symbols) and Lmx1bf/f/p (open symbols) mice; n = 8 for each group. *P < 0.05 between genotype among sexes and between sexes among genotype; †P < 0.05 between sexes for Lmx1bf/f/p mice; #P < 0.05 between genotypes for females; ‡P < 0.05 between sexes for Lmx1bf/f mice. B, latency to achieve each Racine grade in male and female Lmx1bf/f and Lmx1bf/f/p mice. Latencies were determined from the time of the first pilocarpine injection to the time of first appearance of behaviour consistent with each Racine grade. Error bars, ± SEM; *P < 0.001 between genotypes among sexes and between sexes among genotype. C, interval from the time of onset of one Racine grade to the next in male and female Lmx1bf/f and Lmx1bf/f/p mice. Note that the absence of 5-HT neurones causes a decrease in latency to Racine grade 1–3 seizures, but has no effect on progression to generalised seizures (grades 4 and 5); n = 8 for each group. Mean ± SEM are given. *P < 0.001 between genotype among sexes and between sexes among genotype. D, Kaplan–Meier survival curves depicting percentage of male (dotted lines) and female (continuous lines) Lmx1bf/f (black) and Lmx1bf/f/p (grey) mice still alive after each cumulative pilocarpine dose. Log rank P-value = 0.007 for genotype and sex; n = 8 for each group.
Lmx1bf/f/p mice exhibited seizures of Racine grades 3, 4, 5 and 6 following lower cumulative doses of pilocarpine and at a shorter latency from the first pilocarpine injection compared to Lmx1bf/f mice (Fig. 1A, B). Interestingly, once Racine grade 3 was achieved, the latency to reach grade 4 was not significantly different between genotypes for either sex (P = 0.73; Fig. 1C). There was also no significant difference between genotypes or either sex in the latency to reach grade 5 once grade 4 was achieved (P = 0.88; Fig. 1C). These trends were similar in males and females, but males of each genotype had more severe seizures at lower pilocarpine doses compared to their female counterparts (Fig. 1A).
Following chemical induction of seizures with graded pilocarpine dosing, all mice of both genotypes and sexes eventually experienced status epilepticus (Racine grade 5 in Fig. 1A). All Lmx1bf/f/p mice died during status epilepticus (Racine grade 6 in Fig. 1A). In male Lmx1bf/f/p mice, status epilepticus was achieved following relatively low cumulative doses of pilocarpine (168.8 ± 49.8 mg kg−1; range 100–250 mg kg−1; Fig. 1A). Most, but not all, male Lmx1bf/f mice (75%) also ultimately succumbed to status epilepticus under these experimental conditions, although significantly higher cumulative pilocarpine doses (283.3 ± 53.6 mg kg−1; range 200–400 mg kg−1; P < 0.05) were required to induce death compared to male Lmx1bf/f/p mice (Fig. 1D). The cumulative pilocarpine doses required to induce death in females were also lower for Lmx1bf/f/p mice (278.57 ± 27.82 mg kg−1; range 250–300 mg kg−1) compared to Lmx1bf/f mice (450.00 ± 31.62 mg kg−1; range 450–500 mg kg−1; P < 0.05), but were significantly higher for females than for males (P < 0.05). The sex difference was true for both genotypes (P < 0.05). Two Lmx1bf/f mice of each sex remained in status epilepticus but did not die during the recording session. The mortality data from the pilocarpine studies are presented for completeness, but death from status epilepticus is due to multiple complex interacting pathophysiological mechanisms that are likely to be different from those involved in SUDEP.
Lmx1bf/f/p mice were also more susceptible to MES-induced seizures
Seizures were induced in all Lmx1bf/f and Lmx1bf/f/p mice using electrical stimulation with stimulus intensities of 10 mA and higher in a dose-dependent manner (Fig. 2). Two-thirds of Lmx1bf/f/p mice experienced seizures at a stimulus intensity of 5 mA, whereas none of the Lmx1bf/f mice experienced seizures at this intensity. No animals of either genotype or sex had seizures at stimulus intensities of 1 or 3 mA. Seizures induced at any given stimulus intensity were more severe as measured by the E/F ratio in Lmx1bf/f/p compared to Lmx1bf/f mice (Fig. 2A). There was a sex difference, with seizures being more severe in males than females in Lmx1bf/f/p mice at 5 and 10 mA and in both genotypes at 30 and 50 mA (Fig. 2A). Lmx1bf/f/p mice were also more likely to die following MES-induced seizures across stimulus intensities (P < 0.05), and this likelihood increased as stimulus intensity increased (P < 0.05; Fig. 2B). Despite male Lmx1bf/f/p mice displaying more severe seizures (Fig. 2A), there was a sex difference in seizure-related mortality only at the highest stimulus intensity (Fig. 2B). While in general higher severity seizures were associated with death in both genotypes, a larger proportion of less severe seizures resulted in death in Lmx1bf/f/p mice compared to Lmx1bf/f mice. None of the mice of either genotype died following seizures with E/F ratios of 8.5 or lower. Lmx1bf/f/p mice died when the E/F ratio was above 8.5, whereas Lmx1bf/f mice did not die until the E/F ratio increased to at least 10.2 (P = 0.028). This indicates that the increased mortality was not simply due to having more severe seizures at a given stimulus intensity, but that they had cardiorespiratory control systems that were more prone to fail in response to a seizure.
Figure 2. Increased sensitivity to and mortality from electrically induced seizures in Lmx1bf/f/p mice.

A, mean seizure severity observed in female and male Lmx1bf/f (female, black; male, grey) and Lmx1bf/f/p (female, white; male, hatched) mice following seizure induction with 5, 10, 30 and 50 mA of current in Lmx1bf/f and Lmx1bf/f/p mice; n = 6 for each group at 10, 30 and 50 mA, except n = 4 for Lmx1bf/f/p mice of each sex at 10 mA (6 male and 6 female mice were tested; 2 males and 2 females did not have seizures). The absence of black and grey bars at 5 mA indicates that none of the 12 Lmx1bf/f mice tested at 5 mA had seizures. *P < 0.001 between sexes among genotype; #P < 0.001 between genotype among sexes. Error bars, ± SEM. B, percentage female and male Lmx1bf/f (female, black; male, grey) and Lmx1bf/f/p (female, white; male, hatched) mice still alive following seizures that were induced with 5, 10, 30 and 50 mA currents; n = 6 for each group at each intensity. *Logistic regression P-value < 0.05, between genotype and sexes within intensity. The absence of a hatched bar at 50 mA indicates that all six male Lmx1bf/f/p died following seizure induction with 50 mA.
Depletion of 5-HT with PCPA increased seizure severity, but not mortality
To ascertain the effects of subacute depletion of 5-HT instead of lifelong elimination of 5-HT neurones, C57BL/6N mice were treated with vehicle or PCPA for 5 days as described. Five days of PCPA treatment was sufficient to cause maximal reduction in 5-HT levels (Fig. 3A). Treatment with PCPA in this manner reduced 5-HT levels to 5.6 ± 0.5% in the medulla and 11.8 ± 1.9% in the forebrain compared to vehicle-treated controls (PCPA medulla: 766.6 ± 84 pg 5-HT mg–1, forebrain: 415.4 ± 31.5 pg 5-HT mg–1; Fig. 3B). The measured levels of 5-HT in medulla and cortex of untreated Lmx1bf/f/p mice of similar age were less than 1% of C57BL/6N mice (< 30 pg 5-HT (mg tissue)–1; Fig. 3B). The resultant reduction of 5-HT in PCPA-treated animals led to more severe MES-induced (50 mA, 200 ms, 60 Hz) seizures in PCPA-treated mice compared to vehicle-treated mice (Fig. 3C). However, none of the vehicle-treated mice and only one PCPA-treated mouse died.
Figure 3. Depletion of 5-HT with PCPA increased severity of MES-induced seizures.
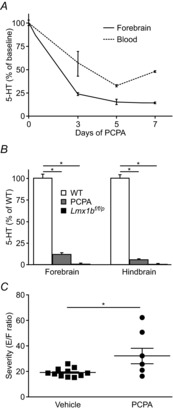
A, mean 5-HT level measured from forebrain (continuous line) or blood (dashed line) without treatment (0 days) or after 3, 5 or 7 days of PCPA (800 mg kg−1 day−1 i.p.). B, bar graph depicting 5-HT levels as measured by HPLC in forebrain and hindbrain in vehicle-treated C57BL/6N mice (WT; white; n = 7), PCPA-treated C57BL/6N mice (grey; n = 6) and untreated Lmx1bf/f/p mice (black; n = 7). *P < 0.0001. C, seizure severity as assessed by the E/F ratio in C57BL/6N mice treated with vehicle (squares; n = 11) versus PCPA (800 mg kg−1 day−1 × 5 days; circles; n = 6). *P < 0.05. Error bars, ± SEM.
Death from MES-induced seizures is due to apnoea that can be prevented by 5-HT2A receptor stimulation
During all MES-induced seizures in both sexes of Lmx1bf/f/p and Lmx1bf/f mice we observed that there was cessation of respiratory activity assessed by whole body plethysmography. To better characterise this we implanted a set of male mice with EEG and ECG electrodes and induced seizures using MES with the animal in the plethysmography chamber (Fig. 4). In animals that did not survive, breathing did not recover and the EEG flattened following the seizure ( Figs 4A, C–E and 5); however, cardiac activity persisted for up to 9 min (mean 7.29 ± 1.39 min; Fig. 4A, F) and became increasingly slow, irregular and lower in amplitude (Fig. 4F). Due to muscle artifacts, cardiac activity could not be accurately assessed during the seizure. In mice that survived the seizure, EEG suppression lasted 164.25 ± 44.29 s and breathing parameters (RR, VT, VE) returned to normal post-ictally ( Figs 5 and 6). To determine whether respiratory arrest was the cause of death, animals were mechanically ventilated via a nose cone at a rate of 150 breaths min−1 and a tidal volume of 300 μl during the tonic phase of the MES-induced seizure (50 mA, 60 Hz, 200 ms in WT; 30 mA, 60 Hz, 200 ms in Lmx1bf/f/p). This resulted in a significant reduction of mortality, with all Lmx1bf/f and 86% of Lmx1bf/f/p mice surviving the seizure (P < 0.05 for both genotypes compared to no ventilation; Fig. 7A).
Figure 4. Death ensued following seizure-induced respiratory arrest.
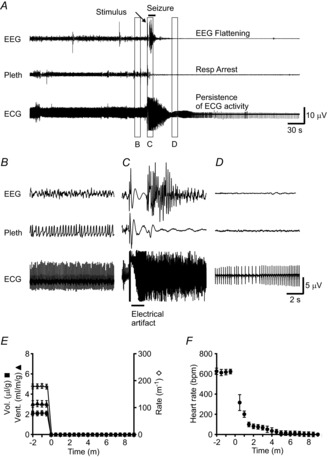
A, 5 min traces of EEG, breathing (pleth) and ECG from an animal that died following a seizure, demonstrating respiratory arrest beginning during seizure, flattening of EEG following seizure and prolonged persistence of ECG activity. The seizure was induced with MES (50 mA, 60 Hz, 200 ms). B–D, 10 s traces of EEG, breathing and ECG before (B), during (C) and after (D) seizure. Temporal position of data for B–D is indicated by the lettered boxes in A. E and F, graphs depicting tidal volume (Vol.), minute ventilation (Vent.), and respiratory rate (E) and heart rate (F) from 2 min before seizure to 9 min after seizure. Each data point represents the average during one 10 s epoch of data averaged for seven male mice. Error bars in E and F are ± SEM.
Figure 5. Recovery of cortical activity in mice that survived seizures.
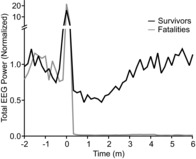
Graph depicting total EEG power between 0.5 and 20 Hz plotted versus time relative to seizure onset (time 0) in 10 s epochs. Fast Fourier power transform is plotted relative to 1 min of baseline EEG during wakefulness prior to seizure induction. Mean data are presented for four mice that survived (black) and seven mice that died (grey).
Figure 6. Time course of respiratory recovery following a non-fatal seizure.

A, 5 min traces of EEG, breathing (pleth) and ECG from an animal that recovered after a seizure, demonstrating respiratory arrest beginning during the seizure, flattening of EEG following the seizure and persistence of ECG activity. The seizure was induced with MES (50 mA, 60 Hz, 200 ms). B–G, 3 s traces of EEG, breathing and ECG before (B), during (C), and 10, 20, 30 and 60 s after (D–G) a seizure. Temporal position of data for B–G is indicated by the lettered boxes in A. H and I, graphs depicting tidal volume (Vol.), minute ventilation (Vent.) and respiratory rate from 90 s before seizure to 3 min after seizure (H) and heart rate from 90 s before seizure to 90 s after seizure (I). Each data point represents the average during one 10 s epoch of data averaged for four male mice. Error bars in H and I are ± SEM.
Figure 7. Death could be prevented with mechanical ventilation, SSRI or 5-HT2A agonist.
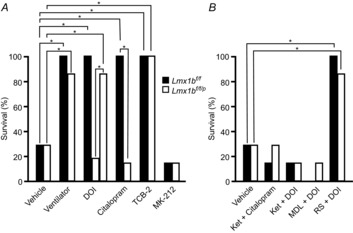
A, percentage of Lmx1bf/f (black) and Lmx1bf/f/p (white) mice surviving MES-induced seizures following vehicle pretreatment, mechanical ventilation during the seizure, or pretreatment with DOI (0.3 mg kg−1 in Lmx1bf/f, 0.3 or 1 mg kg−1 in Lmx1bf/f/p mice), citalopram (20 mg kg−1), TCB-2 (10 mg kg−1) or MK-212 (10 mg kg−1). Seizures were induced with 50 mA in Lmx1bf/f and 30 mA in Lmx1bf/f/p mice to produce seizures of similar severity and mortality rate (as shown in Fig. 2); n = 7 per genotype and condition. *P < 0.001 compared to vehicle. B, percentage of Lmx1bf/f (black) and Lmx1bf/f/p (white) mice surviving MES-induced seizures following pretreatment with vehicle, ketanserin (10 mg kg−1) plus citalopram (20 mg kg−1), ketanserin (10 mg kg−1) plus DOI (1 mg kg−1), MDL11,939 (10 mg kg−1) plus DOI (1 mg kg−1), or RS102221 (10 mg kg−1) plus DOI (1 mg kg−1). *P < 0.001 compared to vehicle.
To determine whether the 5-HT system plays a role in this respiratory-related death, a separate set of male Lmx1bf/f and Lmx1bf/f/p mice were pretreated with the SSRI citalopram (20 mg kg−1), 30 min prior to seizure induction with MES (50 mA Lmx1bf/f, 30 mA Lmx1bf/f/p; 60 Hz; 200 ms). All seven Lmx1bf/f mice treated with citalopram survived the seizure compared to only two of seven that received vehicle prior to MES (P < 0.05; Fig. 7B). Only one of seven Lmx1bf/f/p mice treated with citalopram survived, which is similar to the mortality seen with vehicle treatment (Fig. 7B), and is as expected since Lmx1bf/f/p mice have a significant reduction in 5-HT transporters (Zhao et al. 2006). The 5-HT2 agonist DOI (0.3 or 1 mg kg−1) was given to additional groups of male mice (n = 7 of each genotype) 30 min prior to seizure induction with MES. At a dose of 1 mg kg−1 but not 0.3 mg kg−1, DOI pretreatment prevented seizure-induced respiratory arrest and death in Lmx1bf/f/p mice (P < 0.05). In Lmx1bf/f mice 0.3 mg kg−1 was sufficient to achieve the same effect (P < 0.05; Fig. 7B). The 5-HT2A agonist TCB-2, but not the 5-HT2C agonist MK-212, prevented MES-induced death in both genotypes (Fig. 7A). Pretreatment with the 5-HT2A antagonist MDL 11,939, but not the 5-HT2C antagonist RS102221 prior to citalopram or the 5-HT2 agonist DOI prevented the reduction in mortality from MES-induced seizures seen in Lmx1bf/f mice following treatment with citalopram or DOI alone (Fig. 7A, B). None of these pharmacological treatments had any effect on seizure severity compared to MES with vehicle (Table 2).
Table 2.
Pharmacological manipulations did not affect seizure severity
| Lmx1bf/f | Lmx1bf/f/p | |||
|---|---|---|---|---|
| Not fatal | Fatal | Not fatal | Fatal | |
| Vehicle | 10.0 ± 1.00 | 10.20 ± 1.05 | 10.12 ± 1.06 | 11.25 ± 1.40 |
| Ventilator | 10.29 ± 1.46 | n/a | 11.15 ± 1.56 | 11.13 |
| DOI 0.3 mg kg−1 | 9.56 ± 0.18 | n/a | 11.24 | 10.90 ± 2.40 |
| DOI 1 mg kg−1 | n/a | n/a | 9.99 ± 1.04 | 11.07 |
| Citalopram | 9.90 ± 0.57 | n/a | 11.12 ± 1.82 | 10.73 |
| Ketanserin + Citalopram | 10.96 | 10.10 ± 0.71 | 11.36 ± 0.23 | 11.09 ± 1.29 |
| TCB-2 | 10.51 ± 1.64 | n/a | 11.31 ± 1.12 | n/a |
| MK-212 | 10.27 | 10.14 ± 0.19 | 11.82 | 11.66 ± 0.93 |
| DOI + Ketanserin | 11.04 | 10.35 ± 0.64 | 11.53 | 11.41 ± 1.12 |
| DOI + MDL11,939 | n/a | 10.73 ± 0.87 | 11.36 | 11.11 ± 1.80 |
| DOI + RS102221 | 10.87 ± 1.33 | n/a | 11.38 ± 0.98 | 11.74 |
Values are mean ± SEM; n/a, not applicable.
Discussion
The role of 5-HT in seizure regulation and the mechanisms by which seizures become fatal remain controversial. Some data suggest 5-HT is pro-convulsant, while other data suggest 5-HT is anti-convulsant. Similarly, some evidence indicates that the primary mechanism of death from a seizure is cardiac, while others support a respiratory cause. Using a mouse model employing simultaneous EEG, ECG and breathing measurement with seizure induction, the work presented here more clearly defines a sequence of events that can culminate in death from a seizure. This work supports a critical role for the 5-HT system in regulating seizure threshold, and in reducing seizure severity and mortality, and further demonstrates that 5-HT system dysfunction can contribute to seizure-related respiratory arrest.
Is 5-HT pro-convulsant or anti-convulsant?
Antidepressant medications including SSRIs that increase synaptic 5-HT previously were thought to reduce seizure threshold and promote seizure occurrence (Kondziella & Asztely, 2009). However, most data point to the opposite – that 5-HT is anti-convulsant and that SSRIs are well-tolerated in patients with epilepsy (Hamid & Kanner, 2013). In the current study, the increased sensitivity to acutely induced seizures observed in both Lmx1bf/f/p mice and C57BL/6N mice subjected to subacute depletion of 5-HT with PCPA is consistent with 5-HT being anti-convulsant.
There are 16 different 5-HT receptor subtypes. These can be excitatory, inhibitory or neuromodulatory, and can reside on excitatory or inhibitory neurones (Barnes & Sharp, 1999). Therefore, the net effect of 5-HT on seizure predilection may depend on the site of action and the effect on thalamocortical networks (Bagdy et al. 2007). For instance, activation of hippocampal 5-HT1A receptors reduces network excitability (Wada et al. 1993), whereas activation of 5-HT3 receptors in the hippocampus leads to hyperexcitability (Wada et al. 1997).
Mechanism of death from a seizure: not always cardiac, as is commonly assumed
It is often presumed that death from a seizure is due to cardiac arrest (Schuele, 2009). A subset of patients with temporal lobe epilepsy have profound post-ictal respiratory depression (Langan et al. 2000; Bateman et al. 2008) whose onset coincides with spread to the contralateral temporal lobe (Seyal & Bateman, 2009). Cardiac abnormalities can sometimes be due to hypoxaemia (Bateman et al. 2010b; Seyal et al. 2011). In DBA/1 and DBA/2 mice that are susceptible to respiratory arrest following audiogenic seizures, respiratory arrest and death can be prevented with oxygenation or mechanical ventilation (Venit et al. 2004). In DBA/1 mice, respiratory arrest precedes any cardiac dysfunction (Faingold et al. 2010). In the current study respiratory arrest preceded terminal asystole. Preventing the respiratory consequences prevented cardiac abnormalities, and also prevented death. In a recent large multicentre study, there were both intermittent cardiac and respiratory changes following seizures that were ultimately fatal, although it could not be determined whether the initial abnormalities that occurred were respiratory or cardiac (Ryvlin et al. 2013). The advantage of the mouse model used here is that it was possible to define the events that initiated the fatal cascade.
5-HT is a well-known regulator of breathing (Richerson, 2004). Seizure-related respiratory arrest in DBA mice can be mitigated by pretreatment with SSRIs (Tupal & Faingold, 2006; Faingold et al. 2011; Faingold & Randall, 2013). Mice lacking the 5-HT2C receptor are also sensitive to audiogenic seizures and die from seizure-induced respiratory arrest (Brennan et al. 1997). Patients taking SSRIs have reduced incidence of oxygen desaturation associated with partial seizures compared to patients not taking SSRIs (Bateman et al. 2010a).
Relationship to SUDEP
The current definition of SUDEP allows for death to occur without evidence of an associated seizure (Nashef et al. 2012). It is suspected that at least a subset of SUDEP occurs in association with a seizure. In fact, all SUDEP cases reported in the recent MORTEMUS study (Ryvlin et al. 2013) occurred in association with a precedent seizure. Therefore, understanding how a seizure can lead to death is relevant to SUDEP.
Role of 5-HT in SUDEP
There is no direct evidence of 5-HT involvement in human SUDEP cases, but 5-HT involvement is supported by considerable circumstantial evidence. First, as mentioned 5-HT may be involved with setting seizure threshold and regulating seizure intensity (Bagdy et al. 2007). Second, 5-HT levels can be reduced following seizures in animal models (Ferraz et al. 2002) and this 5-HT reduction may contribute to post-ictal respiratory suppression. Third, seizures have long-range effects on cortical structures that cause unresponsiveness (Englot et al. 2008; Blumenfeld et al. 2009). 5-HT neurones comprise a portion of the ascending activating system and may be involved in this effect (Richerson, 2013). Fourth, previous (Tupal & Faingold, 2006; Bateman et al. 2010a) and current data support that 5-HT system dysfunction might underlie respiratory-mediated death following a seizure. Finally, SUDEP shares many attributes with SIDS (Sowers et al. 2013) and abnormalities in the brainstem 5-HT system have been found in brains of SIDS victims (Kinney et al. 2009).
Lmx1bf/f/p mice exhibited a high mortality rate compared to PCPA-treated C57BL/6N mice, suggesting that either there is a minimum level of 5-HT required to prevent death from a seizure, or another neurotransmitter that is also lost with depletion of 5-HT neurones may similarly act to support peri-ictal ventilation and prevent death. Candidates for the latter include: thyrotrophin-releasing hormone (TRH), substance P, enkephalin, corticotrophin releasing factor (CRF) and galanin (Myers et al. 1977; Yamamoto et al. 1981; Mann et al. 1995; Abbott et al. 2009). Some of these transmitters can also stimulate respiratory neurones and increase breathing (Dekin et al. 1985; Richerson, 2004).
It should be noted that clinically relevant 5-HT deficiencies are likely to be smaller than the profound reductions in the mouse models; however, the models lend insight into the role 5-HT may play.
Post-ictal generalized EEG suppression (PGES): what is it, where does it come from and is it relevant to SUDEP?
In addition to cardiac and respiratory aetiologies for SUDEP, PGES has been proposed to play a role in causing death (Bozorgi & Lhatoo, 2013). The exact mechanisms of PGES are not known, but there are several possibilities. For instance, there could be loss of cortical activity due to prolonged global hypoxia or diffuse excitotoxicity from prolonged activation of excitatory, glutamatergic circuits during seizures. In addition, there could be inhibition of ascending monoamine projections.
It is more likely that PGES could be a marker of SUDEP risk, or in some cases imminent death, and less likely that PGES is a direct cause for SUDEP. For PGES to directly cause SUDEP would suggest that cortical activity is necessary for maintenance of cardiac and respiratory function, which is not true. The cardiac and respiratory systems function autonomously, and can remain active for prolonged periods in the absence of cortical input (e.g. during a persistent vegetative state). Prolonged PGES following non-fatal seizures is associated with a high likelihood of future SUDEP (Lhatoo et al. 2010). The question is why should this be a biomarker of SUDEP? It could be that there is a common underlying mechanism that leads to both PGES and, for instance, respiratory arrest. The aforementioned 5-HT system is a good candidate for this as it is involved in regulation of both sleep–wakefulness and breathing (Richerson, 2013).
Conclusions
SUDEP is probably a disorder with many subtypes. It is unique to patients with epilepsy. Therefore, the only certain way to eliminate SUDEP is to eliminate epilepsy or, for those cases that are seizure-related (possibly all of them), to control seizures. The former is being vehemently pursued, while the latter continues to prove elusive. Despite the existence of over 20 pharmaceutical agents to combat seizures, nearly 25% of patients are categorised as having medically refractory epilepsy (resistant to two or more agents) (Kwan et al. 2011). Less than 50% of patients with epilepsy enjoy relative seizure freedom on a single agent (Brodie & Kwan, 2002). Another approach to eradicating SUDEP is to address individual putative aetiologies and attempt to prevent seizure-related cardiac and respiratory dysfunction that may lead to death.
Studies such as this one help to guide how we manage patients in our epilepsy monitoring units. Clearly there is value in monitoring respiratory function and collecting respiratory data in all patients, but currently this is not done routinely. We should also learn to more rapidly and pre-emptively support breathing in the early post-ictal period. External stimulation may be the easiest and most effective preventive treatment, and may act by increasing respiratory drive by increased arousal. It is too soon to propose that all patients with epilepsy have oxygen available at home to use following a seizure, but it may be reasonable to start doing this in a select subset of patients such as those who are known to have profound ictal oxygen desaturation and/or hypoxaemia. A clear pharmacological intervention is not readily apparent, although it has been proposed that increasing 5-HT availability with an SSRI may be valuable and trials are ongoing. There is a great deal yet to be learned regarding the most salient opportunities for intervention before efforts can be made to implement potential life-saving strategies; however, we believe respiratory support and 5-HT augmentation will have a role in future preventive strategy.
Acknowledgments
The authors thank Xiuqiong Zhou for genotyping assistance.
Glossary
- 2,3-DHBA
2,3-dihydroxybenzoic acid
- 5-HIAA
5-hydroxyindolacetic acid
- DOI
2,5-dimethoxy-4-iodoamphetamine
- E/F
extension/flexion
- MES
maximal electroshock
- PCPA
para-chlorophenylalanine
- RR
respiratory rate
- SIDS
sudden infant death syndrome
- SSRI
selective serotonin reuptake inhibitor
- SUDEP
sudden unexpected death in epilepsy
- VT
tidal volume
- VE
minute ventilation
Additional information
Competing interests
None of the authors has any competing interests to disclose.
Author contributions
The studies were competed in the laboratories of G.F.B. and G.B.R. The experiments were conceived by G.F.B. and G.B.R., and designed by G.F.B., N.M.M., M.A.H. and G.B.R. Data were collected by G.F.B., N.M.M. and M.A.H. Data were analysed and interpreted by G.F.B., N.M.M., M.A.H. and G.B.R. G.F.B. and G.B.R. wrote the paper. All authors have seen and approved the final version of the manuscript.
Funding
This work was supported by K08 NS069667 (G.F.B.), R01 HD052772 (G.B.R.), P20 NS076916 (G.B.R.), the Beth L. Tross Epilepsy Research Fund (G.B.R.), the VAMC (G.B.R.), a summer research fellowship from the Yale SOM (M.A.H.) the Christopher Donnalty and Kyle Coggins Memorial SUDEP Research Award from CURE (G.F.B.), and a VA Special Fellowship in Neuroscience (G.F.B.).
References
- Abbott SB, Burke PG, Pilowsky PM. Galanin microinjection into the PreBotzinger or the Botzinger Complex terminates central inspiratory activity and reduces responses to hypoxia and hypercapnia in rat. Respir Physiol Neurobiol. 2009;167:299–306. doi: 10.1016/j.resp.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Fowler LJ, Whitton PS. Lamotrigine, carbamazepine and phenytoin differentially alter extracellular levels of 5-hydroxytryptamine, dopamine and amino acids. Epilepsy Res. 2005;63:141–149. doi: 10.1016/j.eplepsyres.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Anderson RE, Howard RA, Woodbury DM. Correlation between effects of acute acetazolamide administration to mice on electroshock seizure threshold and maximal electroshock seizure pattern, and on carbonic anhydrase activity in subcellular fractions of brain. Epilepsia. 1986;27:504–509. doi: 10.1111/j.1528-1157.1986.tb03575.x. [DOI] [PubMed] [Google Scholar]
- Bagdy G, Kecskemeti V, Riba P, Jakus R. Serotonin and epilepsy. J Neurochem. 2007;100:857–873. doi: 10.1111/j.1471-4159.2006.04277.x. [DOI] [PubMed] [Google Scholar]
- Banerjee PN, Filippi D, Allen HW. The descriptive epidemiology of epilepsy – a review. Epilepsy Res. 2009;85:31–45. doi: 10.1016/j.eplepsyres.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Bateman LM, Li CS, Lin TC, Seyal M. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010a;51:2211–2214. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]
- Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: report on two deaths in video-EEG-monitored patients. Epilepsia. 2010b;51:916–920. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, Varghese GI, Purcaro MJ, Motelow JE, Enev M, McNally KA, Levin AR, Hirsch LJ, Tikofsky R, Zubal IG, Paige AL, Spencer SS. Cortical and subcortical networks in human secondarily generalized tonic–clonic seizures. Brain. 2009;132:999–1012. doi: 10.1093/brain/awp028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozorgi A, Lhatoo SD. Seizures, cerebral shutdown, and SUDEP. Epilepsy Curr. 2013;13:236–240. doi: 10.5698/1535-7597-13.5.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–390. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58:S2–S8. doi: 10.1212/wnl.58.8_suppl_5.s2. [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Hodges MR. Contribution of chemosensitive serotonergic neurons to interactions between the sleep-wake cycle and respiratory control. In: Monti JM, Pandi-Perumal SR, Jacobs BL, Nutt DJ, Richerson GB, editors. Serotonin and sleep: Molecular, functional and clinical aspects. Switzerland: Birkhauser Verlag; 2008. pp. 529–554. [Google Scholar]
- Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A. 2010;107:16354–16359. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey JW, Reith ME, Yan QS, Li MY, Jobe PC. Carbamazepine increases extracellular serotonin concentration: lack of antagonism by tetrodotoxin or zero Ca2+ Eur J Pharmacol. 1997;328:153–162. doi: 10.1016/s0014-2999(97)83041-5. [DOI] [PubMed] [Google Scholar]
- Dekin MS, Richerson GB, Getting PA. Thyrotropin-releasing hormone induces rhythmic bursting in neurons of the nucleus tractus solitarius. Science. 1985;229:67–69. doi: 10.1126/science.3925552. [DOI] [PubMed] [Google Scholar]
- Englot DJ, Mishra AM, Mansuripur PK, Herman P, Hyder F, Blumenfeld H. Remote effects of focal hippocampal seizures on the rat neocortex. J Neurosci. 2008;28:9066–9081. doi: 10.1523/JNEUROSCI.2014-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL, Randall M. Effects of age, sex, and sertraline administration on seizure-induced respiratory arrest in the DBA/1 mouse model of sudden unexpected death in epilepsy (SUDEP) Epilepsy Behav. 2013;28:78–82. doi: 10.1016/j.yebeh.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Randall M, Tupal S. DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy Behav. 2010;17:436–440. doi: 10.1016/j.yebeh.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011;22:186–190. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Ferraz AC, Anselmo-Franci JA, Perosa SR, de Castro-Neto EF, Bellissimo MI, de Oliveira BH, Cavalheiro EA, Naffah-Mazzacoratti MG, Da CC. Amino acid and monoamine alterations in the cerebral cortex and hippocampus of mice submitted to ricinine-induced seizures. Pharmacol Biochem Behav. 2002;72:779–786. doi: 10.1016/s0091-3057(02)00750-5. [DOI] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groticke I, Hoffmann K, Loscher W. Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp Neurol. 2007;207:329–349. doi: 10.1016/j.expneurol.2007.06.021. [DOI] [PubMed] [Google Scholar]
- Hamid H, Kanner AM. Should antidepressant drugs of the selective serotonin reuptake inhibitor family be tested as antiepileptic drugs? Epilepsy Behav. 2013;26:261–265. doi: 10.1016/j.yebeh.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Wehner M, Aungst J, Smith JC, Richerson GB. Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. J Neurosci. 2009;29:10341–10349. doi: 10.1523/JNEUROSCI.1963-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol. 2009;4:517–550. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondziella D, Asztely F. Don't be afraid to treat depression in patients with epilepsy! Acta Neurol Scand. 2009;119:75–80. doi: 10.1111/j.1600-0404.2008.01088.x. [DOI] [PubMed] [Google Scholar]
- Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med. 2011;365:919–926. doi: 10.1056/NEJMra1004418. [DOI] [PubMed] [Google Scholar]
- Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–213. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, Bird JM. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68:787–796. doi: 10.1002/ana.22101. [DOI] [PubMed] [Google Scholar]
- Lhatoo SD, Sander JW. Cause specific mortality in epilepsy. Epilepsa. 2005;46:36–39. doi: 10.1111/j.1528-1167.2005.00406.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jiang Y, Si Y, Kim JY, Chen ZF, Rao Y. Molecular regulation of sexual preference revealed by genetic studies of 5-HT in the brains of male mice. Nature. 2011;472:95–99. doi: 10.1038/nature09822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann K, Roschke J, Benkert O, Aldenhoff J, Nink M, Beyer J, Lehnert H. Effects of corticotropin-releasing hormone on respiratory parameters during sleep in normal men. Exp Clin Endocrinol Diabetes. 1995;103:233–240. doi: 10.1055/s-0029-1211356. [DOI] [PubMed] [Google Scholar]
- Muller CJ, Groticke I, Hoffmann K, Schughart K, Loscher W. Differences in sensitivity to the convulsant pilocarpine in substrains and sublines of C57BL/6 mice. Genes Brain Behav. 2009;8:481–492. doi: 10.1111/j.1601-183X.2009.00490.x. [DOI] [PubMed] [Google Scholar]
- Myers RD, Metcalf G, Rice JC. Identification by microinjection of TRH-sensitive sites in the cat's brain stem that mediate respiratory, temperature and other autonomic changes. Brain Res. 1977;126:105–115. doi: 10.1016/0006-8993(77)90218-9. [DOI] [PubMed] [Google Scholar]
- Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53:227–233. doi: 10.1111/j.1528-1167.2011.03358.x. [DOI] [PubMed] [Google Scholar]
- Okada M, Kaneko S, Hirano T, Ishida M, Kondo T, Otani K, Fukushima Y. Effects of zonisamide on extracellular levels of monoamine and its metabolite, and on Ca2+ dependent dopamine release. Epilepsy Res. 1992;13:113–119. doi: 10.1016/0920-1211(92)90066-3. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci. 2004;5:449–461. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Buchanan GF. The serotonin axis: shared mechanisms in seizures, depression, and SUDEP. Epilepsia. 2011;52(Suppl 1):28–38. doi: 10.1111/j.1528-1167.2010.02908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson G. Serotonin: the Anti-SuddenDeathAmine. Epilepsy Curr. 2013;13:241–244. doi: 10.5698/1535-7597-13.5.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, Boon P, Crespel A, Dworetzky BA, Hogenhaven H, Lerche H, Maillard L, Malter MP, Marchal C, Murthy JM, Nitsche M, Pataraia E, Rabben T, Rheims S, Sadzot B, Schulze-Bonhage A, Seyal M, So EL, Spitz M, Szucs A, Tan M, Tao JX, Tomson T. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- Schuele SU. Effects of seizures on cardiac function. J Clin Neurophysiol. 2009;26:302–308. doi: 10.1097/WNP.0b013e3181b7f13b. [DOI] [PubMed] [Google Scholar]
- Seyal M, Bateman LM. Ictal apnea linked to contralateral spread of temporal lobe seizures: intracranial EEG recordings in refractory temporal lobe epilepsy. Epilepsia. 2009;50:2557–2562. doi: 10.1111/j.1528-1167.2009.02245.x. [DOI] [PubMed] [Google Scholar]
- Seyal M, Pascual F, Lee CY, Li CS, Bateman LM. Seizure-related cardiac repolarization abnormalities are associated with ictal hypoxemia. Epilepsia. 2011;52:2105–2111. doi: 10.1111/j.1528-1167.2011.03262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers LP, Massey CA, Gehlbach BK, Granner MA, Richerson GB. Sudden unexpected death in epilepsy: fatal post-ictal respiratory and arousal mechanisms. Respir Physiol Neurobiol. 2013;189:315–323. doi: 10.1016/j.resp.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson T, Walczak T, Sillanpaa M, Sander JAW. Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia. 2005;46(Suppl 11):54–61. doi: 10.1111/j.1528-1167.2005.00411.x. [DOI] [PubMed] [Google Scholar]
- Trindade-Filho EM, de Castro-Neto EF, de A Carvalho R, Lima E, Scorza FA, Amado D, Naffah-Mazzacoratti MG, Cavalheiro EA. Serotonin depletion effects on the pilocarpine model of epilepsy. Epilepsy Res. 2008;82:194–199. doi: 10.1016/j.eplepsyres.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21–26. doi: 10.1111/j.1528-1167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Bortolotto ZA, Mello LM, Schwarz M, Turski L. Seizures produced by pilocarpine in mice: a behavioral, electroencephalographic and morphological analysis. Brain Res. 1984;321:237–253. doi: 10.1016/0006-8993(84)90177-x. [DOI] [PubMed] [Google Scholar]
- Venit EL, Shepard BD, Seyfried TN. Oxygenation prevents sudden death in seizure-prone mice. Epilepsia. 2004;45:993–996. doi: 10.1111/j.0013-9580.2004.02304.x. [DOI] [PubMed] [Google Scholar]
- Wada Y, Nakamura M, Hasegawa H, Yamaguchi N. Intra-hippocampal injection of 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) inhibits partial and generalized seizures induced by kindling stimulation in cats. Neurosci Lett. 1993;159:179–182. doi: 10.1016/0304-3940(93)90828-9. [DOI] [PubMed] [Google Scholar]
- Wada Y, Shiraishi J, Nakamura M, Koshino Y. Effects of the 5-HT3 receptor agonist 1-(m-chlorophenyl)-biguanide in the rat kindling model of epilepsy. Brain Res. 1997;759:313–316. doi: 10.1016/s0006-8993(97)00366-1. [DOI] [PubMed] [Google Scholar]
- Walther DJ, Peter JU, Bashammakh S, Hortnagl H, Voits M, Fink H, Bader M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- Winawer MR, Makarenko N, McCloskey DP, Hintz TM, Nair N, Palmer AA, Scharfman HE. Acute and chronic responses to the convulsant pilocarpine in DBA/2J and A/J mice. Neuroscience. 2007;149:465–475. doi: 10.1016/j.neuroscience.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Lagercrantz H, von Euler C. Effects of substance P and TRH on ventilation and pattern of breathing in newborn rabbits. Acta Physiol Scand. 1981;113:541–543. doi: 10.1111/j.1748-1716.1981.tb06935.x. [DOI] [PubMed] [Google Scholar]
- Zhang S, Liu Y, Rao Y. Serotonin signaling in the brain of adult female mice is required for sexual preference. Proc Natl Acad Sci U S A. 2013;110:9968–9973. doi: 10.1073/pnas.1220712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Chiechio S, Sun YG, Zhang KH, Zhao CS, Scott M, Johnson RL, Deneris ES, Renner KJ, Gereau RW, Chen ZF. Mice lacking central serotonergic neurons show enhanced inflammatory pain and an impaired analgesic response to antidepressant drugs. J Neurosci. 2007a;27:6045–6053. doi: 10.1523/JNEUROSCI.1623-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Gao YJ, Sun YG, Zhao CS, Gereau RW, Chen ZF. Central serotonergic neurons are differentially required for opioid analgesia but not for morphine tolerance or morphine reward. Proc Natl Acad Sci U S A. 2007b;104:14519–14524. doi: 10.1073/pnas.0705740104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Scott M, Chiechio S, Wang JS, Renner KJ, Gereau RW, Johnson RL, Deneris ES, Chen ZF. Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci. 2006;26:12781–12788. doi: 10.1523/JNEUROSCI.4143-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]