

Abstract
Although simple steatosis was originally thought to be a pathologically inert histological change, fat accumulation in the liver may play a critical role not only in disease initiation, but also in the progression to nonalcoholic steatohepatitis and cirrhosis. Therefore, prevention of fat accumulation in the liver may be an effective therapy for multiple stages of nonalcoholic fatty liver disease (NAFLD). Promising beneficial effects of betaine supplementation on human NAFLD have been reported in some pilot clinical studies; however, data related to betaine therapy in NAFLD are limited. In this study, we examined the effects of betaine on fat accumulation in the liver induced by high-sucrose diet and evaluated mechanisms by which betaine could attenuate or prevent hepatic steatosis in this model. Male C57BL/6 mice weighing 20 ± 0.5 g (means ± SE) were divided into four groups (8 mice per group) and started on one of four treatments: standard diet (SD), SD+betaine, high-sucrose diet (HS), and HS + betaine. Betaine was supplemented in the drinking water at a concentration of 1% (wt/vol) (anhydrous). Long-term feeding of high-sucrose diet to mice caused significant hepatic steatosis accompanied by markedly increased lipogenic activity. Betaine significantly attenuated hepatic steatosis in this animal model, and this change was associated with increased activation of hepatic AMP-activated protein kinase (AMPK) and attenuated lipogenic capability (enzyme activities and gene expression) in the liver. Our findings are the first to suggest that betaine might serve as a therapeutic tool to attenuate hepatic steatosis by targeting the hepatic AMPK system.
Keywords: fatty liver, triglyceride, methionine, homocysteine, adiponectin
Nonalcoholic Fatty Liver Disease (NAFLD) is a broad spectrum of liver abnormalities ranging from simple hepatic steatosis (accumulation of triglyceride inside hepatocytes) to nonalcoholic steatohepatitis (necrosis and inflammation), with some people ultimately progressing to fibrosis and cirrhosis and liver failure. Because of its high prevalence in conjunction with obesity, diabetes, and insulin resistance, NAFLD is being increasingly appreciated as a hepatic manifestation of the metabolic syndrome, and it represents a major cause of liver-related morbidity and mortality (2, 7, 15).
The pathogenesis of NAFLD has not yet been defined, although several hypotheses exist. One generally accepted theory is the “two-hit” hypothesis, wherein the first hit involves the development of hepatic steatosis, rendering the liver more susceptible to a second undefined hit, resulting in more severe liver damage (9). Fat accumulation in the liver results from an imbalance among the hepatic uptake of free fatty acids, metabolism (synthesis and oxidation) of fatty acids by liver, and export of triglyceride from the liver through very low density lipoproteins. Although simple steatosis was originally thought to be a pathologically inert histological change, there is now convincing evidence that fat accumulation in the liver plays a critical role not only in the initiation, but also in the progression of NAFLD. For instance, steatotic livers are more sensitive to hepatotoxicity caused by agents such as endotoxin (37). Furthermore, the degree of fatty infiltration is predictive of the severity of later stages of NAFLD (i.e., fibrosis and cirrhosis) (31). Therefore, prevention and attenuation of fat accumulation in the liver may lead to new therapies for multiple stages of NAFLD.
Betaine (trimethylglycine) is a naturally occurring metabolite of choline and an essential biochemical component of the methionine-homocysteine cycle (8). It has been reported to be hepatoprotective in a variety of experimental animal models of liver diseases, including alcoholic liver disease and bile acid-induced liver injury (3, 13, 16, 20). The therapeutic effects of betaine on NAFLD have also been investigated and reported in clinical studies. For instance, in a pilot study by Miglio et al. (27), improved aminotransferases and hepatic steatosis were observed when patients with NAFLD were given an oral form of betaine glucuronate combined with diethanolamine glucuronate and nicotinamide ascorbate. A pilot study by Abdelmalek et al. (1) showed that anhydrous betaine supplementation (20 g daily) in 10 nonalcoholic steatosis patients for 1 yr attenuated liver damage and improved overall steatosis, necroinflammatory grade, and fibrosis on repeat biopsy in patients. Although they appear to be promising, data relating betaine to NAFLD are limited, and cellular and molecular mechanisms remain elusive because of the lack of detailed experimental investigations. Thus detailed studies are warranted before any recommendations regarding the use of betaine in NAFLD can be made.
A number of animal models have been developed to study NAFLD (21). Among these are nutritional models that are based on the voluntary access of animals to complete diets containing modified fat or carbohydrate (sucrose) contents. In this study, the experimental model of long-term (16 wk) high-sucrose diet feeding to mice was used to induce fatty liver. Our aims were to investigate the effects of betaine on fat accumulation in the liver induced by high-sucrose diet and to identify mechanisms by which betaine could attenuate or prevent hepatic steatosis in this model.
MATERIALS AND METHODS
Animal model and experimental protocol
Male C57BL/6 mice weighing 20 ± 0.5 g (means ± SE) were obtained from the Jackson Laboratory (Bar Harbor, ME). The mice were housed in the animal quarters at the University of Louisville Research Resources Center, and the studies were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association of Accreditation of Laboratory Animal Care. Initially, all mice were housed in conventional conditions and fed standard diet and water ad libitum at the animal facility (Research Resource Facility) for 1 wk before experiments began. Thereafter, the mice were divided into four groups (n = 8 per group) and started on one of four treatments: standard diet (SD), SD supplemented with betaine (SD+BT), high-sucrose diet (HS), and HS supplemented with betaine (HS+BT). Grain-based standard diet (2018, Harlan Teklad, Madison, WI) contains (by weight) 18.9% crude protein, 5% fat, and 55.7% carbohydrate (5% as simple sugar and 41.24% as starch). The high-sucrose diet was made from purified ingredients and the composition was identical to that used by Feldstein et al. (10) (TD 02366; Harlan Teklad, Madison, WI), which consisted of (by weight) 18% protein, 5% fat, 69.1 carbohydrate (65% as sucrose and 2.6% as corn starch), 4% mineral mixture, and 1% vitamin mixture. Betaine was supplemented in the drinking water at a concentration of 1% (wt/vol) (anhydrous; Sigma, St. Louis, MO). The dosage of betaine utilized in this experiment was based on previous report by Ji et al. (16) showing that betaine at this dose had protective effects on alcohol-induced fatty liver and liver injury. Mice were maintained on the treatments for 16 wk before being killed.
Cells and culture conditions
HepG2 cells, a human hepatoma cell line, were obtained from the American Type Culture Collection (Manassas, VA) and were cultured in DMEM containing 10% (vol/vol) fetal bovine serum, 2 mmol/l glutamine, 5 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in a humidified O2-CO2 (19:1) atmosphere.
Histological examination
At the time of killing, the liver was harvested and small pieces were fixed immediately in 10% buffered formalin. After paraffin embedding, 5-μm sections were deparaffinized in xylene and were rehydrated through a series of decreasing concentrations of ethanol. Sections were stained with hematoxylin and eosin. Alternatively, portions of fresh liver were flash frozen and cryostat sections were cut and prepared for staining with Oil Red O. Photomicrographs were taken on an a Nikon Eclipse E600 microscope (Fryer, Cincinnati, OH) equipped with a digital camera (SPOT; Diagnostic Instruments, Sterling Heights, MI).
Plasma biochemical assays
The plasma biochemical assays were performed with commercially available kits: glucose, triglyceride, cholesterol, alanine aminotransferase (Infinity, Thermo Electron, Melbourne, Australia), free fatty acids (Waco Chemicals, Richmond, VA), TNF-α (R&D System, Minneapolis, MN), adiponectin, and insulin (Linco Research, St. Charles, MO).
Total RNA isolation
Frozen liver samples (~70 mg) were transferred to 1.0 ml of Trizol (Invitrogen, Carlsbad, CA) and homogenized in a Mixer Mill 300 tissue homogenizer (Retsch, Germany). Total RNA was isolated according to the manufacturer’s protocol with an additional phenol-chloroform extraction. Isolated RNA was resuspended in RNA storage solution (Ambion, Austin, TX), quantified (A260), and assessed for purity by determining the A260/A280 ratio and by visual inspection of 1.0 μg on a denaturing gel.
Quantitative real-time RT-PCR
Total RNA was extracted from liver tissue as described in the previous paragraph. For each sample, 1.0 μg of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit as described by the manufacturer (Applied Biosystems, Foster City, CA). The cDNA (1.0 μl) was used as a template in a 25-μl PCR reaction solution containing 10.5 μl, 1 μl 10 μM gene-specific primers, and 12.5 μl 1 × SYBR Green PCR master mix (SuperArray Bioscience, Frederick, MD). PCR amplification was conducted in MicroAmp Optical 96-well reaction plates (Applied Biosystems) on an Applied Biosystems PRISM 7000 sequence detection system under the following conditions: initial denaturation and enzyme activation for 10 min at 95°C, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Each plate contained duplicate standards of purified PCR products of known template concentrations covering seven orders of magnitude to interpolate relative template concentrations of the samples from the standard curves of log copy number vs. threshold cycle (CT). No-template controls (NTC) were also included on each plate. Samples with a CT value within 2 SD of the mean CT values for the NTCs were considered below the limits of detection. The copy number of each unknown sample for each gene was standardized to a house-keeping gene (mouse 18s rRNA) to control for differences in RNA loading, quality, and cDNA synthesis. For graphing purposes, the relative expression levels were scaled such that the expression level of the time-matched control group was equal to 1.
Preparation of nuclear extracts
To isolate nuclear proteins we used a Nuclear Extraction Kit (Imgenex, San Diego, CA) according to manufacturer’s instructions. Briefly, small pieces of liver tissue were washed twice with 5 ml of ice-cold PBS/PMSF buffer, homogenized in 5 ml 1 × hypotonic buffer supplemented with 1 mM DTT and 1% detergent, and put on ice for 30 min. After centrifugation at 10,000 rpm at 4°C for 10 min, supernatants (cytoplasmic fraction) were removed and 500 μl of complete lysis buffer was added to the nuclei and incubated for 30 min at 4°C with rocking. The samples were vortexed and centrifuged at 14,000 rpm for 10 min at 4°C. Supernatants containing nuclear proteins were harvested and transferred to prechilled tubes.
Western blot analysis
Tissues or cells were lysed in Western lysis buffer consisting of the following: 20 mM Tris · HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 2% Nonidet P-40, 1 mM EDTA, pH 8.0, 20 mM sodium fluoride, 30 mM sodium pyrophosphate, 0.2% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 1 mM sodium vanadate, 50 μM leupeptin, and 5 μM aprotinin. Samples were incubated on ice with frequent vortexing for 15 min and centrifuged for 20 min at 18,000 g. The protein content of each supernatant was quantified via a protein assay reagent from Bio-Rad Laboratories (Hercules, CA) in accordance with the manufacturer’s instructions.
Sixty micrograms of each supernatant sample of proteins were separated by electrophoresis through a 12% polyacrylamide gel [8% gel for acetyl-CoA carboxylase (ACC)] and transferred to 0.45 μm Immobilin-P polyvinylidene difluoride membrane (PerkinElmer Life Sciences). After transfer, membranes were blocked in 5% (wt/vol) nonfat dry milk in PBS-0.1% Tween 20 and probed with the antibodies specified. Horseradish peroxidase-conjugated secondary antibodies (Sigma) and enhanced chemiluminescence substrate kit (PerkinElmer Life Science) were used in the detection of specific proteins.
HPLC assay
Deproteinized tissue homogenates (4% metaphosphoric acid) were prepared and hepatic S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), reduced form of glutathione (GSH), and homocysteine levels were determined by a HPLC method using a 5 μm Hypersil C-18 column (250 × 4.6 mm). The mobile phase consisted of 40 mmol/l ammonium phosphate, 8 mmol/l heptane sulfonic acid (ion-pairing reagent, pH 5.0), and 6% acetonitrile and was delivered at a low rate of 1.0 ml/min. SAM and SAH were detected using a Waters 740 UV detector (Milford, MA) at 254 nm. An internal standard, S-adenosylethionine, was added to all samples and standard solutions to a concentration of 100 nmol/ml. For GSH and homocysteine assays, 20 μl samples were injected onto a reversed-phase C18 column (Val-U-Pak HP, fully endcapped octadecyl silane, 5 μm, 250 × 4.6 mm; ChromTech, Apple Valley, IN). The mobile phase, which consisted of a solution of 0.1 M monochloroacetic acid and 2 mmol/l heptane sulfonic acid at pH 2.8 (98%) and acetonitrile (2%), was delivered at a low rate of 1 ml/min. The compounds were detected in the eluant with a Bioanalytical Systems (West Lafayette, IN) dual LC4B amperometric detector using two Au-Hg electrodes in series with potentials of 1.2 and 0.15 V for the upstream and downstream electrodes, respectively. Standard curves for the analytes were plotted as peak area vs. concentration of the analyte. Protein concentrations were measured by a protein assay kit from Bio-Rad Laboratories in accordance with the manufacturer’s instructions.
Intracellular triglyceride measurement
Liver tissues were homogenized and hepatic total lipids were extracted according to Bligh and Dyer (4) and redissolved in 2% Triton X-100 in water. Hepatic triglyceride content was determined by enzymatic colorimetric methods using commercially available kits (Sigma). For the in vitro study, HepG2 cells were plated into a six-well plate for overnight. Twenty hours after treatments, cells were washed twice with 1 ml of cold PBS. Then 0.3 ml of 0.25 M NaOH was added to each well. Samples were collected by scraping cells into a 2-ml tube and total lipids were extracted according to Bligh and Dyer (4). The triglyceride content was determined by enzymatic colorimetric methods using commercially available kits (Sigma).
Statistical analysis
All data were expressed as means ± SD. Statistical analysis was performed using a one-way ANOVA and was analyzed further by Newman-Keuls test for statistical difference. Differences between treatments were considered to be statistically significant at P < 0.05.
RESULTS
Body weight
Absolute body weight of each mouse from each group was measured at the end of the feeding period, and body weight gain was calculated and shown in Fig. 1. No difference in either absolute body weight or body weight gain between the SD and SD+BT groups was observed at the end of feeding period. Compared with the SD group, mice in the HS group had significantly larger body weight and greater body weight gain. Sixteen-week high-sucrose diet feeding was accompanied by 35% (11.36 vs. 15.39 g) more body weight gain than those in the SD group. Supplementation of betaine in the HS group (HS+BT) caused a numerically less body weight gain when compared with the HS group; however, this decrease was not statistically significant.
Fig. 1.
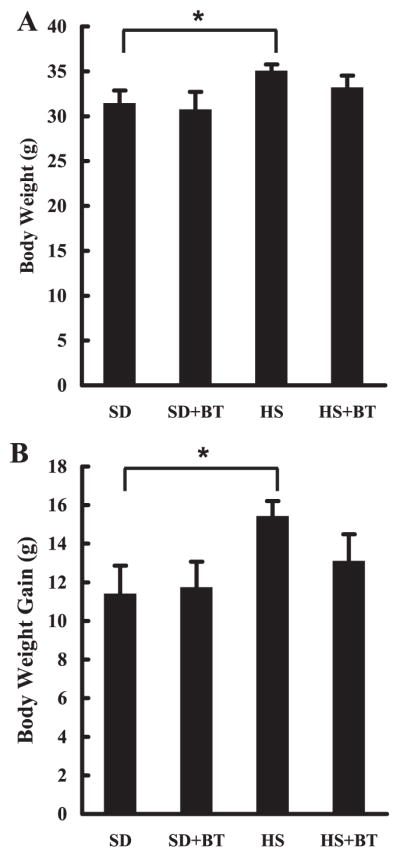
Betaine supplementation had no effect on body weight change in C57BL/6 mice fed high-sucrose diet. A: changes of absolute body weight. C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in drinking water for 16 wk. Data are means ± SD (n = 8), *P < 0.05. SD, standard diet; HS, high-sucrose diet; BT, betaine. B: changes of body weight gain. C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in drinking water (n = 8 mice per group) for 16 wk. Data are means ± SD (n = 8), *P < 0.05.
Fat accumulation in the liver
We next examined changes of hepatic triglyceride content in different groups. As we expected, mice in the HS group displayed significantly increased hepatic triglyceride content when compared with those in the SD group (Fig. 2A). The histological examination confirmed this and showed that the liver from the SD group was normal whereas obvious hepatic fat infiltration was observed in the HS group, but no inflammation or obvious cell death were detected (Fig. 2, B and C). Betaine significantly reduced hepatic triglyceride accumulation induced by high-sucrose feeding, which was confirmed by both hematoxylin and eosin and Oil Red O staining. Since betaine supplementation had no effects on either body weight or hepatic triglyceride content of mice fed standard diet, we excluded this group in all following measurements to simplify our description.
Fig. 2.

Betaine supplementation attenuated fat accumulation in the liver induced by prolonged high-sucrose feeding. A: hepatic triglyceride content. C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in drinking water for 16 wk. Data are means ± SD (n = 8). Bars with different letters differ significantly (P < 0.05). B: formalin-fixed and paraffin-embedded liver sections were stained with hematoxylin and eosin. Livers from SD-fed animals had normal architecture. In contrast, livers from HS-fed animals had obvious fat infiltration with no inflammation or obvious cell death observed. Betaine supplementation groups had significantly less fat accumulation in the liver compared with HS-fed animals. Original magnification ×130. C: flash-frozen liver sections were stained with Oil Red O. SD-fed animals had no obvious fat accumulation in the liver. HS feeding resulted in profound hepatic steatosis, featured by remarkable amount of fat droplet in the liver. Betaine supplementation significantly alleviated hepatic fat accumulation induced by HS diet feeding.
Blood biochemistry
The data in Table 1 showed the effects of high-sucrose diet feeding with and without betaine supplementation on some routine plasma parameters. After 16-wk feeding, mice in the HS group had increased levels of glucose, cholesterol, and insulin in plasma. Unexpectedly, free fatty acids levels in plasma were significantly lowered in the HS group compared with the SD group. No significant changes were observed in alanine aminotransferase, triglyceride, TNF-α, and adiponectin levels between the SD group and the HS group. Betaine supplementation alleviated elevations of plasma glucose, cholesterol, and insulin levels resulting from high-sucrose feeding, with no effects on free fatty acid and triglyceride changes. Plasma adiponectin levels in HS+BT group were slightly increased compared with those in both SD and HS groups.
Table 1.
Biochemical parameters of plasma at the end of the feeding period
| Parameters | SD | HS | HS+BT |
|---|---|---|---|
| ALT, mU/ml | 28.3±3.0 | 31.2±2.9 | 28.6±1.9 |
| Glucose, mM | 8.9±0.3a | 13.7±1.1b | 10.8±0.7c |
| Insulin, ng/ml | 2.0±0.3a | 5.1±0.5b | 2.9±0.9a |
| TG, mg/dl | 96.7±11.7 | 82.2±13.8 | 84.7±15.5 |
| Cholesterol, mg/dl | 33.9±1.8a | 63.8±4.4b | 52.4±5.6c |
| NEFA, meq/l | 0.52±0.04a | 0.35±0.05b | 0.34±0.05b |
| TNF-α, pg/ml | 189.5±18.8 | 186.8±22.3 | 191.3±27.2 |
| Adiponectin, ng/ml | 18.2±2.8a | 16.8±2.1a | 23.8±1.7b |
Values are means ± SD, n = 8. SD, standard diet; HS, high-sucrose diet; BT, betaine supplementation; ALT, alanine aminotransferase; TG, triglyceride; NEFA, nonesterified fatty acid. Means in a row with superscripts without a common letter differ, P ≤ 0.05.
Hepatic methionine metabolism
Since betaine is an essential biochemical component of the hepatic methionine-homocysteine cycle and acts as a donor of methyl groups, the effects of high-sucrose feedings with and without betaine supplementation on hepatic methionine metabolism were examined by measuring concentrations of several metabolites in the hepatic methionine metabolic pathway, particularly SAM, SAH, homocysteine, and GSH levels. The status of transmethylation reactions in the liver was gauged by the SAM-to-SAH ratio. As depicted in Fig. 3, prolonged high-sucrose feeding did not produce significant effects on hepatic SAM (Fig. 3A), SAH, and GSH levels (data not shown); however, it induced a slight increase in hepatic homocysteine levels (Fig. 3B). Betaine markedly increased hepatic SAM levels (Fig. 3A) with no effects on SAH levels (data not shown), leading to a significant elevation in hepatic SAM-to-SAH ratio (data not shown). Furthermore, betaine abolished high-sucrose diet-induced elevation of hepatic homocysteine levels (Fig. 3B). No effects on GSH levels were found following betaine supplementation (data not shown).
Fig. 3.

Changes of hepatic methionine metabolism. Male C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in the drinking water for 16 wk. Liver samples were homogenized with 4% metaphosphoric acid and hepatic S-adenosylmethionine (SAM) and homocysteine contents were assayed by high-performance liquid chromatography. A: changes of hepatic SAM levels. Data are means ± SD (n = 8). Bars with different letters differ significantly (P < 0.05). B: hepatic homocysteine levels. Data are means ± SD (n = 8). Bars with different letters differ significantly (P < 0.05).
Hepatic AMPK and ACC activation
AMP-activated protein kinase (AMPK) has been proposed to act as a fuel gauge in mammalian cells and plays a critical role in the repression of glycolytic and lipogenic pathways and the promotion of fatty acid oxidation in the liver. To determine whether the antisteatotic functions of betaine in our animal model could be ascribed to its effects on AMPK activation, we examined AMPK phosphorylation (activation) in each group by detecting the amount of phosphorylated AMPK proteins by Western blot analysis. As shown in Fig. 4, whereas long-term high-sucrose diet feeding had no obvious effects on the activity of AMPK compared with SD feeding, betaine supplementation to the high-sucrose diet resulted in enhanced activation of AMPK (Fig. 4, A and B), as demonstrated by substantially increased protein phosphorylation. ACC is a rate-limiting enzyme in hepatic de novo fatty acid synthesis and a well-identified protein among the large number of AMPK targets. Therefore, we examined protein levels of ACC in the liver and its phosphorylation (deactivation) status in different groups. As shown in Fig. 4, C and D, high-sucrose feeding resulted in increased ACC protein levels in the liver, which was attenuated by betaine supplementation. Concomitant with its effect on AMPK activity, betaine supplementation markedly alleviated hepatic ACC activation, shown by Western blot as significantly increased protein phosphorylation.
Fig. 4.

Betaine supplementation resulted in increased phosphorylation (p-) of hepatic AMP-activated protein kinase (AMPK) and inhibited acetyl-CoA carboxylase (ACC) activation. Male C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in the drinking water for 16 wk. Total protein extracts from liver tissues were prepared thereafter. Forty micrograms of protein were subjected to Western blot analysis for AMPK or ACC phosphorylation using antibodies specific for AMPK phosphorylated at Thr172 and ACC phosphorylated at Ser79. A: Western blot assay for AMPK. Betaine supplementation increased phosphorylation of liver AMPK (activation). B: quantification of p-AMPK protein levels in different groups. Levels were normalized to total AMPK. Data are means ± SD (n = 6–7). Bars with different letters differ significantly (P < 0.05). C: Western blot assay for ACC. Betaine supplementation attenuated the increase of ACC protein content induced by high-sucrose feeding and increased phosphorylation of ACC (inhibition) in the liver. D: quantification of p-ACC and total ACC protein levels in different groups. Levels were normalized to actin. Data are means ± SD (n = 6–7). Bars with different letters differ significantly (P < 0.05).
SREBP and ChREBP activation in the liver
Sterol regulatory element-binding protein-1c (SREBP-1c) and carbohydrate response element-binding protein (ChREBP) are two critical transcriptional factors for gene expression [e.g., ACC, fatty acid synthase (FAS)] in the hepatic lipogenic pathway. The mature SREBP-1 and ChREBP proteins in nuclear extracts of mouse liver from the three different groups were examined by Western blot. As shown in Fig. 5, long-term high-sucrose diet feeding led to an obvious increase of mature SREBP-1 and ChREBP protein in the nucleus compared with the standard diet feeding. This increase was inhibited by betaine supplementation.
Fig. 5.

Betaine supplementation suppressed increases of sterol regulatory element-binding protein-1 (SREBP-1) and carbohydrate response element-binding protein (ChREBP) in nuclei induced by high-sucrose feeding. Male C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in the drinking water for 16 wk. Nuclear proteins were isolated from fresh liver tissues thereafter. Forty micrograms of protein were subjected to Western blot analysis for SREBP-1 or ChREBP using specific antibodies. A: prolonged high-sucrose feeding caused significant increase of both SREBP-1 and ChREBP levels in the nuclei, which were suppressed by betaine supplementation. m denotes mature form of the protein (in the nuclei). B: quantification of mSREBP-1c and mChREBP protein levels in different groups. Data are means ± SD (n = 6–7). Bars with different letters differ significantly (P < 0.05).
Gene expression of ACC, FAS, and SREBP-1d in the liver
The effects of high-sucrose diet feeding with or without betaine supplementation on gene expression of ACC, FAS, and SREBP-1d in liver tissue were examined by real-time PCR analysis. As shown in Fig. 6, high-sucrose feeding caused significant elevation in the expression of all three genes, which was significantly attenuated by betaine supplementation.
Fig. 6.
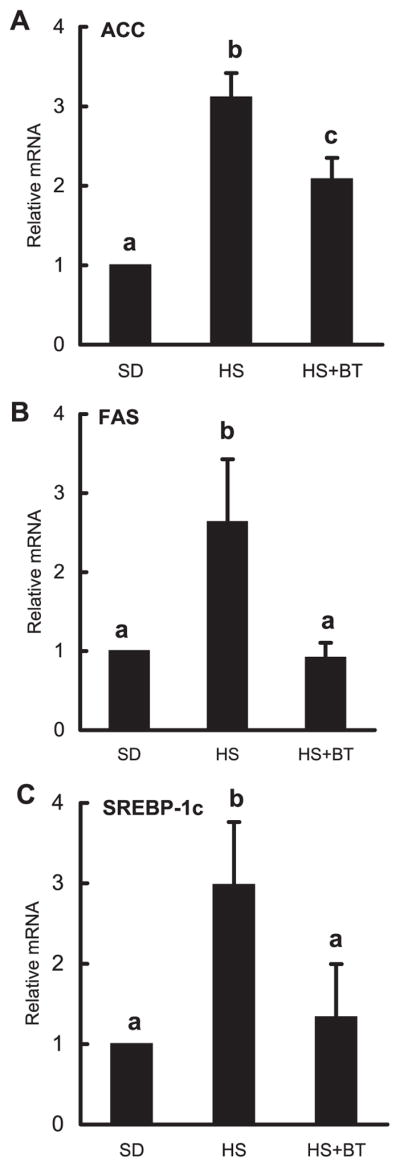
Betaine supplementation suppressed high-sucrose feeding-induced gene expression of ACC (A), fatty acid synthase (FAS; B), and SREBP-1d (C) in the liver. Male C57BL/6 mice were fed with high-sucrose diet in the presence or absence of betaine supplementation (1%) in the drinking water for 16 wk. Total RNA from liver tissues were isolated thereafter and subjected to real-time RT-PCR assay to quantitate mRNAs for ACC, FAS, and SREBP-1c. HS significantly elevated expression of all 3 genes compared with the SD group. Supplementation with betaine suppressed the elevated expression of these genes induced by HS feeding. Data are means ± SD (n = 4). Bars with different letters differ significantly (P < 0.05).
Betaine activated AMPK and reduced lipid accumulation in HepG2 cells
To determine the effect of betaine on the activation of AMPK in vitro, the betaine was added to the culture medium of HepG2 cells. Both time-course and dose-response analyses were performed by Western blot assay using anti-phospho-AMPK antibody. As shown in Fig. 7A, betaine at 2 mM and 5 mM activated AMPK (shown by increased phosphorylation) at the 1-h time point. At 2 h, all levels (0.5, 1, 2, and 5 mM) of betaine activated AMPK, and this activation displayed a dose-dependent pattern. The effects of betaine on AMPK activation subsided after 4 h.
Fig. 7.

Betaine activated AMPK and reduced lipid accumulation in HepG2 cells. A: betaine activated AMPK in vitro. HepG2 cells were cultured in serum-free DMEM medium overnight and incubated in DMEM containing varying concentrations of betaine for 1, 2, and 4 h, respectively. Total cell extracts were subjected to Western blot analysis with phospho-Thr172 AMPK (p-AMPK) and total AMPKα (t-AMPK) antibodies. Western blots shown represent 3 independent experiments. B and C: betaine lowered the lipid accumulation induced by glucose inclusion in the media in HepG2 cells. HepG2 cells were cultured in serum-free DMEM medium overnight and incubated in DMEM containing either normal (5.5 mM) or high (25 mM) glucose in the absence (Con) or presence of 2 mM betaine for an additional 24 h. Data represent a mean of at least 3 experiments ± SD, *P < 0.05.
To determine whether betaine could attenuate lipid accumulation induced by extracellular glucose, HepG2 cells were incubated with either normal (5.5 mM) or high (25 mM) glucose concentrations in the absence or presence of betaine (2 mM) for 24 h. At the normal concentration of glucose, the intracellular contents of triglyceride (Fig. 7B) and cholesterol (Fig. 7C) were lowered ~30% by betaine. A high concentration of glucose in the medium elevated intracellular triglyceride and cholesterol levels, which was also reduced by betaine supplementation.
DISCUSSION
In mammals, the liver is the principal organ responsible for the conversion of excess dietary carbohydrate into triglycerides. Both glucose and fructose are metabolized in the liver to provide the precursor, acetyl CoA, for fatty acid synthesis. Fatty acids are then incorporated into triglycerides that function as a long-term energy reservoir. Using an experimental model with high-sucrose feeding, we demonstrated that long-term (16 wk) feeding of high-sucrose diet to mice induced remarkable hepatic steatosis. Betaine, when supplemented in the drinking water, significantly attenuated hepatic steatosis in this animal model, and this change was associated with increased activation of hepatic AMPK in the liver. Furthermore, betaine supplementation attenuated ACC activation (increased phosphorylation) and increased gene expressions of SREBP-1c, ACC, and FAS in the liver induced by prolonged high-sucrose feeding. Therefore, our results are the first to suggest that betaine might serve as a therapeutic tool to attenuate hepatic steatosis by targeting the hepatic AMPK system.
AMPK is a phylogenetically conserved serine/threonine protein kinase that has been proposed to act as a “metabolic master switch” mediating the cellular adaptation to environmental or nutritional stress factors (6). AMPK has been implicated in the control of hepatic glucose and lipid homeostasis via multiple effects on genes and on short-term regulation of specific enzymes. Activation of AMPK (phosphorylation of its α-subunit at site Thr172) leads to an inhibition of fatty acid synthesis by phosphorylating critical enzyme(s) involved in the process of lipogenesis. Among these is ACC, which is the rate-limiting enzyme for the synthesis of long-chain fatty acids (28, 29). Malonyl-CoA is not only a critical precursor for the biosynthesis of fatty acids, but also a potent inhibitor of mitochondrial fatty acid oxidation through its inhibitory action on carnitine palmitoyltransferase-1 (18). Moreover, the long-term activation of AMPK plays an important role in repressing expression of genes encoding enzymes involved in glycolytic and lipogenic process in the liver. Activation of AMPK inhibits transcriptional stimulation of ACC and FAS induced by insulin or glucose in primary hepatocytes (11). Sustained activation of hepatic AMPK decreases transcriptional activation of SREBP-1c and ChREBP (12). Furthermore, activation of AMPK leads to decreased DNA binding activity of ChREBP and subsequent transcriptional inhibition of lipogenesis in the liver (17). Recent studies suggest that activation of AMPK accounts for some of the antidiabetic and antisteatotic actions of a number of drugs, such as metformin and thiazolidinediones, and is required for the lipid-lowering effects of metformin in cultured hepatocytes (22, 39, 40). Taken together, the overall effects of AMPK activation in the liver are to inhibit lipogenesis and stimulate fatty acid oxidation. Therefore, attempts to find or generate novel, safe, and efficacious therapies for hepatic steatosis via targeting the AMPK system are definitely worthy of pursuit.
Concomitant with substantially attenuated fat accumulation in the liver, betaine supplementation enhanced AMPK activation, inhibited ACC activity, and repressed gene expressions of SREBP-1 and FAS, suggesting that targeting the hepatic AMPK system may play an important role in betaine’s anti-steatotic function and potential beneficial effects in NAFLD. These in vivo observations were further supported by our in vitro experiments using HepG2 cells, in which betaine treatment led to AMPK activation in both time- and dose-dependent manners and decreased fat accumulation following glucose addition.
The finding that betaine can induce hepatic AMPK activation is novel and intriguing, but the mechanism for this remains to be determined. Although AMPK is normally activated in the liver by an intracellular change in AMP-to-ATP ratio (17), recent findings showed that hepatic AMPK could be activated by adiponectin (an adipocyte-derived hormone) and a number of drugs (e.g., metformin), which were believed to be independent of any apparent changes in the AMP-to-ATP ratio (40). In the present study, the effects of betaine on the intracellular AMP-to-ATP ratio were not investigated, nor were possible effects on upstream enzymes modulating AMPK activation, such as AMPKK and PP2A (23); however, a modest increase in the plasma adiponectin concentration in the betaine supplementation group was observed. There is convincing evidence that adiponectin is implicated in the pathogenesis of NAFLD. Recent studies showed that administration of exogenous adiponectin reversed experimental forms of NAFLD (36). Moreover, adiponectin exerts its antisteatotic actions, at least in part, through activation of AMPK in the liver (34). In this context, it is reasonable to extrapolate that betaine may enhance hepatic AMPK activation in part by increasing plasma adiponectin levels in vivo. Therefore, it seems that both direct, as we observed in HepG2 cells, and indirect (via increasing circulating adiponectin levels) mechanisms are involved in the regulation by betaine of hepatic AMPK activation.
In addition to enhancement of hepatic AMPK activation, other metabolic alterations arising from betaine supplementation may also be pertinent to its antisteatotic action. Although the underlying causes of alcoholic fatty liver disease and NAFLD are clearly different, there are histological similarities and mechanistic factors with disturbances in normal pathways of hepatic metabolism (25, 32, 33, 38). The hepatoprotective roles of betaine in alcoholic liver disease have been reported in a number of experimental studies (3, 16, 19, 20). The effects mainly resulted from its ability to rescue abnormalities in hepatic methionine metabolism, as documented by several research groups including our own (14, 24, 30). Depletion of hepatic SAM together with a concomitant elevation of SAH and homocysteine levels have been postulated to be an etiologic contributor of alcohol-induced fatty liver and liver injury (16, 19, 30). In this study, we examined the effects of long-term feeding of high-sucrose diet to mice on hepatic methionine metabolism by measuring hepatic levels of metabolites in the methionine metabolic pathway. Similar to the previous observations in alcoholic liver disease, long-term high-sucrose diet feeding led to significant elevation in hepatic homocysteine levels, which was prevented by betaine supplementation. In addition, betaine supplementation efficiently improved hepatic methylation status, as reflected by a substantially increased SAM-to-SAH ratio. Abnormal lipid metabolism deriving from increased hepatic homocysteine levels has been reported in various studies. Although hepatic homocysteine levels in all these animal models were much higher than observed in our study, a recent study showed that incubation of HepG2 cells with homocysteine at 50–100 μM could induce cAMP response element-binding protein (CREB) phosphorylation and subsequently increased CREB/DNA binding activity (35), implying that modest elevation of hepatic homocysteine levels could lead to prominent changes in intracellular signal transductions involved in lipid metabolism. Therefore, the contribution of abnormal hepatic methionine metabolism resulting from high-sucrose diet feeding in the development of fatty liver should also be taken into account, and further investigations are still required.
Taken together, our data demonstrate that long-term feeding of a high-sucrose diet to mice induced obesity, hepatic steatosis, and insulin resistance. Betaine supplementation in the drinking water resulted in remarkable improvement of hepatic steatosis, which was associated mainly with significant enhancement of hepatic AMPK activation. Moreover, betaine supplementation also rescued abnormalities in hepatic methionine metabolism and alleviated insulin resistance. Unlike other AMPK activators [such as metformin and 5-aminoimi-dazole-4-carboxamide-1-β-D-ribofuranoside (AICAR)] with potential side effects, oral anhydrous betaine solution is safe in doses ranging from 3 to 30 g/day, as has been shown by both experimental and clinical studies. Thus the present study provides strong evidence for further evaluation of the potential therapeutic role of betaine in NAFLD.
Acknowledgments
GRANTS
This research was supported by grants from the National Institutes of Health (Z. Song, C. J. McClain, I. Deaciuc, D. Hill, and Z. Zhou) and the Department of Veterans Affairs (C. J. McClain).
References
- 1.Abdelmalek MF, Angulo P, Jorgensen RA, Sylvestre PB, Lindor KD. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: results of a pilot study. Am J Gastroenterol. 2001;96:2711–2717. doi: 10.1111/j.1572-0241.2001.04129.x. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P, Lindor KD. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2002;17:S186–S190. doi: 10.1046/j.1440-1746.17.s1.10.x. [DOI] [PubMed] [Google Scholar]
- 3.Barak AJ, Beckenhauer HC, Badakhsh S, Tuma DJ. The effect of betaine in reversing alcoholic steatosis. Alcohol Clin Exp Res. 1997;21:1100–1102. [PubMed] [Google Scholar]
- 4.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 5.Caldwell SH, Argo CK, Al-Osaimi AM. Therapy of NAFLD: insulin sensitizing agents. J Clin Gastroenterol. 2006;40:S61–S66. doi: 10.1097/01.mcg.0000168647.71411.48. [DOI] [PubMed] [Google Scholar]
- 6.Carling D. AMP-activated protein kinase: balancing the scales. Biochimie. 2005;87:87–91. doi: 10.1016/j.biochi.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Choudhury J, Sanyal AJ. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:575–894. doi: 10.1016/j.cld.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Craig SA. Betaine in human nutrition. Am J Clin Nutr. 2004;80:539–549. doi: 10.1093/ajcn/80.3.539. [DOI] [PubMed] [Google Scholar]
- 9.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol. 2003;39:978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 11.Ferre P, Azzout-Marniche D, Foufelle F. AMP-activated protein kinase and hepatic genes involved in glucose metabolism. Biochem Soc Trans. 2003;31:220–223. doi: 10.1042/bst0310220. [DOI] [PubMed] [Google Scholar]
- 12.Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes. 2005;54:1331–1339. doi: 10.2337/diabetes.54.5.1331. [DOI] [PubMed] [Google Scholar]
- 13.Graf D, Kurz AK, Reinehr R, Fischer R, Kircheis G, Haussinger D. Prevention of bile acid-induced apoptosis by betaine in rat liver. Hepatology. 2002;36:829–839. doi: 10.1053/jhep.2002.35536. [DOI] [PubMed] [Google Scholar]
- 14.Halsted CH, Villanueva JA, Devlin AM, Niemela O, Parkkila S, Garrow TA, Wallock LM, Shigenaga MK, Melnyk S, James SJ. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc Natl Acad Sci USA. 2002;99:10072–10077. doi: 10.1073/pnas.112336399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James OF, Day CP. Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol. 1998;29:495–501. doi: 10.1016/s0168-8278(98)80073-1. [DOI] [PubMed] [Google Scholar]
- 16.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 17.Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–3835. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- 18.Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- 19.Kharbanda KK, Mailliard ME, Baldwin CR, Beckenhauer HC, Sorrell MF, Tuma DJ. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J Hepatol. 2007;46:314–321. doi: 10.1016/j.jhep.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 20.Kharbanda KK, Rogers DD, 2nd, Mailliard ME, Siford GL, Barak AJ, Beckenhauer HC, Sorrell MF, Tuma DJ. Role of elevated S-adenosylhomocysteine in rat hepatocyte apoptosis: protection by betaine. Biochem Pharmacol. 2005;70:1883–1890. doi: 10.1016/j.bcp.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 21.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 22.LeBrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, Tomas E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291:E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 23.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–1183. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu SC, Huang ZZ, Yang H, Mato JM, Avila MA, Tsukamoto H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in alcoholic rat liver. Am J Physiol Gastrointest Liver Physiol. 2000;279:G178–G185. doi: 10.1152/ajpgi.2000.279.1.G178. [DOI] [PubMed] [Google Scholar]
- 25.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 26.McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40:S17–S29. doi: 10.1097/01.mcg.0000168645.86658.22. [DOI] [PubMed] [Google Scholar]
- 27.Miglio F, Rovati LC, Santoro A, Setnikar I. Efficacy and safety of oral betaine glucuronate in non-alcoholic steatohepatitis. A double-blind, randomized, parallel-group, placebo-controlled prospective clinical study. Arzneimittelforschung. 2000;50:722–727. doi: 10.1055/s-0031-1300279. [DOI] [PubMed] [Google Scholar]
- 28.Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, Saha AK. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- 29.Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem. 2003;253:65–70. doi: 10.1023/a:1026053302036. [DOI] [PubMed] [Google Scholar]
- 30.Song Z, Zhou Z, Uriarte S, Wang L, Kang YJ, Chen T, Barve S, McClain CJ. S-adenosylhomocysteine sensitizes to TNF-alpha hepato-toxicity in mice and liver cells: a possible etiological factor in alcoholic liver disease. Hepatology. 2004;40:989–997. doi: 10.1002/hep.20412. [DOI] [PubMed] [Google Scholar]
- 31.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714–1719. [PubMed] [Google Scholar]
- 32.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;16343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 33.Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis. 2004;24:99–106. doi: 10.1055/s-2004-823104. [DOI] [PubMed] [Google Scholar]
- 34.Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin-a key adipokine in the metabolic syndrome. Diabetes Obes Metab. 2006;8:264–280. doi: 10.1111/j.1463-1326.2005.00510.x. [DOI] [PubMed] [Google Scholar]
- 35.Woo CW, Siow YL, OK Homocysteine activates cAMP-response element binding protein in HepG2 through cAMP/PKA signaling pathway. Arterioscler Thromb Vasc Biol. 2006;26:1043–1050. doi: 10.1161/01.ATV.0000214981.58499.32. [DOI] [PubMed] [Google Scholar]
- 36.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–G6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- 39.Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- 40.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]