

Abstract
Purpose
A2B receptor agonists are studied as possible therapeutic tools for a variety of pathological conditions. Unfortunately, medicinal chemistry efforts have led to the development of a limited number of potent agonists of this receptor, in most cases with a low or no selectivity versus the other adenosine receptor subtypes. Among the developed molecules, two structural families of compounds have been identified based on nucleoside and non-nucleoside (pyridine) scaffolds. The aim of this work is to analyse the binding mode of these molecules at 3D models of the human A2B receptor to identify possible common interaction features and the key receptor residues involved in ligand interaction.
Methods
The A2B receptor models are built by using two recently published crystal structures of the human A2A receptor in complex with two different agonists. The developed models are used as targets for molecular docking studies of nucleoside and non-nucleoside agonists. The generated docking conformations are subjected to energy minimization and rescoring by using three different scoring functions. Further analysis of top-score conformations are performed with a tool evaluating the interaction energy between the ligand and the binding site residues.
Results
Results suggest a set of common interaction points between the two structural families of agonists and the receptor binding site, as evidenced by the superimposition of docking conformations and by analysis of interaction energy with the receptor residues.
Conclusions
The obtained results show that there is a conserved pattern of interaction between the A2B receptor and its agonists. These information and can provide useful data to support the design and the development of A2B receptor agonists belonging to nucleoside or non-nucleoside structural families.
Electronic supplementary material
The online version of this article (doi:10.1186/2193-9616-1-24) contains supplementary material, which is available to authorized users.
Keywords: Purinergic receptors, Adenosine receptors, Adenosine receptor agonists, Nucleosides, Purine derivatives, Pyridine derivatives, Molecular modelling, Homology modelling, Molecular docking
Background
Adenosine (Ado, Figure 1) is a naturally occurring nucleoside that mediates numerous physiological and pathological processes with effects on heart rate and atrial contractility, vascular smooth muscle tone, release of neurotransmitters, lipolysis, as well as renal, platelet, and white blood cell functions (Cristalli and Volpini 2003). Ado activity is mediated by the activation of four receptors that have been cloned (Robeva et al. 1996) and classified as A1, A2A, A2B, and A3 adenosine receptor (AR) subtypes (Fredholm et al. 2001) by considering the respective coupling to second messengers. Furthermore, ARs can be distinguished on the basis of their tissue distribution and unique pharmacological profiles. These membrane proteins, belonging to the G protein-coupled receptor (GPCR) family, are hot targets due to their therapeutic potential even though the lack of potent and selective ligands for all subtypes still represents a weakness for the attribution of specific biological activity and for the development of therapeutic tools.
Figure 1.
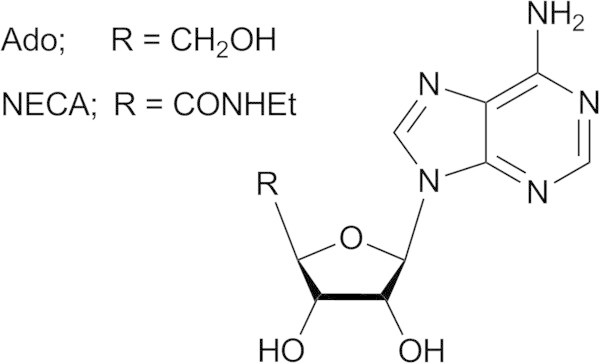
Molecular structure of Ado and NECA.
Among ARs, the A2BAR has been usually defined “low-affinity AR” due to its lower affinity for Ado and for other agonists respect to the other AR subtypes (Feoktistov and Biaggioni 1997; Beukers et al. 2000). Its stimulation leads to activation of phospholipase C and adenylyl cyclase through the coupling to the Gq and Gs proteins, respectively. The A2BAR is widely expressed in the human body and regulates several biological events at cardiovascular, muscular, and central nervous systems, and also in cell growth and during inflammation (Feoktistov and Biaggioni 1997). Hence, this receptor is of therapeutic interest for its targeting in several conditions (Baraldi et al. 2009). In particular, the agonists of this receptor have been evaluated for their cardioprotective effect, due to their reduced bradycardic and hypotensive side effects respect to Ado (Kuno et al. 2007; Philipp et al. 2006; Gao and Jacobson 2007; Eckle et al. 2007), for the treatment of coronary artery disease (Hinschen et al. 2003; Ansari et al. 2007; Kemp and Cocks 1999), and to promote angiogenesis (Feoktistov et al. 2003; Feoktistov et al. 2004). Further applications of A2BAR agonists have been explored on the basis of the anti-inflammatory effect following the activation of this receptor, leading to the suggestion of the use of these molecules in septic shock (Kreckler et al. 2006). Even the use of A2BAR agonists for the treatment of renal diseases, hypertension, cystic fibrosis, diabetes, and pulmonary diseases associated with hyperplasia has been considered (Volpini et al. 2003; Dubey et al. 2005).
Many efforts of medicinal chemistry have been focused to the development of potent and selective AR agonists and antagonists. In the case of the A2BAR, for several years the nucleoside 4’-N-ethylcarboxamidoAdo (NECA, Figure 1, EC50 = 140 nM) has represented and is still used as reference agonist of this AR subtype, despite its lack of subtype selectivity. Further agonists have been developed by modifying the Ado and NECA structures with the insertion of substituents at the 2- and N6-positions. The N6-substituents contain in most cases substituted aromatic rings and polar groups directly bound to 6-amine or to the aromatic function itself. A series of NECA derivatives was developed by inserting at the N6-position a large substituent similar to the one present at the 8-position of xanthine derivatives that behave as potent A2BAR antagonists. Table 1 presents some examples of nucleosides presenting agonist activity at the A2BAR. The activity data are taken from a review article by Baraldi and co-workers (Baraldi et al. 2009). In general, the NECA derivatives present higher potency respect to the corresponding Ado derivatives. In any case, while the obtained potencies at the A2BAR are at nanomolar level, the selectivity versus the other AR subtypes is still low (Baraldi et al. 2009; Volpini et al. 2002; Lambertucci et al. 2003; Gao et al. 2004; Baraldi et al. 2007a; Baraldi et al. 2007b).
Table 1.
Nucleoside A 2B AR agonists analysed in this work (see Additional file 1 for structural details)
| cpd | A1ARK i, nM | A2AARK i, nM | A2BAR EC50, nM | A3ARK i, nM |
|---|---|---|---|---|
| 1 | 0.67 | 1.8 | 920 | 1.4 |
| 2 | 221 | 9.3 | 3490 | 54.2 |
| 3 | 1050 | 1550 | 82 | > 5000 |
| 4 | 2600 | 4100 | 175 | > 5000 |
| 5 | 30.5 | > 1000 | 42.6 | 107 |
| 6 | 2.1 | 2.0 | 220 | 0.75 |
The activity data are taken from Baraldi and co-workers (Baraldi et al. 2009).
A structural novelty on the development of A2BAR agonists has been provided by the publication of a series of pyridine derivatives presenting low nanomolar potency at this AR subtype and in some cases remarkable selectivity versus the other ARs. These compounds present two cyano groups at the 3- and 5-positions, an amino function at the 6-position, and two substituents at the remaining 2- and 4-positions presenting various profiles. In general, the 4-substituent contains an aromatic group with possible substitutions at the 3- and 4- positions of the ring, while the 2-substituent contains a thio-methyl spacer and a polar group (i.e. amide or 2-imidazole) (Rosentreter et al. 2001; Rosentreter et al. 2003). Biological evaluation of these compounds has been reported as functional assays at A2BAR expressed in Chinese Hamster Ovary (CHO) cells and radioligand binding assays at the remaining ARs (Beukers et al. 2004). Among these derivatives, the compound 2-[(6-amino-3,5-dicyano-4-(4-(cyclopropylmethoxy)phenyl)pyridin-2-yl)thio]acetamide (BAY 606583) has been evaluated for the study of the role of the A2BAR in modulating the activity of the immune system (van der Hoeven et al. 2011) and for its possible use in case of heart ischemia (Eckle et al. 2007). Table 2 presents some examples of non-nucleoside A2BAR agonists. The activity data are taken from a review article by Baraldi and co-workers (Baraldi et al. 2009).
Table 2.
Non-nucleoside A 2B AR agonists analysed in this work (see Additional file 1 for structural details)
| cpd | A1ARK i, nM | A2AARK i, nM | A2BAR EC50, nM | A3ARK i, nM |
|---|---|---|---|---|
| 7 | 2.4 | 28 | 19 | 171 |
| 8 | 2.6 | 28 | 12 | 538 |
| 9 | 7.0 | 214 | 9 | 24 |
| 10 | 4.4 | 21 | 10 | 104 |
| 11 | 2.0 | 105 | 34 | 74 |
| 12 | > 10000 | > 10000 | 3 | > 10000 |
The activity data are taken from Baraldi and co-workers (Baraldi et al. 2009).
The aim of this work is to analyse the binding mode of both nucleoside and non-nucleoside A2BAR agonists at 3D models of this AR subtype. The compounds presented in Tables 1 and 2 have been considered for this analysis (see Additional file 1 for structural details). As a preliminary step, the study starts from the rebuilding of homology models of the human A2BAR by using as templates the crystal structures of the human A2AAR that is the member of AR family presenting also the highest sequence conservation with the A2BAR. The binding mode of ligands is then simulated by molecular docking tools, followed by energy minimization and post-docking analysis. In this study, it is not possible to depict any correlation between binding scores or interaction energies and activity data, as the potencies of the compounds have been measured as EC50 with functional studies and not as Ki affinity with radioligand binding assays. Furthermore, the study provides an interpretation of the interaction features for both series of ligands at the A2BAR but do not consider the interaction with the other AR subtypes. Hence, the results of this study may provide useful data for the design of A2BAR agonists but not for the improvement of selectivity versus the other ARs.
Methods
All molecular modelling studies were performed on a Core i7 CPU (PIV 2.20 GHZ) PC workstation. Homology modelling, energy minimization, and docking studies were carried out using Molecular Operating Environment (MOE, version 2010.10) suite (Molecular Operating Environment). All ligand structures were optimized using RHF/AM1 semiempirical calculations and the software package MOPAC (Stewart 1990) implemented in MOE was used for these calculations.
Homology modelling of the human A2BAR
Homology models of the human A2BAR were built using the recently solved X-ray structures of the human A2AAR in complex with Ado and UK-432097 as templates, both structures being retrieved from Protein Data Bank (pdb code: 2YDO; 3.0-Å resolution (Lebon et al. 2011) and pdb code: 3QAK; 2.7-Å resolution (Xu et al. 2011), respectively). A multiple alignment of the AR primary sequences was built within MOE as a preliminary step. For all A2BAR models, the boundaries identified from the used X-ray crystal structure of A2AAR were then applied for the corresponding sequences of the transmembrane (TM) helices of the A2BAR. The missing loop domains were built by the loop search method implemented in MOE. Once the heavy atoms were modelled, all hydrogen atoms were added, and the protein coordinates were then minimized with MOE using the AMBER99 force field (Cornell et al. 1995) until the Root Mean Square (RMS) gradient of the potential energy was less than 0.05 kJ mol-1 Å-1. Reliability and quality of these models were checked using the Protein Geometry Monitor application within MOE, which provides a variety of stereochemical measurements for inspection of the structural quality in a given protein, like backbone bond lengths, angles and dihedrals, Ramachandran φ-ψ dihedral plots, and quality of side chain rotamer and non-bonded contact.
Molecular docking analysis
All compound structures were docked into the binding site of the two A2BAR models using the MOE Dock tool. This method is divided into a number of stages: Conformational Analysis of ligands. The algorithm generated conformations from a single 3D conformation by conducting a systematic search. In this way, all combinations of angles were created for each ligand. Placement. A collection of poses was generated from the pool of ligand conformations using Triangle Matcher placement method. Poses were generated by superposition of ligand atom triplets and triplet points in the receptor binding site. The receptor site points are alpha sphere centres which represent locations of tight packing. At each iteration a random conformation was selected, a random triplet of ligand atoms and a random triplet of alpha sphere centres were used to determine the pose. Scoring. Poses generated by the placement methodology were scored using two available methods implemented in MOE, the London dG scoring function which estimates the free energy of binding of the ligand from a given pose, and Affinity dG scoring which estimates the enthalpic contribution to the free energy of binding. The top 30 poses for each ligand were output in a MOE database.
Post docking analysis
The five top-score docking poses of each compound were then subjected to AMBER99 force field energy minimization until the RMS gradient of the potential energy was less than 0.05 kJ mol-1 Å-1. Receptor residues within 6 Å distance from the ligand were left free to move, while the remaining receptor coordinates were kept fixed. AMBER99 partial charges of receptor and MOPAC output partial charges of ligands were utilized. Once the compound-binding site energy minimization was completed, receptor coordinates were fixed and a second energy minimization stage was performed leaving free to move only compound atoms. MMFF94 force field (Halgren 1996a, b, c, d;Halgren and Nachbar 1996; Halgren 1999a, b) was applied. For each compound, the minimized docking poses were then rescored using London dG and Affinity dG scoring functions and the dock-pKi predictor. The latter tool allows the estimation of the pKi for each ligand using the “scoring.svl” script retrievable at the SVL exchange service (Chemical Computing Group, Inc. SVL exchange: http://svl.chemcomp.com). The algorithm is based on an empirical scoring function consisting of a directional hydrogen-bonding term, a directional hydrophobic interaction term, and an entropic term (ligand rotatable bonds immobilized in binding). For each compound and at each A2BAR model, the top-score docking poses according to at least two out of three scoring functions were selected for final ligand-target interaction analysis.
The interactions between the ligands and the receptors binding site were analysed by using the IF-E 6.0 tool (Shadnia et al. 2009) retrievable at the SVL exchange service. The program calculates and displays the atomic and residue interaction forces as 3D vectors. It also calculates the per-residue interaction energies, where negative and positive energy values (expressed as kcal mol-1) are associated to favourable and unfavourable interactions, respectively. A shell of residues contained within a 10 Å distance from ligand were considered for this analysis.
Results and discussion
To simulate the binding mode of nucleoside and non-nucleoside agonists at A2BAR and to compare the key ligand-target interaction features of the two structural families of compounds, a molecular docking analysis was performed at homology models of the human A2BAR developed by using two recently published crystal structures of the agonist-bound A2AAR as templates (pdb code: 2YDO; 3.0-Å resolution (Lebon et al. 2011) and pdb code: 3QAK; 2.7-Å resolution (Xu et al. 2011), in complex with Ado and UK-432097, respectively). The availability of crystal structure the A2AAR allows to improve the accuracy of AR homology models, due to the high residue conservation in the primary sequences of the AR subtypes (see Figure 2), sharing a sequence identity of ~57% within the TM domains (Dal Ben et al. 2010b). The residues located within the seven TM domains in the upper part of ARs, corresponding to the ligand binding site, present a conservation (identity) at about 71% (Costanzi et al. 2007). The obtained A2BAR homology models were checked by using the Protein Geometry Monitor application within MOE (Environment), which provides a variety of stereochemical measurements for inspection of the structural quality in a given protein, such as backbone bond lengths, angles and dihedrals, Ramachandran φ-ψ dihedral plots, and quality of side chain rotamer and non-bonded contact. The final A2BAR models contain a disulfide bridge given by two cysteine residues belonging to TM3 and extracellular loop (EL) 2 domains (Cys783.25 - where 3.25 indicates the residue position within helix (Ballesteros and Weinstein 1995) - and Cys171, respectively), in agreement with recent mutagenesis studies (Schiedel et al. 2011) and as observed in the case of recently reported modelling analyses on this AR subtype (Sherbiny et al. 2009; Thimm et al. 2013; Inamdar et al. 2013).
Figure 2.
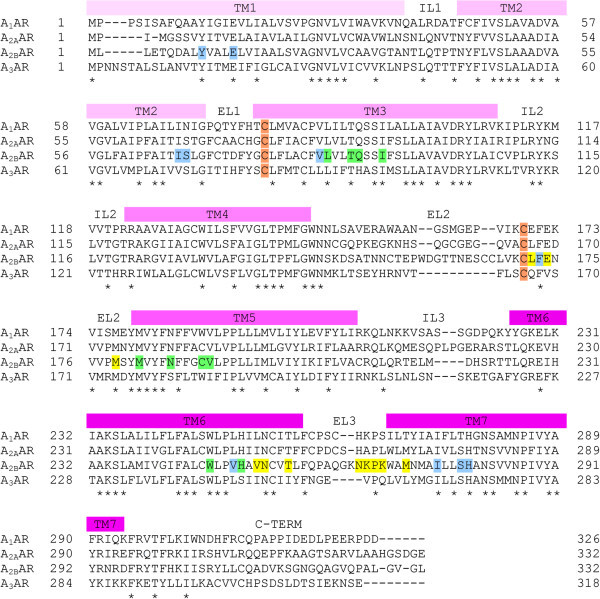
Sequence alignment of the four human AR subtypes. Transmembrane (TM), intracellular loop (IL), extracellular loop (EL), and C-terminal (C-TERM) domains are indicated; * symbols indicate sequence identity in all the four subtypes; C letters indicate cysteine residues involved in the disulfide bridge conserved among the four AR subtypes; letters coloured in yellow, cyan, and green indicate the A2BAR binding site residues involved in ligand interaction (the colour indexing refers to the binding site subdivision described in Figure 5, see its legend for details).
The A2BAR structures were then used as target for the docking analysis of synthesised derivatives. All ligand structures were optimized using RHF/AM1 semi-empirical calculations and the software package MOPAC implemented in MOE was utilized for these calculations (Stewart 1990). The compounds were then docked into the binding site of the A2BAR models by using the MOE Dock tool. Top-score docking poses of each compound were subjected to energy minimization; in this phase the binding site residues within 6 Å proximity were left free to move and to adapt their conformation to the ligand moiety. Once this step was completed, a second minimization phase was performed keeping fixed the receptor coordinates. The obtained ligand-target complexes were then rescored using three available methods implemented in MOE: the London dG scoring function that estimates the free energy of binding of the ligand from a given pose; the Affinity dG scoring tool that estimates the enthalpic contribution to the free energy of binding; the dock-pKi predictor that uses the MOE scoring.svl script to estimate for each ligand a pKi value, which is described by the H-bonds, transition metal interactions, and hydrophobic interactions energy. For each compound, the top-score docking pose at each A2BAR model, according to at least two out of three scoring functions, was selected for final ligand-target interaction analysis.
The binding sites of the two developed A2BAR models are very similar considering both receptor residues orientation and pocket volumes. Slight differences are still observable, due to diverse arrangements of some residues detectable even at the two A2AAR crystal structures templates. For example, a glutamate residue (Glu169 in the A2AAR) located within EL2 segment makes a clear H-bond interaction with the N6-amino group of Ado in the 2YDO crystal structure, while the same residue is oriented in opposite direction when observed within the 3QAK X-ray. This residue presents different orientation even considering four previously reported crystal structures of the A2AAR in complex with ZM241385 antagonist (Jaakola et al. 2008; Dore et al. 2011; Hino et al. 2012; Liu et al. 2012). Furthermore, a recent report on mutagenesis studies at the A2AAR shows that the mutation of this residue does not significantly modify the potency of nucleoside and non-nucleoside agonists at this AR subtype (Lane et al. 2012). These data suggest that the interaction with this residue is important but maybe not critical for the ligand binding to the receptor. Analogously to what observed at the 2YDO and 3QAK crystal structures, comparable arrangements are respectively observed for the corresponding residue (Glu174) at the two A2BAR models. In the second case, this glutamate points towards the side chain of Lys269 (EL3) making a strong polar interaction with this amino acid. In any case, most of differences of binding site residue arrangements are observed at EL domains in peripheral regions of binding site and hence have a marginal impact on the binding site size and chemical-physical properties. As consequence, it is not a surprise that the docking analysis of the synthesised compounds at the two receptor models leads to analogue results.
Considering the nucleoside agonists, the docking conformations share a common motif, presenting the Ado/NECA derivatives located in the binding site similarly to the co-crystallized nucleoside agonists. In detail, adenine scaffold is positioned between TM3, TM6, and TM7, with the 8- and 9-positions pointing towards the core of the receptor, while the 2- and N6-substituents are externally located (Figure 3A-B). The adenine plane is stabilized by an aromatic stacking interaction with the AR conserved phenylalanine residue in EL2 domain (Phe173 in A2BAR), while the N6-amino group and the N7 atom interact through H-bonding with a conserved asparagine (Asn2546.55 in A2BAR). The N6-amino group makes also H-bonding with Glu174 in the case of the 2YDO-based A2BAR model but not in the case of the 3QAK-based model, due to the different orientation of the amino acid side chain. Considering again the 2YDO-based A2BAR model, in the case of the presence of substituents at the N6-position of ligands (compounds 3–5), a rearrangement of Glu174 side chain is observed during post-docking minimization stage. The obtained conformation of the amino acid is comparable to the one of the corresponding residue in the 3QAK-based model and is able to make a polar interaction with Lys269. The N6-substituents contain polar groups able to give polar interactions with residues in their proximity. In particular, compounds 3 and 4 contain a substituted carbonyl-hydrazine at the 6-position, with the two polar hydrogens of hydrazine group oppositely oriented and pointing towards Asn2546.55 and Glu174 polar oxygens. In the case of compound 5, the phenyl ring directly bound to the N6-amine group causes a major rearrangement of Glu174 side chain (2YDO-based A2BAR model) during post-docking minimization step, leading to a conformation highly similar to the same residue in 3QAK-based model. The obtained minimized systems present in both cases the Glu174- Lys269 polar interaction, with an additional H-bonding between Glu174 and the polar hydrogen atom of amide function within the N6-substituent. Further polar interaction is possible between the carbonyl group of the same amide function of 5 and the side chain of Asn266 (EL3). The 2-substituent (compounds 1, 2, 4–6) is located between TM1, TM2, TM7, and EL2 with the position of phenyl ring (compounds 1, 2, 6) roughly corresponding to the one occupied by the urea group within the 2-substituent of co-crystallized UK-432097 agonist. The ribose moiety is located into the TM helices bundle, in a region between TM2, TM3, TM6, and TM7 in close proximity to the conserved tryptophan (Trp2476.48 in A2BAR) side chain. The ribose ring presents the hydroxyl groups at the 2′- and 3′-position giving H-bond interaction with A2BAR His2807.43 and Ser2797.42 side chains, respectively. The Ado derivatives (1–2) present in 4′-position a hydroxymethyl group located between TM3 and TM6 and interacting with His2516.52 side chain, while the 4′-ethylcarboxamido substituent of NECA derivatives (3–6) is located in analogue position and gives H-bond interaction with Thr893.36 and His2516.52.
Figure 3.
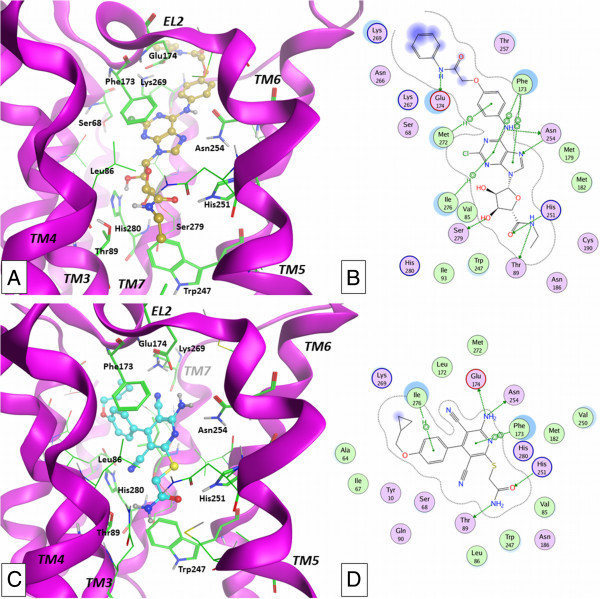
Results of docking studies. Docking conformations of nucleoside (compound 5: A-B; TM4 domain is partially hidden) and non-nucleoside (compound 12: C-D) derivatives at the 2YDO-based A2BAR model and ligand-target interaction plots as computed by MOE software.
Considering the non-nucleoside pyridine derivatives, the lowest score and most populated family of docking conformations shows the pyridine ring located in rough correspondence to the adenine scaffold of nucleoside agonists and forming a π-stacking interaction with Phe173 (EL2, Figure 3C-D). Considering the positioning of the different substituents within the binding cavity, the orientation of the pyridine agonists results somehow comparable to the one obtained and schematically described by Sherbiny and colleagues (Sherbiny et al. 2009) but significantly different respect to the ones obtained in more recent docking studies at the same AR subtype (Thimm et al. 2013) and at the A2AAR (Lane et al. 2012). In detail, the interaction of the scaffold with the A2BAR binding site is given as H-bonding between the N1 atom and the 6-amino group of pyridines and the amine and carbonyl groups of Asn2546.55 amide function, respectively. This double polar interaction is clearly present only for some derivatives, due to the nature of the 4-substituent that slightly modifies in some cases the orientation of the scaffold. Analogously to the nucleoside agonists, the amino group at the 6-position of pyridines gives a H-bond interaction with Glu174 in the case of the 2YDO-based A2BAR model, while in the case of the 3QAK-based model the side chain of the same residue in some cases is not close enough to the ligand amino group and hence not able to provide a clear H-bonding. The thiomethylimidazole (7–11) and thioacetamide (12) groups in 2-position are inserted between TM3, TM5, and TM6 residues. In the case of compounds 7–11, the H-bond donor function of imidazole is oriented towards the oxygen atom of Thr893.36, while the acceptor feature points towards the polar hydrogen of His2516.52. In the case of compound 12, analogue interactions are given by the amine and carbonyl functions of thioacetamide group, respectively. The 3-cyano group is inserted in a sub-cavity between Val853.32, Leu863.33, Thr893.36, Ser2797.42, and His2807.43. No clear interaction with binding site is given by this group, even though the presence of some space between this function and the polar groups of Ser2797.42 and His2807.43 could allow the presence of a water molecule providing a sort of “bridge-interaction” between ligand and binding site residues, as observed, for example, in the case of a crystal structure of A2AAR in complex with ZM241385 (Jaakola et al. 2008). The substituted aromatic group at the 4-position is located in a sub-cavity given by TM1, TM2, TM3, and TM7 residues, in close proximity to Tyr101.35, Ala642.61, Ile672.64, Ser682.65, and Ile2767.39. Further residues in proximity of this group are Val853.32 and Phe173. The interaction is mainly hydrophobic, even if polar interaction could be given by the presence of a hydroxyl function on the aromatic substituent (i.e. compounds 8 and 10). Finally, the 5-cyano group points towards the extracellular environment and is located in proximity to the couple of residues Glu174-Lys269.
To evaluate the possible common features for the interaction with receptor binding site, the docking conformations of nucleoside and non-nucleoside agonists have been subjected to a comparative analysis performed and here presented considering two different points of views. Firstly, a direct comparison can be made by the superimposition of the representative docking conformations of one nucleoside and one non-nucleoside agonist, with the aim of obtaining, if present, a sort of pharmacophore description. In this sense, Figure 4 shows the superimposition of the docking conformations of compounds 5 and 12 (the most potent compounds from the two ligand families) at the two A2BAR models. The results of the superimposition are almost identical at the two receptor models and highlight the presence of a series of common interaction points that can be converted to pharmacophoric features. In details, the 6-amino group of nucleosides and pyridines are located in a similar position, providing a H-bond donor feature able to interact with the H-bond acceptor features given by the carbonyl group of Asn2546.55 and, in some cases, by the carboxyl group of Glu174 (EL2). Similarly, the N7 and N1 atoms of nucleosides and pyridines, respectively, are almost superimposed and represent a H-bond acceptor feature located in close proximity with the donor function given by the amine group belonging to amide function of Asn2546.55. Furthermore, the 4′-N-ethylcarboxamide function of the nucleoside derivative 5 presents the amide carbonyl group and polar hydrogen in a nearly identical position to the analogue functions of 2-thioacetamide group of compound 12, suggesting the importance of the presence in the ligand of a combination of H-bond acceptor and donor features located in that sub-region of the binding site. The carbonyl groups are located in close proximity to a polar hydrogen of His2516.52, while the polar hydrogens of the ligand amide groups are located near the polar oxygen of Thr893.36. While the pyridine derivatives present an amide function (compound 12) or an imidazole ring (7–11) both able to provide the two pharmacophoric features, in the case of nucleoside derivatives the two functions can be present only in the case of NECA derivatives (3–6), while the Ado analogues (1, 2) may fit only one of the two polar features. This data is observable already from the 2YDO and 3QAK crystal structures and could in any case explain the general behaviour at ARs according to which the Ado derivatives are generally less potent respect to the corresponding NECA or MECA (4′-N-methylcarboxamidoAdo) analogues. The docking position of the 3-cyano group of pyridines roughly corresponds to the position of the nucleosides 2′-hydroxyl function. As reported above, the cyano group is inserted in a sub-cavity between Val853.32, Leu863.33, Thr893.36, Ser2797.42, and His2807.43, with the presence of some space between this function and the polar groups of Ser2797.42 and His2807.43. In this sense, the possible presence of a water molecule providing a “bridge-interaction” between ligand and binding site residues could suggest the introduction of a sort of “polar group feature” corresponding to the presence of the 2′-hydroxyl function of nucleosides and the cyano group of pyridines. This polar group could interact with Ser2797.42 and His2807.43 residues directly (like in the case of nucleosides) or with the aid of a bridging water molecule (in the case of pyridines). The presence of a hydrophobic group at the 2-position of nucleosides and at the 4-position of pyridines suggests the presence of a large hydrophobic feature located within TM1, TM2, and TM7 domains. This group seems critical in particular for pyridine derivatives, as only some nucleoside agonists present a 2-hydrophobic substituent. Finally, the position of the N1 atom of nucleosides is almost coincident to the one of the nitrogen atom of the 5-cyano group of pyridines, suggesting a possible H-bond acceptor function in that position. However, it must be noted that these atoms are oriented towards the extracellular environment and the H-bond donor counterpart could be provided only by a special arrangement of Lys269 (EL3) side chain. We underline that the use of a pharmacophore model in this context is aimed only at helping for the description of an interaction pattern that appears conserved between nucleoside and non-nucleoside agonists. For a development of a proper pharmacophore model, a larger set of molecules and Ki affinity data should be employed.
Figure 4.
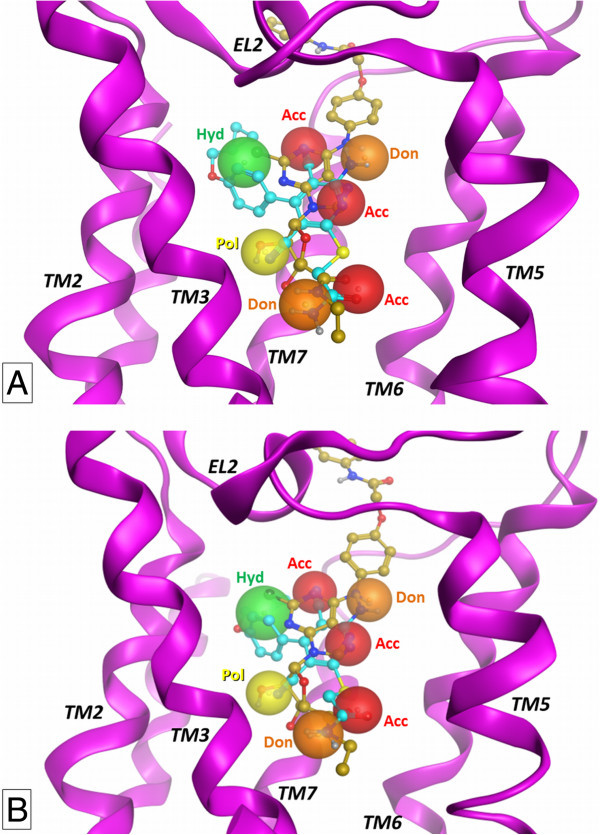
Comparison of docking conformations. Superimposition of docking conformations of nucleoside (compound 5) and non-nucleoside (compound 12) derivatives at A2BAR at the 2YDO- and 3QAK-based A2BAR models (A and B, respectively). TM4 domain is partially hidden. The matching between functional groups presenting analogue properties among the two molecules are represented as pharmacophoric features (see text for details).
A second approach to compare the binding modes of the different families of compounds at A2BAR consists in evaluating the interaction of ligands with each binding site residue, obtaining a set of data representing the different effect of the A2BAR residues during the ligand-target interaction. This analysis has been performed by the use of the IF-E 6.0 (Shadnia et al. 2009) tool retrievable at the SVL exchange service. This method was already used by our research group for the analysis of ligand-target interaction for a series of potent A3AR agonists (Dal Ben et al. 2010a). The script calculates and displays atomic and residue interaction forces as 3D vectors. It also calculates the per-residue interaction energies (values in kcal mol-1), where negative and positive energy values are associated to favourable and unfavourable interactions, respectively. The script has been applied to each compound at both A2BAR models. To display the results, the binding site residues are divided in three sections (Figure 5). The section 1 contains residues belonging to TM1 (Tyr101.35 and Glu141.39), TM2 (Ala642.61, Ile672.64, and Ser682.65), and TM7 (Ile2767.39, Ser2797.42, and His2807.43) domains, with the insertion also of Val853.32, Phe173 (EL2), and Val2506.51. These residues represent the receptor region interacting with ligand adenine and pyridine scaffolds and with the substituents at the 2-position of nucleosides and at the 3- and 4-positions of pyridines. Residues interacting with part of ribose atoms of nucleoside derivatives are also included in this section. Figure 6 represents the obtained results for section 1. The blue and red versions of the plots represent the results obtained at the 2YDO- and 3QAK-A2BAR models, respectively. In agreement with what appears from the superimposition of docking conformations (see above), the effect of these residues is different from nucleoside to non-nucleoside ligands as in the first case the 2-substituent of nucleosides is present only in some cases and presents different structural and chemical profiles, while in the second case the 4-substituent of pyridines is large and forming various interactions with binding site residues. In detail, considering the nucleoside derivatives 1–6, it clearly appears the effect of Phe173, Ser2797.42, and His2807.43 in stabilizing the ligand-target interaction. In particular, Phe173 is the key residue for stabilizing the scaffold position within the binding site through a π-stacking interaction, while Ser2797.42 and His2807.43 are involved in polar interaction with 2′- and 3′-hydroxyl groups of nucleosides. The further contribution of Val2506.51 and Ile2767.39 for scaffold stabilization is more evident at 2YDO-A2BAR model, while the role of the carbonyl group of Ile672.64 backbone atoms in forming H-bond interaction with the hydroxyl group of 2-substituent of 1 and 6 is clearly evident at 3QAK-based model. The latter residue forms also hydrophobic interaction with the alkynyl group of the nucleosides 1 and 6 (3QAK-based model). Considering the non-nucleoside derivatives 7–12, results highlight the role of Val853.32, Phe173, and Val2506.51 in stabilizing the ligand scaffold, while the interaction with Ser2797.42 and His2807.43 is lacking (see above). Tyr101.35 interacts with compound 12 in 3QAK-based model. Ile672.64 forms hydrophobic interaction with the 4-aromatic group of pyridines, while Ala642.61 forms on the one hand a stable H-bond interaction with the para-hydroxyl group of the 4-aromatic substituent of compound 8 (3QAK-based model), on the other hand a hydrophobic interaction with 4-aromatic group of 12. Comparing the results for this section with previously reported data, it can be underlined that Tyr101.35 showed to be critical for compound 12 activity as evidenced from mutagenesis results (Thimm et al. 2013), while Ile672.64 and Ala642.61 were already reported to possibly interact with the 4-substituent of pyridine derivatives (Sherbiny et al. 2009). The effect of Ser2797.42 and His2807.43 is evident for nucleoside agonists but not so well defined for non-nucleoside derivatives. In particular, the mutation of Ser2797.42 to alanine takes to a loss of activity of nucleosides and, on the contrary, to an improvement of non-nucleoside derivative EC50 data (Thimm et al. 2013). These results are confirmed at the A2AAR (Lane et al. 2012). Our study confirms the effect on nucleosides but does not highlight a direct interaction with non-nucleoside derivatives. The histidine residue in position 7.43 of ARs (His2807.43 in A2BAR) is reported to be essential for receptor expression and function (Dal Ben et al. 2010b; Thimm et al. 2013). Considering the interaction of this residue in A2BAR with ligands, it has been recently reported a molecular modelling study consisting in the rebuilding of the three AR models other than A2AAR and the use of the four AR 3D structures for a molecular docking study of antagonists followed by molecular dynamics analysis (Inamdar et al. 2013). Considering the results at the A2BAR, this study suggests a possible direct contact of His2807.43 with a potent thiazole antagonist. Further recent studies report that the mutation of this histidine leads to a loss of interaction with ligands (Thimm et al. 2013). As reported above, our modelling study does not highlight a direct contact of His2807.43 with non-nucleoside derivatives even if we cannot exclude that an interaction of these ligands with this residue and Ser2797.42 could be mediated by a water molecule.
Figure 5.
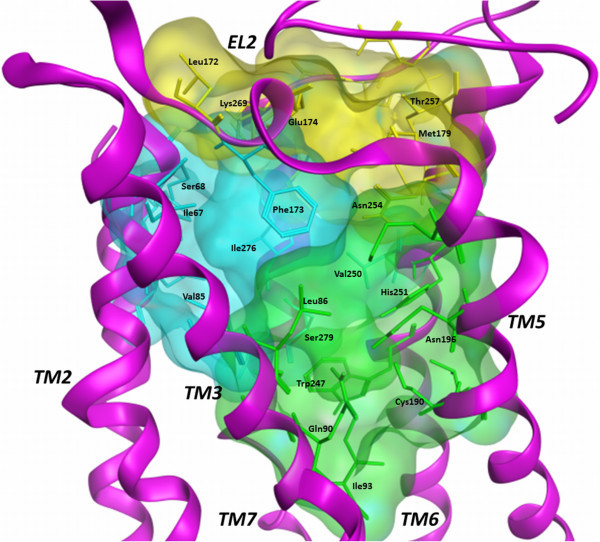
Analysis of the role of binding site residues for the interaction with ligands. The binding site is divided in three sections representing the TM1-TM2-TM7 and the TM3-TM5-TM6 regions of the cavity (cyan and green, respectively) and the entrance of the binding site (yellow). TM4 domain is partially hidden.
Figure 6.
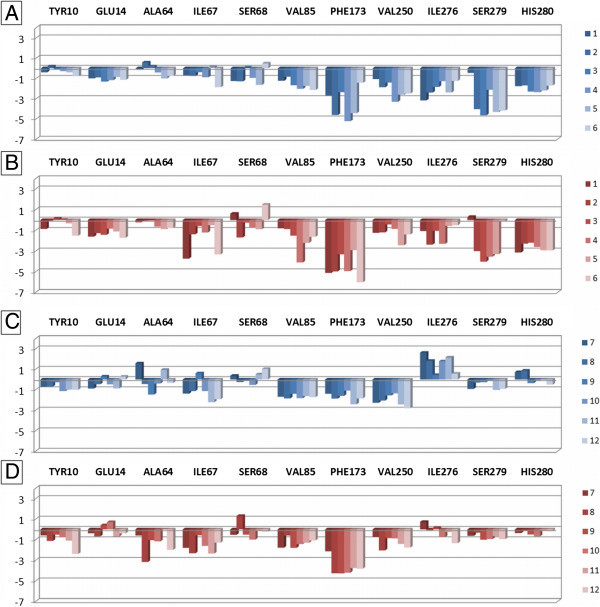
Plot of interaction energies for residues belonging to the section 1 of the A 2B AR binding site, calculated with the MOE IF-E 6.0 tool. Data are represented as kcal mol-1. The blue and red versions of the plots represent the results obtained at the 2YDO- and 3QAK-A2BAR models, respectively. Plots A and B are referred to the interaction energies of nucleoside derivatives 1–6, while plots C and D are referred to the non-nucleoside derivatives 7–12.
The section 2 contains residues belonging to TM3 (Leu863.33, Thr893.36, Gln903.37, and Ile933.40), TM5 (Met1825.38, Asn1865.42, Cys1905.46, and Val1915.47), and TM6 (Trp2476.48 and His2516.52). These residues are involved in interaction with ribose group of nucleoside analogues (in particular the 4′-position) and with 2-substituent of non-nucleoside pyridine derivatives. Figure 7 represents the obtained results for section 2, again with blue and red versions corresponding to the 2YDO- and 3QAK-A2BAR models, respectively. The effect of the receptor residues for the interaction with ligands is comparable considering both nucleoside and non-nucleoside agonists. Firstly, Leu863.33 and Met1825.38 form a hydrophobic interaction with the ribose group of nucleosides and with the 2-substituent of pyridines. Secondly, it appears evident the contribution of Thr893.36 and His2516.52, in accordance with the results of superimposition experiments (see above). In particular, Thr893.36 provide a stabilizing effect for the polar hydrogen of NECAs 5′-amide group from 3–6, of thiomethylimidazole from 7–11, and of thioacetamide from 12. The effect of this residue is largely reduced in the case of the two Ado derivatives 1–2 that do not present a combination of H-bond acceptor and donor features at the 5′-position. His2516.52 cooperates with Thr893.36 in providing a stabilizing effect for ligands. In the case of non-nucleoside derivatives, it is interesting to notate the greater effect of this residue for 12 respect to 7–11 at both A2BAR models, suggesting a better interaction of His2516.52 for the amide function respect to an imidazole ring. The effect of the other residues of this section is not significant, with the exception in some cases of Gln903.37 (polar interaction with non-nucleoside derivatives in 3QAK-A2BAR model) and Trp2476.48. In a previous study (Sherbiny et al. 2009), Thr893.36 was suggested as interacting with 3-cyano substituent of 12 while His2516.52, Gln903.37, and Asn1865.42 were found as interacting (or in proximity) with the amide group of the 2-substituent of the same compound. Asn1865.42 appears as not directly interacting with the compounds of this study. On the other hand, a mutagenesis study has evidenced that the mutation of this residue to alanine leads to an increase of potency of both nucleoside and non-nucleoside derivatives (Thimm et al. 2013). The interaction between 3.37 and 5.42 residues is absent in the antagonist-bound A2AAR crystal structures but present in the agonist-bound 3QAK X-ray data (in the Ado- and NECA-bound A2AAR crystal structures the Gln893.37 is mutated to alanine), hence these residues could have a mechanistic role for the receptor function and could be not necessarily interaction points for ligands (Dal Ben et al. 2013).
Figure 7.
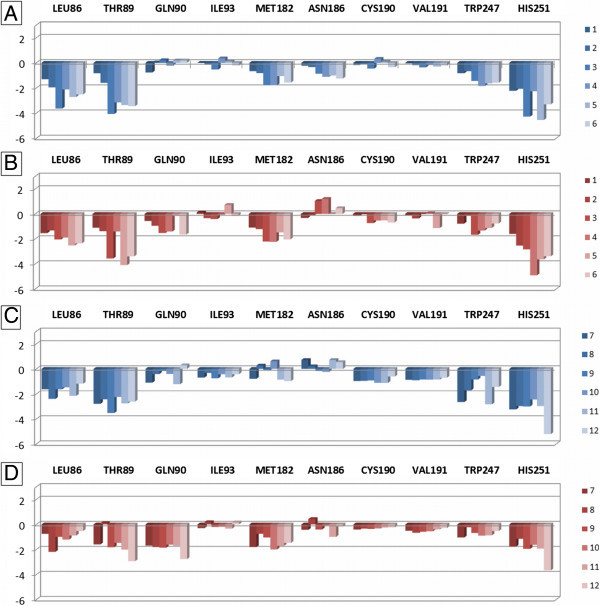
Plot of interaction energies for residues belonging to the section 2 of the A 2B AR binding site, calculated with the MOE IF-E 6.0 tool. Data are represented as kcal mol-1. The blue and red versions of the plots represent the results obtained at the 2YDO- and 3QAK-A2BAR models, respectively. Plots A and B are referred to the interaction energies of nucleoside derivatives 1–6, while plots C and D are referred to the non-nucleoside derivatives 7–12.
The section 3 contains residues located close or at the entrance of the binding site and belonging to EL2 (Leu172, Glu174, and Met179), TM6 (Val2536.54, Asn2546.55, and Thr2576.58), EL3 (Asn266, Lys267, Pro268, and Lys269), and TM7 (Met2727.35). These residues are involved in interaction with the purine and the pyridine scaffold, the C2– and N6-substituents of nucleosides, and the 4-aromatic substituent, the 5-cyano group, and the 6-amino function of pyridines. Even in this case, the representation is divided in plots for nucleoside and non-nucleoside derivatives at the two 2YDO- and 3QAK-A2BAR models (Figure 8). The results confirm the key role of Asn2546.55 for the interaction with both families of ligands at both receptor models. The importance of this residue for the interaction of A2BAR with ligands has been highlighted even in recently reported studies (Sherbiny et al. 2009; Inamdar et al. 2013). Furthermore, it appears a relevant effect of Glu174 for the interaction with nucleoside derivatives 1–6. The carboxyl function of this residue can interact with the unsubstituted 6-amino group (1, 2, 6) or with polar hydrogens present within the N6-substituent (3–5). In the case of pyridine derivatives, the effect of this residue is influenced by its conformation, as in the 2YDO-based A2BAR model its carboxyl group points towards the 6-amino function of ligands providing H-bond interaction, while in the case of 3QAK-based model the side chain is oriented towards the extracellular environment in proximity of Lys269. Analogue consideration can be made for Lys269, whose side chain points towards Glu174 and the N6-substituent of nucleoside derivatives. In the case of compounds 1 and 6, the charged amino group of this lysine interacts also with the hydroxyl group within the 2-substituent. In the case of the non-nucleoside derivatives, the Lys269 charged amino group is located in proximity of 5-cyano function without forming interaction with this group. The effect of the remaining amino acids in this section seems not significant.
Figure 8.
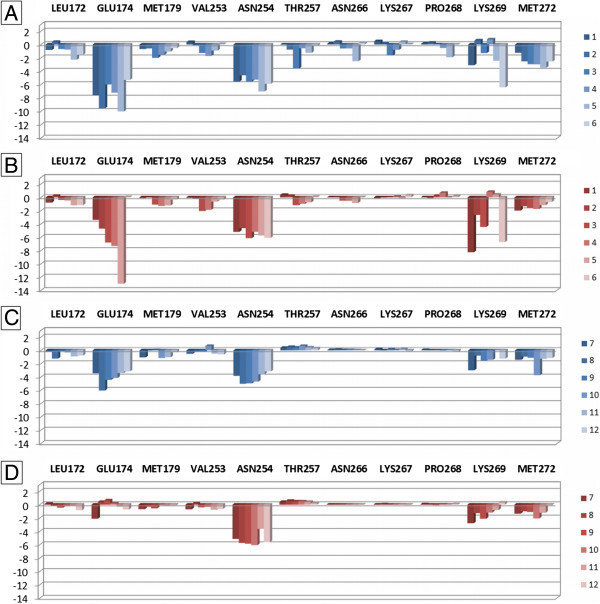
Plot of interaction energies for residues belonging to the section 3 of the A 2B AR binding site, calculated with the MOE IF-E 6.0 tool. Data are represented as kcal mol-1. The blue and red versions of the plots represent the results obtained at the 2YDO- and 3QAK-A2BAR models, respectively. Plots A and B are referred to the interaction energies of nucleoside derivatives 1–6, while plots C and D are referred to the non-nucleoside derivatives 7–12.
Conclusions
In conclusion, this study was aimed at simulating the binding modes of nucleoside and non-nucleoside agonists at two A2BAR homology models developed starting from the X-ray structures of the A2AAR in complex with Ado and UK-432097 as templates (pdb code: 2YDO and 3QAK, respectively). The docking conformations of nucleoside derivatives were quite expected, while the non-nucleoside derivatives demonstrated to bind to these receptor models in a different way respect to previously reported studies at AR models. The generated and minimized docking conformations were compared by superimposition and by analysis of the interaction with the binding site residues located in ligand proximity. Results showed that, beside the evident structural differences among the two ligand families, the nucleoside and non-nucleoside derivatives bind to the A2BAR through a series of conserved interaction points, suggesting a sort of interaction pattern. The findings of this analysis are in agreement with the results of the evaluation of the role of binding site residues for the ligand-target interaction and the conclusions of these studies are in good agreement with mutagenesis and molecular modelling studies reported in the last years. Taken together, these data could be helpful for the design of A2BAR agonists belonging to nucleoside or non-nucleoside structural families.
Authors’ information
DDB, MB, and CL are Assistant Professors of Medicinal Chemistry with interest in molecular modelling and compound synthesis. AT is post-doctoral researcher working in compound synthesis, while RV is Full Professor of Medicinal Chemistry. All authors work at the School of Pharmacy of the University of Camerino, Italy.
Electronic supplementary material
Additional file 1: Table S1: Nucleoside A2BAR agonists analysed in this work. The activity data are taken from Baraldi and co-workers (Baraldi et al. 2009). Table S2. Non-nucleoside A2BAR agonists analysed in this work. The activity data are taken from Baraldi and co-workers (Baraldi et al. 2009). (PDF 48 KB)
Acknowledgments
This work was supported by Fondo di Ricerca di Ateneo (University of Camerino) and by a grant of the Italian Ministry for University and Research (PRIN2010-11 n° 20103W4779_003).
Abbreviations
- Ado
Adenosine
- AR
Adenosine receptor
- GPCR
G Protein-coupled receptor
- CHO
Chinese hamster ovary
- MOE
Molecular operating environment
- pdb
protein data bank
- TM
Transmembrane
- RMS
Root mean square
- EL
Extracellular loop.
Footnotes
Competing interests
All authors declare that they have no competing interest.
Authors’ contributions
DDB and RV designed the research subject and wrote the paper. DDB performed the modelling experiments, analysed the data, and interpreted the results. MB, CL, and AT collaborated on data collection and interpretation. All authors have read and approved the final manuscript.
Contributor Information
Diego Dal Ben, Email: diego.dalben@unicam.it.
Michela Buccioni, Email: michela.buccioni@unicam.it.
Catia Lambertucci, Email: catia.lambertucci@unicam.it.
Ajiroghene Thomas, Email: ajiroghene.thomas@unicam.it.
Rosaria Volpini, Email: rosaria.volpini@unicam.it.
References
- Ansari HR, Nadeem A, Talukder MA, Sakhalkar S, Mustafa SJ. Evidence for the involvement of nitric oxide in A2B receptor-mediated vasorelaxation of mouse aorta. Am J Physiol Heart Circ Physiol. 2007;292:H719–H725. doi: 10.1152/ajpheart.00593.2006. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Baraldi PG, Preti D, Tabrizi MA, Fruttarolo F, Romagnoli R, Carrion MD, Cara LC, Moorman AR, Varani K, Borea PA. Synthesis and biological evaluation of novel 1-deoxy-1-[6-[((hetero)arylcarbonyl)hydrazino]- 9H-purin-9-yl]-N-ethyl-b-D-ribofuranuronamide derivatives as useful templates for the development of A2B adenosine receptor agonists. J Med Chem. 2007;50:374–380. doi: 10.1021/jm061170a. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Preti D, Tabrizi MA, Fruttarolo F, Saponaro G, Baraldi S, Romagnoli R, Moorman AR, Gessi S, Varani K, Borea PA. N6-[(hetero)aryl/(cyclo)alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5′-N-ethylcarboxamido-adenosines: the first example of adenosine-related structures with potent agonist activity at the human A2B adenosine receptor. Bioorg Med Chem. 2007;15:2514–2527. doi: 10.1016/j.bmc.2007.01.055. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Tabrizi MA, Fruttarolo F, Romagnoli R, Preti D. Recent improvements in the development of A2B adenosine receptor agonists. Purinergic Signal. 2009;5:3–19. doi: 10.1007/s11302-009-9140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beukers MW, den Dulk H, van Tilburg EW, Brouwer J, IJzerman AP. Why are A2B receptors low-affinity adenosine receptors? Mutation of Asn273 to Tyr increases affinity of human A2B receptor for 2-(1-Hexynyl)adenosine. Mol Pharmacol. 2000;58:1349–1356. doi: 10.1124/mol.58.6.1349. [DOI] [PubMed] [Google Scholar]
- Beukers MW, Chang LC, von Frijtag Drabbe Kunzel JK, Mulder-Krieger T, Spanjersberg RF, Brussee J, IJzerman AP. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J Med Chem. 2004;47:3707–3709. doi: 10.1021/jm049947s. [DOI] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. doi: 10.1021/ja00124a002. [DOI] [Google Scholar]
- Costanzi S, Ivanov AA, Tikhonova IG, Jacobson KA. Structure and function of g protein-coupled receptors studied using sequence analysis, molecular modelling and receptor engineering: adenosine receptors. Front Drug Des Discov. 2007;3:63–79. [Google Scholar]
- Cristalli G, Volpini R. Adenosine receptors: chemistry and pharmacology. Curr Top Med Chem. 2003;3:355–469. doi: 10.2174/1568026033392165. [DOI] [PubMed] [Google Scholar]
- Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Thomas A, Volpini R, Cristalli G. Molecular modelling study on potent and selective adenosine A3 receptor agonists. Bioorg Med Chem. 2010;18:7923–7930. doi: 10.1016/j.bmc.2010.09.038. [DOI] [PubMed] [Google Scholar]
- Dal Ben D, Lambertucci C, Marucci G, Volpini R, Cristalli G. Adenosine receptor modelling: what does the A2A crystal structure tell us? Curr Top Med Chem. 2010;10:993–1018. doi: 10.2174/156802610791293145. [DOI] [PubMed] [Google Scholar]
- Dal Ben D, Buccioni M, Lambertucci C, Kachler S, Falgner N, Marucci G, Thomas A, Cristalli G, Volpini R, Klotz K-N. Biochem Pharmacol. 2013. Different efficacy of adenosine and NECA derivatives at the human A3 adenosine receptor: insight into the receptor activation switch. [DOI] [PubMed] [Google Scholar]
- Dore AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey RK, Gillespie DG, Mi Z, Jackson EK. Adenosine inhibits PDGF-induced growth of human glomerular mesangial cells via A2B receptors. Hypertension. 2005;46:628–634. doi: 10.1161/01.HYP.0000178464.63393.88. [DOI] [PubMed] [Google Scholar]
- Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I. Adenosine A2B receptors. Pharmacol Rev. 1997;49:381–402. [PubMed] [Google Scholar]
- Feoktistov I, Ryzhov S, Goldstein AE, Biaggioni I. Mast cell-mediated stimulation of angiogenesis: cooperative interaction between A2B and A3 adenosine receptors. Circ Res. 2003;92:485–492. doi: 10.1161/01.RES.0000061572.10929.2D. [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Ryzhov S, Zhong H, Goldstein AE, Matafonov A, Zeng D, Biaggioni I. Hypoxia modulates adenosine receptors in human endothelial and smooth muscle cells toward an A2B angiogenic phenotype. Hypertension. 2004;44:649–654. doi: 10.1161/01.HYP.0000144800.21037.a5. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz K-N, Linden J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Jacobson KA. Emerging adenosine receptor agonists. Expert Opin Emerg Drugs. 2007;12:479–492. doi: 10.1517/14728214.12.3.479. [DOI] [PubMed] [Google Scholar]
- Gao ZG, Mamedova LK, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P. [DOI] [Google Scholar]
- Halgren TA. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J Comput Chem. 1996;17:520–552. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<520::AID-JCC2>3.0.CO;2-W. [DOI] [Google Scholar]
- Halgren TA. Merck molecular force field. III. Molecular geometries and vibrational frequencies for MMFF94. J Comput Chem. 1996;17:553–586. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<553::AID-JCC3>3.0.CO;2-T. [DOI] [Google Scholar]
- Halgren TA. Merck molecular force field. IV. Conformational energies and geometries for MMFF94. J Comput Chem. 1996;17:587–615. [Google Scholar]
- Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Halgren TA. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comput Chem. 1999;20:730–748. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Nachbar R. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J Comput Chem. 1996;17:616–641. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<616::AID-JCC5>3.0.CO;2-X. [DOI] [Google Scholar]
- Hino T, Arakawa T, Iwanari H, Yurugi-Kobayashi T, Ikeda-Suno C, Nakada-Nakura Y, Kusano-Arai O, Weyand S, Shimamura T, Nomura N, Cameron AD, Kobayashi T, Hamakubo T, Iwata S, Murata T. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482:237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinschen AK, Rose’Meyer RB, Headrick JP. Adenosine receptor subtypes mediating coronary vasodilation in rat hearts. J Cardiovasc Pharmacol. 2003;41:73–80. doi: 10.1097/00005344-200301000-00010. [DOI] [PubMed] [Google Scholar]
- Inamdar GS, Pandya AN, Thakar HM, Sudarsanam V, Kachler S, Sabbadin D, Moro S, Klotz K-N, Vasu KK. New insight into adenosine receptors selectivity derived from a novel series of [5-substituted-4-phenyl-1,3-thiazol-2-yl] benzamides and furamides. Eur J Med Chem. 2013;63:924–934. doi: 10.1016/j.ejmech.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp BK, Cocks TM. Adenosine mediates relaxation of human small resistance-like coronary arteries via A2B receptors. Br J Pharmacol. 1999;126:1796–1800. doi: 10.1038/sj.bjp.0702462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreckler LM, Wan TC, Ge ZD, Auchampach JA. Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;317:172–180. doi: 10.1124/jpet.105.096016. [DOI] [PubMed] [Google Scholar]
- Kuno A, Critz SD, Cui L, Solodushko V, Yang XM, Krahn T, Albrecht B, Philipp S, Cohen MV, Downey JM. Protein kinase C protects preconditioned rabbit hearts by increasing sensitivity of adenosine A2B-dependent signaling during early reperfusion. J Mol Cell Cardiol. 2007;43:262–271. doi: 10.1016/j.yjmcc.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertucci C, Volpini R, Costanzi S, Taffi S, Vittori S, Cristalli G. 2-Phenylhydroxypropynyladenosine derivatives as high potent agonists at A2B adenosine receptor subtype. Nucleos Nucleot Nucl Acids. 2003;22:809–812. doi: 10.1081/NCN-120022640. [DOI] [PubMed] [Google Scholar]
- Lane JR, Klein Herenbrink C, van Westen GJ, Spoorendonk JA, Hoffmann C, IJzerman AP. A novel nonribose agonist, LUF5834, engages residues that are distinct from those of adenosine-like ligands to activate the adenosine A2A receptor. Mol Pharmacol. 2012;81:475–487. doi: 10.1124/mol.111.075937. [DOI] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, Cherezov V, Stevens RC. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment C.C.G., Inc: 1255 University St., Suite 1600. Montreal, Quebec, Canada: H3B 3X3;
- Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2B receptor cascade. Cardiovasc Res. 2006;70:308–314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Robeva AS, Woodard RL, Jin X, Gao Z, Bhattacharya S, Taylor HE, Rosin DL, Linden J. Molecular characterization of recombinant human adenosine receptors. Drug Dev Res. 1996;39:243–252. doi: 10.1002/(SICI)1098-2299(199611/12)39:3/4<243::AID-DDR3>3.0.CO;2-R. [DOI] [Google Scholar]
- Rosentreter U, Henning R, Bauser M, Krämer T, Vaupel A, Hübsch W, Dembowsky K, Salcher-Schraufstätter O, Stasch JP, Krahn T, Perzborn E. Substituted 2-Thio-3,5-Dicyano-4-Aryl-6-Aminopyridines and the use Thereof as Adenosine Receptor Ligands. 2001. [Google Scholar]
- Rosentreter U, Kraemer T, Shimada M, Huebsch W, Diedrichs N, Krahn T, Henninger K, Stasch JP. Substituted 2-Thio-3,5-Dicyano-4-Phenyl-6-Aminopyridines and their use as Adenosine Receptor-Selective Ligands. 2003. [Google Scholar]
- Schiedel AC, Hinz S, Thimm D, Sherbiny F, Borrmann T, Maass A, Müller CE. The four cysteine residues in the second extracellular loop of the human adenosine A2B receptor: role in ligand binding and receptor function. Biochem Pharmacol. 2011;82:389–399. doi: 10.1016/j.bcp.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Shadnia H, Wright JS, Anderson JM. Interaction force diagrams: new insight into ligand-receptor binding. J Comput Aided Mol Des. 2009;23:185–194. doi: 10.1007/s10822-008-9250-3. [DOI] [PubMed] [Google Scholar]
- Sherbiny FF, Schiedel AC, Maass A, Müller CE. Homology modelling of the human adenosine A2B receptor based on X-ray structures of bovine rhodopsin, the β2-adrenergic receptor and the human adenosine A2A receptor. J Comput Aided Mol Des. 2009;23:807–828. doi: 10.1007/s10822-009-9299-7. [DOI] [PubMed] [Google Scholar]
- Stewart JJ. MOPAC: a semiempirical molecular orbital program. J Comput Aided Mol Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- Thimm D, Schiedel AC, Sherbiny FF, Hinz S, Hochheiser K, Bertarelli DCG, Maass A, Müller CE. Ligand-specific binding and activation of the human adenosine A2B receptor. Biochemistry. 2013;52:726–740. doi: 10.1021/bi3012065. [DOI] [PubMed] [Google Scholar]
- van der Hoeven D, Wan TC, Gizewski ET, Kreckler LM, Maas JE, Van Orman J, Ravid K, Auchampach JA. A role for the low-affinity A2B adenosine receptor in regulating superoxide generation by murine neutrophils. J Pharmacol Exp Ther. 2011;338:1004–1012. doi: 10.1124/jpet.111.181792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J Med Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- Volpini R, Costanzi S, Vittori S, Cristalli G, Klotz K-N. Medicinal chemistry and pharmacology of A2B adenosine receptors. Curr Top Med Chem. 2003;3:427–443. doi: 10.2174/1568026033392264. [DOI] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Nucleoside A2BAR agonists analysed in this work. The activity data are taken from Baraldi and co-workers (Baraldi et al. 2009). Table S2. Non-nucleoside A2BAR agonists analysed in this work. The activity data are taken from Baraldi and co-workers (Baraldi et al. 2009). (PDF 48 KB)