

Abstract
Backgruond
Since their first description, activating epidermal growth factor receptor (EGFR) mutations identify a distinct clinical entity of patients with non-small-cell lung cancer (NSCLC).
Findings
New targeted therapies for molecularly selected NSCLC are changing the natural history of the disease, with results superior to standard chemotherapy as demonstrated in large phase III studies with first generation EGFR tyrosine kinase inhibitors (TKIs) erlotinib and gefitinib. However, after an initial response, all patients inevitably progress and several mechanisms including a secondary mutation in exon 20 of the EGFR gene (T790M) or MET or HER2 amplifications are responsible for acquired resistance (AR). In clinical practice few options are available for patients with AR, and several new agents or strategies are currently under investigation, including second generation TKIs.
Conclusions
Aim of the present review is to present available data on new EGFR-TKIs and to discuss how these agents could overcome AR to erlotinib or gefitinib.
Keywords: Afatinib, Dacomitinib, Erlotinib, Neratinib, CO-1686, AP26113, BMS-690514, XL647 EGFR, NSCLC
Findings
In the last few years, the treatment of advanced non-small-cell lung cancer (NSCLC) has radically changed, due to advances in cancer biology. The old-fashioned ‘one size fits all’ chemotherapeutic approach is nowadays replaced by a careful selection mainly based on tumor histology and, most importantly, on biological characteristics. Specifically, the discovery of the biologic and therapeutic importance of acquired genetic alterations in two genes that encode pharmacologically targetable tyrosine kinases, the Epidermal Growth Factor Receptor (EGFR) and Anaplastic Lymphoma Kinase (ALK) has changed the way these cancers are treated.
In 2004, EGFR gene mutations were firstly identified: classical-activating EGFR mutations are localized in exon 19, mainly consisting of an in-frame deletion (45-50%), and in exon 21, consisting of the L858R point mutation (40-45%), even if there are less common mutations localized in other exons [1–3]. Since their identification, it was clear that EGFR mutations, more frequently observed in never smokers, adenocarcinoma histology, women and Asiatic patients, outline a distinct subgroup of NSCLC. During the last years, six phase III trials (Table 1) established that patients harboring activating EGFR mutations benefit more from a first line treatment with an EGFR tyrosine kinase inhibitor (TKI), such as erlotinib or gefitinib, than from standard chemotherapy, at least in terms of response rate (RR), progression-free survival (PFS) and quality of life [4–9]. On the basis of these solid results, regulatory agencies have progressively approved EGFR-TKIs for the first line treatment of NSCLC harbouring activating EGFR mutations.
Table 1.
First line trials of 1 st generation TKI in EGFR mutated patients
| Study | N | OR% | mPFS (mo) | mOS (mo) |
|---|---|---|---|---|
| First Signal 4 | ||||
| Gefitinib | 26 | 85 | 8 | 27.2 |
| Cis/Gem | 16 | 38 | 2.1 | 25.6 |
| IPASS 5 | ||||
| Gefitinib | 132 | 71 | 9.5 | 19 |
| Carbo/Taxol | 129 | 47 | 6.5 | 18 |
| WJTOG 6 | ||||
| Gefitinib | 86 | 62 | 9.2 | 30.9 |
| Cis/Doce | 86 | 31 | 6.3 | NR |
| NEJOG 7 | ||||
| Gefitinib | 114 | 74 | 10.8 | 27.7 |
| Carbo/Taxol | 110 | 31 | 5.4 | 26.6 |
| OPTIMAL 8 | ||||
| Erlotinib | 83 | 83 | 13.1 | NR |
| Carbo/Gem | 72 | 36 | 4.6 | NR |
| EURTAC 9 | ||||
| Erlotinib | 86 | 58 | 9.7 | NR |
| Plat Doublet | 87 | 15 | 5.2 | NR |
N, number of patients; OR, objective response rate; mPFS, median progression free servival; mOS, median overall survival; mo, months; cis, cisplatin; gem, gemcitabine; carbo, carboplatin; doce, docetaxel; plat, platinum; NR, not reached.
Unfortunately, after a median of 8–10 months, all responding patients develop acquired resistance (AR) to EGFR-TKI therapy, with the inevitable consequence of disease progression. Recent studies demonstrated that several mechanisms are responsible for AR (Figure 1), with approximately 30% of patients in which resistance factors are not yet identified [10]. The firstly described and the most common event responsible for resistance is the acquisition of the T790M missense mutation, which is found in ≈ 50% of patients progressing after an initial response to erlotinib or gefitinib [11, 12]. Other less frequent mechanisms include secondary mutations within EGFR[13, 14], MET amplification [15], HER2 amplification [16, 17], small cell histologic transformation [18]. Identification of mechanisms responsible for AR has therapeutic implications, and several agents are currently under investigation particularly for individuals with the secondary T790M mutation.
Figure 1.
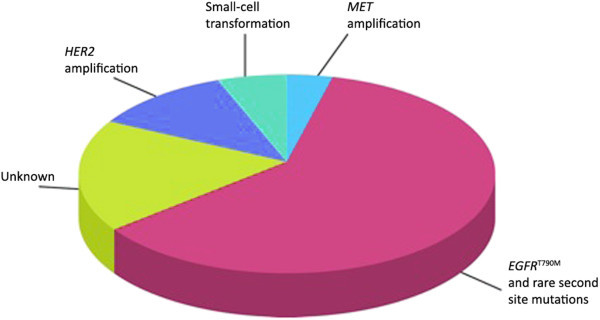
Mechanisms responsible for acquired resistance (adapted from http://cancerdiscovery.aacrjournals.org ).
The family of 2nd generation TKIs exploit three basic approaches aimed at increasing the efficacy over first-generation EGFR-TKIs and overcoming the AR, such as intensification of EGFR inhibition, targeting a specific alteration of the EGFR downstreaming signaling and finally combining the EGFR plus alternate pathway inhibition. This review will discuss the mechanism of action, the available data and the future implications of second-generation EGFR-TKIs for the treatment of advanced NSCLC.
Intensification of EGFR inhibition
EGFR mutations identify a distinct subgroup of NSCLC characterized by oncogene addiction, for which cell’s growth and survival signals are dependent upon EGFR activation. In this scenario, cells would develop resistance mechanisms that reactivate EGFR despite the presence of an inhibitor, as the acquired T790M second site mutation in the exon 20 of EGFR gene. The T790M missense mutation could be classified as the gatekeeper mutation, occurring within the ATP-binding site and interfering with the first-generation TKI’s binding by steric hindrance, causing a bulky methionine side chain in the receptor kinase domain [19]. In vitro studies showed that exposing EGFR-mutant lung cancer cell lines to a mutagen and culturing them in the presence of an EGFR-TKI, the resistant clones with the T790M mutation maintained a persistent phosphorylation [20]. Given this role of persistent EGFR signaling, many trials evaluated the intensification of EGFR inhibition through the use of drug molecules with additional activity against other receptors in the EGFR family, as the second-generation neratinib, dacomitinib and afatinib [21]. These inhibitors are mainly different from erlotinib and gefitinib for two features: each forms a covalent and irreversible attachment to the EGFR kinase domain, and each also inhibits other members of the ERBB family (Figure 2).
Figure 2.

Epidermal Growth Factor Receptor Family and intracellular pathway (adapted from http://nature.com ).
Neratinib
Neratinib is an oral, irreversible inhibitor of both EGFR and HER2; in preclinical studies conducted on cell lines with both an activating EGFR mutation and the T790M, neratinib was more effective at suppressing cell proliferation than gefitinib [22]. After a phase I trial, recruiting heavily pretreated patients with NSCLC in which the maximum tolerated dose (MTD) was at 320 mg daily [23], neratinib was studied in a phase II study. During the study, due to the development of grade 3 diarrhea in more than 50% of patients at MTD, the dose was decreased to 240 mg orally daily. Unfortunately the drug showed modest clinical efficacy with only 3% response rate in 91 EGFR mutant patients included in a phase II study and with no response in patients EGFR T790M mutated [24]. The lack of efficacy could probably be related to the high concentrations of neratinib required in preclinical studies to inhibit EGFR T790M and the limitations of clinical dosing.
Dacomitinib
Dacomitinib is an irreversible EGFR, HER2 and HER4 inhibitor with a higher kinase inhibition than gefitinib/erlotinib in both gefitinib/erlotinib-sensitive and in EGFR-T790M and HER2-mutated cell lines [25]. After a phase I trial, recruiting pretreated NSCLC patients and establishing the MTD at 45 mg orally daily [26], a phase II study, performed in patients with NSCLC after failure of ≥ 1 chemotherapy regimen and erlotinib, showed a promising activity and a meaningful improvements in the patient-reported outcomes (PROs) [27]. A large phase II study randomly assigned 188 pretreatred NSCLC to dacomitinb or erlotinib. In the overall population PFS, the primary end-point of the study was 12.4 weeks for dacomitinib arm and 8.3 weeks for erlotinib arm; the PFS benefit was consistent across several subgroups and particularly remarkable in patients with KRAS wild-type tumors, with a median PFS of 16.1 weeks versus 8.3 weeks in the experimental and control arm respectively [28]. The ongoing ARCHER 1009 study, a phase III, multicenter, double-blinded trial comparing dacomitinib to erlotinib in pretreatred NSCLC will clarify whether new generation EGFR-TKIs are superior to first-generation agents particularly in the KRAS wild-type population [29]. A recent study evaluated the efficacy of dacomitinib in front-line setting in NSCLC patients with activating EGFR mutations (Table 2). The study showed that dacomitinib is particularly effective with a response rate of 74% and a median PFS of 16.8 months [30]. Based on these data, the ARCHER 1050, a phase III, open label trial has been designed to compare the efficacy of first line dacomitinib versus gefitinib in 440 EGFR mutant patients with stage IIIB/IV NSCLC. The primary endpoint is PFS by Independent Radiologic Review. This trial is ongoing, with no data currently available [31].
Table 2.
First line trials of 2 nd generation TKI in EGFR mutated patients
| N | OR% | mPFS (mo) | mOS (mo) | |
|---|---|---|---|---|
| Dacomitinib | ||||
| Phase II trial30 | 92 | 74 | 17 | NR |
| Afatinib | ||||
| LUX LUNG II trial 34 | ||||
| All EGFR mutations | 129 | 61 | 10.1 | 24.8 |
| Exon 19–21 mutations | 106 | 66 | 13.7 | 38.7 |
| LUX LUNG III trial 35 | ||||
| All EGFR mutations | ||||
| Afatinib | 230 | 56 | 11.1 | NR |
| Cis/Gem | 115 | 23 | 6.9 | NR |
| Exon 19–21 mutations | ||||
| Afatinib | 204 | - | 13.6 | NR |
| Cis/Gem | 104 | - | 6.9 | NR |
| LUX LUNG VI trial 39 | ||||
| All EGFR mutations | ||||
| Afatinib | 242 | 66.9 | 11.1 | |
| Cis/Gem | 122 | 23 | 5.6 | |
| XL647 | ||||
| Phase II trial53 | ||||
| All EGFR mutations | 41 | 20 | 5.3 | 19 |
| Exon 19–21 mutations | 14 | 57 | 9.3 | NR |
Afatinib
Afatinib is an orally, irreversible EGFR, HER2 and HER4 inhibitor, showing preclinical activity against cancer cells harboring common activating EGFR mutations and the T790M mutation, albeit with a lower potency [32]. Phase 1 studies established the MTD at 50 mg orally daily, with diarrhea and rash as most common adverse event [33]. The role of afatinib in first line setting has been investigated in three studies (Table 2). The first, the LUX-LUNG 2 trial, was a phase 2 trial exploring the efficacy of afatinib in patients with stage IIIb/IV, EGFR mutated NSCLC. The study, enrolling patients untreated or previously exposed to chemotherapy, was subsequently amended to allow only untreated patients. The primary endpoint was objective response rate by Independent Radiologic Review. Among the 129 enrolled patients, 61 received afatinib as first-line treatment and 68 in second line, 99 received 50 mg orally daily as starting dose and 30 received 40 mg orally daily; 106 patients presented the common exon 19 or 21 EGFR mutations and 23 the other less common mutations. The objective response rate in the overall population was 61% by independent review and 60% by investigator assessment. In the EGFR-mutated patients, the objective response rate by independent review was 66%, whereas was 39% in patients with uncommon EGFR mutations. Drug-related adverse event, mainly diarrhea and skin rush were observed in the vast majority of cases, with about a quarter of patients experiencing grade 3 adverse events with 50 mg dose. These data indicated that the daily dose of 40 mg was preferable for additional studies [34]. Two other phase III trials, in the same setting, have been subsequently designed. The LUX-LUNG 3 study [35], a multicenter, randomized, open-label phase III study compared afatinib with cisplatin plus pemetrexed in patients with lung adenocarcinoma, stage IIIb/IV harboring EGFR mutations [36, 37]. Among the 1,269 screened patients, 345 resulted eligible and were randomized in a two-to-one fashion to afatinib 40 mg daily or chemotherapy up to a maximum of six cycles (without any maintenance therapy). As expected, patients were mainly East Asian, never-smokers and women; EGFR mutations were predominantly exon 19 deletions and L858R point mutations. The PFS assessed by independent review (primary endpoint) has been significantly prolonged in the afatinib arm compared to chemotherapy arm, with median PFS of 11.1 and 6.9 months, and 13.6 versus 6.9 months in patients with classical (exon 19 deletion or exon 21) EGFR mutations. Afatinib has achieved a higher response rates compared with chemotherapy according to both independent (56% versus 23%) and investigator (69% versus 44%) assessment, and higher disease control rate (90% versus 81% by independent review). The most frequent (≥20% incidence) adverse reactions from afatinib were diarrhea, rash/dermatitis acneiform, stomatitis, paronychia, dry skin, decreased appetite and pruritus; treatment-related adverse events grade ≥ 3 occurred in 112 patients (49%) receiving afatinib but therapy was discontinued just in 8%; predose plasma samples on days 1 and 8 of cycle two and day 1 of cycle three demonstrate that dose modification, due to individual tolerability, optimized the exposure to afatinib, holding efficacious plasma levels [38]. In the third study, the LUX LUNG 6 [39], Asian patients harbouringEGFR mutations have been randomized in a two-to-one fashion to afatinib 40 mg daily or cisplatin plus gemcitabine. The study showed that patients treated with afatinib had a significantly longer PFS than individuals receiving chemotherapy (median PFS 11.0 versus 5.6 months, p < 0.0001), as well as higher response rate (66.9% versus 23.0%, p < 0.0001) and higher disease control rate (92.6% versus 60.2%, p < 0.0001).
Two prospective studies have investigated the role of afatinib in patients with acquired resistance to first generation EGFR-TKIs (Table 3). The first reported trial, the LUX LUNG 1 [40, 41], randomly allocated 585 NSCLC patients to afatinib 50 mg daily or placebo. Eligible patients had received one or two previous chemotherapy regimens and had disease progression after at least 12 weeks of treatment with erlotinib or gefitinib. The median OS (primary endpoint) was not different even if a significant difference in PFS was observed (3.3 versus 1.1 months, p < 0.0001 by independent review). In the same setting, the LUX LUNG 4 trial [42] enrolled 62 Japanese patients showing 8.2% response rate and a median PFS of only 4.4 months. Overall these data indicate that afatinib is modestly effective in patients with EGFR-TKI acquired resistance and the best setting for using the agent is in front-line only in patients harbouring EGFR mutations.
Table 3.
Trials of 2 nd generation TKI in acquired resistance setting
| N | OR% | mPFS (mo) | mOS (mo) | |
|---|---|---|---|---|
| Afatinib | ||||
| LUX LUNG I trial 40 | ||||
| Afatinib | 390 | 7 | 3.3 | 10.8 |
| Placebo | 195 | <1 | 1.1 | 12.0 |
| LUX LUNG IV trial 42 | 61 | 8.2 | 4.4 | 19 |
| XL647 | ||||
| Phase II trial52 | 41 | 3 | 3.6 | 16.1 |
<, less than.
In EGFR-TKI pretreatred patients, preclinical models suggested that combination of afatinib with cetuximab, a monoclonal antibody against the extracellular domain of the EGFR, is particularly effective in presence of acquired resistance [43].The superiority of the combination is hypothesized to be due to cetuximab’s ability to cause down-regulation of EGFR and afatinib’s ability to block its kinase activity, leading to significantly lower level EGFR pathway signaling, even with a T790M mutation. Based on these findings, a large phase Ib/II study showed a promising 32% response rate among the 53 individuals with EGFR-T790M mutation [44, 45]. Although cetuximab-afatinib combination showed promising results, current data do not justify its use outside clinical trials.
Targeting a specific alteration of the EGFR downstreamingsignalling
Recently, investigators have identified covalent pyrimidine EGFR inhibitors, which are 30–100 fold more potent than quinazoline based EGFR inhibitors against EGFR T790M cells, and up to 100 fold less potent against wild type EGFR cells, as CO-1686 and AP26113 [46].
CO-1686, an oral covalent TKI, has been investigated in a phase I/II study [47], enrolling T790M-mutated patients pretreated with first generation TKIs. Preliminary results suggested a relevant activity and satisfactory tolerability, with the absence of typical adverse events derived from EGFR inhibition. AP26113, a novel TKI that potently inhibits mutant activated forms of ALK, EGFR and TKI-resistant forms including T790M positive, is nowadays testing in a phase I dose finding study [48] with initial evidence of activity in EGFR mutated patients progressed to prior TKI therapy.
Considering the high selective target, this class of inhibitors could likely represent a real chance of treatment for patients with EGFR-TKI acquired resistance, after the necessary confirmation derived from large, prospective, randomized trials.
Combinating the EGFR plus alternate pathways inhibition
Vascular endothelial growth factor (VEGF) pathway has been extensively studied as therapeutic target in NSCLC. Several studies demonstrated the existence of a cross-talk between VEGF and EGFR pathways, being the EGFR a regulator of VEGF expression, in turn associated with resistance to EGFR inhibition [49, 50]. Therefore, there is a strong rationale for using strategies inhibiting both targets as the multi-targeted TKI agents XL647 and BMS-690514.
XL647, an oral small-molecule inhibitor of EGFR, VEGFR-2, HER2 and Ephrin type-B receptor 4 (EphB4), in preclinical studies showed efficacy against EGFR-driven tumors, including those harboring T790M[51]. However, a phase II trial enrolling patients with EGFR-TKI acquired resistance, demonstrated a disappointing 3% RR, not supporting further investigation [52], Table 3. At the same time, another phase II trial of first-line XL647 in a population enriched for EGFR mutations (i.e. lung adenocarcinoma, never-smokers, females, EGFR mutated patients) (Table 2) showed a RR of 19.6% in the overall population and a RR of 57% among the 14 patients harbouring an activating EGFR mutation. These data suggested that the efficacy of XL647 is confined to the EGFR mutated population [53]. In a subsequent exploratory study, Chmielecki et al. investigated the clinical characteristics of eight patients with metastatic EGFR-mutant lung adenocarcinoma who were treated first-line with XL647 and then progressed. The study showed that, among the 5 tumor samples collected at the time of XL647 failure, only one harbored the T790M mutation and that three patients treated with second line erlotinib derived additional long-term benefit from the EGFR TKI [54]. Overall, the study emphasize the potential role of XL647 as an agent that do not necessarily select for T790M-mediated resistance and allow the sequential use of non-cross-resistant EGFR TKIs.
BMS-690514, a reversible oral inhibitor of EGFR, HER-2 and −4, VEGFRs-1 to −3, showed antitumour activity in tumour xenograft models and in cell lines containing the EGFR T790M mutation, suggesting a role against erlotinib-resistant tumours [55]. A phase I/II trial has recently evidenced a modest activity and disease control in both erlotinib-naıve and erlotinib-resistant NSCLC patients [56]. A randomised phase II trial comparing BMS 690514 with erlotinib is currently ongoing.
Conclusion
In the last years, the treatment of NSCLC has dramatically changed. Currently, the treatment’s choice is based on a careful assessment of tumor histology and, most importantly, of biological characteristics, specifically of the EGFR and ALK status. EGFR mutations identify a distinct subgroup of NSCLC which benefit from a first line treatment with first generation EGFR-TKIs, as clearly evidenced by several phase III trials. Second generation TKIs are new agents currently under investigation with the intent to improve the efficacy in the first line setting and to provide a valid treatment option in the acquired resistance setting. New inhibitors such as dacomitinib and afatinib differ from first generation EGFR-TKIs because they form irreversible link to the EGFR kinase domain and inhibit other receptors (i.e. HER2 and HER4). The potential superiority of dacomitinib versus erlotinib in KRAS wild-type NSCLC is currently under investigation in a large phase III trial. Afatinib demonstrated superiority versus standard chemotherapy in two phase III studies and based on the results of the LUX LUNG 3 trial, Food and Drug Administration has recently approved the drug for the first-line treatment of patients with NSCLC whose tumors harbored EGFR exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test, specifically the therascreen EGFR RGQ PCR Kit (QIAGEN). Subsequently, the European Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for afatinib.
From the clinical point of view there are many questions about new EGFR-TKIs including efficacy and toxicity over erlotinib or gefitinib. At the present time there is no phase III study directly comparing new versus old EGFR-TKIs. Based on indirect comparison it seems that new generation EGFR-TKIs produce a similar response rate to erlotinib or gefitinib with a possible improvement in terms of progression-free survival and with an increased risk of side effects particularly skin rash and diarrhea. Available data with afatinib in NSCLC with acquired resistance to erlotinib or gefitinib indicate that this agent is modestly effective in such setting and new drugs and new strategies are urgently needed.
Disclosure of potential conflicts of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was utilized in the production of this manuscript.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
IS and FC carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Irene Stasi, Email: stasi.irene@gmail.com.
Federico Cappuzzo, Email: f.cappuzzo@gmail.com.
References
- 1.Pao RW, Miller V, Zakowsky M, et al. Proc Natl Acad Sci U S A. 2004. ECF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tokumo M, Toyooka S, Trak KI, et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non-small cell lung cancers. Clin Cancer Res. 2005;ll:1167–1173. [PubMed] [Google Scholar]
- 3.Sequist LV, Bell D\7, Lynch TJ, Herb DA. Molecular predictors of response to epidermal growth facror receptor antagonists in non-small-cell lung. J Clin Oncol. 2007;25:587–595. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 4.Han JY, Park K, Kim SW, et al. First-SIGNAL: fırst-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;30:1122–1128. doi: 10.1200/JCO.2011.36.8456. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Thongprasert S, et al. Gefıtinib or carboplatin paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.Mitsudomi T, Morita S, Yatabe Y, et al. Gefıtinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 7.Maemondo M, Inoue A, Kobayashi K, et al. Gefıtinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 8.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as fırstline treatment for patients with advanced EGFR mutation-positive nonsmall- cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 9.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as fırst-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;3:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 10.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75–126. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005;2:73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 13.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 14.Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–7525. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 16.Pellegrini C, Falleni M, Marchetti A, et al. HER-2/Neu alterations in non-small cell lung cancer A comprehensive evaluation by real time reverse transcription-PCR, fluorescence in situ hybridization, and immunohistochemistry. Clin Cancer Res. 2003;9:3645–3652. [PubMed] [Google Scholar]
- 17.Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 Amplifi cation: A Potential Mechanism of Acquired Resistance to EGFR Inhibition in EGFR -Mutant Lung Cancers That Lack the Second-Site EGFR T790M Mutation. Cancer Discovery. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci Transl Med. 2011;3(75):75. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14(10):2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 20.Godin-Heymann N, Ulkus L, Brannigan BW, et al. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7(4):874–879. doi: 10.1158/1535-7163.MCT-07-2387. [DOI] [PubMed] [Google Scholar]
- 21.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to EGFR tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17(17):5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 23.Wong KK, Fracasso PM, Bukowski RM. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15:2552–2558. doi: 10.1158/1078-0432.CCR-08-1978. [DOI] [PubMed] [Google Scholar]
- 24.Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J clinical oncology. 2010;28:3076–3083. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 25.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 26.Janne PA, Schellens JH, Engleman JA, et al. Preliminary activity and safety results from a phase I clinical trial of PF-00299804, an irreversible pan-HER inhibitor, in patients (pts) with NSCLC. J Clin Oncol. 2008;26:8027. doi: 10.1200/JCO.2007.14.7611. [DOI] [Google Scholar]
- 27.Campbell A, Reckamp KL, Camidge DR, et al. PF-00299804 (PF299) patient-reported outcomes and efficacy in adenocarcinoma and nanadeno non-small cell lung cancer: a phase 2 trial in advanced NSCLC after failure of chemotherapy and erlotinib. J Clin Oncol. 2010;15(28):7602. [Google Scholar]
- 28.Ramalingam S, Blackhall F, Krzakowski M, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30(27):3337–3344. doi: 10.1200/JCO.2011.40.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyer M, Janne PA, Mok T, et al. J Clin Oncol. 2012. ARCHER: Dacomitinib (D; PF-00299804) versus erlotinib (E) for advanced (adv) non-small cell lung cancer (NSCLC)—A randomized double-blind phase III study. [Google Scholar]
- 30.Kris MG, Mok T, Ou SH, et al. First-line dacomitinib (PF-00299804), an irreversible pan-HER tyrosine kinase inhibitor, for patients with EGFR-mutant lung cancers. J Clin Oncol. 2012;30:7602. [Google Scholar]
- 31.Mok T, Nakagawa K, Rosell R, et al. Phase III randomized, open label study (ARCHER 1050) of first-line dacomitinib (D) versus gefitinib (G) for advanced (adv) non-small cell lung cancer (NSCLC) in patients (pts) with epidermal growth factor receptor (EGFR) activating mutation (s) J Clin Oncol. 2013;31:abstr TPS8123. doi: 10.1200/JCO.2012.43.0652. [DOI] [Google Scholar]
- 32.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yap TA, Vidal L, Adam J, et al. Phase I trial of the irreversible ErbB1 (EGFR) and ErbB2 (HER2) kinase inhibitor BIBW 2992 in patients with advanced solid tumours. J Clin Oncol. 2010;28:3965–3972. doi: 10.1200/JCO.2009.26.7278. [DOI] [PubMed] [Google Scholar]
- 34.Yang JC, Shih JY, Su WC, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): A phase 2 trial. Lancet Oncol. 2012;13:539–548. doi: 10.1016/S1470-2045(12)70086-4. [DOI] [PubMed] [Google Scholar]
- 35.Sequist L, Yang JC, Yamamoto N, et al. J Clin Oncol. 2013. Phase III study of afatinib or cispaltin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. [DOI] [PubMed] [Google Scholar]
- 36.Scagliotti G, Hanna N, Fossella F, et al. The differential efficacy of pemetrexed according to NSCLC histology: A review of two phase III studies. Oncologist. 2009;14:253–263. doi: 10.1634/theoncologist.2008-0232. [DOI] [PubMed] [Google Scholar]
- 37.Al-Saleh K, Quinton C, Ellis PM. Role of pemetrexed in advanced non-small-cell lung cancer: Meta-analysis of randomized controlled trials, with histology subgroup analysis. Curr Oncol. 2012;19:e9–e15. doi: 10.3747/co.19.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang JC, Hirsh V, Schuler M, et al. Symptom Control and Quality of Life in LUX-Lung 3: A Phase III Study of Afatinib or Cisplatin/Pemetrexed in Patients With Advanced Lung Adenocarcinoma With EGFR Mutations. J Clin Oncol. 2013;31:1764. doi: 10.1200/JCO.2012.46.1764. [DOI] [PubMed] [Google Scholar]
- 39.Wu YL, Zhou C, Hu CP, et al. J Clin Oncol. 2013. LUX-Lung 6: A randomized, open-label, phase III study of afatinib (A) versus gemcitabine/cisplatin (GC) as first-line treatment for Asian patients (pts) with EGFR mutation-positive (EGFR M+) advanced adenocarcinoma of the lung. [Google Scholar]
- 40.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 41.Jackman D, Pao W, Riely GJ, et al. Clinical defi nition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katakami N, Atagi S, Goto K, et al. J Clin Oncol. 2013. LUX-Lung 4: A Phase II Trial of Afatinib in Patients With Advanced Non–Small-Cell Lung Cancer Who Progressed During Prior Treatment With Erlotinib, Gefitinib, or Both. [DOI] [PubMed] [Google Scholar]
- 43.Regales L, Gong Y, Shen R, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119:3000–3010. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janjigian Y, Groen H, Horn L. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol. 2011;29:482s. [Google Scholar]
- 45.Janjigian YY, Smit EF, Horn L, et al. Ann Oncol. 2012. Activity of afatinib/cetuximab in patients (pts) with EGFR mutant non-small cell lung cancer (NSCLC) and acquired resistance (AR) to EGFR inhibitors. [Google Scholar]
- 46.Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sequist LV, Soria JC, Gadgeel SM, et al. J Clin Oncol. 2013. First-in-human evaluation of CO-1686, an irreversible, selective, and potent tyrosine kinase inhibitor of EGFR T790M. [Google Scholar]
- 48.Camidge R, Bazhenova L, Salgia R, et al. J Clin Oncol. 2013. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: updated results. [Google Scholar]
- 49.Ciardiello F, Troiani T, Bianco R, Orditura M, Morgillo F, et al. Interaction between the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor (VEGF) pathways: A rational approach for multi-target anticancer therapy. Ann Oncol. 2006;17:vii109–vii114. doi: 10.1093/annonc/mdl962. [DOI] [PubMed] [Google Scholar]
- 50.Naumov GN, Nilsson MB, Cascone T, Briggs A, Straume O, et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res. 2009;15:3484–3494. doi: 10.1158/1078-0432.CCR-08-2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gendreau SB, Ventura R, Keast P, et al. Inhibition of the T790M gatekeeper mutant of the epidermal growth factor receptor by EXEL-7647. Clin Cancer Res. 2007;13:3713–3723. doi: 10.1158/1078-0432.CCR-06-2590. [DOI] [PubMed] [Google Scholar]
- 52.Pietanza MC, Lynch TJ, Jr, Lara PN, Jr, et al. XL647 a multitargeted tyrosine kinase inhibitor: results of a phase II study in subjects with non-small cell lung cancer who have progressed after responding to treatment with either gefitinib or erlotinib. J thoracic oncology. 2012;7:219–226. doi: 10.1097/JTO.0b013e31822eebf9. [DOI] [PubMed] [Google Scholar]
- 53.Pietanza MC, Gadgeel SM, Dowlati A, et al. Phase II study of the multitargeted tyrosine kinase inhibitor XL647 in patients with non-small-cell lung cancer. J thoracic oncology. 2012;7:856–865. doi: 10.1097/JTO.0b013e31824c943f. [DOI] [PubMed] [Google Scholar]
- 54.Chmielecki J, Pietanza MC, Aftab D, et al. EGFR mutant lung adenocarcinomas treated first-line with the novel EGFR inhibitor, XL647, can subsequently retain moderate sensitivity to erlotinib. J Thorac Oncol. 2012;7(2):434–442. doi: 10.1097/JTO.0b013e31823c5aee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong TV, Lee FY, Emanuel S, et al. Antitumor and Antiangiogenic Activities of BMS-690514, an Inhibitor of Human EGF and VEGF Receptor Kinase Families. Clinical Cancer Res. 2011;17(12):4031–4041. doi: 10.1158/1078-0432.CCR-10-3417. [DOI] [PubMed] [Google Scholar]
- 56.Soria JC, Baselga J, Hanna N, et al. Phase I–IIa study of BMS-690514, an EGFR, HER-2 and −4 and VEGFR-1 to −3 oral tyrosine kinase inhibitor, in patients with advanced or metastatic solid tumours. Eur J Cancer. 2013;49(8):1815–1824. doi: 10.1016/j.ejca.2013.02.012. [DOI] [PubMed] [Google Scholar]