

Abstract
The complex tasks of making a confident diagnosis of a specific form of interstitial lung disease (ILD) and formulating a patient-centered, personalized management plan in an attempt to achieve remission or stabilization of the disease process can pose formidable challenges to clinicians. When patients are evaluated for suspected ILD, an accurate diagnosis of the specific form of ILD that a patient has developed must be made to provide the patient with useful prognostic information and to formulate an appropriate management plan that can relieve symptoms and restore or significantly improve quality of life. A well-performed patient history and physical examination provides invaluable information that can be combined with appropriate laboratory testing, imaging, and, if needed, tissue biopsy to reach a confident ILD diagnosis, and high-resolution computed tomography (HRCT) of the thorax is usually a key component of the diagnostic evaluation. If treatment is indicated, many forms of ILD can respond significantly to immunosuppressive anti-inflammatory therapies. However, ILD accompanied by extensive fibrosis may be difficult to treat, and the identification of an effective pharmacologic therapy for idiopathic pulmonary fibrosis (IPF) has remained elusive despite the completion of many phase 3 clinical trials over the past decade. Nonetheless, patients with IPF or advanced forms of non-IPF ILD can benefit significantly from detection and treatment of various co-morbid conditions that are often found in patients (especially the elderly patient), and supportive care (oxygen therapy, pulmonary rehabilitation) can have a beneficial impact on quality of life and symptom palliation. Finally, lung transplantation is an option for patients with progressive, advanced disease that does not respond to other therapies, but only a relatively small subset of patients with end-stage ILD are able to meet wait listing requirements and eventually undergo successful lung transplantation.
Keywords: Interstitial lung disease, Idiopathic pulmonary fibrosis, Therapeutics, Diagnosis
Introduction
Well over one hundred different forms of interstitial lung disease (ILD) have been described (see Table 1 for major categories). These diffuse infiltrative lung disorders are typically characterized by the presence of inflammation and altered lung interstitium, and specific forms of ILD can be differentiated from one another when clinical data, radiologic imaging, and pathologic findings (if lung biopsy is needed) are combined to reach a confident diagnosis [1, 2]. The histopathologic changes in the lungs of patients with ILD can range from granulomatous inflammation without parenchymal fibrosis in patients with sarcoidosis to extensive pulmonary fibrosis with architectural distortion of the lung in patients with idiopathic pulmonary fibrosis (IPF). Some forms of ILD have been linked to specific genetic abnormalities (e.g. Hermansky-Pudlak syndrome, familial pulmonary fibrosis), and a number of gene variants have been associated with an increased risk to develop ILD disorders such as IPF, sarcoidosis, or chronic beryllium disease (CBD). Interstitial lung disease can also complicate connective tissue disorders (CTD), and lung histopathologic changes can have features of usual interstitial pneumonia (UIP) or non-specific interstitial pneumonia (NSIP) patterns in CTD-associated ILD [3].
Table 1.
Interstitial lung disorders: major categories
| • Idiopathic interstitial pneumonia | • Sarcoidosis |
| o Idiopathic pulmonary fibrosis (IPF) | • Hypersensitivity pneumonitis |
| o Non-specific interstitial pneumonia (NSIP) | • Iatrogenic pneumonitis/fibrosis (drug-induced ILD, radiation injury) |
| o Cryptogenic organizing pneumonia (COP) | • Eosinophilic ILD (e.g. eosinophilic pneumonia) |
| o Respiratory bronchiolitis interstitial lung disease (RBILD) | • Occupational lung disease |
| o Desquamative interstitial pneumonia (DIP) | • Inherited disorders (e.g. familial pulmonary fibrosis, Hermansky-Pudlak syndrome) |
| o Acute interstitial pneumonia (AIP) | • Primary disorders (e.g. pulmonary Langerhans cell histiocytosis) |
| o Lymphoid interstitial pneumonia (LIP) | |
| • Connective tissue disease-associated interstitial lung disease (CTD-ILD) |
Successful management of patients with ILD is dependent upon establishing an accurate and specific diagnosis [1, 2]. Because various forms of ILD such as IPF, non-IPF forms of idiopathic interstitial pneumonia (IIP), CTD-ILD, and hypersensitivity pneumonitis (HP) can have similar clinical presentations, patients with suspected ILD must undergo an evaluation that adequately establishes a confident diagnosis of a specific ILD, as treatment and various management decisions are diagnosis-specific and may vary considerably according to the specific form of ILD that is diagnosed. This manuscript will focus on [1] the appropriate steps that are required to make an accurate diagnosis of specific types of ILD, [2] general approaches to disease monitoring and management, and (3) therapies for specific disorders such as IPF.
Review
Clinical evaluation
A thorough and comprehensive history may provide invaluable information that can suggest certain entities and provide suspicion that a patient may have a specific diagnosis such as hypersensitivity pneumonitis (HP) or CTD-ILD (Table 2). Additional clues to an ultimate diagnosis can be provided by pulmonary and extra-pulmonary physical examination findings (Table 3), and the differential diagnosis can be considerably narrowed when key elements of the patient interview and physical examination findings are combined with appropriate measurements of lung function, specific blood tests (Table 4) such as autoimmune serologies to assist in the detection of CTD if such are indicated, extra-pulmonary tissue sampling (e.g. lymph node or skin biopsy), and thoracic imaging.
Table 2.
Clues from the initial evaluation that suggest specific types of ILD
| History elicited | Frequently associated ILD or complications of ILD |
|---|---|
| Rapid onset and worsening | AIP |
| Infection | |
| Acute HP, acute EP | |
| Drug reaction | |
| COP | |
| CTD (e.g. acute lupus pneumonitis) | |
| DAH (e.g. GPS) | |
| Smoking | RB-ILD, DIP, PLCH |
| Occupation: Pipefitter, foundry worker, coal miner, | Pneumoconiosis |
| Pneumotoxic drug exposure | Drug-induced ILD |
| Hemoptysis | DAH, pulmonary capillaritis, pulmonary venoocclusive disease, LAM |
| Superimposed complications (e.g. pulmonary emboli, lung neoplasm) | |
| Pleurisy | CTD (SLE, RA) |
| Wheezing | HP, EP |
| Eye symptoms | CTD, sarcoidosis, PAG |
| Impaired vision combined with albinism & Puerto Rican heritage | HPS |
| Rash | Sarcoidosis, CTD |
| Exposure to organic antigens at home or at work (e.g. birds, grain dust, humidifiers, visible molds, hot tubs,etc.) | HP |
| Occupational ILD | |
| Abnormal GER, GERD, dysphagia | CTD (especially scleroderma), IPF |
| Sicca symptoms | Sjögren’s disease |
| Raynaud’s phenomenon | CTD |
| Arthralgias, arthritis | CTD, sarcoidosis |
| Myalgias, muscle weakness | DM-PM |
| Morning stiffness | RA, CTD |
| Age >70 years | IPF > other ILD if HRCT suspicious for IIP |
AIP = acute interstitial pneumonia; COP = cryptogenic organizing pneumonia; CTD = connective tissue disease; DAH = diffuse alveolar hemorrhage; DM-PM = dermatopolymyositis; EP = eosinophilic pneumonia; GER = gastroesophageal reflux; GERD = gastroesophageal reflux disease; GPS = Goodpasture’s syndrome; HP = hypersensitivity pneumonitis; HPS = Hermansky-Pudlak syndrome; IIP = idiopathic interstitial pneumonia; IPF = idiopathic pulmonary fibrosis; LAM = lymphangioleiomyomatosis; PAG = polyangiitis with granulomatosis; PLCH = pulmonary Langerhans cell histiocytosis; SLE = systemic lupus erythematosus; RA = rheumatoid arthritis.
Reprinted with permission from Interstitial Lung Disease: A Practical Approach. Meyer KC, Raghu G: Patient evaluation. Second Edition, New York: Springer; 2011:3–16.
Table 3.
Clues from the physical examination and their disease associations
| Organ system | Finding | Associated ILD or its complications |
|---|---|---|
| Lung | Velcro crackles | IPF, asbestosis > > other |
| Squeaks | HP, bronchiolitis | |
| Pleural rub | RA, SLE | |
| Skin | Erythema nodosum | Sarcoidosis, CTD, Behçet’s disease |
| Maculopapular rash | CVD, drugs, sarcoidosis, amyloid | |
| Heliotrope rash | DM-PM | |
| Gottron’s papules | DM-PM | |
| Café-au-lait spots | Neurofibromatosis | |
| Albinism | HPS | |
| Telangiectasia | Scleroderma | |
| Calcinosis | Scleroderma, DM-PM | |
| Subcutaneous nodules | RA, neurofibromatosis, vasculitis | |
| Cutaneous vasculitis | PAG, RA, MPA, SLE, drug reaction | |
| Mechanic’s hands | DM-PM | |
| Tight skin/ulcerations over digits | Scleroderma | |
| Eyes | Uveitis | Sarcoidosis, Behçet’s disease, AS |
| Scleritis | SLE, scleroderma, sarcoidosis, PAG | |
| Keratoconjunctivitis sicca | Sjögren’s disease, CTD | |
| Lacrimal gland enlargement | Sarcoidosis | |
| Horizontal nystagmus | HPS | |
| Salivary glands | Enlarged | Sjögren’s disease, sarcoidosis |
| Lymphatic | Lymphadenopathy | Sarcoidosis, lymphangitic CA, lymphoma |
| Reticuloendothelial | Hepatosplenomegaly | Sarcoidosis, LIP, CTD, EG, amyloid, lymphoma |
| Musculoskeletal | Muscle weakness, myositis | CTD (especially DM-PM), sarcoidosis |
| Synovitis, arthritis | CTD, sarcoidosis | |
| Nervous system | Neurologic dysfunction | Sarcoidosis, lymphangitic CA, NF, TS, CTD, PAG, MPA |
| Cardiovascular | Systemic hypertension | CTD, GPS, PAG, MPA, NF |
| Prominent P2 | Suggests secondary PH (IPF, CTD, end stage sarcoidosis) | |
| Pericardial rub | Sarcoidosis, SLE | |
| Extremities | Digital clubbing | IPF, asbestosis > chronic HP, DIP > other fibrotic ILD |
| Raynaud’s phenomenon | CTD |
AS = ankylosing spondylitis; CA = cancer; CTD = connective tissue disease; DAH = diffuse alveolar hemorrhage; DM-PM = dermatopolymyositis; GPS = Goodpasture’s syndrome; HP = hypersensitivity pneumonitis; HPS = Hermansky-Pudlak syndrome; LAM = lymphangioleiomyomatosis; LCH = Langerhans cell histiocytosis; LIP = lymphoid interstitial pneumonia; MPA = microscopic polyangiitis; NF = neurofibromatosis; P2 = auscultated pulmonic valve closure sound; PAG = polyangiitis with granulomatosis; PH = pulmonary hypertension; RA = rheumatoid arthritis; SLE = systemic lupus erythematosus; TS = tuberous sclerosis.
Reprinted with permission from Interstitial Lung Disease: A Practical Approach. Meyer KC, Raghu G: Patient evaluation. Second Edition, New York: Springer; 2011:3–16.
Table 4.
Clues for specific diagnoses from blood and urine testing
| Laboratory test | Abnormal result | Suggested disorder |
|---|---|---|
| CBC | Microcytic anemia | Occult pulmonary hemorrhage |
| Normocytic anemia | CTD, chronic disease | |
| Leukocytosis | Infection, hematologic malignancy | |
| Eosinophilia | Eosinophilic pneumonia, drug toxicity | |
| Thrombocytopenia | CTD, sarcoidosis | |
| Calcium | Hypercalcemia | Sarcoidosis |
| Creatinine | ↑ | CTD, pulmonary-renal syndrome, sarcoidosis; amyloidosis |
| Liver function | ↑ GGT, ALT, AST | Sarcoidosis, amyloidosis, CTD (polymyositis) |
| Urine | Abnormal sediment with RBC casts and/or dysmorphic RBCs | Vasculitis (CTD, PAG, GPS, MPA) |
| Muscle enzymes | ↑Increased CK, aldolase | PM, DM-PM |
| Angiotensin Converting Enzyme (ACE) | ↑ | Sarcoidosis (non-specific; can be increased in other ILD) |
| Lymphocyte proliferation | Stimulated by beryllium | CBD |
| Serum antibodies | ↓ Quantitative immunoglobulins | Immunodeficiency (CVID) |
| ↑ ANA, RF, anti-CCP | CTD, RA | |
| ↑ C-ANCA | PAG | |
| ↑ P-ANCA | CTD, vasculitis | |
| ↑ anti-GBM | GPS | |
| Positive specific precipitin | Supportive of HP | |
| ↑ anti-Jo-1 or other anti-synthetase autoantibodies | PM, DM-PM | |
| ↑ SS-A, SS-B | Sjögren’s syndrome |
CBD = chronic beryllium diseases; COP = cryptogenic organizing pneumonia; CTD = connective tissue disease; CVID = common variable immunodeficiency; DAH = diffuse alveolar hemorrhage; DM-PM = dermatopolymyositis; DIP = desquamative interstitial pneumonia; GPS = Goodpasture’s syndrome; HP = hypersensitivity pneumonitis; MPA = microscopic polyangiitis; PM = polymyositis; PAG = polyangiitis with granulomatosis; RA = rheumatoid arthritis;
Reprinted with permission from Interstitial Lung Disease: A Practical Approach. Meyer KC, Raghu G: Patient evaluation. Second Edition, New York: Springer; 2011:3–16.
The routine postero-anterior chest X-ray (CXR) can be highly suggestive of specific ILD entities (Table 5), and previous CXRs, if available, should be sought to determine whether the disease is acute versus chronic. Other studies should not be overlooked; as specific examples, abdominal computed tomographic (CT) scans usually show a substantial portion of the lower lung regions and thorax, and cervical spine CT scans can show extensive areas of the upper chest and lungs.
Table 5.
Thoracic imaging patterns
| Imaging modality | Pattern | Consistent ILD diagnoses, mimics of ILD, and/or complications of ILD |
|---|---|---|
| Routine CXR | Hilar lymphadenopathy | Sarcoidosis, silicosis, CBD, infection, malignancy |
| Septal thickening | CHF, malignancy, infection, PVOD | |
| Lower lung zone predominance | IPF, asbestosis, DIP, CTD, NSIP | |
| Mid/upper lung zone predominance | Sarcoidosis, silicosis, acute HP, LCH, CBD, AS, chronic EP | |
| Peripheral lung zone predominance | COP, chronic EP, IPF | |
| Honeycomb change | IPF, asbestosis, chronic HP, sarcoidosis, fibrotic NSIP, CTD | |
| Small nodules | Sarcoidosis, HP, infection | |
| Cavitating nodules | PAG, mycobacterial infection, CA | |
| Migratory or fluctuating opacities | HP, COP, DIP | |
| Pneumothorax | PLCH, LAM, neurofibromatosis, TS | |
| Pleural involvement | Asbestosis, CTD, acute HP, malignancy, sarcoidosis, Radiation fibrosis | |
| Kerley B line prominence | Lymphangitic carcinomatosis, CHF | |
| HRCT | Nodules | Sarcoidosis HP, CBD, pneumoconiosis, RA, malignancy |
| Septal thickening | Edema, malignancy, infection, drug toxicity, PVOD | |
| Cyst formation | LAM, LCH, LIP, DIP, SS | |
| Reticular lines | IPF, asbestosis, chronic EP, chronic HP, CTD, NSIP | |
| Traction bronchiectasis | IPF, other end-stage fibrosis | |
| Honeycomb change | IPF, chronic EP and HP, asbestosis, sarcoidosis | |
| Ground-glass opacity | AIP, acute EP, PAP, chronic EP, COP, lymphoma, sarcoidosis, NSIP, infection, hemorrhage |
AIP = acute interstitial pneumonia; AS = ankylosing spondylitis; CA = cancer; CBD = chronic beryllium diseases; COP = cryptogenic organizing pneumonia; CTD = connective tissue disease; DAH = diffuse alveolar hemorrhage; DM-PM = dermatopolymyositis; DIP = desquamative interstitial pneumonia; EP = eosinophilic pneumonia; HP = hypersensitivity pneumonitis; HPS = Hermansky-Pudlak syndrome; IPF = idiopathic pulmonary fibrosis; LAM = lymphangioleiomyomatosis; LCH = Langerhans cell histiocytosis; LIP = lymphoid interstitial pneumonia; NF = neurofibromatosis; NSIP = non-specific interstitial pneumonia; PAG = polyangiitis with granulomatosis; PAP = pulmonary alveolar proteinosis; P2 = auscultated pulmonic valve closure sound; PH = npulmonary hypertension; PVOD = pulmonary veno-occlusive disease; RA = rheumatoid arthritis; SLE = systemic lupus erythematosus; SS = Sjögren’s syndrome; TS = tuberous sclerosis.
Reprinted with permission from Interstitial Lung Disease: A Practical Approach. Meyer KC, Raghu G: Patient evaluation. Second Edition, New York: Springer; 2011:3–16.
Although the combination of history, physical examination, CXR, and other appropriate laboratory testing (peripheral blood tests and lung physiologic testing) may provide a likely diagnosis, additional testing is usually needed to reach a confident diagnosis of a specific ILD. HRCT of the thorax can provide invaluable information that strongly supports a specific diagnosis (Table 5) and may be diagnostic (e.g. typical changes of UIP) such that further testing with bronchoscopy or surgical lung biopsy is not required (Figure 1). Indeed, the HRCT has become a standard test for the evaluation of patients with possible ILD [4]. In general, a complete lack of pulmonary parenchymal changes on HRCT imaging virtually excludes a diagnosis of ILD, although ILD may rarely still be present with the lungs having microscopic involvement that does not reach the threshold for the detection of an abnormality that is detectable by HRCT. Multi-detector computed tomography (MDCT) scanning can scan the entire thorax with a single breath-hold maneuver and allow even better imaging than traditional HRCT, and an algorithmic approach can be utilized that facilitates differentiation among UIP, NSIP, and chronic HP patterns [5].
Figure 1.
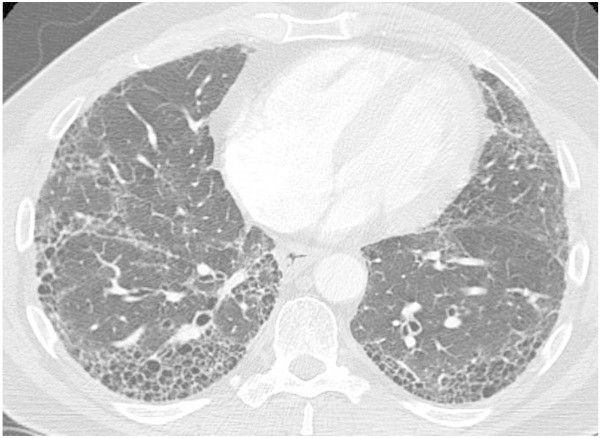
HRCT cross-sectional view showing a pattern of peripheral reticulation and honeycomb change that is diagnostic of the presence of UIP.
Invasive diagnostic procedures
Bronchoscopy and/or surgical lung biopsy may be required to make a confident diagnosis of a specific ILD (Figure 2). Bronchoscopy is generally a very safe procedure if performed by an experienced bronchoscopist [6], and the most serious potential complications are pneumothorax or excessive bleeding that may occur as a consequence of transbronchial biopsy (TBLBx). Bronchoalveolar lavage (BAL) can be readily performed, and the recently published, ATS Task Force Report on BAL for the diagnosis of ILD recommends using recently obtained HRCT imaging to choose an appropriate segment of the lung in which to perform BAL from a wedge position [7]. The right middle lobe or lingula of the left upper lobe are likely the best regions to perform lavage when diffuse disease is present, and areas with ground-glass opacification or profuse nodular change are more likely to provide useful diagnostic information (e.g. differential cell count of nucleated immune cells) than areas with extensive fibrosis. In addition to total and differential BAL cell counts, BAL fluid and sediment can be analyzed for infection or the presence of malignant cells, and the gross appearance of freshly retrieved BAL fluid may provide diagnosistic information (e.g. progressively increasing blood in sequential aliquots that is seen with diffuse alveolar hemorrhage or white-tan discoloration of the BAL fluid with rapidly settling tan sediment [due to gravity] that can be seen with pulmonary alveolar proteinosis). Significant BAL lymphocytosis or eosinophilia may provide strong support for a specific diagnosis when combined with imaging and clinical data, but routine determination of BAL lymphocyte subsets is unlikely to provide additional useful information [7, 8].
Figure 2.
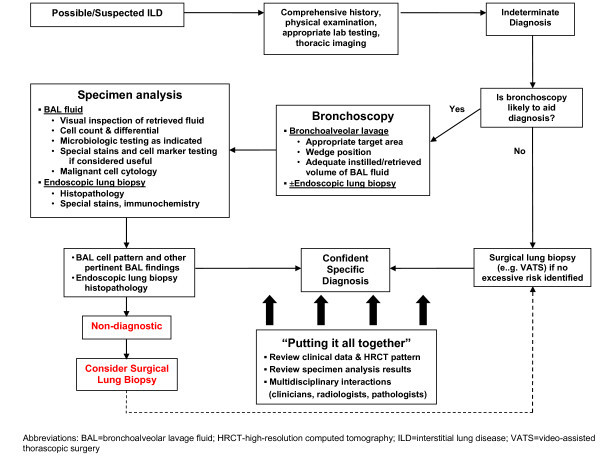
Suggested approach to the diagnosis of ILD. Abbreviations: BAL = bronchoalveolar lavage fluid; HRCT-high-resolution computed tomography; ILD = interstitial lung disease; VATS = video-assisted thorascopic surgery.
Endobronchial biopsy may provide useful information if endobronchial abnormalities are present (e.g. superficial nodules, mucosal ulceration). Similar to performing BAL, TBLBx is best performed away from areas of advanced fibrosis (as identified by HRCT). Multiple biopsies performed with an adequately sized forceps can provide good tissue sampling for some forms of ILD (e.g. sarcoidosis), but TBLBx is likely to be non-diagnostic when extensive/advanced fibrotic disease is present.
A surgical lung biopsy (SLB) obtained via video-assisted thoracic surgery (VATS) or open biopsy is likely to provide an excellent specimen (if properly performed) that shows a histopathologic pattern that can usually be considered to be definitively diagnostic of a specific disease entity. However, one must weigh risks and benefits when performing a SLB is considered, especially in frail elderly patients, patients with ventilatory compromise, patients with moderate to severe pulmonary hypertension, or patients with multiple co-morbidities. Open lung biopsy in patients with suspected ILD has approximately a 4.3% 30-day mortality rate, whereas VATS biopsy appears to be safer with an associated 30-day mortality that is somewhat lower than open biopsy but not negligible at approximately 2.1% [9]. Additionally, the highest mortality risk may occur in patients whose ultimate diagnosis is IPF [10, 11], and SLB in patients with IPF has been reported to trigger an acute exacerbation of IPF [12]. However, confirming the diagnosis and differentiating among specific forms of IIP may not be possible without performing SLB.
There is increasing awareness that an abnormal degree of gastroesophageal reflux (GER) combined with aspiration may play a significant role in the pathogenesis of a number of forms of ILD [13, 14]. GER disease (GERD) has been associated with IPF and with pulmonary fibrosis in patients with systemic sclerosis (scleroderma), and GER with aspiration may play a role in triggering and/or driving lung inflammation and fibrosis in IPF and scleroderma, and it has been linked to acute exacerbations in patients with IPF [15, 16]. Additionally, esophageal dysmotility may play a role in predisposition to aspiration and is usually found in patients with scleroderma and may be present in patients with other forms of CTD with associated ILD [14, 17, 18]. Esophageal manometry to detect esophageal functional abnormalities plus impedance/pH testing to detect and characterize GER may provide evidence of foregut functional abnormalities that may play a role in IPF and possibly CTD-ILD pathogenesis, and obtaining such information by placing esophageal probes may provide findings that support the employment of strategies to prevent or blunt pathologic GER [19]. More research is needed in this area to better understand the role of GER and microaspiration in the pathogenesis of IPF and other forms of ILD.
Making an accurate and confident diagnosis of a specific form of ILD
Patients with suspected ILD should have information from the history, physical examination, thoracic imaging, and other testing (e.g. peripheral blood tests, pulmonary function testing) carefully and thoughtfully reviewed to determine whether or not additional procedures are needed and whether such procedures are likely to be helpful in reaching a confident diagnosis. If one needs to obtain invasive testing (bronchoscopy with BAL and/or TBLBx, VATS biopsy), all findings should be reviewed (preferably in a multi-disciplinary fashion) to identify the ultimate diagnosis that best fits with the combination of clinical information, imaging, and invasive testing results [20]. Risks and potential benefit of invasive testing should be carefully considered, especially for frail, elderly patients.
Some specific combinations of clinical data with imaging results and other findings can strongly support specific ILD diagnoses. A younger patient with nodular changes along bronchovascular structures and bilateral hilar lymphadenopathy on HRCT imaging is highly likely to have a diagnosis of sarcoidosis, and the presence of a significant lymphocytosis on a BAL cell count determination would be highly supportive of this diagnosis. A patient with clearly abnormal autoimmune serologies and either a NSIP or UIP pattern on HRCT imaging is likely to have CTD-ILD and may have a specific CTD diagnosis such as rheumatoid arthritis or scleroderma that can be detected and confirmed via peripheral blood serologic testing. A patient with a significant exposure history to potential organic antigens (e.g. bird fancier or farmer) with a HRCT findings of upper lung field dominant centrilobular ground glass nodules, acute or subacute onset of symptoms (e.g. dyspnea, myalgias) is quite likely to have acute HP, and this diagnosis is strongly supported by the findings of significant BAL lymphocytosis. Similarly, a patient with such an exposure history plus a subacute or chronic symptom onset and a HRCT that shows ground glass opacities or fibrotic changes with extensive mosaic attenuation due to air-trapping is likely to have chronic HP. Lastly, an older patient who presents with subacute or chronic disease onset and has bibasilar Velcro crackles on chest auscultation is highly likely to have IPF as their specific ILD diagnosis, and this diagnosis can be confidently confirmed if the HRCT shows a typical UIP pattern (peripheral and basilar predominance of fibrotic changes with reticulation and honeycomb change (Figure 1) and very little or no ground glass opacities) and alternative etiologies are lacking (e.g. presence of CTD, asbestosis, drug reaction with fibrosis).
Key management decisions
Once a confident diagnosis has been reached, a plan to treat and monitor disease activity can be put in place (Table 6). Key management decisions include whether to administer pharmacologic agents, how the disease will be monitored to determine whether it has stabilized or improved versus progressive deterioration, whether a patient should be referred for lung transplantation, and whether the disease is end-stage and unlikely to respond to therapies such that providing supportive, palliative care is the best approach. A treatment plan should not only consist of pharmacologic agents that are prescribed to prevent progression and/or induce remission (if the specific disorder can respond to such) but should also include supportive therapies (e.g. supplemental oxygen if indicated, pulmonary rehabilitation), measures to relieve symptoms (e.g. cough, anxiety, depression, dyspnea) and treatment of co-morbid conditions (e.g. anemia, sleep-disordered breathing, GER, infectious complications).
Table 6.
Management of the patient with ILD/IPF
| ▪ Establish a partnership with the patient to provide a patient-centered, personalized medicine care plan | ▪ Provide supplemental oxygen if indicated (keep SpO2 ≥90%) |
| ▪ Provide patients with: | · During exertion |
| · Nocturnal during sleep | |
| • Useful information concerning the nature of their disease and its prognosis | · Continuous if indicated |
| • Treatment options accompanied by thoughtful counseling | ▪ Detect and treat co-morbidities and complications: |
| · Enrollment in clinical trials | · Gastroesophageal reflux disease |
| · Off-label therapies (e.g. corticosteroids, cytoxic drugs, other agents) | · Cardiovascular disease |
| · Lung transplantation | · Drug toxicity (if treated) |
| · Best supportive care | · Sleep-disordered breathing |
| ▪ Use disease-specific monitoring (for prognostication and treatment decisions) | · Secondary pulmonary hypertension |
| · Pulmonary function testing (FVC, DLCO, 6-MWT) | · Metabolic bone disease (osteopenia, osteoporosis) |
| · Thoracic imaging | · Anemia |
| · Dyspnea score | · Anxiety & depression |
| ▪ Pulmonary rehabilitation (optimal exercise program, patient education) | ▪ Maintain ideal body-mass index (weight reduction if obese, improved nutrition if cachectic) |
| ▪ Vaccinations (pneumococcal vaccine, seasonal influenza, others as indicated) |
DLCO = diffusion capacity of the lung for CO; FVC = forced vital capacity; 6-MWT = six-minute walk test; SpO2 = oxyhemoglobin percent saturation.
Measurements that can be made periodically to objectively assess changes in physiologic function over time include formal dyspnea assessment tools, the forced vital capacity (FVC), diffusion capacity of the lung for carbon monoxide (DLCO), and the 6-minute walk test (6-MWT) distance and oxyhemoglobin saturation change [21–26]. The baseline FVC value has not been shown to correlate well with disease course for patients with IPF, but change in FVC over time has been show to correlate well with stable versus progressive disease with greater than 10% decline considered to be significant and indicative of disease progression [27, 28]. A decline of ≥15% in DLCO has also been correlated with disease progression in IPF [28], and declining 6-MWT distance or oxyhemoglobin saturation are also associated with disease progression [25, 29]. More recent analyses suggest that changes in FVC that are less than 10% may represent important clinical change [30], and using relative change rather than absolute change in FVC values may provide a better indication of clinical response [31]. In addition to its utility in diagnosis, HRCT can be scored for the extent/severity of fibrosis, and the fibrosis severity scoring has been shown to correlate with prognosis [32, 33]. However, the use of serial HRCT scanning has not been validated as a useful gauge of disease progression over time for IPF and presents a significant radiation risk to the patient. Biomarkers that reflect disease severity and correlate with prognosis have been reported for IPF [34, 35], but these have yet to be validated for use in the clinical setting.
Immunosuppressive anti-inflammatory agents are used to treat various forms of ILD (Table 7) [36, 37]. Although treatment of any form of ILD with immunosuppressive therapy is off-label in the U.S. and anti-inflammatory/immunosuppressive pharmacologic therapy has not been validated in placebo-controlled clinical trials, there is reasonably compelling evidence that the administration of agents such as corticosteroids is strongly associated with improvement or even clearing of lung pathology for many forms of ILD. This is particularly the case for disorders such as cryptogenic organizing pneumonia (COP), eosinophilic pneumonia, sarcoidosis, or cellular non-specific interstitial pneumonia (NSIP) [36].
Table 7.
Therapies for select types of ILD
| ILD type | Key features of immunopathogenesis | Current therapy* | Additional and/or alternative therapies |
|---|---|---|---|
| IPF | • Prominent fibroblast proliferation and matrix deposition | Supportive care | Anti-reflux therapy |
| • Patchy, temporally heterogeneous changes | Consider anti-reflux measures | N-acetylcysteine | |
| • Architectural distortion of tissue | - Anti-reflux surgery | Clinical trials | |
| • Epithelial injury, microvascular remodeling | - Acid suppressants (e.g. PPI) | (experimental) | |
| • Variable inflammatory component (usually minimal/mild) | Pirfenidone (not approved in US) | ||
| • Areas of NSIP- and DIP-like change often present | |||
| • PH frequently present with advanced disease | Lung transplantation | ||
| Sarcoidosis | • Well-formed non-caseating granulomata in tissues | Observation (mild/stable disease) | Infliximab |
| • Extra-pulmonary disease may be present | Other IS agent | ||
| • May be asymptomatic; may resolve spontaneously without therapy | Corticosteroids (oral or inhaled) | Lung transplantation | |
| Methotrexate | |||
| NSIP | • Homogeneous, diffuse involvement of the lung | Corticosteroids | Other IS drugs |
| • Histopathologic subtypes include cellular (prominent lymphocyte influx; best prognosis), mixed (cellular & fibrotic), & fibrotic (worst prognosis) | Mycophenolate | Lung transplantation | |
| • Usually responsive to IS (less likely to respond if advanced fibrosis is established) | |||
| COP | • Prominent inflammatory cell infiltrate (↑ lymphocytes, neutrophils, and/or eosinophils can all be present) | Corticosteroids | Other IS drugs |
| Macrolides | |||
| • Usually responds to IS therapy; relapse frequently occurs | |||
| HP | • Prominent lymphocyte influx with formation of loose granulomata | Exposure cessation | Other IS drugs |
| • Can have appearance of cellular NSIP or OP | Corticosteroids | Lung transplantation | |
| • Can progress to advanced fibrosis (and masquerade as IPF or fibrotic NSIP) | |||
| Eosinophilic pneumonia | • Prominent influx of eosinophils | Corticosteroids | Other IS drugs |
| • Usually responsive to IS therapy | |||
| CTD-ILD | • Lung histopathology can reveal NSIP (common), UIP (less common); other ILD (e.g. OP, DIP, RBILD – very uncommon) | Corticosteroids | Anti-reflux therapy |
| Mycophenolate | Lung transplantation | ||
| • PH often present (with or without ILD) | Other DMARD agent(s) | Treatment of PH | |
| AIP/DAD | • Intense inflammation and alveolar damage | Corticosteroids | Cytotoxic drugs |
| • Hyaline membrane formation | |||
| • Prominent neutrophil influx early |
*Therapies that are usually administered on the basis of expert opinion and clinical trial results; none have received US Food and Drug Administration approval for the indication of ILD/IPF (but pirfenidone is approved for treatment of IPF in some countries, and many DMARD agents are approved for treatment of CTD).
Abbreviations: AIP acute interstitial pneumonia, COP cryptogenic organizing pneumonia, CTD-ILD connective tissue disease-associated ILD, DAD diffuse alveolar damage, DIP desquamative interstitial pneumonia, DMARD disease-modifying anti-rheumatic drug, HP hypersensitivity pneumonitis, IPF idiopathic pulmonary fibrosis, ILD interstitial lung disease, IS immunosuppression, NSIP non-specific interstitial pneumonia, OP organizing pneumonia, PH pulmonary hypertension, RBILD respiratory bronchiolitis with interstitial lung disease.
When extensive fibrosis is present, such therapies may be less efficacious, especially for patients with IPF, for whom currently available immunosuppressive or anti-fibrotic therapies are not recommended [38]. However, some forms of CTD-associated ILD (NSIP or UIP pathologies) have been reported to respond to mycophenolate therapy, which also allowed a significant lowering of corticosteroid dosing [39]. If immunosuppressive agents are used to treat patients with ILD, treating clinicians should be adequately familiar with potential adverse reactions and drug-drug interactions, and guideline precautions (including recommended monitoring) should be followed [40]. Anti-fibrotic pharmacologic therapies are being increasingly brought to clinical trials [41, 42], and patients should be encouraged to enroll in clinical trials if they are found to have IPF or other forms of advanced ILD for which effective therapies have yet to be identified and clinical trials for their specific form of ILD are open to enrollment.
Treatment of IPF
The prognosis of IPF is generally poor, and the majority of patients have progressive loss of lung function and may suffer acute exacerbations with acceleration of lung function loss that often leads to death [43, 44]. Traditional therapies that were suggested to benefit patients with IPF included corticosteroids and cytotoxic drugs (e.g. azathioprine, cyclophosphamide) [45]. However, these agents have never been shown to have significant benefit in any adequately powered, prospective, randomized, placebo-controlled clinical trial. Furthermore, it was recently demonstrated that azathioprine, an agent that has been suggested to have efficacy for the treatment of IPF [45–47], was associated with significant harm compared to placebo when administered to patients with IPF [48]. This observation triggered the termination of the azathioprine/N-acetylcysteine (NAC)/prednisone arm of the NIH-sponsored IPF PANTHER clinical trial when it became obvious that excess mortality and other complications occurred in this cohort versus the other study arms of either NAC alone or placebo. There are no treatment options for patients with IPF that have been approved by the U.S. Food and Drug Administration, and any pharmacologic treatment given in the US would be considered off-label.
Many new agents that target fibrogenesis have been evaluated in Phase 3 clinical trials (Table 8), but some of these agents (e.g. bosentan, macitentan, ambrisentan, interferons gamma and beta) have not shown benefit despite pre-clinical studies or phase 2 clinical trials that suggested potential efficacy. Indeed, there can be considerable inter-individual variability in genetic abnormalities that have predisposed an individual to develop the disease, in pathophysiologic characteristics of the disease process, and in responses to specific drugs. It should be recognized that a subset of patients that may benefit from a promising drug are very unlikely to be identified in a prospective, double-blind, randomized phase 3 clinical trial in which these patients are combined with a much larger number of enrolled subjects for whom the drug has little or no effect, and the conclusion may be reached that the drug lacks benefit despite its potential to help a subset of patients. Nonetheless, the results of some recently completed clinical trials suggest that pirfenidone [49, 50] or nintedanib (BIBF 1120) [51] may have a significant impact on disease progression versus placebo, and pirfenidone has been licensed and is clinically available in Japan, Europe, and Canada. Stem cell therapy, specifically the use of mesenchymal stem cells (MSC), has shown potential benefit in pre-clinical trials [52], and early results of a phase 1 clinical trial with adipose-derived MSC were recently reported [53].
Table 8.
Pharmacotherapy for IPF: agents in current clinical trials
| Agent | Target | Rationale |
|---|---|---|
| Pirfenidone | TGF-β, PDGF | Down-regulation of TGF-β-stimulated collagen synthesis and extracellular matrix accumulation and PDGF proliferative effects on fibroblasts |
| Tyrosine kinase inhibitors | Tyrosine kinase receptors | Inhibit fibrinogenic pathways by inhibiting receptor tyrosine kinase (RTK) binding by ligands (e.g. TGF-β, PDGF-B, CTGF, FGF, VEGF) |
| N-acetylcysteine | ROI (oxidant-antioxidant imbalance) | Replenish pulmonary glutathione stores and thereby antagonize signaling and tissue damaging effects of oxygen radicals (e.g. stimulatory effects of ROI on myofibroblasts) |
| Anti-TGF-β | TGF-β | Block TGF-β-induced fibroblast migration & proliferation, differentiation of myofibroblasts into fibroblasts, epithelial-mesenchymal transition, and resistance of myofibroblasts to apoptosis |
| Anti-CTGF | CTGF | Suppress fibroblast stimulation by CTGF |
| Anti-IL-13 | IL-13 | Inhibit induction of profibrotic cytokines (e.g. TGF-β, PDGF, IGF-1, PDGF, MMP-9) |
| Anti-LPA | Lysophosphatidic acid (LPA) | Prevent fibroblast recruitment into lung interstitium that can occur via the G protein-coupled LPA1 receptor |
| Anti-CCL2 | CCL-2 | Inhibit cell (e.g. lymphocytes, monocytes, fibrocytes) chemotaxis/influx to lung tissue and TGF-β expression |
| Anti-LOXL2 | Lysyl oxidase-like protein-2 (LOXL-2) | Inhibit LOXL2-mediated fibroblast activation and deposition/accumulation of collagen |
| Plasma exchange, rituximab, steroids | Immune/inflammatory mediators | Suppress inflammation associated with an episode of acute exacerbation of IPF |
Abbreviations: TGF-β transforming growth factor-β, TNF-α tumor necrosis factor-α, ROI reactive oxygen intermediates, CTGF connective tissue growth factor, PH pulmonary hypertension.
Comorbidities can have a significant impact on disease course and quality of life for patients with IPF and other fibrotic lung diseases [54, 55]. These include secondary pulmonary hypertension, coronary artery disease, venous thromboembolism, obstructive sleep apnea, coexistent emphysema, osteoporosis, diabetes mellitus, anxiety, and depression. Coronary artery disease is highly prevalent in patients with IPF [56, 57], and a significantly increased risk of developing primary lung cancer has been observed [58]. An increased risk of venous thromboembolism has also been observed [59], and sleep-disordered breathing is frequently present [60]. An IPFNet phase 3 clinical trial was performed to assess the effect of sildenafil in patients with idiopathic pulmonary fibrosis, but despite a trend toward improvement, a significant increase in 6MWT distance (the primary endpoint) was not attained [61], although a recent analysis of these data suggests that a subset of patients with right heart dysfunction may benefit from sildenafil therapy [62]. Similarly, anticoagulation, when given to disrupt the contribution of the coagulation cascade to the fibrotic process, provided no benefit and was associated with increased risk of significant adverse events [63].
An abnormal degree of GER, which is present in a majority of IPF patients and has been linked to the presence of pepsin and/or bile acids in BAL fluid [64], has also been considered to be an IPF-associated comorbidity. It has been suggested that reflux of foregut contents into the proximal esophagus via a dysfunctional lower esophageal sphincter (e.g. presence of a hiatal hernia) can predispose to (micro)aspiration, which may initiate and/or drive lung inflammation that can progress to pulmonary fibrosis in a susceptible individual, and accumulating evidence has linked GER with aspiration to IPF pathogenesis [13]. Use of medical therapy that inhibits acid production or having undergone a Nissen fundoplication has been associated with significantly improved survival for IPF patients [65], and an analysis of combined, placebo-arm cohorts from three IPFNet-sponsored studies has shown less FVC decline in subjects who were using acid-suppression therapy [16]. Additionally, high pepsin levels in BAL fluid have been linked to some cases of acute exacerbation of IPF [15], and a significantly reduced incidence of acute exacerbations of IPF was observed for subjects enrolled in combined placebo cohorts from the IPFNet phase 3 clinical trials if they were taking anti-reflux medication [66].
Lung transplantation
Lung transplantation is an accepted form of treatment for patients with ILD that is progressive, clearly leading to respiratory failure, and refractory to other therapies [66, 67]. The number of lung transplants performed for patients with IPF surpassed that for COPD in 2007, when IPF became the leading indication for lung transplants performed in the United States [68]. Lung transplantation is the only form of therapy that may improve quality of life and survival for patients with IPF, and 5-year survival following lung transplantation for IPF or other forms of pulmonary fibrosis is approximately 50% [69].
Key decisions that pertain to a potential lung transplant recipient include timing of referral, whether criteria are met that allow a patient to be listed for the procedure, and whether to perform a single versus bilateral lung transplant. International Society for Heart and Lung Transplantation (ISHLT) guidelines [70] state that referral to a transplant center should be considered when the diagnosis of IPF or fibrotic NSIP is made due to the relatively poor prognosis for patients with fibrotic lung disease and, especially, for those with IPF. These guidelines also recommend that transplantation thresholds for patients with IPF include DLCO <40% predicted, >10% decline in FVC, or desaturation below 88% on pulse oximetry during a 6MWT. Potential candidates must be evaluated very carefully to detect issues that are contraindications to being allowed to proceed to the point of being placed on a lung transplant waitlist [71], and if a candidate is placed on a waitlist, the type of transplant that the candidate could potentially receive (e.g. single only, bilateral only, single or bilateral) must be determined. Single lung transplantation is a simpler operation that may be better tolerated by patients with ILD, and bilateral lung transplant has not been shown to have a better survival outcome than single lung transplant for patients with ILD (or the subset of patients with IPF) at our center [72]. Although lung transplant recipients are at considerable risk to develop a considerable number of complications [71, 73], patients can achieve good quality of life and enhanced survival following lung transplant [69].
Future directions
Our understanding of the natural history and pathobiology of various forms of ILD continues to evolve, and classification systems, such as that for the IIPs [74], must be periodically revised to incorporate new knowledge. It is also clear from decades of research that the etiology and pathogenesis of IPF is highly complex [15, 75, 76] and likely involves an exposure stimulus (e.g. inhaled agents, aspirated gastrointestinal refluxate), genetic predisposition to consequent lung injury, and gene/genomics-directed aberrant repair responses that lead to sustained inflammation and matrix disruption/remodeling. Many clinical investigations with single agents have not shown benefit, and clinical trials with various agents have not simultaneously targeted multiple pathways (e.g. immune-mediated inflammatory responses plus abnormal myofibroblast behavior with progressive matrix deposition). Targeting only a single pathway or process rather than using a combination of agents may represent the “Achilles heel” of using monotherapy to treat IPF (and possibly other ILD with progressive fibrosis). While a single agent may have no significant impact on the clinical course of IPF, combination therapy may have an impact (e.g. combination therapy that includes an anti-fibrotic agent, immunomodulatory agent, anti-reflux therapy, and potent antioxidant). Selection of the appropriate endpoint measures for clinical trials may be key to identifying therapies that are clearly of benefit to patients with IPF [77, 78], and additional clinical research is much needed to identify effective therapies for IPF and other ILD associated with progressive fibrosis and early mortality. An improved understanding of the genetics and genomics of ILD will likely lead to the identification of new therapies that may have a significant treatment effect that relieves symptoms and restores quality of life for patients with significant, progressive ILD, but such therapies should have minimal risk of precipitating adverse reactions that can abrogate the benefits of pharmacotherapy.
Conclusions
The diagnosis and treatment of the various types of ILD present a considerable challenge to clinicians. Nonetheless, a comprehensive clinical evaluation combined with appropriate imaging and diagnostic procedures can achieve a confident diagnosis of a specific type of ILD, and invasive testing with bronchoscopy or SLB may not be required. Treatment of ILD entities that are characterized by lung inflammation without the presence of extensive fibrosis can be quite successful when anti-inflammatory immunosuppressive agents are administered. However, when extensive fibrosis has become established, such therapies may have little or no impact on disease progression, especially in patients with IPF. Patients with progressive disease for which effective therapies are lacking should be encouraged to enroll in clinical trials if such are available, and lung transplantation can be considered for appropriate candidates. The diagnosis and treatment of comorbid conditions may also provide significant benefit to patients, and treating clinicians should focus on optimizing quality of life and symptom palliation for patients with advanced, progressive disease.
Acknowledgments
Supported in part by the George and Julie Mosher Pulmonary Research Fund
Abbreviations
- ATS
American thoracic society
- BAL
Bronchoalveolar lavage
- COPD
Chronic obstructive pulmonary disease
- CT
Computed tomography
- CTD
Connective tissue disease
- CTD-ILD
Connective tissue disease-associated interstitial lung disease
- CXR
Chest x-ray
- DLCO
Diffusion capacity of the lung for carbon monoxide
- FVC
Forced vital capacity
- GER
Gastroesophageal reflux
- GERD
Gastroesophageal reflux disease
- HP
Hypersensitivity pneumonitis
- HRCT
High-resolution computed tomography
- IIP
Idiopathic interstitial pneumonia
- ILD
Interstitial lung disease
- IPF
Idiopathic pulmonary fibrosis
- ISHLT
International society for heart and lung transplantation
- MDCT
Multi-detector computed tomography
- MSC
Mesenchymal stem cells
- 6-MWT
6-minute walk test
- NAC
N-acetylcysteine
- NSIP
Non-specific interstitial pneumonia
- SLB
Surgical lung biopsy
- TBLBx
Transbronchial lung biopsy
- UIP
Usual interstitial pneumonia
- VATS
Video-assisted thorascopic surgery.
Footnotes
Competing interests
Dr. Meyer has been an investigator in clinical trials sponsored by Abbott, Actelion, Altana, Amgen, Asthmatx, Bayer, Boehringer-Ingelheim, Bristol Meyers Squibb, Chiron, Discovery Labs, DuPont Merck, Fibrogen, Genentech, Gilead, GlaxoSmithKline, Inspire. InterMune, Johnson & Johnson, Novartis, Nycomed, Pfizer, Pharmaxis, PreAnalytiX, Roche, Ross Laboratories, Vertex, and Wyeth. Dr. Meyer also serves on a Clinical Advisory Board for InterMune. The author does not report any other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Authors’ information
Dr. Keith C. Meyer, MD, MS, FACP, FCCP, is Professor of Medicine at the University of Wisconsin School of Medicine and Public Health and a faculty member of the Section of Allergy, Pulmonary, and Critical Care Medicine in the Department of Medicine. He is a transplant pulmonologist with the University of Wisconsin Lung Transplant and Advanced Pulmonary Disease Program (formerly Medical Director of Lung Transplantation), Director of the UW Interstitial Lung Disease Clinic, and Director of the UW Adult Cystic Fibrosis Program. His e-mail address is kcm@medicine.wisc.edu.
References
- 1.Meyer KC, Raghu G. Patient evaluation. In: Baughman RP, Du Bois RM, editors. Interstitial Lung Disease: A Practical Approach. Second. New York: Springer; 2011. pp. 3–16. [Google Scholar]
- 2.Meyer KC. Interstitial lung disease in the elderly: pathogenesis, diagnosis and management. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28:3–17. [PubMed] [Google Scholar]
- 3.Swigris JJ, Brown KK, Flaherty KR. The idiopathic interstitial pneumonias and connective tissue disease-associated interstitial lung disease. Curr Rheum Rev. 2010;6:91–98. doi: 10.2174/157339710791330731. [DOI] [Google Scholar]
- 4.Kanne JP. Interstitial lung disease (ILD): imaging finding, and the role of imaging in evaluating the patient with known or suspected ILD. Semin Roentgenol. 2010;45:3. doi: 10.1053/j.ro.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Hodnett PA, Naidich DP. Fibrosing Interstitial Lung Disease. A Practical High-Resolution Computed Tomography-based Approach to Diagnosis and Management and a Review of the Literature. Am J Respir Crit Care Med. 2013;188:141–149. doi: 10.1164/rccm.201208-1544CI. [DOI] [PubMed] [Google Scholar]
- 6.Joos L, Patuto N, Chhajed PN, Tamm M. Diagnostic yield of flexible bronchoscopy in current clinical practice. Swiss Med Wkly. 2006;136:155–159. doi: 10.4414/smw.2006.11344. [DOI] [PubMed] [Google Scholar]
- 7.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, Du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P, Saltini C, Selman M, Strange C, Wood B. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004–1014. doi: 10.1164/rccm.201202-0320ST. [DOI] [PubMed] [Google Scholar]
- 8.Meyer KC, Raghu G. Bronchoalveolar lavage for the evaluation of interstitial lung disease: is it clinically useful? Eur Respir J. 2011;38:761–769. doi: 10.1183/09031936.00069509. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen W, Meyer KC. Surgical lung biopsy for the diagnosis of interstitial lung disease: a review of the literature and recommendations for optimizing safety and efficacy. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30:3–16. [PubMed] [Google Scholar]
- 10.Utz JP, Ryu JH, Douglas WW, Hartman TE, Tazelaar HD, Myers JL, Allen MS, Schroeder DR. High short-term mortality following lung biopsy for usual interstitial pneumonia. Eur Respir J. 2001;17:175–179. doi: 10.1183/09031936.01.17201750. [DOI] [PubMed] [Google Scholar]
- 11.Park JH, Kim DK, Kim DS, Koh Y, Lee SD, Kim WS, Kim WD, Park SI. Mortality and risk factors for surgical lung biopsy in patients with idiopathic interstitial pneumonia. Eur J Cardiothorac Surg. 2007;31:1115–1119. doi: 10.1016/j.ejcts.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 12.Kondoh Y, Taniguchi H, Kitaichi M, Yokoi T, Johkoh T, Oishi T, Kimura T, Nishiyama O, Kato K, Du Bois RM. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med. 2006;100:1753–1759. doi: 10.1016/j.rmed.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Raghu G, Meyer KC. Silent gastro-oesophageal reflux and microaspiration in IPF: mounting evidence for anti-reflux therapy? Eur Respir J. 2012;39:242–245. doi: 10.1183/09031936.00211311. [DOI] [PubMed] [Google Scholar]
- 14.Meyer KC, Raghu G. GER and aspiration in interstitial lung disease. In: Meyer KC, Raghu G, editors. Gastroesophageal Reflux and the Lung. New York: Springer; 2012. pp. 175–198. [Google Scholar]
- 15.Lee JS, Song JW, Wolters PJ, Elicker BM, King TE, Jr, Kim DS, Collard HR. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J. 2012;39:352–358. doi: 10.1183/09031936.00050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JS, Collard HR, Anstrom KJ, Martinez FJ, Noth I, Roberts RS, Yow E, Raghu G, for the IPFnet Investigators Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomized controlled trials. Lancet Respiratory Medicine. 2013;1:369–376. doi: 10.1016/S2213-2600(13)70105-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savarino E, Bazzica M, Zentilin P, Pohl D, Parodi A, Cittadini G, Negrini S, Indiveri F, Tutuian R, Savarino V, Ghio M. Gastroesophageal reflux and pulmonary fibrosis in scleroderma: a study using pH-impedance monitoring. Am J Respir Crit Care Med. 2009;179:408–413. doi: 10.1164/rccm.200808-1359OC. [DOI] [PubMed] [Google Scholar]
- 18.Soares RV, Forsythe A, Hogarth K, Sweiss NJ, Noth I, Patti MG. Interstitial lung disease and gastroesophageal reflux disease: key role of esophageal function tests in the diagnosis and treatment. Arq Gastroenterol. 2011;48:91–97. doi: 10.1590/S0004-28032011000200002. [DOI] [PubMed] [Google Scholar]
- 19.Oelschlager BK, Chang L, Pope CE, 2nd, Pellegrini CA. Typical GERD symptoms and esophageal pH monitoring are not enough to diagnose pharyngeal reflux. J Surg Res. 2005;128:55–60. doi: 10.1016/j.jss.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 20.Flaherty KR, King TE, Jr, Raghu G, Lynch JP, 3rd, Colby TV, Travis WD, Gross BH, Kazerooni EA, Toews GB, Long Q, Murray S, Lama VN, Gay SE, Martinez FJ. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170:904–910. doi: 10.1164/rccm.200402-147OC. [DOI] [PubMed] [Google Scholar]
- 21.Swigris JJ, Yorke J, Sprunger DB, Swearingen C, Pincus T, Du Bois RM, Brown KK, Fischer A. Assessing dyspnea and its impact on patients with connective tissue disease-related interstitial lung disease. Respir Med. 2010;104:1350–1355. doi: 10.1016/j.rmed.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swigris JJ, Han M, Vij R, Noth I, Eisenstein EL, Anstrom KJ, Brown KK, Fairclough D. The UCSD shortness of breath questionnaire has longitudinal construct validity in idiopathic pulmonary fibrosis. Respir Med. 2012;106:1447–1455. doi: 10.1016/j.rmed.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collard HR, King TE, Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168:538–542. doi: 10.1164/rccm.200211-1311OC. [DOI] [PubMed] [Google Scholar]
- 24.Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, Colby TV, Du Bois RM, Hansell DM. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167:962–969. doi: 10.1164/rccm.2111053. [DOI] [PubMed] [Google Scholar]
- 25.Lama VN, Flaherty KR, Toews GB, Colby TV, Travis WD, Long Q, Murray S, Kazerooni EA, Gross BH, Lynch JP, 3rd, Martinez FJ. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2005;168:1084–1090. doi: 10.1164/rccm.200302-219OC. [DOI] [PubMed] [Google Scholar]
- 26.Flaherty KR, Andrei AC, Murray S, Fraley C, Colby TV, Travis WD, Lama V, Kazerooni EA, Gross BH, Toews GB, Martinez FJ. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute walk test. Am J Respir Crit Care Med. 2006;174:803–809. doi: 10.1164/rccm.200604-488OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King TE, Jr, Safrin S, Starko KM, Brown KK, Noble PW, Raghu G, Schwartz DA. Analyses of efficacy end points in a controlled trial of interferon-gamma1b for idiopathic pulmonary fibrosis. Chest. 2005;127:171–177. doi: 10.1378/chest.127.1.171. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt SL, Nambiar AM, Tayob N, Sundaram B, Han MK, Gross BH, Kazerooni EA, Chughtai AR, Lagstein A, Myers JL, Murray S, Toews GB, Martinez FJ, Flaherty KR. Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J. 2011;38:176–183. doi: 10.1183/09031936.00114010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, Lancaster L, Noble PW, Sahn SA, Szwarcberg J, Thomeer M, Valeyre D, King TE., Jr Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med. 2011;183:1231–1237. doi: 10.1164/rccm.201007-1179OC. [DOI] [PubMed] [Google Scholar]
- 30.Du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, King TE, Jr, Lancaster L, Noble PW, Sahn SA, Thomeer M, Valeyre D, Wells AU. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184:1382–1389. doi: 10.1164/rccm.201105-0840OC. [DOI] [PubMed] [Google Scholar]
- 31.Richeldi L, Ryerson CJ, Lee JS, Wolters PJ, Koth LL, Ley B, Elicker BM, Jones KD, King TE, Jr, Ryu JH, Collard HR. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax. 2012;67:407–411. doi: 10.1136/thoraxjnl-2011-201184. [DOI] [PubMed] [Google Scholar]
- 32.Lynch DA, Godwin JD, Safrin S, Starko KM, Hormel P, Brown KK, Raghu G, King TE, Jr, Bradford WZ, Schwartz DA, Richard Webb W, Idiopathic Pulmonary Fibrosis Study Group High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med. 2005;172:488–493. doi: 10.1164/rccm.200412-1756OC. [DOI] [PubMed] [Google Scholar]
- 33.Edey AJ, Devaraj AA, Barker RP, Nicholson AG, Wells AU, Hansell DM. Fibrotic idiopathic interstitial pneumonias: HRCT findings that predict mortality. Eur Radiol. 2011;21:1586–1593. doi: 10.1007/s00330-011-2098-2. [DOI] [PubMed] [Google Scholar]
- 34.Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, Li K, Choi J, Vuga LJ, Lindell KO, Klesen M, Zhang Y, Gibson KF. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J RespirCrit Care Med. 2012;185:67–76. doi: 10.1164/rccm.201101-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143:1422–1429. doi: 10.1378/chest.11-2735. [DOI] [PubMed] [Google Scholar]
- 36.Kim R, Meyer KC. Therapies of interstitial lung disease—past, present, and future. Therapeutic Advances Respir Dis. 2008;2:319–338. doi: 10.1177/1753465808096948. [DOI] [PubMed] [Google Scholar]
- 37.Meyer KC, Bierach J. Immunosuppressive therapy for autoimmune lung diseases. Immunol Allergy Clin North Am. 2012;32:633–639. doi: 10.1016/j.iac.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer A, Brown KK, Du Bois RM, Frankel SK, Cosgrove GP, Fernandez-Perez ER, Huie TJ, Krishnamoorthy M, Meehan RT, Olson AL, Solomon JJ, Swigris JJ. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol. 2013;40:640–646. doi: 10.3899/jrheum.121043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baughman RP, Meyer KC, Nathanson I, Angel L, Bhorade SM, Chan KM, Culver D, Harrod CG, Hayney MS, Highland KB, Limper AH, Patrick H, Strange C, Whelan T. Executive summary: monitoring of nonsteroidal immunosuppressive drugs in patients with lung disease and lung transplant recipients: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;142:1284–1288. doi: 10.1378/chest.12-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lota HK, Wells AU. The evolving pharmacotherapy of pulmonary fibrosis. Expert Opin Pharmacother. 2013;14:79–89. doi: 10.1517/14656566.2013.758250. [DOI] [PubMed] [Google Scholar]
- 42.Adamali HI, Maher TM. Current and novel drug therapies for idiopathic pulmonary fibrosis. Drug Design Develop Ther. 2013;6:261–272. doi: 10.2147/DDDT.S29928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE, Jr, Lasky JA, Loyd JE, Noth I, Olman MA, Raghu G, Roman J, Ryu JH, Zisman DA, Hunninghake GW, Colby TV, Egan JJ, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kondoh Y, Lynch DA, Müller-Quernheim J, Myers JL, Nicholson AG, Selman M, Toews GB, Wells AU, Martinez FJ, Idiopathic Pulmonary Fibrosis Clinical Research Network Investigators Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim DS. Acute exacerbations in patients with idiopathic pulmonary fibrosis. Respir Res. 2013;14:86. doi: 10.1186/1465-9921-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 46.Raghu G, Depaso WJ, Cain K, Hammar SP, Wetzel CE, Dreis DF, Hutchinson J, Pardee NE, Winterbauer RH. Azathioprine combined with prednisone in the treatment of idiopathic pulmonary fibrosis: a prospective double-blind, randomized, placebo-controlled clinical trial. Am Rev Respir Dis. 1991;144:291–296. doi: 10.1164/ajrccm/144.2.291. [DOI] [PubMed] [Google Scholar]
- 47.Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M, IFIGENIA Study Group High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 48.Idiopathic Pulmonary Fibrosis Clinical Research Network Prednisone, azathioprine, and nacetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, Taguchi Y, Takahashi H, Nakata K, Sato A, Takeuchi M, Raghu G, Kudoh S, Nukiwa T, Pirfenidone Clinical Study Group in Japan Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- 50.Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE, Jr, Lancaster L, Sahn SA, Szwarcberg J, Valeyre D, Du Bois RM, CAPACITY Study Group Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 51.Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Klüglich M, Du Bois RM. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 52.Toonkel RL, Hare JM, Matthay MA, Glassberg MK. Mesenchymal stem cells and idiopathic pulmonary fibrosis. Potential for clinical testing. Am J Respir Crit Care Med. 2013;188:133–140. doi: 10.1164/rccm.201207-1204PP. [DOI] [PubMed] [Google Scholar]
- 53.Tzouvelekis A, Paspaliaris V, Koliakos G, Ntolios P, Bouros E, Oikonomou A, Zissimopoulos A, Boussios N, Dardzinski B, Gritzalis D, Antoniadis A, Froudarakis M, Kolios G, Bouros D. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J Transl Med. 2013;11:171. doi: 10.1186/1479-5876-11-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 55.King C, Nathan SD. Identification and treatment of comorbidities in idiopathic pulmonary fibrosis and other fibrotic lung diseases. Curr Opin Pulm Med. 2013;19:466–473. doi: 10.1097/MCP.0b013e328363f460. [DOI] [PubMed] [Google Scholar]
- 56.Kizer JR, Zisman DA, Blumenthal NP, Kotloff RM, Kimmel SE, Strieter RM, Arcasoy SM, Ferrari VA, Hansen-Flaschen J. Association between pulmonary fibrosis and coronary artery disease. Arch Intern Med. 2004;164:551–556. doi: 10.1001/archinte.164.5.551. [DOI] [PubMed] [Google Scholar]
- 57.Nathan SD, Basavaraj A, Reichner C, Shlobin OA, Ahmad S, Kiernan J, Burton N, Barnett SD. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010;104:1035–1041. doi: 10.1016/j.rmed.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 58.Ozawa Y, Suda T, Naito T, Enomoto N, Hashimoto D, Fujisawa T, Nakamura Y, Inui N, Nakamura H, Chida K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology. 2009;14:723–728. doi: 10.1111/j.1440-1843.2009.01547.x. [DOI] [PubMed] [Google Scholar]
- 59.Sprunger DB, Olson AL, Huie TJ, Fernandez-Perez ER, Fischer A, Solomon JJ, Brown KK, Swigris JJ. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. Eur Respir J. 2012;39:125–132. doi: 10.1183/09031936.00041411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lancaster LH, Mason WR, Parnell JA, Rice TW, Loyd JE, Milstone AP, Collard HR, Malow BA. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest. 2009;136:772–778. doi: 10.1378/chest.08-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW, Idiopathic Pulmonary Fibrosis Clinical Research Network A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363:620–628. doi: 10.1056/NEJMoa1002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han MK, Bach DS, Hagan PG, Yow E, Flaherty KR, Toews GB, Anstrom KJ, Martinez FJ, IPFnet Investigators Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest. 2013;143:1699–1708. doi: 10.1378/chest.12-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noth I, Anstrom KJ, Calvert SB, De Andrade J, Flaherty KR, Glazer C, Kaner RJ, Olman MA, Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:88–95. doi: 10.1164/rccm.201202-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Savarino E, Carbone R, Marabotto E, Furnari M, Sconfienza L, Ghio M, Zentilin P, Savarino V. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J. 2013;42:1322–1331. doi: 10.1183/09031936.00101212. [DOI] [PubMed] [Google Scholar]
- 65.Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, King TE, Jr, Collard HR. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:1390–1394. doi: 10.1164/rccm.201101-0138OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kotloff RM, Thabut G. Lung transplantation. Am J Respir Crit Care Med. 2011;184:159–171. doi: 10.1164/rccm.201101-0134CI. [DOI] [PubMed] [Google Scholar]
- 67.Meyer KC. Lung transplantation. F1000Prime Rep. 2013;5:16. doi: 10.12703/P5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Organ Procurement and Transplantation Network (OPTN) and Scientific Registry of Transplant Recipients (SRTR). OPTN/SRTR 2010 Annual Data Report. Rockville, MD: Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation. 2011. http://srtr.transplant.hrsa.gov/annual_reports/2010/. Accessed 02 Sep 2012
- 69.Christie JD, Edwards LB, Kucheryavaya AY, Benden C, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI. The registry of the international society for heart and lung transplantation: twenty-eighth adult lung and heart-lung transplant report–2011. J Heart Lung Transplant. 2011;30:1104–1122. doi: 10.1016/j.healun.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Orens JB, Estenne M, Arcasoy S, Conte JV, Corris P, Egan JJ, et al. International guidelines for the selection of lung transplant candidates: 2006 update—a consensus report from the pulmonary scientific council of the international society for heart and lung transplantation. J Heart Lung Transplant. 2006;25:745–755. doi: 10.1016/j.healun.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 71.McCartney J, Meyer KC. Optimizing post-transplant outcomes in lung transplantation. Expert Rev Respir Med. 2008;2:183–199. doi: 10.1586/17476348.2.2.183. [DOI] [PubMed] [Google Scholar]
- 72.De Oliveira NC, Osaki S, Maloney JD, Cornwell RD, Meyer KC. Lung transplant for interstitial lung disease: outcomes for single versus bilateral lung transplantation. Interact Cardiovasc Thor Surg. 2012;14:263–267. doi: 10.1093/icvts/ivr085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Todd JL, Palmer SM. Bronchiolitis obliterans syndrome: the final frontier for lung transplantation. Chest. 2011;140:502–508. doi: 10.1378/chest.10-2838. [DOI] [PubMed] [Google Scholar]
- 74.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, ATS/ERS Committee on Idiopathic Interstitial Pneumonias et al. An official american thoracic society/european respiratory society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Günther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21:152–160. doi: 10.1183/09059180.00001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012;5:11. doi: 10.1186/1755-1536-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raghu G, Collard HR, Anstrom KJ, Flaherty KR, Fleming TR, King TE, Jr, Martinez FJ, Brown KK. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med. 2012;185:1044–1048. doi: 10.1164/rccm.201201-0006PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C, IPFnet investigators Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res. 2013;14:73. doi: 10.1186/1465-9921-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]