

Abstract
Pseudomonas aeruginosa produces the peptide siderophore pyoverdine, which is used to acquire essential Fe3+ ions from the environment. PvdQ, an Ntn hydrolase, is required for the biosynthesis of pyoverdine. PvdQ knockout strains are not infectious in model systems, suggesting that disruption of siderophore production via PvdQ inhibition could be exploited as a target for novel antibacterial agents, by preventing cells from acquiring iron in the low iron environments of most biological settings. We have previously described a high-throughput screen to identify inhibitors of PvdQ that identified inhibitors with IC50 values of ∼100 μM. Here, we describe the discovery of ML318, a biaryl nitrile inhibitor of PvdQ acylase. ML318 inhibits PvdQ in vitro (IC50 = 20 nM) by binding in the acyl-binding site, as confirmed by the X-ray crystal structure of PvdQ bound to ML318. Additionally, the PvdQ inhibitor is active in a whole cell assay, preventing pyoverdine production and limiting the growth of P. aeruginosa under iron-limiting conditions.
The growing prevalence of drug-resistant bacterial pathogens is of significant concern in the United States and worldwide. Of particular concern are the multidrug resistant Gram-negative bacteria including Klebsiella pneumoniae, Acinetobacter species, Enterobacter species, and Pseudomonas aeruginosa.(1,2) Gram-negative human pathogens such as P. aeruginosa typically require intracellular iron levels in the micromolar range for growth and infectivity.3,4 The low abundance of iron in a typical host environment has provided a selective pressure for P. aeruginosa to develop a mechanism to extract iron from the extracellular milieu.
Targeting siderophore biosynthesis as a strategy to reduce virulence5 has received much attention recently. Salicyl-AMS (5′-O-(N-salicylsulfamoyl)adenosine), a nM inhibitor of the myocobactin biosynthetic enzyme MbtA,6−8 reduces the growth of M. tuberculosis in mouse lungs.9 Importantly, this work validates the approach that preventing pathogen access to essential nutrients and demonstrates the bioavailability of the Salicyl-AMS inhibitor and the primary importance of mycobactin over other iron-acquisition pathways.
P. aeruginosa produces pyoverdine, a peptide siderophore that scavenges extracellular iron.10 Secreted pyoverdine binds to Fe3+ with high affinity (Kf ∼ 1024 M–1 at pH 7.0) and the resulting complex is taken into the bacterial cell through a specific receptor.11 Pyoverdine also plays a role in the regulation of other P. aeruginosa virulence factors12−14 and biofilm formation.15−17
It has been shown that pyoverdine-deficient mutant strains are not infectious in the mouse lung,18 plant,19 and C. elegans.20 Pyoverdine is biosynthesized by four nonribosomal peptide synthetases (NRPSs) and 10 additional modifying enzymes.10,21 The modular NRPS enzymes contain multiple catalytic domains joined in a single protein that catalyze peptide production in an assembly line fashion. During synthesis, the nascent peptide is covalently bound to an integrated peptidyl carrier protein domain and delivered to the neighboring catalytic domains. In this modular architecture, each NRPS module catalyzes the incorporation of a single substrate into the final peptide product.22
Pyoverdine is composed of a conserved dihydroquinoline-type chromophore and a peptide tail that varies among different Pseudomonas species (Figure 1A).11,21 Additionally, most strains produce variable pyoverdine isoforms with N-terminal succinate, succinamide, or glutamate moieties bound to the chromophore. PvdL, the first NRPS protein of the pyoverdine pathway, is shared among all sequenced pseudomonads and generates the peptide backbone that is converted into this chromophore.21 Interestingly, PvdL contains a N-terminal module with homology to fatty acyl-CoA ligases.23 We recently24 showed that this unusual NRPS architecture incorporates a myristate molecule, subsequently identified as either myristic or myristoleic acid,25 at the N-terminus of an intermediate in pyoverdine biosynthesis. Additionally, we demonstrated that the incorporated fatty acid, which is not present on mature pyoverdine, is removed by PvdQ,24 one of the 10 auxiliary proteins necessary for pyoverdine synthesis (Figure 1B).10 PvdQ belongs to a family of N-terminal nucleophile (Ntn) hydrolases that catalyze the cleavage of amide bonds via an acylated enzyme intermediate.26 PvdQ exhibits promiscuity in activity and also cleaves acyl-homoserine lactones that are involved in quorum signaling.27,28
Figure 1.

Structure of pyoverdine and the role of PvdQ in biosynthesis. (A) The mature pyoverdine siderophore is an undecapeptide containing an N-terminal sidearm, the chromophore, and a species-specific peptide chain. (B) PvdQ catalyzes the removal of the myristoyl group from the pyoverdine precursor.
To examine the role of PvdQ in pyoverdine maturation, we developed a high-throughput biochemical assay to find inhibitors of the PvdQ acylase activity.24 The assay monitored the hydrolysis of p-nitrophenyl myristate (Figure 2A) and showed good reproducibility and signal-to-noise parameters, with Z′ scores of 0.7–0.9 within one plate and 0.6 overall. In this proof-of-concept study, we screened 1280 compounds, identifying aryl bromides 1 and 2 (Figure 2B), which exhibit IC50 values of 130 μM and 65 μM and bind in the fatty acid binding pocket.24 This success with a small library suggested that a more thorough effort might lead to compounds with higher affinity. We therefore conducted a high-throughput screen with a larger chemical library and 4-methylumbelliferyl laurate (4-MU laurate), a fluorogenic substrate with improved signal-to-noise properties, to identify more potent scaffolds for PvdQ inhibition that can serve as tool compounds for understanding pyoverdine maturation and therapeutic leads for P. aeruginosa infection.
Figure 2.

High-throughput screen for PvdQ inhibitors. (A) PvdQ hydrolysis of either p-nitrophenyl myristate or 4-MU-laurate substrates result in production of compounds that can be detected through absorbance or fluorescence. (B) Comparison of biochemical PvdQ inhibition activities of prior art compounds (1, 2) to the high-throughput screening lead 3 and the optimized probe 4.
Results
High Throughput Screening of PvdQ
Our previous screening with the LOPAC1280 chemical library used the chromogenic substrate. Optimization and miniaturization of this assay resulted in improved signal-to-noise with the fluorogenic substrate 4-MU laurate. A high-throughput biochemical assay with the fluorogenic substrate was screened with the NIH MLSMR (National Institutes of Health Molecular Libraries Small Molecule Repository) collection of 337 488 compounds at 10 μM. Fluorescence measurements at 0 and 60 min were read and a total of 213 compounds were deemed active (inhibition of >20% fluorescence in both replicates) and 198 compounds were considered inconclusive (inhibition of >20% in one replicate). Of these 411 compounds, 396 were readily available and were rescreened at 9 concentrations ranging from 0.003 to 19.5 μM to assess preliminary potency. Here, 89 had IC50 values lower than 10 μM. The most potent inhibitors of PvdQ acylase were tested in a preliminary whole-cell P. aeruginosa (PAO1 strain) in the presence of metal chelator ethylenediamine-N,N′-bis(2-hydroxyphenyl-acetic acid) (EDDHA) and in a HeLa cell toxicity counter screen.29 The whole cell assay with P. aeruginosa had two readouts; absorption at 600 nm was measured as a reporter of growth inhibition, while absorption at 405 nm was measured as a reporter of pyoverdine production.30
Biarylnitrile 3 was selected for further development on the basis of activity against PvdQ, activity in the whole cell assay, specificity as determined by lack of activity in other PubChem Bioassays, lack of toxicity with HeLa cells, and chemical tractability for generation of compound analogues. The hit compound 3 exhibited an IC50 of 40 nM against PvdQ acylase in biochemical assays (Figure 2). In preliminary growth assays, the hit compound displayed an IC50 of 59 μM against P. aeruginosa PAO1.29 In addition, 3 was not active in any other assay submitted to PubChem at the time of analysis, including toxicity studies with other bacteria, including M. tuberculosis and E. coli.
Synthesis of Biaryl Nitrile Analogues
Based on scaffold 3, a panel of analogues was synthesized and tested for PvdQ inhibition activity. The racemates of most analogues were conveniently synthesized via arylations of benzylacetonitriles with halopyridines31 or alkylations with haloalkanes.32 (We note that separation of the enantiomers was not successful due to racemization of the benzylic position at ambient temperatures and neutral conditions.) A representative example of the arylation is shown for the synthesis of biaryl nitrile 4 (Figure 3). Coupling of 2-bromo-5-trifluoromethylpyridine 5 with 4-fluorobenzyl nitrile 6 using potassium tert-butoxide gave the desired biaryl nitrile 4 in 69% yield. Tetrazoles were synthesized from nitriles via [3 + 2] cycloaddition and amino nitriles were synthesized from 4-chlorobenzaldehyde by a Strecker reaction (not shown).33,34
Figure 3.

Synthesis of biaryl nitrile 4 from bromopyridine 5 and benzyl nitrile 6.
Structure–Activity Relationship Analysis
Modifications to the phenyl ring comprising the eastern half of lead 3 were analyzed in the biochemical assay (Table 1). Relative to the 4-chlorophenyl (3), the unsubstituted phenyl ring (7) and the addition of a hydrogen bond acceptor at the ortho position (8,11) led to at least 7-fold decreased in vitro potency. By comparison, analogues with an electron-withdrawing group at the para position (9, 10, 12, 13) performed better.
Table 1. SAR Analysis of Eastern Analogues.
| analog | R | μM |
| 3 | 4-Cl | 0.04 ± 0.01 |
| 7 | H | 0.30 ± 0.02 |
| 8 | 2-OCH3 | 0.40 ± 0.08 |
| 9 | 4-F | 0.07 ± 0.02 |
| 10 | 4-CF3 | 0.39 ± 0.10 |
| 11 | 2-OCH3 | 1.7 ± 0.6 |
| 12 | 2-Cl, 4-Cl | 0.25 ± 0.04 |
| 13 | 2-F, 4-Cl | 0.06 ± 0.02 |
Average of at least three replicates ± standard deviation.
With regards to the western half of the scaffold, synthetic efforts focused on removing the potentially labile 2-chloro substituent on the pyridine ring without sacrificing potency (Table 2). Removal of the 2-chloro (24) resulted in a 7-fold increase in IC50. The 3-chloro analogue (14) showed reduced potency relative to lead 3. Meanwhile, the 5-chloro analogue 15 increased potency 4-fold in vitro relative to 3, as did replacement of the 2-chloro substituent on the pyridine ring with trifluoromethyl (16). We note that the IC50 of 15 and 16 were at or slightly below half the concentration of PvdQ used in the biochemical assays, indicating that these are tight-binding inhibitors. Potentially, the true binding affinity is even better for these and the most potent compounds identified than is reflected by their IC50 values. All other substitutions on the pyridine resulted in lower inhibitory efficiency (17–23) when the eastern aryl ring retained the 4-chlorophenyl.
Table 2. SAR of Pyridine with a 4-Substituted Phenyl Ring.

| analog | R1 | R2 | μM |
| 3 | 2-Cl | Cl | 0.04 ± 0.01 |
| 14 | 3-Cl | Cl | 0.13 ± 0.04 |
| 15 | 5-Cl | Cl | 0.01 ± 0.004 |
| 16 | 2-CF3 | Cl | 0.01 ± 0.005 |
| 17 | 2-CN | Cl | 0.18 ± 0.05 |
| 18 | 2-CH3 | Cl | 0.25 ± 0.04 |
| 19 | 3-CH3 | Cl | 0.25 ± 0.09 |
| 20 | 3-CF3 | Cl | 2.4 ± 1.2 |
| 21 | 2-Cl, 3-CF3 | Cl | 0.62 ± 0.08 |
| 22 | 2-CN, 3-CH3 | Cl | 0.18 ± 0.02 |
| 23 | 3-CN | Cl | 2.5 ± 0.5 |
| 24 | H | Cl | 0.27 ± 0.03 |
| 4 | 2-CF3 | F | 0.02 ± 0.01 |
| 25 | 2-CH | F | 0.13 ± 0.02 |
| 26 | 2-CN, 3-CH3 | F | 0.13 ± 0.07 |
| 27 | 2-Cl, 3-CF3 | F | 0.74 ± 0.28 |
| 28 | 2-Cl, 3-CF3 | CF3 | 2.3 ± 1.9 |
| 29 | 2-CF3 | CF3 | 0.14 ± 0.06 |
Average of at least three replicates ± standard deviation.
Replacement of the 4-chloro substituent with 4-fluoro on the eastern phenyl ring led to a series of analogues (4,25–27) with similar potencies to those observed in the 4-chloro series. Notably, this 4-fluoro series included the 2-trifluoromethylpyridine analogue (4) with very similar potency to 2-trifluoromethylpyridine analogue (16) of the 4-chloro series. Substitution at carbon four on the eastern ring with trifluoromethyl was not beneficial (28–29).
Incorporation of multiple halogens into the phenyl ring (Table 3) demonstrated that fluoride is tolerated in the 2-position of the phenyl ring (30–32), and the 2-CF3 pyridine analogue (31) showed improvement over the HTS lead (3). A 2-chloro substituent on the phenyl ring is less tolerated, as shown by the decrease in potency for 2,4-dichloro analogues (33–36) compared to the monochloride analogues described in Table 2.
Table 3. SAR of Pyridine Analogues with 2,4-Halide Substituted Phenyl Ring.

| analog | R1 | R2 | μM |
| 3 | 2-Cl | H | 0.04 ± 0.01 |
| 30 | 2-CN | F | 0.11 ± 0.01 |
| 31 | 2-CF3 | F | 0.01 ± 0.001 |
| 32 | 2-Cl, 3-CF3 | F | 0.63 ± 0.05 |
| 33 | 2-CN | Cl | 0.29 ± 0.03 |
| 34 | 3-CN | Cl | 1.8 ± 0.34 |
| 35 | 5-Cl | Cl | 0.21 ± 0.03 |
| 36 | 2-Cl, 3-CF3 | Cl | 0.97 ± 0.46 |
Average of at least three replicates ± standard deviation.
To address the chemical liability of the benzylic nitrile group and to mimic the natural, lipophilic myristate ligand of PvdQ, the pyridyl ring was removed and replaced with various hydrophobic alkyl chains (Table 4, 37–40, 46), which resulted in lower in vitro potency. Efforts to enhance the solubility of these alkyl analogues through the incorporation of heteroatoms in either the alkyl chain (41–45, 47, 48) or the aryl ring (49–51) afforded analogues that did not perform well (Table 4).
Table 4. SAR of Alkylated Nitriles.

| analog | R1 | R2 | X | μM |
| 3 | 2-Cl pyridine | Cl | CH | 0.04 ± 0.01 |
| 37 | (CH2)2CH3 | Cl | CH | 5.9 ± 1.3 |
| 38 | (CH2)3CH3 | Cl | CH | 1.4 ± 0.8 |
| 39 | CH2CH(CH3)2 | Cl | CH | 0.29 ± 0.09 |
| 40 | CH2CH(CH2)2 | Cl | CH | 0.54 ± 0.17 |
| 41 | (CH2)2OCH3 | Cl | CH | 4.8 ± 0.4 |
| 42 | ((CH2)2O)2CH3 | Cl | CH | 4.7 ± 3.0 |
| 43 | (CH2)3N(CH3)2 | Cl | CH | >10 |
| 44 | NH(CH2)2OCH3 | Cl | CH | >10 |
| 45 | 1-pyrrolidine | Cl | CH | 0.26 ± 0.29 |
| 46 | (CH2)3CH3 | F | CH | 0.32 ± 0.16 |
| 47 | (CH2)2OCH3 | F | CH | 0.29 ± 0.12 |
| 48 | (CH2)3CH3 | CF3 | CH | >10 |
| 49 | (CH2)3CH3 | Cl | N | 3.8 ± 3.5 |
| 50 | (CH2)3CH3 | F | N | 0.37 ± 0.08 |
| 51 | (CH2)3CH3 | H | N | 8.4 ± 3.2 |
Average of at least three replicates ± standard deviation.
Other compounds that were examined included analogues in which the nitrile was replaced with a tetrazole (Table 5, 52–53), an analogue in which the benzylic center was quaternarized (54), and an analogue in which the western and eastern aromatic rings were tethered in a seven-membered ring (55) (Table 5). These compounds all showed significantly worse activity.
Table 5. SAR of Bis-benzylic Nitrile Replacements.

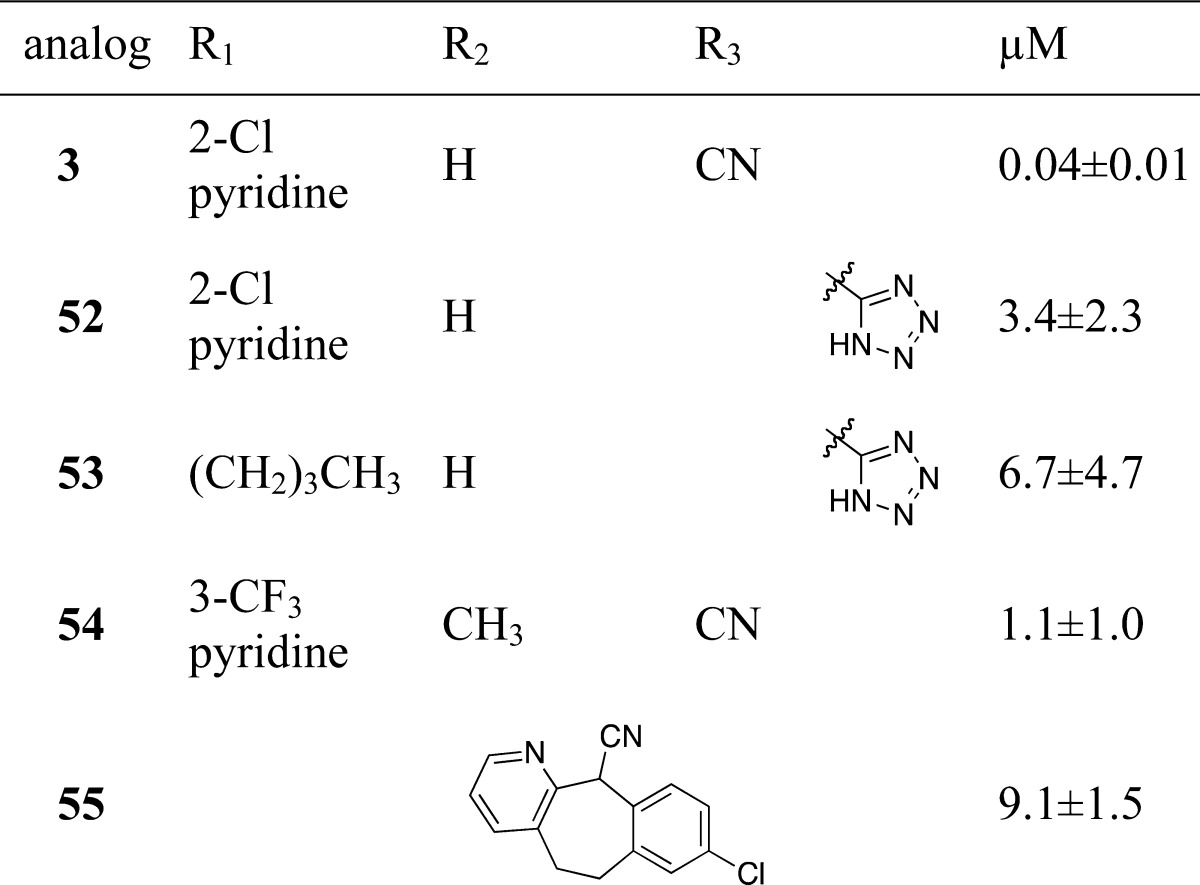
Average of at least three replicates ± standard deviation.
Crystal Structure Analysis
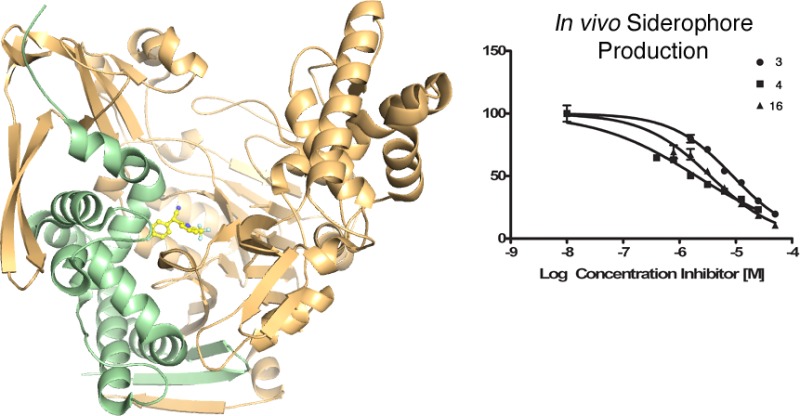
We examined the binding of the new compounds with X-ray crystallography. The initial HTS lead 3 and the fluorinated analogue 4 were selected for structural studies. The binding mode of biaryl nitriles to PvdQ was first studied by solving the structure of PvdQ bound to 3 at a resolution of 2.0 Å (Figure 4). Hit compound 3 sits in the acyl-binding site with the 4-chlorophenyl directed into the pocket away from the catalytic nucleophile (Figure 4). Hydrophobic residues Leu169, Leu269, Val374, Leu375, Trp378, and Pro401, as well as the methyl group of Thr166 surround the 4-chlorophenyl ring. The 2-chloropyridine lies in a hydrophobic pocket created by Met245, Phe248, Phe240, and His284. Leu266 fits between the two aromatic rings of and contributes to the hydrophobic cavities for both rings. Hydrophobic interactions dominate both spaces that accommodate the aromatic rings of the biarylnitrile scaffold.
Figure 4.

Structure of PvdQ bound to HTS hit compound 3. (A) Electron density is shown, calculated with coefficients of the form Fo-Fc generated prior to building the ligand in the active site. Density is contoured at 3σ (gray) and 8σ (red). (B) Active site of the enzyme is shown of the PvdQ bound to 3. Superposed on the structure is the fatty acid chain from covalently acylated structure from PDB 3L94.
A parallel-displaced π-stacking interaction between the 4-chlorophenyl and Trp378, and a T-shaped π-stacking interaction between the 2-chloropyridine and Phe240 could account for improved binding and the corresponding increase in potency for the biaryl nitrile analogues versus analogues containing aliphatic chains (Figure 4, Table 4). Interestingly, an overlay of the natural myristate ligand extends across the synthetic substrate with maximal overlap at the 2-position on the pyridine and the 4-position on the phenyl ring. This suggests that substitutions at these positions offer the best fit in the active site. The electrophilic 2-chloropyridine is approximately 7.4 Å from the catalytic nucleophile Ser217 and is not poised for covalent attachment. Therefore, the expectation is that nonelectrophilic substitutions will not reduce potency and these inhibitors would be competitive with substrate binding.
The nitrile moiety is directed into a ring formed by the main chain atoms of Pro401, Trp402, and Val403; the main chain amine of Trp402 hydrogen bonds to the nitrogen atom of the ligand nitrile (Figure 4). The side chains of all three of these residues contribute to this hydrophobic pocket. A π-stacking interaction between the nitrile moiety of the ligand and the indole ring of Trp402 stabilizes the binding orientation.
Co-crystallization of PvdQ with the more potent biaryl nitrile 4 offered additional insights (Figure 5). Crystals diffracted at 2.3 Å and the density of 4 was positioned in the PvdQ acyl-binding pocket analogously to 3, allowing facile placement of the ligand. Residues Thr166, Leu266, Leu269, Val374, and Trp378 make contacts to 4-fluorophenyl as with 4-chlorophenyl of 3. Again, the nitrile was surrounded by Pro401, Trp402, and Val403 residues, which provided key side chain and π-stacking interactions. Parallel-displaced and T-shaped π-stacking to Phe240 and Trp378, respectively, also enforced strong binding of the aromatic rings within the active site. Interestingly, the replacement of 2-chloro substituent on pyridine with trifluoromethyl provided the largest structural change between the binding of ligands 3 and 4. Whereas the only residue within 4 Å of the pyridyl chlorine atom of 3 is His284, each fluorine atom from the CF3 interacts with hydrophobic functionality on Phe240, Ile274, Trp378, Trp402, and Val403 side chains. These hydrophobic interactions are along the same vector observed for the natural myristate ligand and these interactions could account for the increase in potency observed for 4.
Figure 5.

Structure of PvdQ bound to 4. (A) Final probe compound ML318 is shown with electron density, calculated with coefficients of the form Fo-Fc generated prior to building the ligand in the active site, also included. Density is contoured at 3σ (gray) and 8σ (red). (B) Active site of the enzyme is shown of the PvdQ bound to 4. As in Figure 4, the fatty acid chain from covalently acylated structure from PDB 3L94 is also shown.
Analysis of the mostly hydrophobic fatty acid binding pocket in the context of the inhibitor-bound crystal structures informs the SAR of the analogues. The 2-chloropyridine group binds in the pocket closer to the catalytic serine while the 4-chlorophenyl group sits more deeply in the pocket. Compounds 3 and 4 both bind tightly in the pocket; except for the pyridyl nitrogen, each atom on the two aryl groups is less than 4 Å from a protein atom. While one edge of the phenyl ring points directly toward the peptide bond that joins Leu169 and Leu170, the other edge of the ring points toward a relatively large pocket that ends at Asn273. The presence of this pocket likely accommodates the variations in the C2 and C4 positions of the phenyl ring (Table 1). The 4-chlorophenyl could be replaced with a 2-fluoro-4-chlorophenyl group without a dramatic loss of activity, as in compounds 13 and 31.
Binding at the wider side of the hydrophobic binding pocket, the pyridine ring is more accepting of substitutions. Indeed, a wide variety of pyridine analogues retained activity with IC50 values below 200 nM (Table 2). As a whole, the alkyl substituted analogues (Table 4) that replaced the pyridine ring with the alkyl group showed weaker activity. This suggests that the π–π stacking interactions with Phe240 may be an important feature of binding the bisaryl inhibitor.
The nitrile replacements (Table 5) show the importance of the inhibitor framework to binding affinity. The replacement of the nitrile with a tetrazole in 52 and 53 increased the IC50 above 3 μM. The tight binding of the nitrile to the pocket formed by the peptide backbone of Pro401 through Val403 provides an explanation for this result. The ring positions shows that the geometry of binding can not accommodate the bridged seven-membered ring of 55.
As noted above, we were unable to purify enantiopure compounds due to the racemization of the chiral carbon at ambient conditions. The structure demonstrates, however, that the enzyme bound specifically to the S-isomer of both 2 and 3, which results in the nitrile positioned in the pocket formed by the main chain of Pro401-Val403 and the pyridine ring deeper in the acyl binding pocket. Given the approximate symmetry of the bisbenzylic compounds, it is possible that the R-isomers also could bind in the pocket with the result of switching the positions of the phenyl and pyridine rings. However, substitutions at the C3 position of the pyridyl ring result in drops of at least 10-fold in potency and indeed are some of the worst compounds identified—20, 23, 28, and 34, for example—suggesting that substitutions para to the benzylic carbon are not well-accommodated and thus the R-enantiomer may not bind well in the PvdQ binding pocket.
Inhibitor Activity in Whole-Cell Assays
Despite exhibiting an IC50 of 40 nM in the biochemical assay, the primary hit compound 3 showed only μM activity in the preliminary whole cell assay.29 We reasoned that this observation may be due partly to the nonlethality of the PvdQ knockout14,35 and the known difficulty of small molecules to effectively permeate P. aeruginosa.36 We therefore initiated a study of compound stability and the impact on pyoverdine production in whole cells.
We first screened compounds 3 and 4 for stability (Supporting Information). After 48 h in neutral PBS buffer, about 30–40% of 3 remains. After 6 h in the presence of 50 μM glutathione, about 90% remains. These values are modestly improved for the probe compound 4, but overall the modest stability is a liability for further biological examination and the potential to develop these lead compounds for pharmaceutical activities.
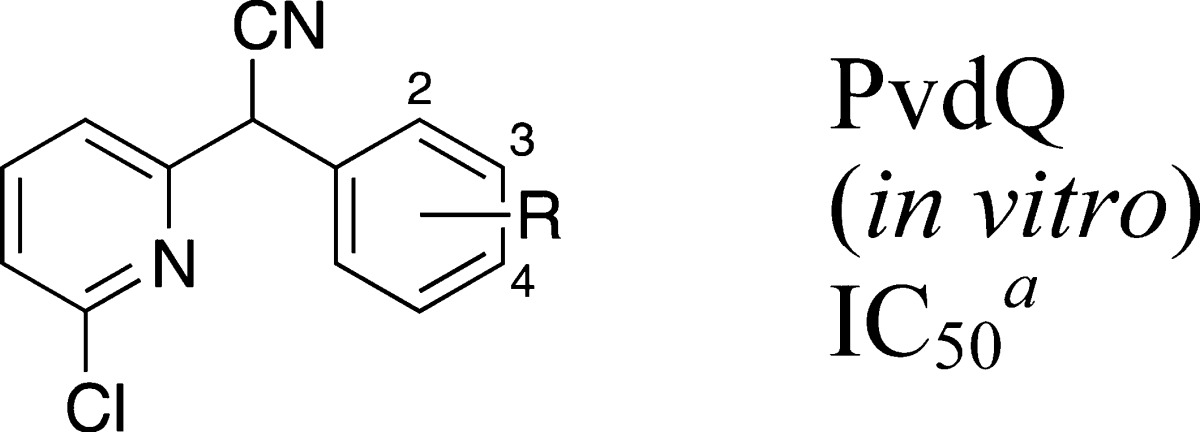
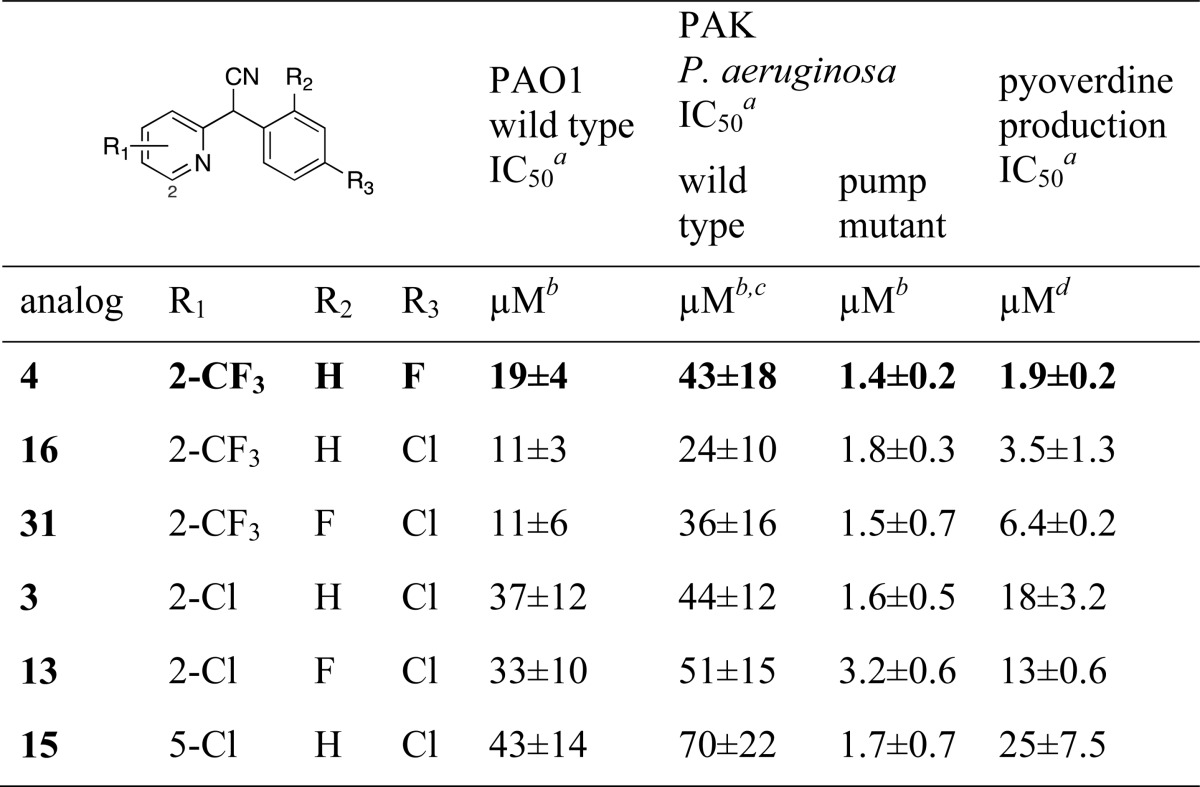
Because of the limited stability of the compounds, we first developed a short-term assay to monitor the effect of several of the best inhibitors (those with IC50 < 0.06 μM) upon pyoverdine production in a whole cell assay. To demonstrate an on-target effect, P. aeruginosa (PAO1) was grown for 4 h in the presence of compounds and the amount of pyoverdine produced was directly measured using HPLC. Pyoverdine migrates as a cluster of HPLC peaks that were integrated for IC50 calculation (Table 6). This direct readout of pyoverdine production demonstrates the ability of these compounds to inhibit PvdQ in P. aeruginosa. Analogue 4 proved to be the most potent inhibitor of pyoverdine production with a whole cell IC50 of 1.9 μM.
Table 6. Activity of Analogues in Other P. aeruginosa Strains and Toxicity Counter Screen.
Average of at least three replicates ± standard deviation.
405 nm wavelength measuring effect on pyoverdine production.
Measurement taken as AbsAC–35 (absolute active concentration at 35% inhibition), the predetermined activity threshold chosen for comparison because not all compounds reached a plateau of activity at 50% inhibition.
Quantified by analysis of the isolated pyoverdine HPLC peaks present in culture media of cells treated with varying concentrations of each inhibitor.
We then tested these compounds against P. aeruginosa in 48 h growth cultures, understanding that differences in compound stability may influence the ability to rigorously compare the impact on pyoverdine production (Table 6). We first analyzed the compounds against the PA01 strain used earlier and monitored absorbance of culture media at 405 nm. The IC50 values for six biaryl nitriles ranged from 11 to 43 μM. We expected that the difference between the biochemical (nM) and whole cell (μM) activities are likely related to the ability of P. aeruginosa to export the compounds through nonspecific exporters.37 We therefore screened the same compounds against a second P. aeruginosa strain (PAK) and a pump mutant (mexAB-oprM) of the PAK strain (Table 6). The most potent compounds retained similar activity in the PAK wild type strain and the potency of these small molecules was augmented in the pump mutant mexAB-oprM (Table 6). These data illustrate that these compounds are efficacious across two P. aeruginosa strains (PAO1 and PAK) and that the difference between the in vitro and in vivo activities is partly due to the efflux mechanisms.
Complementation with Inhibitors of Pyoverdine Synthesis Genes
Finally, we asked whether reduction in the expression of pyoverdine production enzymes such as PvdQ could further sensitize P. aeruginosa. Recently, a library of U.S. FDA-approved compounds were screened for molecules that block expression of pyoverdine synthetic genes.38 These efforts identified 5-fluorocytosine (5-FC), which blocks transcription of pvdS, a σ-factor that is involved in activating a variety of genes in iron-depleted conditions. The effectiveness of 5-fluorocytosine was investigated in vivo and reduced P. aeruginosa growth and virulence in a mouse pulmonary infection. We therefore examined pyoverdine production in the HPLC assay for cells treated with either 4 or 5-FC alone, or with equal concentrations of 4 and 5-FC. Whereas the IC50 value for 4 and for 5-FC alone were 7.9 and 8.3 μM, the IC50 for the combined treatment was 1.7 μM. This demonstrates that reducing the expression level of the pyoverdine synthesis proteins sensitized the cells to the effects of the PvdQ inhibitor and could be an effective strategy for reducing pyoverdine production.
Despite the active nitrile moiety on the compounds identified herein, there is no evidence of a covalent interaction between PvdQ and either 3 or 4. Recently, Fast and colleagues designed a covalent inhibitor of PvdQ using a tridecylboronic acid that binds covalently to the catalytic serine.39 Kinetic and structural evidence show it is a competitive inhibitor that binds in the fatty acyl binding pocket. Growth of P. aeruginosa was inhibited by the boronic acid inhibitor when cotreated with phenylalanine-arginine-β-naphthylamide, a wide-spectrum inhibitor of multidrug resistance exporters. Given our results with the mexA-oprM mutant PAK strain, we are examining whether the inclusion of the exporter inhibitor may further enhance the efficacy of the biaryl nitrile inhibitors of PvdQ.
Summary
A high-throughput screen against the pyoverdine maturation enzyme PvdQ with the National Institutes of Health (NIH) MLSMR collection identified a biaryl nitrile scaffold (3) amenable to medicinal chemistry studies due to its modular nature. Numerous analogues were synthesized to investigate SAR at three regions of the scaffold leading to a more potent fluorinated biaryl nitrile 4. Compound 4 was the most potent in the whole cell assays for inhibition of pyoverdine production directly measured by HPLC and has been designated as Probe Compound ML318.29 On the pyridyl portion of the scaffold, electron-withdrawing groups, particularly in the 2-position were found in the most active analogues. On the phenyl portion, a halide in the 4-position was optimal. Crystallographic analyses of lead 3 and the optimized probe 4 in the acyl-binding pocket of PvdQ suggest a competitive binding mode for this class of biaryl nitriles. Distinctive hydrophobic, π-stacking and nitrile hydrogen bonding interactions rationalized the potent activities observed. The hit and probe compounds were shown to be active in whole cell assays, reducing the production of mature pyoverdine with IC50 values in the low μM range. The stability of the compounds limited effectiveness, as did the presence of Pseudomonas export machinery that likely explained the difference between the compounds in the whole cell vs biochemical assays. Nonetheless, the compounds confirm that targeting PvdQ specifically and siderophore synthesis proteins in general can be a suitable strategy to reduce the production of these important virulence determinants.
Methods
High-Throughput Screening of PvdQ Activity
The enzymatic substrate 4-methylumbelliferyl (4-MU) laurate (Research Organics) solution was formulated using 1 volume isopropanol (Sigma), 0.1 volume Triton X-100 (Sigma), and 15 volumes of TNT buffer. The PvdQ protein preparation and 4-MU laurate solution were mixed together at the time of the screen using a BioRaptr FRD microfluidic workstation (Beckman Coulter) to a final concentration 0.02 μM PvdQ and 0.8 mM 4-MU laurate at RT in 1536-well high base black bar-coded square well assay plates (Aurora Biotechnologies) with compounds added previously, using an Echo acoustic liquid handler (Labcyte). The primary assay was performed with 337 488 library compounds at a concentration of 10 μM. Active compounds were rescreened at 8 concentrations of 3-fold dilutions starting at a highest concentration of 10 μM. The positive control, isopropyl dodecylfluorophosphonate (IDFP) (Cayman chemicals), was added in 24 selective wells to a final concentration of 200 μM. After the addition steps, the assay plates were first read for fluorescence at time = 0, then incubated at RT for 60 min, and then were read again at time = 60 min. Fluorescence was measured using the ViewLux uHTS Microplate Imager (PerkinElmer) with 303–367 nm excitation filter and 440–460 nm emission filter. The fluorescence generated by the enzymatic reaction was calculated as the difference between the two reads (60 min vs 0 min). Each dose response curve was normalized to a DMSO control set at 0% activity and the positive control (IDFP) set at −100% activity. The raw value attributed to each compound was converted to a percent effect based on these controls.
IC50 values were determined using the same fluorogenic assay with nine compound concentrations that ranged from 3 nM to 19.5 μM with PvdQ concentration of 20 nM. Additional details concerning the assay and the compounds determined to be active, as well as initial characterization of probe 4, designated ML318, are available in the NIH Molecular Probe Report.29
Chemical Synthesis of Biaryl Nitriles
Biaryl nitriles were synthesized for SAR studies of inhibition of PvdQ acylase activity from phenylacetonitriles and substituted pyridines. Specific synthesis procedures, NMR characterization, and yields are described in the Supporting Information.
Determination of the Structure of PvdQ Bound to 3 and 4
The structure determination of PvdQ bound to 3 and 4 along with diffraction and refinement statistics are presented in the Supporting Information.
In Vivo Inhibition of Pyoverdine Production
Cultures of Pseudomonas aeruginosa PAO1 wild-type (ATCC 15692) were grown overnight (16 h) in 50 mL rich LB media. A pellet containing the bacteria was generated through centrifugation at 1725g for 10 min at 20 °C. The cells were washed once with 25 mL of 5% CAA media and centrifuged again at 20 °C (1725g, 10 min). The bacteria were resuspended in CAA media with 0.5% DMSO to calculated OD600 = 1.25. PAO1 wildtype and PvdQ deletion mutant cells (1 mL, OD600 = 0.2) were inoculated into seven serial dilutions of PvdQ small molecule inhibitors (100 μM to 1.5 μM final concentrations) in triplicate. Each bacteria and PvdQ compound inhibitor dilution/combination was grown for 4 h at 37 °C with shaking (250 rpm). An aliquot of 200 μL was transferred into a Costar clear bottom black side where the bacterial growth was measured at 600 nm. The remaining 800 μL of the bacterial culture was poured into a 1.5 mL eppendorf tube and centrifuged at 16 200 g for 5 min at 4 °C. The supernatant (500 μL) was removed and filtered through a 0.45 μM filter. The eluted solution was centrifuged under vacuum at 16 200 g at 20 °C to dryness (2 min). The dry pellet was reconstituted in 350 μL of a solution containing 2% acetonitrile, 98% H2O, and 0.1% formic acid. The pyoverdine peaks were analyzed using the GraphPad Prism software on a transformed (x = log(x)) versus (no inhibitor = 100%). Dose response curves were calculated using a variable slope with constraints at 0 and 100. Details regarding chromatography are in the Supporting Information
Pseudomonas aeruginosa PAO1 Growth Delay Assay
Pseudomonas aeruginosa PAO1 wild-type cells were grown overnight (16 h) in SM9 media. Prior to the addition of the bacteria, 30 μL of SM9 media with or without 2 μM (final concentration) of EDDHA was added to the 384-well black clear bottom assay plates (Aurora Biotechnologies). Compounds were added by pin tool transfer at 200 nL, to the final concentrations starting from 67 μM, with 8 concentrations at 3-fold dilutions. P. aeruginosa PAO1 cells (10 μL, diluted to a calculated OD600 = 2 × 10–6) with or without 2 μM EDDHA was dispensed in the same wells. After incubation for 18–30 h at 37 °C, the absorbance at 600 and 405 nm was measured using an EnVision (PerkinElmer) plate reader. The data were reported at the concentration where the normalized dose response curve crosses a predetermined threshold since no positive control exists to set the −100% value.
Pseudomonas aeruginosa PAK Growth Delay Assay
Pseudomonas aeruginosa PAK wild-type or pump mutant mexAB-OprM were grown overnight (16 h) in SM9 media at 37 °C. SM9 media was prepared ± 2 μM EDDHA and 20 μL was added to each well of 384-well black, clear bottom plates. Compounds (8 concentrations, 3 fold dilutions starting at 10 mM) were transferred via pin tool at 300 nL, giving final compound concentrations from 100 μM to 15 nM. P. aeruginosa wild-type or pump mutant mexAB-OprM culture (10 μL) diluted at OD600 = 0.05 in SM9 media (±2 μM EDDHA) was added to each well and the plates were incubated for 28 h at 30 °C, in a humidified environment. After 28 h incubation, the absorbance values were read with an EnVision plate reader at 600 nm (cell growth) and 405 nm (pyoverdine production). The data were again reported at the concentration where the normalized dose response curve crosses a predetermined threshold since no positive control exists to set the −100% value.29 For the wild-type PAK cells, the data were reported at a slightly higher threshold since not all compounds reached 50% inhibition at the concentrations tested. This would make the impact of the mexAB-OprM mutation even larger.
Acknowledgments
We gratefully acknowledge the National Institutes of Health (NIH) (U54HG005032 awarded to S.L.S.) for funding and the Broad Institute Analytical team, especially C. Mosher, T. Anthoine, and S. Johnston for expert mass spectral analysis and compound stability testing. We also acknowledge N. Dasgupta (Massachusetts General Hospital) and M. Brannon (University of Washington, Seattle) for bacterial strain construction. We thank C. Scherer for helpful discussions. This work was additionally supported by grants GM-068440 and MH-092076 (A.M.G.), by grant AI-067653 (S.M.M.), and TATRC cooperative agreement W81XWH-11-2-0218 (A.M.G.). Portions of this work were carried out at SSRL, which is supported by the DOE and NIGMS (including P41GM103393). S.L.S. is an Investigator with the Howard Hughes Medical Institute.
Supporting Information Available
Detailed description of bacterial media, protein production, biological assays, analytical assays, pump mutant strain construction, NMR spectra, and crystal structures. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The final crystal structures and structure factors have been deposited in the PDB: PvdQ plus 3,4K2F; PvdQ plus 4, 4K2G.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kanj S. S.; Kanafani Z. A. (2011) Current concepts in antimicrobial therapy against resistant gram-negative organisms: Extended-spectrum β-lactamase-producing Enterobacteriaceae, carbapenem-resistant Enterobacteriaceae, and multidrug-resistant Pseudomonas aeruginosa. Mayo Clinic Proceedings 86, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson L. R. (2009) Bad bugs, no drugs: No ESCAPE revisited. Clin. Infect. Dis. 49, 992–993. [DOI] [PubMed] [Google Scholar]
- Mekalanos J. J. (1992) Environmental signals controlling expression of virulence determinants in bacteria. J. Bacteriol. 174, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy M.; Butler A. (2009) Microbial iron acquisition: Marine and terrestrial siderophores. Chem. Rev. 109, 4580–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke M.; Marahiel M. A. (2007) Siderophore-based iron acquisition and pathogen control. Microbiol Mol. Biol. Rev. 71, 413–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras J. A.; Ryu J. S.; Di Lello F.; Tan D. S.; Quadri L. E. (2005) Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 1, 29–32. [DOI] [PubMed] [Google Scholar]
- Miethke M.; Bisseret P.; Beckering C. L.; Vignard D.; Eustache J.; Marahiel M. A. (2006) Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 273, 409–419. [DOI] [PubMed] [Google Scholar]
- Qiao C.; Gupte A.; Boshoff H. I.; Wilson D. J.; Bennett E. M.; Somu R. V.; Barry C. E. 3rd; Aldrich C. C. (2007) 5′-O-[(N-Acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: An adenylation enzyme required for siderophore biosynthesis of the mycobactins. J. Med. Chem. 50, 6080–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun S.; Guo H.; Adamson J.; Cisar J. S.; Davis T. D.; Chavadi S. S.; Warren J. D.; Quadri L. E.; Tan D. S.; Bishai W. R. (2013) Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother. 57, 5138–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner U. A.; Wilderman P. J.; Vasil A. I.; Vasil M. L. (2002) GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: Identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 45, 1277–1287. [DOI] [PubMed] [Google Scholar]
- Visca P.; Imperi F.; Lamont I. L. (2007) Pyoverdine siderophores: From biogenesis to biosignificance. Trends Microbiol. 15, 22–30. [DOI] [PubMed] [Google Scholar]
- Lamont I. L.; Beare P. A.; Ochsner U.; Vasil A. I.; Vasil M. L. (2002) Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 99, 7072–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. M.; Neely A.; Stintzi A.; Georges C.; Holder I. A. (1996) Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect. Immun. 64, 518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal Jimenez P.; Koch G.; Papaioannou E.; Wahjudi M.; Krzeslak J.; Coenye T.; Cool R. H.; Quax W. J. (2009) Role of PvdQ in Pseudomonas aeruginosa virulence under iron-limiting conditions. Microbiology 156, 49–59. [DOI] [PubMed] [Google Scholar]
- Banin E.; Vasil M. L.; Greenberg E. P. (2005) Iron and Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 102, 11076–11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton J. W.; Stewart P. S.; Greenberg E. P. (1999) Bacterial biofilms: A common cause of persistent infections. Science 284, 1318–1322. [DOI] [PubMed] [Google Scholar]
- Sauer K.; Cullen M. C.; Rickard A. H.; Zeef L. A.; Davies D. G.; Gilbert P. (2004) Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 186, 7312–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux D. E.; Sanschagrin F.; Levesque R. C. (2000) Genomics of the 35-kb pvd locus and analysis of novel pvdIJK genes implicated in pyoverdine biosynthesis in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 190, 141–146. [DOI] [PubMed] [Google Scholar]
- Taguchi F.; Suzuki T.; Inagaki Y.; Toyoda K.; Shiraishi T.; Ichinose Y. (2010) The siderophore pyoverdine of Pseudomonas syringae pv. tabaci 6605 is an intrinsic virulence factor in host tobacco infection. J. Bacteriol. 192, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaioannou E.; Wahjudi M.; Nadal-Jimenez P.; Koch G.; Setroikromo R.; Quax W. J. (2009) Quorum-quenching acylase reduces the virulence of Pseudomonas aeruginosa in a Caenorhabditis elegans infection model. Antimicrob. Agents Chemother. 53, 4891–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalk I. J.; Guillon L. (2013) Pyoverdine biosynthesis and secretion in Pseudomonas aeruginosa: implications for metal homeostasis. Environ. Microbiol. 15, 1661–1673. [DOI] [PubMed] [Google Scholar]
- Challis G. L.; Naismith J. H. (2004) Structural aspects of non-ribosomal peptide biosynthesis. Curr. Opin Struct Biol. 14, 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossialos D.; Ochsner U.; Baysse C.; Chablain P.; Pirnay J. P.; Koedam N.; Budzikiewicz H.; Fernandez D. U.; Schafer M.; Ravel J.; Cornelis P. (2002) Identification of new, conserved, non-ribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol. Microbiol. 45, 1673–1685. [DOI] [PubMed] [Google Scholar]
- Drake E. J.; Gulick A. M. (2011) Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6, 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannauer M.; Schafer M.; Hoegy F.; Gizzi P.; Wehrung P.; Mislin G. L.; Budzikiewicz H.; Schalk I. J. (2012) Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586, 96–101. [DOI] [PubMed] [Google Scholar]
- Artymiuk P. J. (1995) A sting in the (N-terminal) tail. Nat. Struct. Biol. 2, 1035–1037. [DOI] [PubMed] [Google Scholar]
- Bokhove M.; Jimenez P. N.; Quax W. J.; Dijkstra B. W. (2010) The quorum-quenching N-acyl homoserine lactone acylase PvdQ is an Ntn-hydrolase with an unusual substrate-binding pocket. Proc. Natl. Acad. Sci. U.S.A. 107, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sio C. F.; Otten L. G.; Cool R. H.; Diggle S. P.; Braun P. G.; Bos R.; Daykin M.; Camara M.; Williams P.; Quax W. J. (2006) Quorum quenching by an N-acyl-homoserine lactone acylase from Pseudomonas aeruginosa PAO1. Infect. Immun. 74, 1673–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault J. R., Wurst J., Jewett I., Verplank L., Perez J. R., Gulick A. M., Drake E. J., Palmer M., Moskowitz S., Dasgupta N., Brannon M. K., Dandapani S., Munoz B., and Schreiber S. (2013) Identification of a small molecule inhibitor of Pseudomonas aeruginosa PvdQ acylase, an enzyme involved in siderophore pyoverdine synthesis., In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotchnology Information (U.S.), Bethesda, MD. [PubMed] [Google Scholar]
- Meyer J. M., Halle F., Hohnadel D., Lemanceau P., and Ratefiarivelo H. (1987) Siderophores of Pseudomonas–biological properties, In Iron Transport in Microbes, Plants, and Animals (Winkelmann G., Van der Helm D., and Neilands J. B., Eds.), pp 189–205, VCH, Weinheim Germany. [Google Scholar]
- Sommer M. B.; Begtrup M.; Bogeso K. P. (1990) Displacement of halogen of 2-halogeno-substituted benzonitriles with carbanion. Preparation of (2-cyanoaryl)arylacetonitriles. J. Org. Chem. 55, 4817–4821. [Google Scholar]
- Hino K.; Nagai Y.; Uno H. (1988) Agents acting on the central nervous system. Synthesis of 3-phenyl-2-piperazinyl-1-benzazocines, 3-substituted-2-piperazinyl-1-benzazepines, and related compounds. Chem. Pharmaceut. Bull. 36, 2386–2400. [DOI] [PubMed] [Google Scholar]
- Gutmann B.; Roduit J. P.; Roberge D.; Kappe C. O. (2010) Synthesis of 5-substituted 1H-tetrazoles from nitriles and hydrazoic acid by using a safe and scalable high-temperature microreactor approach. Angew. Chem., Int. Ed. Engl. 49, 7101–7105. [DOI] [PubMed] [Google Scholar]
- Tran V. H.; Kantharaj R.; Roufogalis B. D.; Duke C. C. (2006) An efficient and facile synthesis of capsaicin-like compounds as agonists of the TRPV1 receptor. Eur. J. Org. Chem. 2006, 2970–2976. [Google Scholar]
- Clatworthy A. E.; Pierson E.; Hung D. T. (2007) Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3, 541–548. [DOI] [PubMed] [Google Scholar]
- Lomovskaya O.; Warren M. S.; Lee A.; Galazzo J.; Fronko R.; Lee M.; Blais J.; Cho D.; Chamberland S.; Renau T.; Leger R.; Hecker S.; Watkins W.; Hoshino K.; Ishida H.; Lee V. J. (2001) Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 45, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages J. M.; Amaral L. (2009) Mechanisms of drug efflux and strategies to combat them: Challenging the efflux pump of Gram-negative bacteria. Biochim. Biophys. Acta 1794, 826–833. [DOI] [PubMed] [Google Scholar]
- Imperi F.; Massai F.; Facchini M.; Frangipani E.; Visaggio D.; Leoni L.; Bragonzi A.; Visca P. (2013) Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 110, 7458–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger K. D.; Wu R.; Er J. A.; Liu D.; Fast W. (2013) Rational design of a transition state analogue with picomolar affinity for Pseudomonas aeruginosa PvdQ, a siderophore biosynthetic enzyme. ACS Chem. Biol. 8, 2192–2200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.