

Abstract
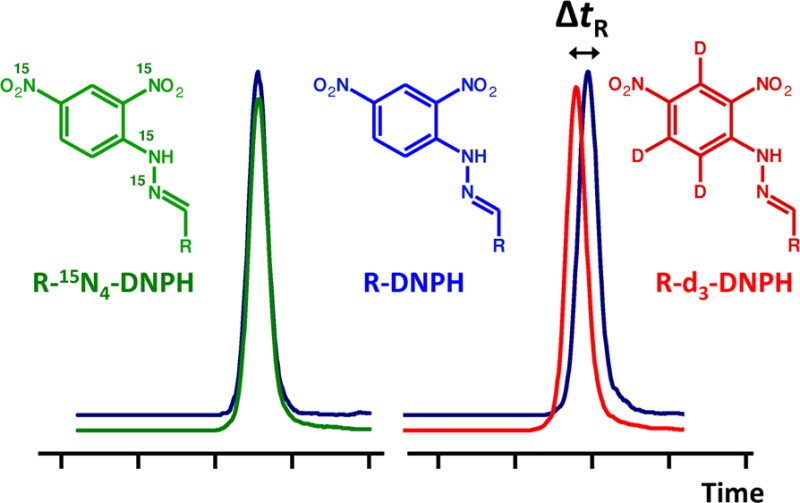
Isotope effect may cause partial chromatographic separation of labeled (heavy) and unlabeled (light) isotopologue pairs. Together with a simultaneous matrix effect, this could lead to unacceptable accuracy in quantitative liquid chromatography–mass spectrometry assays, especially when electrospray ionization is used. Four biologically relevant reactive aldehydes (acrolein, malondialdehyde, 4-hydroxy-2-nonenal, and 4-oxo-2-nonenal) were derivatized with light or heavy (d3-, 13C6-, 15N2-, or 15N4-labeled) 2,4-dinitrophenylhydrazine and used as model compounds to evaluate chromatographic isotope effects. For comprehensive assessment of retention time differences between light/heavy pairs under various gradient reversed-phase liquid chromatography conditions, major chromatographic parameters (stationary phase, mobile phase pH, temperature, organic solvent, and gradient slope) and different isotope labelings were addressed by multiple-factor screening using experimental designs that included both asymmetrical (Addelman) and Plackett–Burman schemes followed by statistical evaluations. Results confirmed that the most effective approach to avoid chromatographic isotope effect is the use of 15N or 13C labeling instead of deuterium labeling, while chromatographic parameters had no general influence. Comparison of the alternate isotope-coded derivatization assay (AIDA) using deuterium versus 15N labeling gave unacceptable differences (>15%) upon quantifying some of the model aldehydes from biological matrixes. On the basis of our results, we recommend the modification of the AIDA protocol by replacing d3-2,4-dinitrophenylhydrazine with 15N- or 13C-labeled derivatizing reagent to avoid possible unfavorable consequences of chromatographic isotope effects.
High-performance liquid chromatography coupled with tandem mass spectrometric (LC–MS/MS) detection is a frequently used technique for the identification and quantification of a wide range of chemical species in biological matrixes. The superior specificity of MS allows the simplification of sample preparation, and the application of fast gradient methods, where a baseline separation of all components is not required. However, coeluting matrix components can significantly affect the accuracy and precision of a method.1−4 The use of a stable-isotope-labeled internal standard (IS), presumed to have identical physicochemical properties to those of the corresponding unlabeled analyte, is considered to correct for the variability arising from sample preparations and instrumental analyses. Generally, deuterium (d-) labeled ISs are available commercially; however, they may elute at different time points than their analytes due to chromatographic isotope effect.5−7 The shifts in the retention times can be large enough to cause differential matrix effect in LC–MS, especially when electrospray ionization (ESI) is used.2 Therefore, the change in the analyte to IS peak area ratios may be significant enough to influence the accuracy of quantitation.8−13
Several approaches have been recommended to overcome caveats associated with the use of d-labeled ISs.12−17 Additional sample preparation steps, the use of shallow gradient profiles to achieve adequate separation of analytes from major matrix components eluting with the solvent front, application of atmospheric pressure chemical ionization (APCI) which is less prone to matrix effect than ESI,2 or using other than d- (i.e., 13C-, 15N-, or 18O-) labeled ISs are examples of these efforts. Unfortunately, all of these practices have limitations; for instance, multiple sample preparation steps and longer chromatography using shallow gradients decrease sample throughput without actually eliminating problems related to matrix effect.4 In many cases, the APCI technique results in sensitivity loss compared to ESI.18,19 It has also been reported that data obtained through the use of the “minimal labeling approach” require corrections by an isotope pattern deconvolution algorithm.16,17 On the other hand, the limited availability and/or high price of other than d-labeled analogues may be a practical hurdle that prevents their widespread applications.
Despite earlier reports,5−9,13,20 the influence of operating parameters (e.g., stationary phase, mobile phase pH, temperature, organic solvent, and gradient slope) relevant to most gradient LC–MS-based bioassays on chromatographic isotope effects has not been assessed and understood. Non-deuterium-labeled species also have not been studied extensively in this context, especially when the most widely applied analytical gradient reversed-phase HPLC (RPLC) conditions are considered.21 Therefore, we evaluated the chromatographic isotope effect brought about by stable isotope labeling on different atoms in the framework of isotope-coded derivatization (ICD) that represents an emerging method for quantitative metabolite profiling.22,23 We focused on its implementation known as alternate isotope-coded derivatization assay (AIDA) and applied it to the LC–MS/MS analysis of biologically relevant aldehydes.24 AIDA uses 2,4-dinitro-3,5,6-trideuterophenylhydrazine (d3-DNPH) for isotope-coded “heavy” labeling, and has been validated for malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE) quantifications. Here we also included 15N- and 13C-labeled DNPH reagents, and extended our study to acrolein (ACR) and 4-oxo-2-nonenal (ONE), as additional lipid peroxidation (LPO) end-products (Figure 1).
Figure 1.

Chemical structure of selected reactive aldehydes and their DNPH derivatives.
Overall, we aspired to a comprehensive assessment of the subject through experimental screening designs25,26 for the first time to address the contributions of operating parameters relevant to most gradient LC–MS-based bioassays and stable isotope labeling to the chromatographic isotope effect that could compromise the accuracy and precision of quantitation by RPLC–ESI-MS. These multivariate approaches allow for predicting the effect of a given factor at several levels of combinations with other factors.
Experimental Section
Chemicals and Reagents
Butylated hydroxytoluene (BHT), ACR, DNPH, MDA-tetrabutylammonium salt, [15N]KNO3 (98% isotope purity), [15N2]hydrazine sulfate (98% isotope purity), and d5-chlorobenzene (99% isotope purity) were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.). [13C6]Chlorobenzene (99% isotope purity) was from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, U.S.A.). HNE (10 mg/mL in ethanol) and ONE (5 mg/mL in methyl acetate) were purchased from Cayman Chemicals (Ann Arbor, MI, U.S.A.). Solvents used for LC–MS/MS measurements were of HPLC grade and supplied by Fisher Scientific (Atlanta, GA, U.S.A.). Preparative TLC plates (silica gel G, 20 cm × 20 cm, UNIPLATE-T taper plate) were from Analtech Inc. (Newark, DE, U.S.A.), and analytical TLC silica gel 60 F254 plates were from AMD Millipore (Billerica, MA, U.S.A.). Charcoal-stripped human serum was ordered from Innovative Research (Novi, MI, U.S.A.).
Synthesis of Stable Isotope Labeled DNPH Analogues
The isotope-labeled “heavy” hydrazines, d3-DNPH, [13C6]DNPH, [15N4]DNPH, and [15N2]DNPH, were synthesized according to routine procedures based on the nucleophile substitution reaction between hydrazine and 2,4-dinitrochlorobenzene, as reported before.14 First, the appropriately labeled 2,4-dinitrochlorobenzenes were synthesized; d3-2,4-dinitrochlorobenzene and [13C6]2,4-dinitrochlorobenzene were prepared from d5-chlorobenzene and [13C6]chlorobenzene, respectively, in a mixture of KNO3/H2SO4. The 15N-labeled 2,4-dinitrochlorobenzene was obtained from chlorobenzene in a mixture of [15N]KNO3/H2SO4. Then, the individual 2,4-dinitrochlorobenzenes were reacted with hydrazine in acetic acid, with the exception of the synthesis leading to [15N4]DNPH where [15N2]hydrazine sulfate was used. Each product was then purified on preparative TLC using hexane/ethyl acetate (6:1, v/v). On analytical TLC, we detected only one UV–vis spot, with Rf = 0.63, using hexane/ethyl acetate (6:1, v/v).
For the derivatization of the carbonyls, individual DNPH reagents (light and heavy: d3-, [15N2]-, [15N4]-, or [13C6]-labeled) were dissolved in formic acid/acetonitrile 1:50 (v/v) to obtain 1 mg/mL of derivatizing stock solutions.
Sample Solutions for the Evaluation of Isotope Effect
Stock solutions of MDA, ACR, HNE, and ONE, respectively, were prepared freshly in acetonitrile at 1 mg/mL concentration for each aldehyde. Aliquots of 10 μL of each aldehyde were added into 160 μL of derivatizing solution containing the light or individual heavy (d3-, [15N2]-, [15N4]-, or [13C6]-labeled) DNPH, respectively. The samples were incubated at 40 °C for 2 h in the dark. Then, equal aliquots (10 μL) of the light hydrazones’ mixture and each of their heavy hydrazones’ mixture were brought together, resulting in four heavy–light labeled solutions of the derivatized aldehydes. These samples were then diluted with 50% (v/v) acetonitrile to obtain 0.1 and 1.0 μg/mL solutions (expressed in terms of free carbonyl content), respectively, for isotope effect screenings.
Evaluation of Isotope Effect Using Screening Experimental Designs
A Surveyor LC system (Thermo Electron Corporation, Trace Chemical Analysis, Austin, TX, U.S.A.) was employed throughout the studies. A Phenomenex Kinetex phenyl-hexyl silica column (50 mm × 2.1 mm i.d., 5 μm particles) protected by a Phenomenex 2 mm × 2.1 mm i.d. SecurityGuard phenyl-hexyl silica guard column (PhenHex) and a Phenomenex Kinetex C18 column (50 mm × 2.1 mm i.d., 5 μm particles) protected by a Phenomenex 2 mm × 2.1 mm i.d. SecurityGuard C18 guard column (C18) were used to assess the isotope effect. RPLC conditions, including mobile phase pH, column temperature, and gradient time, are summarized in Table 1. The k* gradient retention factors were within the recommended optimal range (0.5 < k* < 20) for analytical HPLC.21 The selected seven factors to study their effects on the chromatographic isotope effects are shown in Table S-1 (Supporting Information). An asymmetrical experimental design (Table S-2 in the Supporting Information) was constructed from Addelman’s basic plan using a template proposed by Hund et al.25 The experiments were sorted according to RP analytical columns and mobile phases used, from practical reasons. The Plackett–Burman experimental design (Table S-3 in the Supporting Information) was set up according to Vander Heyden et al.26 To investigate the influence of uncontrolled factors such as time effects due to response drifts, we repeated selected experiments, e.g., experiment 1 in Supporting Information Table S-2 and experiment 12 in Supporting Information Table S-3, before and after executing the sequences specified in these tables. Statistical and graphical interpretations of data included visual inspection of half-normal probability plots and estimation of the critical effects such as negligible factor effects (Ecritical), and margin of error (ME), and simultaneous margin of error (SME).26
Table 1. Gradient RPLC Conditions Selected for the Evaluation of Chromatographic Isotope Effects.
| column | |
| PhenHex | Phenomenex Kinetex phenyl-hexyl (50 mm × 2.1 mm i.d., 5 μm particles) with SecurityGuard (2 mm × 2.1 mm i.d.) |
| C18 | Phenomenex Kinetex C18 (50 mm × 2.1 mm i.d., 5 μm particles) with SecurityGuard (2 mm × 2.1 mm i.d.) |
| mobile phase | |
| eluent A1 | 0.1% (v/v) acetic acid in water at pH 3.3 |
| eluent A2 | 5 mM ammonium acetate in water at pH 8.2 |
| eluent B1 | ACN |
| eluent B2 | MeOH |
| gradient profile | 50% to 100% B |
| gradient time (min) | |
| tg1 | 2 (k* = 3.5a) |
| tg2 | 4 (k* = 7a) |
| tg3 | 8 (k* = 14a) |
| flow rate (mL/min) | 0.4 |
| column oven temperature (°C) | |
| T1 | 25 |
| T2 | 40 |
| T3 | 50 |
| injection volume (μL) | 5 |
k* denotes gradient retention factor.
RPLC Conditions for the Quantification of Hydrazones
Analyses were done using the same PhenHex column (50 mm × 2.1 mm i.d., 5 μm particles) as specified above with gradient elution at 30 °C. The mobile phase was a mixture of (A1) 0.1% (v/v) acetic acid in water and (B1) acetonitrile (see Table 1), with linear gradient from 40% B to 100% B in 4 min. Then, the column was flushed with 100% B for 1 min and, finally, was re-equilibrated with 40% B for 3 min. Flow rate was maintained constant at 0.4 mL/min, and the injection volume was 5 μL.
Mass Spectrometry
LC–MS/MS analysis of the light and heavy DNPH derivatives was performed on a TSQ Quantum Ultra (Thermo Electron Corporation) mass spectrometer equipped with heated ESI (H-ESI) probe. With the exception of DNPH–MDA, negative ion mode was applied. H-ESI spray voltage, H-ESI temperature, and capillary temperature were maintained at 3.5 and −3.0 kV (positive and negative ionization, respectively), 350, and 325 °C, respectively. Nitrogen sheath gas and auxiliary gas flow rates were 30 and 25 arbitrary units (corresponding to approximately 0.45 and 7.5 L/min according to the manufacturer’s specification), respectively. Collision-induced dissociation was performed with argon at 1.5 mTorr pressure. Selected reaction monitoring (SRM) with unit mass resolution for the precursor and product ions was used for quantification (Table S-4 in the Supporting Information). Tube lens voltage was optimized for MDA–DNPH and HNE–DNPH in positive and negative mode, respectively. The optimal collision energy determined for each hydrazone was obtained by direct infusion experiments. Data acquisition and processing were carried out by the Xcalibur software (version 2.1).
Direct infusion and isocratic (acetonitrile/water/acetic acid, 50:50:0.1, v/v; all other LC parameters are specified above in the previous section) RPLC–MS scan and MS/MS product ion scan (Figure S-1 in the Supporting Information) analysis was applied for the confirmation of identity and for assessment of purity of the labeled DNPHs. We estimated that the purity was >98% after preparative TLC purification for each reagent.
Animals and Tissue Collection
Ovariectomized CD-1 mice were purchased from Harlan Laboratories (Indianapolis, IN, U.S.A.). All procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the UNT Health Science Center. Animals were euthanized by intraperitoneal administration of 60 mg/kg ketamine and 10 mg/kg xylazine. Brain and liver were collected and processed immediately.
Sample Preparation for Quantification
Tissue extracts were prepared in a BeadBug microtube homogenizer D1030 (Benchmark Scientific, Edison, NJ, U.S.A.). Brain and liver (100 mg) were mixed with 400 μL of acetonitrile/water (3:1, v/v) containing 25 μg/mL BHT, a strong phenolic antioxidant, in a 2 mL homogenizer tube prefilled with zirconium beads (1.5 mm i.d.) and homogenized for 3 min at 3300 rpm. The tissue homogenate was centrifuged (5 min at 14 500 rpm), and the clean supernatant was separated. It was then spiked with different amounts (ranging from 20 to 2000 ng/g fresh tissue) of MDA, ACR, and HNE to obtain quality control (QC) samples for quantification according to the AIDA protocol.24 Briefly, to a 100 μL aliquot of a QC sample 100 μL of light or heavy derivatizing stock solution was added. The reaction was carried out as specified above. ICD standards (i.e., heavy- and light-labeled DNPH–aldehyde mixtures) were prepared in 100 μL of charcoal stripped human serum precipitated with 300 μL acetonitrile containing BHT (25 μg/mL). After centrifugation, the clear supernatant was spiked with 200 ng of MDA, 100 ng of ACR, and 100 ng of HNE and derivatized as specified above. After derivatization, the samples were diluted with 800 μL of acetonitrile. An amount of 200 μL of heavy-labeled ICD standard was then added to 200 μL of light-labeled QC sample, and 200 μL of light-labeled ICD standard was added to 200 μL of heavy-labeled QC sample. Next, the resultant mixtures were diluted with 1.6 mL water and subjected to solid-phase extraction (SPE). Briefly, Supelclean LC-18 SPE cartridges (1 mL, 100 mg; Sigma-Aldrich, St. Louis, MO, U.S.A.) were conditioned with 1 mL of acetonitrile, then with 1 mL of 0.1% formic acid in water. After sample loading, the cartridges were washed with 2 × 1 mL of water/acetonitrile/formic acid (70:30:0.1, v/v), and the hydrazones were then eluted with 2 × 200 μL of acetonitrile. The effluents were dried at room temperature in an Eppendorf Vacufuge (Hauppauge, NY, U.S.A.) and reconstituted in 50 μL of 50% acetonitrile for subsequent LC–MS/MS analysis. All samples were analyzed within 3 days.24 No aldehydes were detected in the unspiked blank QC samples; therefore, blank correction24 was not applied in AIDA calculations.
Statistical Analysis
Statistical evaluations were carried out with the XLSTAT (version 2014.1.01) and the Primer of Biostatistics (version 5.0; McGraw-Hill, New York, NY, U.S.A.) software. Statistical differences between means of data sets were determined with one-way ANOVA at p < 0.05. Statistical interpretation of the method comparison test results was achieved following the guidelines proposed in previous reports.27−29 Linear regression lines were obtained by using the Deming regression model.28 Percentile differences were visualized in Bland–Altman plots.27 Normal distribution of the data was confirmed with the Shapiro–Wilk test.
Results and Discussion
Multivariate approaches employed by screening experimental designs25,26 have several advantages over the classically applied one-variable-at-a-time methodologies, where only one factor at a time is changed. Specifically, screening experimental designs calculate the influence of a given factor at several levels through combinations with other factors, which permits general conclusions.25,26,30 Moreover, they permit the evaluation of relatively large number of factors in a relatively small number of experiments, and thus, the strategy can be used in high-throughput assays.
First, we employed asymmetrical screening experimental design (Supporting Information Table S-2) using factor nos. 1–6 from Supporting Information Table S-1. The levels were chosen to cover a wide range of conditions applied in practical gradient RPLC separations.21 Four biologically relevant reactive aldehydes (ACR, MDA, HNE, and ONE) were selected as model compounds for these studies, and ICD involved conversion to hydrazones with unlabeled (“light”) and various heavy (d3-, 15N2-, 15N4-, or 13C6-) labeled DNPH. The derivatized MDA represented an early eluting compound, retention of ACR–DNPH was intermediate, while the derivatized HNE and ONE were late-eluting compounds upon simultaneously analyzing these model aldehydes by gradient RPLC. As shown in Table 2 and Figure 2, both statistical and graphical data interpretation revealed that only deuterium isotope substitution had a significant effect on the retention time shifts, with the exception of MDA. The latter may be due to the poor retention (apparent retention factor, kapp < 4) of the first-eluting MDA–DNPH under the chromatographic conditions applied. The calculated absolute effect (0.302) was slightly larger than the margin of error (ME = 0.301) for HNE, when the 2 and 8 min gradients were compared. At the same time, it was smaller than the simultaneous margin of error (SME = 0.490). Since there is an increased chance for false positive decisions using ME and significance was not confirmed by other statistical and graphical interpretation methods, this effect may only be considered as “possibly significant” according to Vander Heyden et al.26 Nevertheless, the results obtained by our asymmetrical design experiment (Table 2) pointed out that neither 13C nor 15N labeling had a significant effect on retention time difference; only deuterium isotope substitution ended up as a significant factor. However, two-factor or higher-order interaction effects can confound the main effects, and therefore, individual RPLC factors affecting chromatographic deuterium isotope effect may be underestimated by the approach. To overcome this potential problem, the asymmetric design was deconstructed and a Plackett–Burman design was adopted using only light- and d3-DNPH-derived hydrazones. Results of this method are listed in Table 3 and plotted in Figures 3 and 4.
Table 2. Absolute Effects of Various Factors Calculated from the Retention Time Shift between Light and Heavy Pairs of DNPH Derivatives Obtained from the Asymmetrical Experimental Designa.
| factor | MDA | ACR | HNE | ONE |
|---|---|---|---|---|
| isotope labeling | ||||
| d3-15N4 | 0.539 | 1.099b,c,d | 0.697b,c,d | 0.947b,c,d |
| d3-15N2 | 0.619 | 1.000b,c,d | 0.746b,c,d | 1.044b,c,d |
| d3-13C6 | 0.659 | 1.025b,c,d | 0.669b,c,d | 1.020b,c,d |
| 15N4-15N2 | 0.081 | 0.098 | 0.049 | 0.098 |
| 15N4-15C6 | 0.120 | 0.074 | 0.028 | 0.073 |
| 15N2-13C6 | 0.039 | 0.024 | 0.077 | 0.025 |
| temperature | ||||
| 25–50 °C | 0.479 | 0.344 | 0.149 | 0.248 |
| 25–40 °C | 0.550 | 0.382 | 0.188 | 0.284 |
| 40–50 °C | 0.071 | 0.039 | 0.039 | 0.036 |
| gradient time | ||||
| 2–4 min | 0.010 | 0.137 | 0.087 | 0.086 |
| 2–8 min | 0.480 | 0.396 | 0.302e | 0.395 |
| 4–8 min | 0.470 | 0.260 | 0.215 | 0.309 |
| stationary phase | 0.443 | 0.062 | 0.114 | 0.112 |
| pH | 0.143 | 0.112 | 0.063 | 0.209 |
| organic solvent | 0.290 | 0.186 | 0.086 | 0.185 |
| dummies | ||||
| dummy 1 | 0.191 | 0.135 | 0.086 | 0.136 |
| dummy 2 | 0.250 | 0.087 | 0.189 | 0.136 |
| dummy 3 | 0.190 | 0.187 | 0.135 | 0.186 |
| critical effects | ||||
| Ecritical | 0.675 | 0.452 | 0.455 | 0.491 |
| ME | 0.791 | 0.439 | 0.301 | 0.419 |
| SME | 1.300 | 0.715 | 0.490 | 0.683 |
Critical effects were estimated according to Vander Heyden et al. (ref (26)).
Significant effect compared to Ecritical.
Significant effect compared to simultaneous margin of error (SME).
Significant effects from the half-probability normal plot.
Possibly significant effect compared to margin of error (ME).
Figure 2.

Half-normal probability plots of the absolute effects on the retention time difference measured between light and heavy labeled hydrazones in the asymmetrical design with identification of margin of error (ME) and simultaneous margin of error (SME) as critical effects. A, B, and C factors denote stable isotope labeling, column temperature, and gradient time, respectively.
Table 3. Absolute Effects of Various Factors on the Retention Time Difference between Light and d3-Labeled DNPH Derivatives Obtained from the Plackett–Burman Designa.
| factor | MDA | ACR | HNE | ONE |
|---|---|---|---|---|
| temperature | 0.376 | 0.366 | 0.131 | 0.189 |
| gradient time | 0.106 | 0.436 | 0.464b,c,d | 0.655b,d,e |
| stationary phase | 0.589 | 0.098 | 0.167b | 0.153 |
| pH | 0.494 | 0.032 | 0.162b | 0.224 |
| organic solvent | 0.392 | 0.560b | 0.065 | 0.189 |
| analyte concn | 0.249 | 0.299 | 0.228b | 0.287b |
| dummies | ||||
| dummy 1 | 0.310 | 0.161 | 0.067 | 0.009 |
| dummy 2 | 0.380 | 0.300 | 0.030 | 0.022 |
| dummy 3 | 0.036 | 0.168 | 0.073 | 0.189 |
| dummy 4 | 0.450 | 0.168 | 0.066 | 0.048 |
| dummy 5 | 0.319 | 0.164 | 0.032 | 0.087 |
| critical effects | ||||
| Ecritical | 0.849 | 0.513 | 0.146 | 0.247 |
| ME | 0.758 | 0.642 | 0.267 | 0.556 |
| SME | 1.214 | 1.032 | 0.427 | 0.894 |
Critical effects were estimated according to Vander Heyden et al. (ref (26)).
Significant effect compared to Ecritical.
Significant effect compared to simultaneous margin of error (SME).
Significant effects from the half-probability normal plot.
Possibly significant effect compared to margin of error (ME).
Figure 3.

Half-normal probability plots of the absolute effects on the retention time difference measured between light and d3-labeled aldehyde hydrazones in the Plackett–Burman experimental design with identification of margin of error (ME) and simultaneous margin of error (SME) as critical effects. C factor denotes gradient time.
Figure 4.
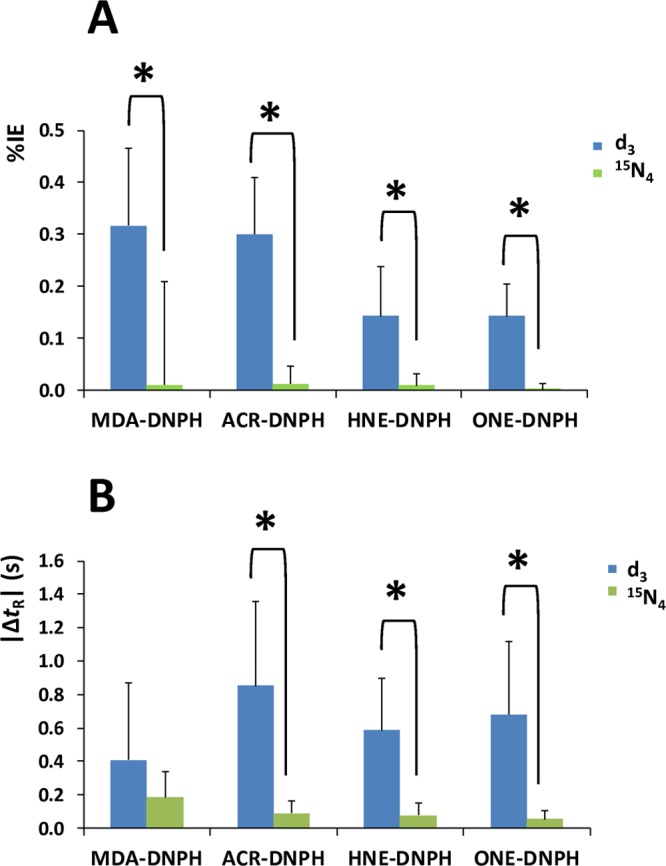
Mean (A) percentile single isotope effect (%IE) and (B) absolute retention time differences (|ΔtR|) obtained by the Plackett–Burman experimental designs. Error bars represent standard deviation; asterisks (∗) indicate statistically significant difference determined by one-way ANOVA (p < 0.05).
In agreement with findings concluded from the asymmetric experimental plan (Table 2), no significant effects were identified for MDA–DNPH (Table 3) with the Plackett–Burman approach. For ACR, we found only one factor (the organic modifier in the mobile phase) that had significant effect on the retention time difference between the ACR–DNPH and d3-ACR–DNPH upon comparing the absolute effect (0.560) with critical effect derived from negligible factor effects (Ecritical = 0.513). On the other hand, the gradient time had significant effect for the two late-eluting analytes (HNE–DNPH and ONE–DNPH). All three data interpretation methods (i.e., comparison to Ecritical and SME, as well as inspection of the half-normal probability plot) confirmed this finding for the HNE derivative (Table 3), while for ONE–DNPH only “possibly significant” effect can be concluded in terms of SME. As such, the isotope effect can be significantly reduced for these compounds by applying a steeper gradient slope (data not shown). It is interesting to note that the analyte’s concentration also had significant effect, when the absolute effects (0.228 and 0.287) were compared to the corresponding Ecritical values (0.146 and 0.247) for HNE–DNPH and ONE–DNPH, respectively. Although the significance of analyte concentration was not confirmed by alternative interpretation methods, such as the considering SME or the visual inspection of the half-normal probability plots (Figure 3), this finding may need to be considered in terms of accuracy and precision of quantitation. Additionally, small but significant effects of the stationary phase and the pH of the mobile phase were also revealed for HNE–DNPH by one of interpretation methods we employed (Table 3).
To validate findings summarized in Table 3, we repeated the Plackett–Burman design using [15N4]DNPH. This setup, however, did not yield any statistically significant main effects on chromatographic isotope effect (Table S-5 and Figure S-2 in the Supporting Information). As expected, the use of [15N4]DNPH brought about a statistically significant decrease in percentile single isotope effect (%IE, Figure 4A) compared to d3 labeling, along with a significant decrease in the retention time difference (ΔtR) for all aldehydes except for MDA (Figure 4B). The observed large standard deviation for the d3-labeled MDA derivative suggests that this compound is more sensitive to changes in RPLC conditions than the later-eluting hydrazones; we found a strong linear correlation (R2 = 0.9178) between kapp and ΔtR (Figure S-3 in the Supporting Information). Accordingly, negligible ΔtR can only be achieved for MDA–DNPH when kapp < 3, while kapp ≥ 3 is recommended for bioanalytical LC–MS applications.21
Taken together, the only way to eliminate universally the retention time difference between light and heavy isotopologues is when ICD applies reagents labeled on heavy atoms (C, N, O, etc.). 15N4-labeled DNPH could be safely used for correcting LC–ESI-MS detector response under a wide range of RPLC conditions without the risk of manifesting significant ΔtR. Therefore, we propose the modification of the AIDA protocol24 to replace d3-DNPH with 15N4- or 13C6-DNPH for the derivatization of carbonyls.
Results, such as critical resolution, analysis time, and detector response obtained by the asymmetrical design experiment, were also used to select the optimal RPLC conditions21 for the reliable and fast simultaneous analysis of the chosen model aldehydes in mixtures. Satisfactory separation of HNE–DNPH and ONE–DNPH was a very important aspect of method optimization. The molecular masses of these hydrazones differ only by 2 Da, and such a small difference can cause significant cross-talk effect if these peaks overlap. As shown in Figure S-4 in the Supporting Information, we achieved acceptable resolution (Rs ≥ 2)21 by using acetonitrile as organic mobile phase, while the pH of the aqueous phase and the stationary phase chemistry had only little effect. On the other hand, acceptable separation was not obtained at all when methanol was used. Moreover, acetonitrile considerably decreased the analysis time, consequently increasing assay throughput. The detector sensitivity was dramatically influenced by the mobile phase pH. As expected, MDA–DNPH response significantly decreased upon high pH (5 mM ammonium acetate at pH 8.2) chromatography, when positive ion formation is not favored. The other aldehyde derivatives produced abundant negative ions during ESI; thus, negative ionization mode is preferred for the monitoring of these species.24,31,32 Contrary to our expectation, high pH decreased the signal even in the negative ion mode. This unique phenomenon, which is known as the “wrong-way-round ionization,”18,19 is actually advantageous, since efficient ionization can be obtained using both polarity modes within one assay.
On the basis of the results obtained by the asymmetric screening experiment, we propose an LC–MS/MS method for quantification of MDA, ACR, HNE, and ONE by ICD. We suggest gradient separation to be performed using PhenHex column with low pH (0.1% acetic acid at pH 3.3) aqueous phase and acetonitrile as the organic modifier. When the organic content of the initial mobile phase is reduced to 40%, adequate retention (kapp ≥ 3) for the first-eluting MDA–DNPH can be achieved. Gradient time of 4 min and column temperature set at 30 °C also allow for the excellent separation of the critical peak pair (RsHNE–DNPH/ONE–DNPH = 3.3) within only 3.2 min (Figure S-5 in the Supporting Information).
In order to reveal potential accuracy differences, a series of sample pairs were quantified based on this optimized LC–MS/MS assay using both the previously used d3-DNPH24 and our own [15N4]DNPH reagent14 in AIDA. To compare the proposed modification to the original method,24 we used mouse brain and liver homogenates as biological matrixes to generate QC samples focusing on the quantification of selected LPO end-products. The accumulation of these compounds in various human organs and body fluids has been linked to major tissue and cellular dysfunction believed to play a role in aging and most age- and oxidative stress-related diseases.33 However, high lipid content make the brain and liver, besides exposing them to well-documented LPO-related pathology34−36 and thereby putting AIDA in the context of application to high-relevance biological studies, ideal as complex matrixes for a comparative evaluation of the previously developed method and the modification we propose herein. Because we did not detect ONE upon screening the chosen mouse tissue homogenates by LC–MS/MS using DNPH derivatization, therefore, we conducted this evaluation with MDA, ACR and HNE as analytes. Leaving out ONE also avoided addressing its potential conversion to E-1-hydroxynon-2-en-4-one in biological samples,37 which was beyond the scope of our work primarily focusing on chromatographic isotope effects and its potential impact on bioanalyses using ICD followed by LC–MS/MS.
The results obtained by the two labeling approaches that relied on d3-DNPH24 and [15N4]DNPH,14 respectively, as reagents are visualized on scatterplots in Figure S-6 (Supporting Information). The statistically significant differences in the slopes of Deming regression lines from the lines of equality indicate that a proportional error exists between the data generated by the use of d3-DNPH versus [15N4]DNPH. The confidence intervals for the biases and limits of agreement (±1.96 SD) could be estimated, because the percentile differences followed normal distribution (data not shown). These data are included in the Bland–Altman plots shown in Figure 5. The measurement bias values were calculated as 3.0%, −1.3%, and 0.4% for MDA, ACR, and HNE, respectively. The 95% confidence intervals for the bias of these aldehydes also include zero; therefore, measurement biases were not significant. The limits of agreement show whether 95% of the differences would lie between the limits, if the differences were normally distributed. These intervals ranged from −13% to 19%, −23% to 21%, and −9% to 10% for MDA, ACR, and HNE, respectively (Figure 5). Accordingly, the two methods in both matrixes afforded similar quantitative results only for HNE. For the quantification of MDA and ACR, the limit of agreement values exceeded the maximum tolerable inaccuracy (±15%).38,39 Therefore, these methods may not be considered equivalent or interchangeable for the quantitative analysis of these aldehydes by AIDA from the given matrixes.
Figure 5.
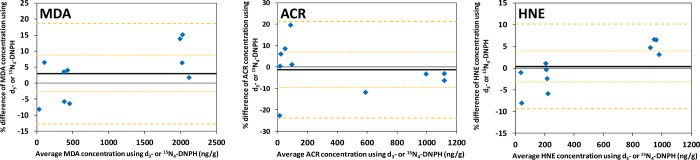
Bland–Altman plots of quantities of the selected aldehydes measured in fortified mouse tissue extracts by AIDA using d3 vs 15N4 labeling. Bold solid lines show the measurement bias between the two methods, dotted lines represent the 95% confidence limits of the bias, and the dashed lines indicate the limits of agreement values.
Conclusions
The synthesis of DNPH is relatively simple. Isotope labeling can be customized, because several different stable isotope atoms (such as d, 13C, 15N, and/or 18O) can be incorporated in various numbers, even at specific positions, into the structure of this carbonyl-specific reagent. Therefore, it provided a convenient model for comprehensive assessment of chromatographic isotope effect, when AIDA is used as a typical ICD to analyze a subset of the metabolome (specifically, the LPO end-products that possess at least one carbonyl group) using LC–MS/MS.
The main finding of this study, relying for the first time on experimental screening designs to evaluate chromatographic isotope effects, is that the most effective approach to avoid a chromatographic isotope effect is through the use of non-deuterium-labeled reagents in AIDA. When AIDA relies on deuterium-labeled reagents, one has limited options to reduce this isotope effect universally. A steep gradient program appeared to be the only operational parameter that reliably decreased the retention time difference between the unlabeled and the d3-labeled hydrazones for the two late-eluting aldehydes (HNE and ONE) selected for our study. On the other hand, adequate separation can be impaired seriously by such conditions, which can result in the loss of specificity for isobaric derivatives due to cross-talk effects. A comparison of our model ICD-based quantification using d3 or 15N4 labeling has demonstrated unacceptable inaccuracy for MDA and ACR, even without measuring significant bias. Therefore, we recommend the modification of the AIDA protocol24 by including either [15N4]- or [13C6]DNPH instead of the originally proposed d3-DNPH to abolish chromatographic isotope effects and their potential unfavorable consequences on quantitative measurements.
Acknowledgments
We thank Dr. Vien Nguyen and Mr. Kurt Kamrowski for their excellent technical assistance. This work was supported in part by the National Institutes of Health (Grant No. AG025384) and the Robert A. Welch Foundation (endowment BK-0031 to L.P.).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Matuszewski B. K.; Constanzer M. L.; Chavez-Eng C. M. Anal. Chem. 2003, 75, 3019–3030. [DOI] [PubMed] [Google Scholar]
- Taylor P. J. Clin. Biochem. 2005, 38, 328–334. [DOI] [PubMed] [Google Scholar]
- Bakhtiar R.; Majumdar T. K. J. Pharmacol. Toxicol. Methods 2007, 55, 227–243. [DOI] [PubMed] [Google Scholar]
- Hewavitharana A. K. J. Chromatogr., A 2011, 1218, 359–361. [DOI] [PubMed] [Google Scholar]
- Wade D. Chem.-Biol. Interact. 1999, 117, 191–217. [DOI] [PubMed] [Google Scholar]
- Filer C. N. J. Labelled Compd. Radiopharm. 1999, 42, 169–197. [Google Scholar]
- Turowski M.; Yamakawa N.; Meller J.; Kimata K.; Ikegami T.; Hosoya K.; Tanaka N.; Thornton E. R. J. Am. Chem. Soc. 2003, 125, 13836–13849. [DOI] [PubMed] [Google Scholar]
- Wieling J. Chromatographia 2002, 55, S107–S113. [Google Scholar]
- Jemal M.; Schuster A.; Whigan D. B. Rapid Commun. Mass Spectrom. 2003, 17, 1723–1734. [DOI] [PubMed] [Google Scholar]
- Wang S.; Cyronak M.; Yang E. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [DOI] [PubMed] [Google Scholar]
- Lindegardh N.; Annerberg A.; White N. J.; Day N. P. J. J. Chromatogr., B 2008, 862, 227–236. [DOI] [PubMed] [Google Scholar]
- Berg T.; Strand D. H. J. Chromatogr., A 2011, 1218, 9366–9374. [DOI] [PubMed] [Google Scholar]
- Lee S.; Kim B.; Kim J. J. Chromatogr., A 2013, 1277, 35–41. [DOI] [PubMed] [Google Scholar]
- Prokai L.; Szarka S.; Wang X.; Prokai-Tatrai K. J. Chromatogr., A 2012, 1232, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szarka S.; Nguyen V.; Prokai L.; Prokai-Tatrai K. Anal. Bioanal. Chem. 2013, 405, 3399–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Antuña A.; Domínguez-Romero J. C.; García-Reyes J. F.; Rodríguez-González P.; Centineo G.; Alonso J. I. G.; Molina-Díaz A. J. Chromatogr., A 2013, 1288, 40–47. [DOI] [PubMed] [Google Scholar]
- Fabregat-Cabello N.; Castillo Á.; Sancho J. V.; González F. V.; Roig-Navarro A. F. J. Chromatogr., A 2013, 1301, 19–26. [DOI] [PubMed] [Google Scholar]
- Kostiainen R.; Kauppila T. J. J. Chromatogr., A 2009, 1216, 685–699. [DOI] [PubMed] [Google Scholar]
- Thurman E. M.; Ferrer I.; Barceló D. Anal. Chem. 2001, 73, 5441–5449. [DOI] [PubMed] [Google Scholar]
- Valleix A.; Carrat S.; Caussignac C.; Léonce E.; Tchapla A. J. Chromatogr., A 2006, 1116, 109–126. [DOI] [PubMed] [Google Scholar]
- Snyder L. R.; Kirkland J. J.; Dolan J. W.. Introduction to Modern Liquid Chromatography, 3rd ed.; Wiley: Hoboken, NJ, 2010. [Google Scholar]
- Toyo’oka T. J. Pharm. Biomed. Anal. 2012, 69, 174–184. [DOI] [PubMed] [Google Scholar]
- Bruheim P.; Kvitvang H. F. N.; Villas-Boas S. G. J. Chromatogr., A 2013, 1296, 196–203. [DOI] [PubMed] [Google Scholar]
- Manini P.; Andreoli R.; Sforza S.; Dall’Asta C.; Galaverna G.; Mutti A.; Niessen W. M. A. J. Chromatogr., B 2010, 78, 2616–2622. [DOI] [PubMed] [Google Scholar]
- Hund E.; Vander Heyden Y.; Haustein M.; Massart D. L.; Smeyers-Verbeke J. J. Chromatogr., A 2000, 874, 167–185. [DOI] [PubMed] [Google Scholar]
- Vander Heyden Y.; Nijhuis A.; Smeyers-Verbeke J.; Vandeginste B. G. M.; Massart D. L. J. Pharm. Biomed. Anal. 2001, 24, 723. [DOI] [PubMed] [Google Scholar]
- Altman D. G.; Bland J. M. Statistician 1983, 32, 307–317. [Google Scholar]
- Linnet K. Clin. Chem. 1999, 45, 882–894. [PubMed] [Google Scholar]
- Hanneman S. K. AACN Adv. Crit. Care 2008, 19, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejaegher B.; Vander Heyden Y. J. Pharm. Biomed. Anal. 2011, 56, 141–158. [DOI] [PubMed] [Google Scholar]
- Andreoli R.; Manini P.; Corradi M.; Mutti A.; Niessen W. M. A. Rapid Commun. Mass Spectrom. 2003, 17, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Salomon R. G. Free Radical Biol. Med. 2012, 52, 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre-Salvayre A.; Auge N.; Ayala V.; Basaga H.; Boada J.; Brenke R.; Chapple S.; Cohen G.; Feher J.; Grune T.; Lengyel G.; Mann G. E.; Pamplona R.; Poli G.; Portero-Otin M.; Riahi Y.; Salvayre R.; Sasson S.; Serrano J.; Shamni O.; Siems W.; Siow R. C. M.; Wiswedel I.; Zarkovic K.; Zarkovic N. Free Radical Res. 2010, 44, 1125–1171. [DOI] [PubMed] [Google Scholar]
- Adibhatla R. M.; Hatcher J. F. Antioxid. Redox Signaling 2010, 12, 125–169. [DOI] [PubMed] [Google Scholar]
- Macdonald G. A.; Bridle K. R.; Ward P. J.; Walker N. I.; Houglum K.; George D. K.; Smith J. L.; Powell L. W.; Crawford D. H. G.; Ramm G. A. J. Gastroenterol. Hepatol. 2001, 16, 599–606. [DOI] [PubMed] [Google Scholar]
- Guo J.; Prokai-Tatrai K.; Nguyen V.; Rauniyar N.; Ughy B.; Prokai L. J. Proteomics 2011, 74, 2370–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnke M. M.; Wanigasekara E.; Singhal S. S.; Singhal J.; Awasthi S.; Armstrong D. W. Anal. Bioanal. Chem. 2008, 392, 1325–1333. [DOI] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services; FDA; CDER; CVM . Guidance for Industry: Bioanalytical Method Validation; U.S. Food and Drug Administration: Rockville, MD, 2001. [Google Scholar]
- EMEA; CHMP; EWP . Guideline on Bioanalytical Method Validation; EMEA: London, 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.