

Abstract
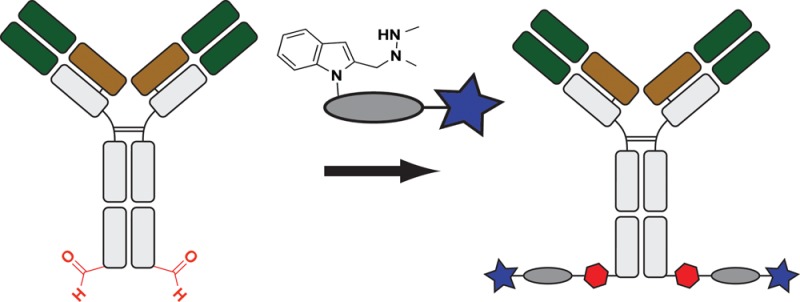
It is becoming increasingly clear that site-specific conjugation offers significant advantages over conventional conjugation chemistries used to make antibody–drug conjugates (ADCs). Site-specific payload placement allows for control over both the drug-to-antibody ratio (DAR) and the conjugation site, both of which play an important role in governing the pharmacokinetics (PK), disposition, and efficacy of the ADC. In addition to the DAR and site of conjugation, linker composition also plays an important role in the properties of an ADC. We have previously reported a novel site-specific conjugation platform comprising linker payloads designed to selectively react with site-specifically engineered aldehyde tags on an antibody backbone. This chemistry results in a stable C–C bond between the antibody and the cytotoxin payload, providing a uniquely stable connection with respect to the other linker chemistries used to generate ADCs. The flexibility and versatility of the aldehyde tag conjugation platform has enabled us to undertake a systematic evaluation of the impact of conjugation site and linker composition on ADC properties. Here, we describe the production and characterization of a panel of ADCs bearing the aldehyde tag at different locations on an IgG1 backbone conjugated using Hydrazino-iso-Pictet-Spengler (HIPS) chemistry. We demonstrate that in a panel of ADCs with aldehyde tags at different locations, the site of conjugation has a dramatic impact on in vivo efficacy and pharmacokinetic behavior in rodents; this advantage translates to an improved safety profile in rats as compared to a conventional lysine conjugate.
Introduction
Conceptually, antibody–drug conjugates (ADCs) represent an elegant solution to the problems of systemic toxicity encountered during treatment with conventional chemotherapeutics.1 By linking a cytotoxic payload to an antibody that specifically recognizes antigens expressed on target cells, one could, in principle, achieve potent efficacy without unwanted side effects. However, in practice, translating this idea into clinical treatments has been difficult, as exemplified by the fact that it has been more than 30 years since cytotoxins were first conjugated to antibodies, and yet to date only three ADCs have successfully cleared the FDA approval process. In spite of the selective targeting potential of the antibody, systemic toxicity remains one of the key challenges encountered during ADC development. Indeed, Mylotarg, the first ADC to win FDA approval, was voluntarily pulled from the market after 11 years due to safety concerns. Nonetheless, the field has made significant progress, garnering two recent FDA approvals for Adcetris (brentuximab vedotin) in 2011 and Kadcyla (ado-trastuzumab emtansine) in 2013. Furthermore, the ADC drug development pipeline has exploded, with more than 30 conjugates currently in clinical trials.2
The first-generation ADCs, including Kadcyla and Adcetris, are produced using nonselective conjugation chemistries that exploit the reactivities of native lysines or cysteines, respectively.3 The ADCs made by these methods are characterized by extreme heterogeneity at the molecular level. Multiple surface-accessible reactive residues result in a spectrum of conjugation sites and ADCs with drug-to-antibody ratios (DARs) ranging from 0 to 8. The products are typically assessed in aggregate, with the measured efficacy, pharmacokinetic (PK), and toxicity values describing the mean of the population. This heterogeneity manifests across all stages of ADC development as complex mixtures of ADC isoforms plague analytical characterization, individual ADC batches can be difficult to reproduce, and, most importantly, an ADC with heterogeneous DAR populations results in suboptimal pharmacological profiles.
The importance of DAR in defining the properties of an ADC has been highlighted in experiments where conventional ADC mixtures were separated into subpopulations of defined DARs and their biological properties were analyzed. These experiments have shown that drug loading plays a dominant role in PK, efficacy, and the therapeutic index.4 Specifically, ADCs with higher DARs (e.g., DAR 8) were cleared faster and demonstrated reduced efficacy, in spite of higher drug loading, as compared to ADCs with lower DARs (e.g., DAR 2).4 As a result, the past few years have seen the emergence of site-specific conjugation methods for making ADCs that promise to produce homogeneous products with controlled drug loading, simplified analytics, and, most importantly, an improved therapeutic index. In principle, these methods rely on introducing reactive amino acids at defined locations on the antibody backbone and using chemical or enzymatic approaches to exploit the unique reactivity of these amino acids to conjugate payloads.5−8 We have pioneered a novel chemoenzymatic approach to the site-selective modification of proteins that uses the naturally occurring formylglycine-generating enzyme (FGE) to introduce a formylglycine (fGly) residue into protein backbones. We have designed modular linker systems that selectively react with the aldehyde side chain of fGly to form a stable C–C bond with the protein. Here, we demonstrate the application of this method to create site-specifically conjugated ADCs with controlled stoichiometry and clean analytics that permit structure–activity relationship (SAR) studies of ADCs at the conjugate level that enable the design of ADCs with excellent potency, minimal toxicity, and wide therapeutic windows.
Our platform for site-specific ADC production is built upon the incorporation of formylglycine (fGly), a non-natural amino acid, into the protein sequence. To install fGly (Figure 1), a short consensus sequence, CXPXR—where X is usually serine, threonine, alanine, or glycine—is inserted at the desired location in the conserved regions of antibody heavy or light chains using standard molecular biology cloning techniques.9 This “tagged” construct is produced recombinantly in cells that coexpress the formylglycine-generating enzyme (FGE), which cotranslationally converts the cysteine within the tag into an fGly residue, generating an antibody expressed with two aldehyde tags per molecule.10 The aldehyde functional group serves as a chemical handle for bioorthogonal conjugation.11 We developed the Hydrazino-iso-Pictet-Spengler (HIPS) ligation to connect the payload to fGly, resulting in the formation of a stable, covalent C–C bond between the cytotoxin payload and the antibody.12 This C–C bond is expected to be stable to physiologically relevant challenges encountered by the ADC during circulation and FcRn recycling, e.g., proteases, low pH, and reducing reagents. Due to the modular nature of our platform, we can produce antibodies bearing the aldehyde tag at a variety of locations, enabling us to empirically discover ideal conjugation sites. In this work, we tested the effects of inserting the aldehyde tag at one site in the light chain and seven sites in the heavy chain. We present in-depth biophysical and functional characterization of three of the resulting ADCs made by conjugation to maytansine payloads via a HIPS linker.6,13,14 We observed that HIPS conjugation produces physiologically stable conjugates; however, modulating the conjugation site had a pronounced effect on antibody efficacy and PK, and resulted in ADCs with an improved safety profile as compared to a conventional lysine-conjugated ADC.
Figure 1.

Aldehyde tag coupled with HIPS chemistry yields site-specifically modified antibodies carrying a payload attached through a stable C–C bond. (A) A formylglycine-generating enzyme (FGE) recognition sequence is inserted at the desired location along the antibody backbone using standard molecular biology techniques. Upon expression, FGE, which is endogenous to eukaryotic cells, catalyzes the conversion of the Cys within the consensus sequence to a formylglycine residue (fGly). (B) Antibodies carrying aldehyde moieties (in red, 2 per antibody) are reacted with a Hydrazino-iso-Pictet-Spengler (HIPS) linker and payload to generate a site-specifically conjugated ADC. (C) The HIPS chemistry proceeds through an intermediate hydrazonium ion followed by intramolecular alkylation with a nucleophilic indole to generate a stable C–C bond.
Results and Discussion
Development and Initial Screening of an Antibody Tag-Placement Library
As a first step toward generating a library of antibodies carrying the aldehyde tag at various locations, we surveyed the human IgG1 crystal structure15 to identify exposed, relatively unstructured areas within the heavy and light chain constant regions. Our design principle was to install the tag at locations that minimally perturbed the native IgG structure but remained accessible for conjugation. We incorporated each tag once into either the heavy or light chain, such that each antibody would bear two aldehyde groups. Specifically, we chose one internal site in the light chain, one at the C-terminus and seven internal sites (three in the CH1, two in the CH2, one in the CH3 domains) of the heavy chain (Figure 2, top). For the purpose of this discussion we have represented these heavy chain sites in alphabetical order according to their occurrence from N- to C-terminus (Tags A-G), while the light chain tag is designated as “LC”. The selected tag sites were cloned into the constant regions of a prototype human IgG1 heavy chain and kappa light chain. Proteins were produced transiently in bulk pools of cells overexpressing human FGE to ensure efficient conversion of Cys to fGly within the consensus sequence and resulting tagged antibodies were purified using Protein A affinity columns and stored in PBS.
Figure 2.
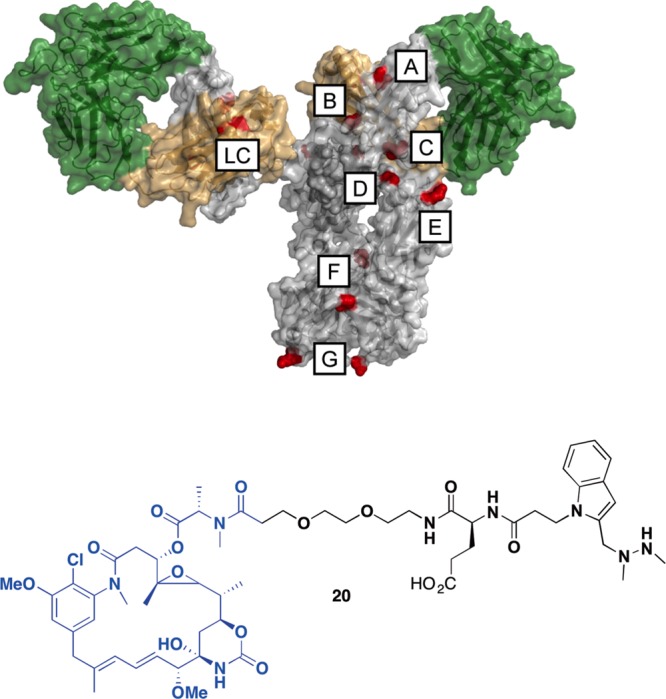
Together, the aldehyde tag and HIPS chemistry allow for stable cytotoxic payload conjugation at precise locations across the antibody surface. (Top) We inserted the aldehyde tag (red) at one location in the light chain (LC) and seven locations (labeled A–G) in the heavy chain. Antibodies bearing these tags were produced and analyzed as the first step in making ADCs conjugated at different sites. (Bottom) HIPS-Glu-PEG2-maytansine 20 served as the linker (in black) and the cytotoxic payload (in blue) for ADCs used in these studies.
We tested the propensity for immunogenicity of our aldehyde tagged antibodies by in silico analysis in order to assess whether they could be viable as ADCs. Specifically, we used both known MHC class II peptide binding motifs (iTope) and previously identified immunogenic sequences (TCED) to identify peptides that may bind promiscuously in a number of MHC contexts with high and moderate affinity.16,17 Of the eight aldehyde tag placements that we screened, only one (Tag B) was determined to generate peptides that were likely to be immunogenic. Next, to address the effect of tag placement on antibody stability, we looked at aggregation. By size-exclusion chromatography (SEC) it was apparent that out of the eight tagged antibodies that we screened, six exhibited no to very little aggregation (Table 1). Two antibodies, containing Tags D and F, in the CH2 and CH3 domains, respectively, demonstrated significant aggregation.
Table 1. Aldehyde Tag Is Well-Tolerated when Inserted into a Variety of Locations along the Antibody Backbone.
| tag designation | tag domain | residues bordering taga | % aggregation |
|---|---|---|---|
| A | CH1 | G118, V121 | 0 |
| B | CH1 | P123, S128 | 0 |
| C | CH1 | A165, G169 | 2.3 |
| D | CH2 | D283, E285 | 31.5 |
| E | CH2 | N344, A349 | 4.5 |
| F | CH3 | G361, E366 | 76 |
| G | C-terminus | K478 | 7 |
| LC | LC | A153, Q155 | 0 |
Kabat numbering.
We decided to focus our attention on generating ADCs with a representative panel of tag sites using a well-characterized ADC reference made using conventional conjugation that could serve as a good comparator for our studies. We decided to focus on Her2-targeting ADCs and generate site-specific ADCs that were equivalent to T-DM1 (Kadcyla), in terms of the underlying antibody (trastuzumab) and the cytotoxin payload (maytansine).18 We chose this system because T-DM1 is a clinically approved drug, with good preclinical models and literature benchmarks.19,20 Thus, we expressed trastuzumab, the antibody component of T-DM1, with aldehyde tags at the LC, Tag C, or Tag G positions, which represent conjugation sites distributed across antibody domains. These tag placements produced antibodies with high titers, had low aggregation, and underwent facile conjugation to produce well-behaved ADCs, as determined by chromatographic analysis, as well as biophysical and functional tests. In addition, we generated an ADC (α-HER2-DM1) made by conjugating trastuzumab through conventional lysine chemistry to SMCC-DM1 that served as a positive control in our experiments.
Site-Specific Conjugation of a Cytotoxic Payload to Three Different Locations on Aldehyde-Tagged α-HER2 Antibodies Yields Stable ADCs
Trastuzumab antibodies modified to contain the aldehyde tag in either the light chain (LC), the CH1 domain (Tag C), or at the heavy chain C-terminus (CT, Tag G) were produced in bulk pools of cells overexpressing human FGE. In terms of Cys to fGly conversion efficiency, we achieved 86%, 92%, and 98% conversion at the LC, CH1, and CT aldehyde tag sites, respectively, as measured by a mass spectrometric method.9 The conjugation reaction was carried out by treating the fGly-tagged antibody with 8–10 equiv of HIPS-Glu-PEG2-maytansine in 50 mM sodium citrate, 50 mM NaCl pH 5.5 containing 0.85% DMA and 0.085% Triton X-100 at 37 °C, and the progress of the reaction was tracked by analytical hydrophobic interaction chromatography (HIC). Upon completion, the excess payload was removed by tangential flow filtration and the unconjugated antibody was removed by preparative HIC. These reactions were high yielding, with >90% conjugation efficiency at the CH1 and CT tag sites, and 75% conjugation efficiency at the LC tag site. HIC analysis of the final product highlights the facile analytics (Figure 3), which are a major benefit of site-specific conjugation approaches as compared to global conjugation strategies. SEC analysis of the conjugates demonstrated minimal aggregation (SI Figure S1).
Figure 3.

Hydrophobic interaction chromatography analysis demonstrates the clean conversion of LC-, CH1-, and CT-tagged antibodies into homogeneous ADCs. Unconjugated antibody (black) elutes as one peak. After conjugation to HIPS-Glu-PEG2-maytansine, the ADC (green) elutes as a diconjugated material (right). This clean separation of conjugated from unconjugated material allows for conjugate enrichment and simple determination of DAR. α-HER2-DM1 was included as a comparator.
In order to assess the impact of tag incorporation on antibody structure, the thermal stability of these antibodies was examined by thermofluorescence. There were no detectable differences in Tm1 (the lowest observed thermal transition) among the α-HER2 antibodies tested (range 67.6–68 °C), which included the untagged sequence as well as antibodies tagged at the LC, CH1, or CT locations (SI Table S1). Furthermore, conjugation of the CT-tagged antibody with HIPS-Glu-PEG2-maytansine had only a minor effect on Tm1, decreasing the melting temperature by only one degree as compared to the untagged antibody. Next, we determined the effect of tag placement and payload conjugation on FcRn binding as determined by surface plasmon resonance analysis. Previous reports have documented the significant role of this receptor in antibody pharmacokinetics.21 Specifically, FcRn is broadly expressed on vascular endothelium, where it is poised to capture internalized IgG (by binding at acidic pH in endosomes) and return it to the circulation (by releasing the antibody at neutral pH). Interestingly, both association at pH 6.0 and dissociation at pH 7.3 correlate with an antibody’s circulating half-life, with the latter value having a greater influence than the former.22 We measured both parameters (Table 2). Our controls included the untagged α-HER2 and α-HER2-DM1. We found no effect of aldehyde tag placement or payload conjugation on the FcRn KD at pH 6.0. By contrast, the dissociation at pH 7.3 did reveal differences in the percent of antibody that remained bound after 5 s. Specifically, trastuzumab had the smallest amount of retained antibody, and inclusion of the aldehyde tag increased the retention slightly, but not significantly. Conjugating the antibodies did somewhat affect dissociation at pH 7.3, although the aldehyde-tagged ADCs were less impacted as compared to the α-HER2-DM1. Retention of the latter conjugate was significantly different from all other measured analytes. These trends suggest that insertion of the aldehyde tag into the antibody does not significantly modulate FcRn binding, and that aldehyde-mediated site-specific conjugation yields ADCs with FcRn dissociation characteristics that are more similar to the wild-type antibody as compared to the nonspecifically conjugated α-HER2-DM1.
Table 2. Aldehyde Tag Insertion and Payload Conjugation Minimally Affect Antibody FcRn Binding Characteristics, and Show Improved Dissociation at pH 7.3 Relative to α-HER2-DM1.
| measured value | α-HER2 untagged | α-HER2 CH1 tag | α-HER2 CT tag | α-HER2 LC tag | α-HER2 CH1 ADC | α-HER2 CT ADC | α-HER2 LC ADC | α-HER2-DM1 ADC |
|---|---|---|---|---|---|---|---|---|
| KDa (association [nM] at pH 6.0) | 5.2 ± 1.3 | 5.2 ± 1.9 | 5.8 ± 0.7 | 4.7 ± 1.2 | 4.8 ± 1.0 | 4.4 ± 1.1 | 4.8 ± 1.0 | 5.0 ± 0.2 |
| % Bound after 5 s at pH 7.3 | 8.6 ± 0.8 | 9.9 ± 1.0 | 9.9 ± 1.7 | 11.0 ± 1.2 | 9.5 ± 0.9 | 12.0 ± 1.4b | 10.9 ± 0.6b | 14.9 ± 0.8c |
Mean KD values are not statistically significantly different as determined by one way ANOVA.
Significantly different from αHER2 untagged, p < 0.03, Two-tailed t-test.
Significantly different from all of the other analytes, p < 0.04, Two-tailed t-test.
The immunogenicity profiles of the LC, CH1, and CT tags were explored by conducting an ex vivo human T-cell assay (EpiScreen) in which both the unconjugated and ADC versions of these constructs were incubated with leukocytes from 50 healthy donors representing the world population of HLA allotypes23−25 and compared with unmodified trastuzumab. T-cell responses were measured by assessing proliferation and IL-2 cytokine secretion. By this functional measure, the unconjugated and ADC versions of LC-, CH1-, and CT-tagged antibodies were found not to be immunogenic (SI Table S2). Specifically, the analytes induced T-cell proliferation in only 2–10% of donor leukocytes for trastuzumab and all other tagged antibody samples, as compared to proliferation in 22% of samples induced by a positive control—a relatively immunogenic, monoclonal antibody for which clinical immunogenicity data are available.26−28
In a parallel set of experiments using a different antibody backbone but the same three tag placements, we tested the stability of aldehyde-tagged HIPS conjugates in plasma at 37 °C. We tested antibodies carrying the HIPS-Glu-PEG2 linker attached to either a fluorophore (Alexa Fluor 488, AF488) or cytotoxin payload (maytansine). The purpose of testing two payloads was to explore how differences in payload attachment to the linker (e.g., ester vs aryl amide bond, see SI Figure S2) can affect stability. The results indicated that the HIPS conjugation chemistry is highly stable; specifically, for the AF488 conjugates, we saw no loss of payload signal over 12 d at 37 °C in rat plasma, regardless of tag placement (Figure 4A). However, this stability did not completely translate to the maytansine conjugates, which did show some loss of payload signal over time (Figure 4B). The amount of payload loss differed according to tag placement, with the CT site showing the greatest stability, followed by the CH1 and LC sites. We hypothesize that the differences in stability between the AF488 and maytansine conjugates are related to the differences in the chemical linkages connecting the payload to the PEG2 portion of the linker (SI Figure S2). While the AF488 is attached to the PEG2 by a stable aryl amide bond, the ester bond that connects the maytansine payload is a known chemical liability at high pH. Therefore, the differences that we observed in the stability of the three maytansine ADCs might reflect distinct microenvironments at the three attachment sites that influence the differential hydrolysis of the ester bond.
Figure 4.
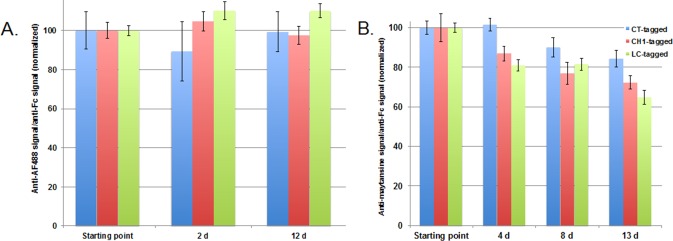
Aldehyde-tagged HIPS conjugates are stable in plasma at 37 °C, but payload attachment plays a role. We tested the plasma stability of LC-, CH1-, and CT-tagged antibodies conjugated using HIPS-Glu-PEG2 to either (A) Alexa Fluor 488 (AF488) or (B) maytansine. Conjugates were incubated in rat plasma at 37 °C for up to 13 d. When analyzed by ELISA for total payload and total antibody, we observed no loss of total payload signal relative to total antibody signal for the AF488 conjugates, regardless of tag placement. For the maytansine conjugates, we observed evidence that some deconjugation occurred over time at 37 °C. The stability differed according to tag placement, with the CT-tag showing the highest conservation of payload-to-antibody signal (84%), followed by CH1 (72%), and LC (65%).
LC-, CH1-, and CT-Tagged ADCs Demonstrate Potent Activity against Tumor Targets in Vitro and in Vivo
As a first measure of efficacy, the LC, CH1, and CT tagged α-Her2 ADCs were tested in vitro against the HER2-overexpressing cell line, NCI-N87. Free maytansine and α-HER2-DM1 (DAR 3.4) were used as comparators. All three of the α-HER2 HIPS-Glu-PEG2-maytansine ADC conjugates showed excellent in vitro cytotoxicity that was on par with free maytansine and α-HER2-DM1 (Figure 5). By contrast, the isotype control CT-tagged conjugate showed essentially no activity, as expected.
Figure 5.
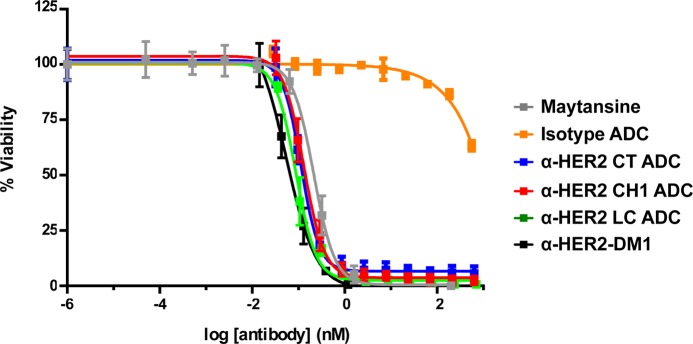
Payload location does not influence in vitro potency of aldehyde-tagged α-HER2 ADCs against NCI-N87 target cells. NCI-N87 cells, which overexpress HER2, were used as targets for in vitro cytotoxicity in a 6 day assay. Free maytansine (gray line) was included as a positive control, and an isotype control ADC (orange line) was used as a negative control to indicate specificity. α-HER2 HIPS-Glu-PEG2-maytansine ADCs bearing the aldehyde tag on the light chain (LC, green), or on the CH1 (red) or C-terminal (CT, blue) regions of the heavy chain showed comparable activity. α-HER2-DM1 was included as a comparator. IC50 values (reflecting the antibody concentrations except in the case of the free drug) were measured as follows: free maytansine, 214 pM; isotype control ADC, could not be determined; LC ADC 87 pM; CH1 ADC, 132 pM; CT ADC 114 pM, α-HER2-DM1, 54.7 pM.
The in vivo efficacy of LC-, CH1-, and CT-tagged α-HER2 ADCs was explored using a staged NCI-N87 xenograft model in SCID mice. Trastuzumab alone and an isotype control CT-tagged HIPS-Glu-PEG2-maytansine ADC were used as negative controls, and α-HER2-DM1 (DAR 3.4) was included as a comparator. All compounds were administered as a single 5 mg/kg dose at the onset of the study. While the tumors continued to grow in mice treated with either trastuzumab or the isotype control ADC, a single dose of α-HER2-targeted ADC was sufficient to stop tumor growth for ∼30 days in treated animals (Figure 6A). When tumors did eventually begin to grow back, it became clear that the tumor sizes were larger in animals treated with the CH1-tagged ADC as compared to those treated with LC- or CT-tagged ADCs (p < 0.026 and 0.016, respectively, two-tailed t-test at day 67). In order to investigate this effect, we looked at the log10 cell kill for tumors dosed with the various treatments (Table 3). Indeed, the results suggested that treatment with the CH1-tagged ADC killed fewer tumor cells as compared to treatment with the other ADCs. Furthermore, the CT-tagged ADC appeared to be the most efficacious conjugate resulting in the highest log10 cell kill. This increased potency translated into a significant survival advantage for animals treated with the CT-tagged ADC (Figure 6B).
Figure 6.
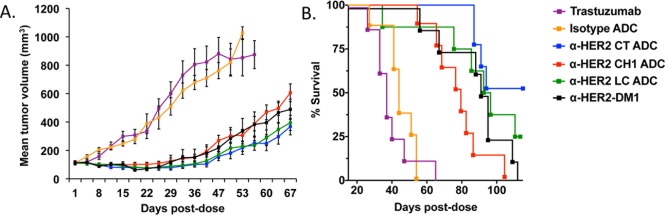
Payload placement modifies the in vivo efficacy of aldehyde-tagged α-HER2 ADCs against NCI-N87 xenografts in mice. CB.17 SCID mice (8/group) were implanted subcutaneously with NCI-N87 cells. When the tumors reached ∼113 mm3, the animals were given a single 5 mg/kg dose of trastuzumab alone, an isotype ADC, or an α-HER2 HIPS-Glu-PEG2-maytansine ADC conjugated to either the light chain (LC), or the CH1 or C-terminal (CT) regions of the heavy chain. α-HER2-DM1 was included as a comparator. (A) Tumor growth was monitored twice weekly. (B) The differences in efficacy among the tag placements tested were reflected in survival curves. Animals were euthanized when tumors reached 800 mm3.
Table 3. In Vivo Log10 Cell Kill of NCI-N87 Tumor Cells Achieved by a Single 5 mg/kg ADC Dose.
| treatment | log10 cell kill |
|---|---|
| αHER2 CT ADC | 1.24 |
| αHER2 CH1ADC | 0.83 |
| αHER2 LC ADC | 1.08 |
| αHER2-DM1 ADC | 1.03 |
ADCs Carrying the Payload at Different Locations on the Antibody Demonstrate Distinct Pharmacokinetics
We wanted to determine if the differential efficacy of our ADC panel was a function of distinct pharmacokinetic profiles. Mice were dosed with 5 mg/kg of LC-, CH1-, or CT-tagged ADC, or with trastuzumab or α-HER2-DM1 as comparators. Plasma was collected from the mice and analyzed by ELISA to quantitate the total ADC and total antibody concentrations. To measure total ADC, analytes were captured with an anti-human Fab-specific antibody and detected with an anti-maytansine antibody. To measure total antibody, analytes were captured with an anti-human IgG-specific antibody and detected with an anti-human Fc-specific antibody. The measured concentrations over time were fit to a two-compartment model by nonlinear regression to determine half-lives (Table 3). The total antibody half-life for each aldehyde-tagged ADC was the same as, or longer than, trastuzumab, suggesting that aldehyde tag insertion and HIPS conjugation did not change the basic PK properties of the antibody. By contrast, the total antibody half-life of the α-HER2-DM1 conjugate was significantly shorter, suggesting that the nonspecific conjugation chemistry (which leads to overconjugated species) had a negative effect on PK. We also measured the conjugated antibody (total ADC) half-lives, which showed that the CT-tagged ADC, which conferred the biggest survival benefit to tumor-bearing mice, also demonstrated longest total half-life. The conjugate half-lives of the α-HER2-DM1, and the CH1- and LC-tagged ADCs, were shorter than the CT-tagged conjugate. These numbers clearly indicate that the conjugation site played a key role in governing ADC half-lives. In all cases, the aldehyde-tagged conjugates were stable in the circulation, with percent area under the curve ratios of total ADC to total antibody concentrations comparable to or better than the α-HER2-DM1 conjugate (Figure 7).
Figure 7.

α-HER2 HIPS-Glu-PEG2-maytansine ADCs are highly stable in vivo regardless of tag placement. BALB/c mice were dosed with 5 mg/kg of aldehyde-tagged α-HER2 HIPS-Glu-PEG2-maytansine ADCs conjugated to either the light chain (LC), or to the CH1 or C-terminal (CT) regions of the heavy chain. α-HER2-DM1 was included as a comparator. Plasma was sampled at the time points indicated and assayed by ELISA. Area under the curve (AUC) was determined using GraphPad Prism and is reported in Table 4.
Table 4. Pharmakinetic Parameters Are Influenced by Tag Placement and Conjugation Chemistry.
| analyte | total ADC half-life (days) | total antibody half-life (days) | total ADC AUCa | total antibody AUCa |
|---|---|---|---|---|
| α-HER2 CTADC | 7.8 ± 0.5 | 16.6 ± 2 | 366 880 | 695 913 |
| α-HER2 LC ADC | 5.2 ± 0.2 | 14.13 ± 1.5 | 289 607 | 678 971 |
| α-HER2 CH1 ADC | 5.7± 0.3 | 15.0 ±1.9 | 275 677 | 617 428 |
| α-HER2-DM1 ADC | 6 ± 0.3 | 10.7 ± 0.7 | 240 082 | 482 220 |
| Trastuzumab | n.a.b | 13.65 ± 1 | n.a.b | 809 674 |
Area under the curve (day × ng/mL) for the beta phase, measured from 2 to 28 d.
Not applicable.
CT-Tagged ADC Exhibits an Improved Nonclinical Safety Profile as Compared to a Conventional Lysine-Conjugated ADC
Finally, we wanted to get an initial assessment of the safety profile of our site-specific conjugates. Accordingly, we conducted a single dose exploratory toxicology study in Sprague–Dawley rats. Animals (5/group) received a 6, 20, or 60 mg/kg dose of CT-tagged α-HER2 followed by a 12 day observation period. As a comparator, we used the conventional α-HER2-DM1 at the same doses. Body weight and food intake were assessed at days 0, 1, 4, 8, and 11. Blood was drawn on days 5 and 12 for clinical chemistry, hematology, and toxicokinetic (TK) analysis. Additional TK blood samples were drawn at 8 h and on day 9. All of the animals in the α-HER2-DM1 60 mg/kg group died on day 5 (Table 5). This mortality was consistent with the known preclinical safety profile of the analogue, T-DM1.29 By contrast, no mortality was observed in the α-HER2 CT ADC groups, even at 60 mg/kg. With respect to body weight (Figure 8A), treatment with 6 or 20 mg/kg of α-HER2 CT ADC had no effect, while treatment with 60 mg/kg of α-HER2 CT ADC reduced rat body weight to a similar extent as treatment with 20 mg/kg of α-HER2-DM1. With respect to clinical chemistry, levels of both alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were essentially unchanged in rats treated with 6 or 20 mg/kg of α-HER2 CT ADC, but were somewhat elevated in rats treated with 20 mg/kg of α-HER2-DM1 (Figure 8B and C). Levels of both enzymes were elevated at the 60 mg/kg dose for both compounds, although to a lesser extent with the α-HER2 CT ADC. Increased ALT and AST levels are indicators of liver toxicity, the former a much more specific marker than the latter. Increases in these enzymes are consistent with known toxicity profiles of maytansine and maytansine conjugates. With respect to hematology, platelet counts were essentially unchanged in rats treated with 6 or 20 mg/kg of α-HER2 CT ADC, but were decreased in rats treated with 20 mg/kg of α-HER2-DM1 (Figure 8D). Platelet counts were decreased at the 60 mg/kg dose for both compounds, although to a lesser extent with the α-HER2 CT ADC. Decreases in platelet counts at day 5 postdose are generally indicative of localized tissue damage, rather than bone marrow toxicity, which takes longer to manifest due to the long half-life of platelets. For all surviving animals, the clinical chemistry and hematology indicators of toxicity observed at day 5 were essentially resolved by day 12 (SI Figure S3). In total, the data indicated that the α-HER2 CT ADC was less toxic to rats than the α-HER2-DM1 at the same doses; however, as expected, the target organs were the same. Furthermore, toxicokinetic analyses of the total ADC and total antibody concentrations showed that the α-HER2 CT ADC was stable in vivo, showing similar profiles in the rat as were observed in the mouse. Notably, the α-HER2-DM1 total ADC was cleared faster than the α-HER2 CT ADC total ADC (Figure 9).
Table 5. No Mortality Was Observed in α-HER2 CTADC Groups, even at 60 mg/kg, Which Was a Lethal Dose for α-HER2-DM1.
| group | test article | dose (mg/kg) | mortality |
|---|---|---|---|
| 1 | Vehicle | 0 | 0/5 |
| 2 | α-HER2-DM1 | 6 | 0/5 |
| 3 | α-HER2-DM1 | 20 | 1/5a |
| 4 | α-HER2-DM1 | 60 | 5/5 |
| 5 | α-HER2 CTADC | 6 | 0/5 |
| 6 | α-HER2 CT ADC | 20 | 0/5 |
| 7 | α-HER2 CT ADC | 60 | 0/5 |
Animal euthanized for reasons not related to treatment.
Figure 8.

α-HER2 CT ADC is less toxic than the α-HER2-DM1 at the same doses in Sprague–Dawley rats. Animals (5/group) received a 6, 20, or 60 mg/kg dose of α-HER2 CT ADC (shown in blue) or α-HER2-DM1 (shown in red), followed by a 12 day observation period. Body weight (A) was monitored at the times indicated. Alanine aminotransferase (B), aspartate aminotransferase (C), and platelet counts (D) were assessed at day 5 postdose. Due to premature death (for 2 animals in the α-HER2-DM1 60 mg/kg group) or sample collection errors (platelet-specific, for animals in the vehicle control and both 6 mg/kg groups), some groups comprise fewer than 5 data points.
Figure 9.
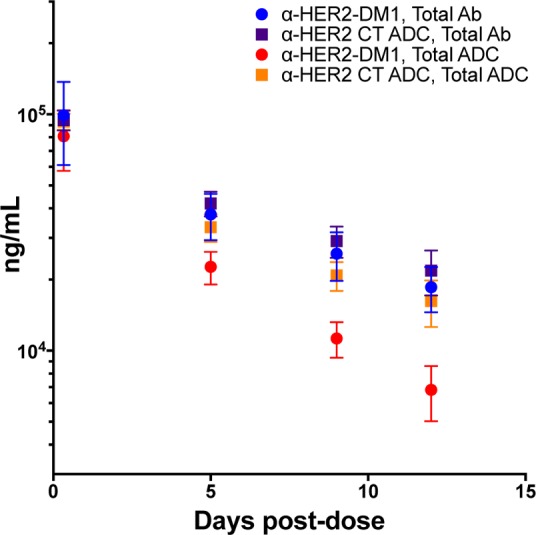
Toxicokinetic analysis demonstrated that the α-HER2 CT ADC was more stable in rats than the α-HER2-DM1. The same animals that were analyzed for indicators of toxicity (Figure 8) were used to assess the toxicokinetic profiles of the α-HER2 CT ADC and α-HER2-DM1 analytes. Plasma was sampled at the time points indicated and assayed by ELISA.
ADC Structure–Activity Relationship Mapping
In this study, we demonstrated that the aldehyde tag coupled with HIPS chemistry could be used to site-specifically conjugate cytotoxic payloads to antibody heavy and light chains. Critically, this included varying the conjugation placement at internal, as well as N- or C-terminal sites,30 allowing wide flexibility in terms of exploring the SAR space and optimizing the ADC structure. Furthermore, we demonstrated that our approach generated ADCs with improved PK, equivalent or better efficacy, and improved safety profiles as compared to a conventional conjugate. The aldehyde-tagged ADCs were also highly stable in vivo as shown by the pharmacokinetic and toxicokinetic studies. This observed stability is likely a combination of HIPS chemistry, which results in a C–C bond between antibody and payload, and the ability to conjugate at a site that minimizes the hydrolysis of the payload from the linker.
Our data add to a growing body of evidence supporting the functional advantages of site-specific ADCs.5,13,31 Beyond the overall influence of stoichiometry, whereby ADCs with higher DARs are cleared faster from the circulation,4 less is known about how conjugation chemistries and locations affect ADC properties. The conjugation site can be considered both from a macro viewpoint—the targeted antibody domain—and a micro viewpoint—the local chemical and structural environment, including contributions from the antibody backbone and from the payload linker. To date, our data presented here along with some recent examples suggest that the latter perspective may be more instructive in influencing the design and control over the biological disposition of the resulting ADCs.6,7,14,32
We tested three aldehyde-tagged ADCs with conjugation sites in distinct antibody domains and observed that the CT-tagged ADC had superior PK and in vivo efficacy as compared to the CH1- and LC-tagged ADCs. The serum stability studies with the AF88 amide conjugates showed comparable stability at the three tag sites, whereas the maytansine ester conjugates demonstrated greater stability with the CT conjugate. These observations indicate that the HIPS conjugation to the antibody is inherently stable with little difference across the three sites, and that the difference in the stability of the maytansine conjugates at the three sites is a manifestation of the differences in the protein microenvironment of the three sites and its influence on the ester bond hydrolysis.33 Although it will need to be demonstrated empirically, we surmise that conjugation sites that work well in the context of an IgG1 ADC (explored here) might also work well in the context of other, related antibody fragments. The likely drivers determining tag site efficacy are protein conformation and the local chemical microenvironment. To the extent that these remain the same or are similar across antibodies or related proteins, our observations may hold true.
The HIPS chemistry is less subject to environmental influences at different conjugation sites, as it results in a chemically stable C–C bond formation between the protein and the payload. However, other portions of the linker may well influence stability and efficacy.13 The ability to vary linker composition in order to test the affects of length, hydrophobicity, and conformation remains an important aspect of optimizing in vivo properties of ADCs. The flexibility and versatility of the aldehyde tag conjugation platform has enabled us to undertake a systematic evaluation of the impact of conjugation site and linker composition on ADC properties. Our novel and practical chemoenzymatic bioconjugation platform enables exploring bioconjugate SAR space in order to find optimal combinations of conjugation site and linker chemistry to produce ADCs with enhanced stability, safety, and efficacy.
Experimental Procedures
General
The murine anti-maytansine antibody was made and validated in-house. The rabbit anti-AF488 antibody was purchased from Life Technologies. The goat anti-human IgG-specific and goat anti-human Fab-specific antibodies, and the donkey anti-rabbit, goat anti-mouse IgG subclass I-specific, and goat anti-human Fc-specific HRP-conjugates were from Jackson Immunoresearch.
Cloning, Expression, and Purification of Tagged Antibodies
The aldehyde tag sequence was inserted at various points in the light and heavy chain consensus regions using standard molecular biology techniques. For small-scale production, CHO-S cells were transfected with human FGE expression constructs and pools of FGE-overexpressing cells were used for the transient production of antibodies. For larger-scale production, GPEx technology (Catalent Pharma Solutions, LLC) was used to generate a pooled cell line overexpressing human FGE.34 Then, the FGE pool was used to generate bulk stable pools of antibody-expressing cells. Antibodies were purified from the conditioned medium using Protein A chromatography (MabSelect, GE Healthcare Life Sciences). Purified antibodies were flash frozen and stored at −80 °C until further use.
Bioconjugation, Purification, and HPLC Analytics
Aldehyde-tagged antibodies (15 mg/mL) were conjugated to HIPS-Glu-PEG2-maytansine (8 mol equiv drug:antibody) for 72 h at 37 °C in 50 mM sodium citrate, 50 mM NaCl pH 5.5 containing 0.85% DMA and 0.085% Triton X-100. Free drug was removed using tangential flow filtration. Unconjugated antibody was removed using preparative-scale hydrophobic interaction chromatography (HIC; GE Healthcare 17–5195–01) with mobile phase A: 1.0 M ammonium sulfate, 25 mM sodium phosphate pH 7.0, and mobile phase B: 25% isopropanol, 18.75 mM sodium phosphate pH 7.0. An isocratic gradient of 33% B was used to elute unconjugated material, followed by a linear gradient of 41–95% B to elute mono- and diconjugated species. To determine the DAR of the final product, ADCs were examined by analytical HIC (Tosoh #14947) with mobile phase A: 1.5 M ammonium sulfate, 25 mM sodium phosphate pH 7.0, and mobile phase B: 25% isopropanol, 18.75 mM sodium phosphate pH 7.0. To determine aggregation, samples were analyzed using analytical size exclusion chromatography (SEC; Tosoh #08541) with a mobile phase of 300 mM NaCl, 25 mM sodium phosphate pH 6.8.
In Vitro Stability
Antibody–fluorophore and antibody–drug conjugates were spiked into rat plasma at ∼1 pmol (payload)/mL. The samples were aliquoted and stored at −80 °C until use. Aliquots were placed at 37 °C under 5% CO2 for the indicated times, and then were analyzed by ELISA to assess the anti-maytansine and anti-Fab signals. As a first step for the analysis, a dilution series of the analyte into 1% bovine serum albumin was performed to ensure that the analyte concentration was within the linear range of the assay (20–40 ng/mL). Once the appropriate dilution was determined, samples were removed from the incubator and tested. A freshly thawed aliquot was used as a reference starting value for conjugation. All analytes were measured together on one plate to enable comparisons across time points. Analytes were captured on plates coated with an anti-human Fab-specific antibody. Then, the payload was detected with either an anti-AF488 or an anti-maytansine antibody followed by an HRP-conjugated secondary; the total antibody was detected with a directly conjugated anti-human Fc-specific antibody. Bound secondary antibody was visualized with TMB substrate. The colorimetric reaction was stopped with H2SO4, and the absorbance at 450 nm was determined using a Molecular Devices SpectraMax M5 plate reader. Data analysis was performed in Excel. Each sample was analyzed in quadruplicate, and the average values were used. The ratio of anti-maytansine signal to anti-Fab signal was used as a measure of antibody conjugation.
In Vitro Cytotoxicity
The HER2-positive gastric carcinoma cell line, NCI-N87, was obtained from ATCC and maintained in RPMI-1640 medium (Cellgro) supplemented with 10% fetal bovine serum (Invitrogen) and Glutamax (Invitrogen). Twenty-four hours prior to plating, cells were passaged to ensure log-phase growth. On the day of plating, 5000 cells/well were seeded onto 96-well plates in 90 μL normal growth medium supplemented with 10 IU penicillin and 10 μg/mL streptomycin (Cellgro). Cells were treated at various concentrations with 10 μL of diluted analytes, and the plates were incubated at 37 °C in an atmosphere of 5% CO2. After 6 d, 100 μL/well of Cell Titer-Glo reagent (Promega) was added, and luminescence was measured using a Molecular Devices SpectraMax M5 plate reader. GraphPad Prism software was used for data analysis, including IC50 calculations.
Xenograft Studies
Female C.B-17 SCID mice were inoculated subcutaneously with 1 × 107 NCI-N87 tumor cells in 50% Matrigel. When the tumors reached an average of 113 mm3, the animals were given a single 5 mg/kg dose of ADC, trastuzumab antibody (untagged), or vehicle alone. The animals were monitored twice weekly for body weight and tumor size. Tumor volume was calculated using the formula
where w = tumor width and l = tumor length.
Tumor doubling times were obtained by averaging the tumor growth rate curves from four groups of mice. Then, log10 cell kill was estimated using the formula
Pharmacokinetic Analysis
Male BALB/c mice were dosed intravenously with a single 5 mg/kg bolus of antibody conjugate. Plasma was collected at 1, 8, and 20 h, and 2, 4, 6, 8, 10, 14, 21, and 28 days postdose, with three animals per time point. No single animal was sampled more than twice per week. Plasma samples were stored at −80 °C, and the concentrations of total antibody and total ADC were quantified by ELISA. For the former, conjugates were captured with an anti-human IgG-specific antibody and detected with an HRP-conjugated anti-Fc-specific antibody. For the latter, conjugates were captured with an anti-human Fab-specific antibody and detected with a mouse anti-maytansine primary antibody, followed by an HRP-conjugated anti-mouse IgG-subclass 1-specific secondary antibody. Bound secondary antibody was detected using Ultra TMB One-Step ELISA substrate (Thermo Fisher). After quenching the reaction with sulfuric acid, signals were read by taking the absorbance at 450 nm on a Molecular Devices Spectra Max M5 plate reader equipped with SoftMax Pro software. Data were analyzed using GraphPad Prism software. The measured concentrations over time were fit to a two-compartment model by nonlinear regression of the mean of the Y values (weighted by 1/Y2) with the following equation
The resulting exponential decay constant (τβ) was used to calculate t1/2.
Rat Toxicology Study and Toxicokinetic Analysis
Male Sprague–Dawley rats (8–9 wk old at study start) were given a single intravenous dose of 6, 20, or 60 mg/kg of either the α-HER2 CT ADC or α-HER2-DM1 (5 animals/group). Animals were observed for 12 days postdose. Body weights were recorded on days 0, 1, 4, 8, and 11. Blood was collected from all animals at 8 h and at 5, 9, and 12 d for toxicokinetic analyses (all time points) and for clinical chemistry and hematology analyses (days 5 and 12). Toxicokinetic analyses were performed by ELISA, using the same conditions and reagents described for the pharmacokinetic analyses.
Acknowledgments
Both the in silico and ex vivo immunogenicity assessments were performed by Antitope Ltd. This work was funded in part by grants to DR from the NIH (GM096494) and the NSF (1151234).
Glossary
Abbreviations
- HIPS
Hydrazino-Pictet-Spengler
- HIC
hydrophobic interaction chromatography
- SEC
size-exclusion chromatography
- FGE
formylglycine-generating enzyme
- fGly
formylglycine
- LC
light chain
- CT
C-terminal
- ANOVA
analysis of variance
- AF488
Alexa Fluor 488
Supporting Information Available
Size-exclusion chromatography traces corresponding to the LC-, CH1-, and CT-α-HER2 HIPS-Glu-PEG2-maytansine ADCs shown in Figure 2. Experimental methods for thermofluorescence, FcRn-binding, and ex vivo immunogenicity experiments, and tables (S1–S3) of the results. Synthetic route for and analytical data describing the HIPS-Glu-PEG2-maytansine payload. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): All authors are employees of Redwood Bioscience and hold financial interest in the company.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sievers E. L.; Senter P. D. (2013) Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 64, 15–29. [DOI] [PubMed] [Google Scholar]
- Mullard A. (2013) Maturing antibody-drug conjugate pipeline hits 30. Nat. Rev. Drug Discovery 12, 329–332. [DOI] [PubMed] [Google Scholar]
- Panowksi S., Bhakta S., Raab H., Polakis P. & Junutula J. R.. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014,6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblett K. J.; et al. (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 10, 7063–7070. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; et al. (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 26, 925–932. [DOI] [PubMed] [Google Scholar]
- Strop P.; et al. (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 20, 161–167. [DOI] [PubMed] [Google Scholar]
- Axup J. Y.; et al. (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. U. S. A. 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung Sutherland M. S.; Walter R. B.; Jeffrey S. C.; Burke P. J.; Kostner H. A.; Stone I.; Ryan M. C.; Sussman D.; Lyon R. P.; Zeng W.; Harrington K. H.; Klussman K.; Westendorf L.; Meyer D.; Bernstein I. D.; Senter P. D.; Benjamin D. R.; Drachman J. G.; McEarchern J. A. (2013) SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 122, 1455–1463. [DOI] [PubMed] [Google Scholar]
- Rabuka D.; Rush J. S.; deHart G. W.; Wu P.; Bertozzi C. R. (2012) Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 7, 1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson B. L.; et al. (2008) Function and structure of a prokaryotic formylglycine-generating enzyme. J. Biol. Chem. 283, 20117–20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. Engl. 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; et al. (2013) Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjugate Chem. 24, 846–851. [DOI] [PubMed] [Google Scholar]
- Tian F.; et al. (2014) A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. U. S. A. 111, 1766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B.-Q.; et al. (2012) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 30, 184–189. [DOI] [PubMed] [Google Scholar]
- Ramsland P. A.; Farrugia W. (2002) Crystal structures of human antibodies: a detailed and unfinished tapestry of immunoglobulin gene products. J. Mol. Recognit. 15, 248–259. [DOI] [PubMed] [Google Scholar]
- Bryson C. J.; Jones T. D.; Baker M. P. (2010) Prediction of immunogenicity of therapeutic proteins: validity of computational tools. BioDrugs 24, 1–8. [DOI] [PubMed] [Google Scholar]
- Perry L. C. A.; Jones T. D.; Baker M. P. (2008) New approaches to prediction of immune responses to therapeutic proteins during preclinical development. Drugs R & D 9, 385–396. [DOI] [PubMed] [Google Scholar]
- LoRusso P. M.; Weiss D.; Guardino E.; Girish S.; Sliwkowski M. X. (2011) Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 17, 6437–6447. [DOI] [PubMed] [Google Scholar]
- Girish S.; et al. (2012) Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother. Pharmacol. 10.1007/s00280-011-1817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila T. T.; Li G.; Parsons K.; Phillips G. L.; Sliwkowski M. X. (2011) Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 128, 347–356. [DOI] [PubMed] [Google Scholar]
- Roopenian D. C.; Akilesh S. (2007) FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 7, 715–725. [DOI] [PubMed] [Google Scholar]
- Wang W.; et al. (2011) Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab. Dispos. 39, 1469–1477. [DOI] [PubMed] [Google Scholar]
- Jones T. D.; et al. (2005) Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J. Thromb. Haemost. 3, 991–1000. [DOI] [PubMed] [Google Scholar]
- Jones T. D.; et al. (2004) The development of a modified human IFN-alpha2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J. Interferon Cytokine Res. 24, 560–572. [DOI] [PubMed] [Google Scholar]
- Baker M. P.; Jones T. D. (2007) Identification and removal of immunogenicity in therapeutic proteins. Curr. Opin Drug Discov Devel 10, 219–227. [PubMed] [Google Scholar]
- Ritter G.; et al. (2001) Serological analysis of human anti-human antibody responses in colon cancer patients treated with repeated doses of humanized monoclonal antibody A33. Cancer Res. 61, 6851–6859. [PubMed] [Google Scholar]
- Scott A. M.; et al. (2005) A phase I trial of humanized monoclonal antibody A33 in patients with colorectal carcinoma: biodistribution, pharmacokinetics, and quantitative tumor uptake. Clin. Cancer Res. 11, 4810–4817. [DOI] [PubMed] [Google Scholar]
- Welt S.; et al. (2003) Phase I study of anticolon cancer humanized antibody A33. Clin. Cancer Res. 9, 1338–1346. [PubMed] [Google Scholar]
- Poon K. A.; et al. (2013) Preclinical safety profile of trastuzumab emtansine (T-DM1): Mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol. Appl. Pharmacol. 273, 298–313. [DOI] [PubMed] [Google Scholar]
- Carrico I. S.; Carlson B. L.; Bertozzi C. R. (2007) Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 3, 321–322. [DOI] [PubMed] [Google Scholar]
- Boswell C. A.; et al. (2011) Impact of drug conjugation on pharmacokinetics and tissue distribution of Anti-STEAP1 antibody–drug conjugates in rats. Bioconjugate Chem. 22, 1994–2004. [DOI] [PubMed] [Google Scholar]
- Jackson D.; et al. (2014) In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLoS One 9, e83865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Phillips G. D.; et al. (2008) Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290. [DOI] [PubMed] [Google Scholar]
- Bleck G. T. (2007) An alternative method for the rapid generation of stable, high-expressing mammalian cell lines. BioProcess. J. 5, 36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.