

Abstract
The reducing substrates 4-thiolumazine and 2,4-dithiolumazine have been used to form MoIV-product complexes with xanthine oxidase (XO) and xanthine dehydrogenase. These MoIV-product complexes display an intense metal-to-ligand charge-transfer (MLCT) band in the near-infrared region of the spectrum. Optical pumping into this MLCT band yields resonance Raman spectra of the Mo site that are devoid of contributions from the highly absorbing FAD and 2Fe2S clusters in the protein. The resonance Raman spectra reveal in-plane bending modes of the bound product and low-frequency molybdenum dithiolene and pyranopterin dithiolene vibrational modes. This work provides keen insight into the role of the pyranopterin dithiolene in electron-transfer reactivity.
Short abstract
The reducing substrates 4-thiolumazine and 2,4-dithiolumazine have been used to form MoIV-product complexes in xanthine oxidase (XO) and xanthine dehydrogenase (XDH). The MoIV-product complexes absorb in the near-infrared (NIR) region of the spectrum. Optical pumping into this NIR metal-to-ligand charge-transfer band reveals in-plane bending modes of the bound product in addition to low-frequency molybdenum dithiolene and pyranopterin dithiolene (pyranopterin ditholene) vibrational modes. The work provides insight into how pyranopterin ditholene is coupled to redox changes at the Mo site and how pyranopterin ditholene functions as an electron-transfer conduit in the oxidative half-reaction of XO/XDH.
Mammalian xanthine oxidoreductase (XOR) and R. capsulatus xanthine dehydrogenase (XDH) are molybdenum hydroxylases with broad substrate specificities.1,2 These enzymes possess a high degree of sequence homology and virtually identical coordination geometries.4,5 Unlike monooxygenases, the oxygen atom incorporated into substrate C–H bonds derives from metal-activated water, and the enzymes generate rather than consume reducing equivalents.2 These reducing equivalents are transferred sequentially from the reduced MoIV center via an apparent electron-transfer (ET) chain consisting of the pyranopterin ditholene (Figure 1), two 2Fe2S clusters, and FAD.5,9 The ultimate electron acceptor for the oxidase form of XOR (xanthine oxidase, XO) is O2, and this results in the formation of reactive oxygen species that have been implicated in reperfusion injury following ischemia.2 The ultimate electron acceptor for XDH and the dehydrogenase form of XOR is NAD.4 Integral to the ET regeneration of the catalytically competent MoVI site is the pyranopterin ditholene chelate,9−11 which has been hypothesized to facilitate ET and modulate the molybdenum reduction potential.12,13 The pyranopterin ditholene is one of the most electronically complex ligands in biology,1,9−11,14 containing a redox noninnocent dithiolene,14,15 a pyran ring that can exist in both ring-opened16,17 and ring-closed forms, and a redox-active pterin ring system. The Mo ion is not covalently linked to the protein but is anchored via the pyranopterin dithiolene through an extensive hydrogen-bonding network with the protein. Recently, we showed that pyranopterin ditholene distortions can be correlated with enzyme function.11 As a result of this analysis, XO family enzymes are proposed to possess a tetrahydropyranopterin ditholene (Figure 1) that is intimately involved in the transfer of redox equivalents from Mo to the proximal 2Fe2S center. In spite of the intense interest in metallodithiolenes10 and, more specifically, in the complexity of the pyranopterin ditholene,9,10 there is a dearth of spectroscopic studies that have been directed toward understanding how the pyranopterin ditholene facilitates ET in XO/XDH. Although XO has been studied by resonance Raman (rR) spectroscopy,18,19 modes attributed to the pyranopterin ditholene have not been assigned. In order to address this issue, we have synthesized new XO/XDH reducing substrates that, when oxidized, bind tightly to the MoIV form of the enzyme. The MoIV–product bonding interaction results in the appearance of an intense near-infrared (NIR) metal-to-ligand (product) charge-transfer (MLCT) band in the electronic absorption spectrum.18 Specifically, we generated MoIV-product charge-transfer complexes for bovine XO and R. capsulatus XDH by the enzyme-catalyzed oxidation of 4-thiolumazine and 2,4-dithiolumazine to 4-thioviolapterin (4-TV) and 2,4-dithioviolapterin (2,4-TV), respectively, in a manner similar to that used for the seminal studies on violapterin.18,20 Alternatively, enzymatically generated product collected and concentrated by centrifugation/filtration, then incubated with reduced XO/XDH, generates the same MoIV-product MLCT complex, as evidenced by electronic absorption spectroscopy.
Figure 1.
Left: oxidized and reduced XO/XDH. Right: Reduced “tetrahydro” structure proposed for the pyranopterin dithiolene ligand in the XO family of enzymes. The metalated form of pyranopterin dithiolene is often referred to as the molybdenum cofactor, or Moco.
We anticipated that heavy-atom congeners of lumazine would result in bathochromic shifts of the MLCT absorption maximum relative to the analogous complex formed with lumazine.18,20 The bathochromic MLCT shift would result in high-quality rR data because it effectively eliminates the dominant higher-energy absorption contributions from the 2Fe2S and FAD centers and deleterious contributions from free FAD fluorescence. The electronic absorption spectra for Mo-product complexes with 4-TV (758 nm) and 2,4-TV (778 nm) possess NIR absorbance maxima that are red-shifted by ∼3000 and ∼4000 cm–1, respectively, relative to the lumazine MLCT complex (Figure 2).18,20 Because the MLCT transition derives from a Mo(xy) → product(π*) (HOMO → LUMO) one-electron promotion (Figure S3 in the Supporting Information, SI), optical pumping of this transition creates an excited state with appreciable MoV-P•– character, and interrogation of this MLCT state by rR spectroscopy (Figure 3 and Table 1) provides important information regarding the nature of low-frequency Mo-(pyranopterin ditholene) distortions that are coupled to a one-electron oxidation of the MoIV site. These are the same distortions anticipated for the MoIV → MoV ET event in the oxidative half-reaction of the enzyme, providing new insight into the extent to which the pyranopterin dithiolene is coupled into ET regeneration processes in XO/XDH.
Figure 2.
Room temperature electronic absorption difference spectra (Eproduct – Ered) for XOred-product complexes in N,N-bis(2-hydroxyethyl)glycine buffer at pH = 8.3. Inset: Structure of the enzyme–product complexes. E = O for 4-TV, and E = S for 2,4-TV.
Figure 3.
rR spectra for MLCT complexes of XDH (top) and XO (bottom). The rR spectra with 4-TV are depicted in red, and the 2,4-TV spectra are in black.
Table 1. Selected Vibrational Frequencies.
| MoIV-4-TV |
MoIV-2,4-TV |
|||||
|---|---|---|---|---|---|---|
| mode | expa | calca | expa | calca | ||
| dithiolene fold + Mo≡O rocking + pyranopterin ditholene | ν26 | 234 | 234 | ν28 | 234 | 242 |
| S–Mo–S symmetric core stretch | ν36 | 328 | 333 | ν36 | 326 | 331 |
| product in-plane ring stretching | ν50 | 493 | 487 | ν45 | 411 | 423 |
| ν51 | 513 | 501 | ||||
Vibrational frequencies in wavenumbers (cm–1).
Low-frequency rR spectra for reduced XOR and XDH product-bound species, collected on resonance with the MoIV-product MLCT band (780 nm excitation), are essentially identical (Figure 3). However, the spectra are dependent on the nature of the product molecule bound to molybdenum and display multiple resonantly enhanced vibrations in the 200–600 cm–1 region. Beause the sulfur heteroatoms are part of the heterocyclic π system of the product and the mass of sulfur is approximately twice that of oxygen, vibrations with appreciable product character will display frequency shifts relative to other resonantly enhanced modes due to force constant and reduced mass changes. This heavy-atom-congener approach, coupled with vibrational-frequency calculations, represents a powerful method for the assignment of vibrational modes that are localized on either the product half or the pyranopterin ditholene half of the MoIV-product complex. Product-dependent spectral differences are clearly apparent at Raman shifts greater than ∼375 cm–1. The experimental dependence of these vibrational bands on the nature of the product allows us to assign the MoIV-4-TV resonantly enhanced vibration at 493 cm–1 (Figure 4A) and the MoIV-2,4-TV enhanced vibrations at 411 and 513 cm–1 (Figure S2 in the SI) as in-plane product bending modes localized on the product.
Figure 4.
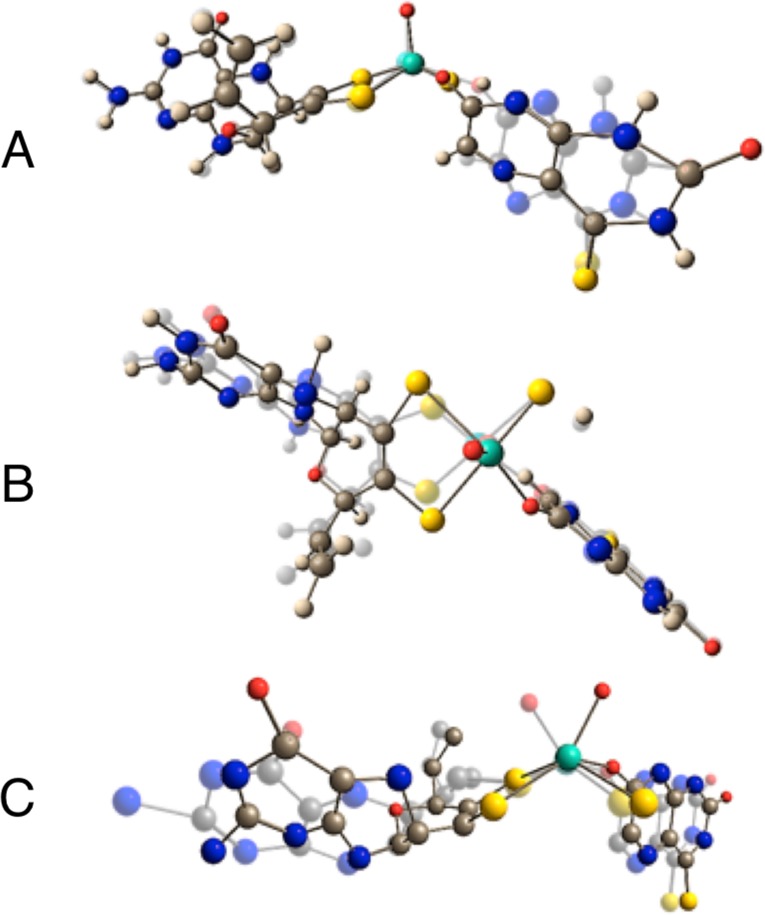
Key vibrational modes computed for MoIV-4-TV: (A) product in-plane ring bending; (B) S–Mo–S symmetric core stretch; (C) dithiolene fold + Mo≡O rocking + pyranopterin ditholene. Other normal modes for MoIV-2,4-TV and MoIV-4-TV are in the SI.
The rR spectra at frequencies lower than ∼375 cm–1 are virtually identical. Thus, these vibrational bands derive from modes localized primarily on the Mo-(pyranopterin ditholene) half of the MoIV-product complex because they are not dependent on the nature of the product. We assign the resonantly enhanced bands at 328 and 326 cm–1 in MoIV-4-TV and MoIV-2,4-TV, respectively, as the Mo-dithiolene core vibration that possesses S–Mo–S symmetric stretching and bending character (Figure 4B). To our knowledge, these are the first definitive assignments of a core Mo-dithiolene vibrational mode in a Mo-hydroxylase enzyme. Additional support for this assignment is based on polarized rR spectra of the benchmark Mo-ditholene complex Tp*Mo(bdt) (bdt = benzene-1,2-dithiolate), where the S–Mo–S symmetric stretch and bend are observed at 393 and 362 cm–1, respectively.21,22 Although low-frequency rR data for pyranopterin molybdenum enzymes are sparse, vibrational data for dimethyl sulfoxide reductase (DMSOR) have been collected and analyzed.23−27 In contrast to XO/XDH, DMSOR family enzymes possess two pyranopterin ditholenes bound to the Mo ion.28 In the reduced form, DMSORred displays S–Mo–S core vibrations at 352 and 383 cm–1, while for DMSORox, these vibrations have been assigned at 350 and 370 cm–1.25,29 Thus, the lower S–Mo–S frequencies for XO/XDH suggest a modified Mo–dithiolene bonding interaction compared with the DMSOR enzymes, and this may be associated with their different functions.
The vibrational band at 234 cm–1 (Figure 4C) is assigned as a dithiolene folding + Mo≡O rocking mode, with appreciable pyranopterin ditholene character. The observation of pyranopterin ditholene character in this mode indicates that the effect of instantaneous hole generation on Mo, induced by photoexcitation into the MLCT band, is felt at long distances from the Mo center. This supports the hypothesis that MoIV → MoV oxidation in the oxidative half-reaction of XO/XDH is also coupled to pyranopterin ditholene vibrational distortions, providing strong evidence for the direct involvement of the pyranopterin ditholene in enzymatic ET. A similarly large number of low-frequency vibrational modes have also been observed in the rR spectra of the blue copper proteins plastocyanin and azurin upon photoexcitation into the intense SCys → Cu ligand-to-metal charge-transfer band.30 This has been explained by kinematic coupling of the coordinated cysteine side chain with the Cu–SCys stretching coordinate, underscoring the importance of an ET pathway that involves the coordinated cysteine. Similarly, the four less intense vibrational modes in XO/XDH that are observed at frequencies below ∼375 cm–1 all likely possess Mo-(pyranopterin ditholene) character (Figures S1 and S2 in the SI) and further support the hypothesis that the pyranopterin ditholene functions as an effective conduit for ET between the reduced Mo center and the 2Fe2S clusters in the protein.
In summary, we have obtained high-quality rR spectra of two new MoIV-product complexes by exciting into the NIR MoIV → product MLCT band. The use of S/O substitution for the violapterin product carbonyl oxygen atoms enables the assignment of resonantly enhanced low-frequency Mo-(pyranopterin ditholene) modes. The observation of these modes provides evidence for the pyranopterin ditholene being coupled to redox changes at the Mo site and serving as an ET conduit in the oxidative half-reaction of XO/XDH.
Acknowledgments
M.L.K. acknowledges the National Institutes of Health for financial assistance (Grant GM 057378).
Supporting Information Available
Synthesis, spectroscopic and computational details, vibrational modes, and rR and absorption spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kirk M. L.; Stein B. In Comprehensive Inorganic Chemistry II, 2nd ed.; Jan R., Kenneth P., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; p 263. [Google Scholar]
- Hille R.; Hall J.; Basu P. Chem. Rev. 2014, 114, 3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enroth C.; Eger B.; Okamoto K.; Nishino T.; Nishino T.; Pai E. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truglio J.; Theis K.; Leimkuhler S.; Rappa R.; Rajagopalan K.; Kisker C. Structure 2002, 10, 115. [DOI] [PubMed] [Google Scholar]
- Basu P.; Burgmayer S. J. N. Coord. Chem. Rev. 2011, 255, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Electronic Structure and Spectroscopy of Metallo-Dithiolene Complexes; Kirk M. L., Helton M. E., McNaughton R. L., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, 2004; Vol. 52. [Google Scholar]
- Rothery R. A.; Stein B.; Solomonson M.; Kirk M. L.; Weiner J. H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. M.; Inscore F. E.; Hille R.; Kirk M. L. Inorg. Chem. 1999, 38, 4963. [DOI] [PubMed] [Google Scholar]
- Mtei R. P.; Lyashenko G.; Stein B.; Rubie N.; Hille R.; Kirk M. L. J. Am. Chem. Soc. 2011, 133, 9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz K. G.; Mtei R. P.; Rothstein R.; Kirk M. L.; Burgmayer S. J. N. Inorg. Chem. 2011, 50, 9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz K. G.; Mtei R. P.; Leung B.; Burgmayer S. J. N.; Kirk M. L. J. Am. Chem. Soc. 2010, 132, 7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloer D. P.; Hagel C.; Heider J.; Schulz G. E. Structure 2006, 14, 1377. [DOI] [PubMed] [Google Scholar]
- Bertero M. G.; Rothery R. A.; Palak M.; Hou C.; Lim D.; Blasco F.; Weiner J. H.; Strynadka N. C. J. Nat. Struct. Biol. 2003, 10, 681. [DOI] [PubMed] [Google Scholar]
- Hemann C.; Ilich P.; Stockert A. L.; Choi E. Y.; Hille R. J. Phys. Chem. B 2005, 109, 3023. [DOI] [PubMed] [Google Scholar]
- Maiti N. C.; Tomita T.; Kitagawa T.; Okamoto K.; Nishino T. J. Biol. Inorg. Chem. 2003, 8, 327. [DOI] [PubMed] [Google Scholar]
- Oertling W. A.; Hille R. J. Biol. Chem. 1990, 265, 17446. [PubMed] [Google Scholar]
- Inscore F. E.; McNaughton R.; Westcott B. L.; Helton M. E.; Jones R.; Dhawan I. K.; Enemark J. H.; Kirk M. L. Inorg. Chem. 1999, 38, 1401. [Google Scholar]
- Inscore F. E.; Knottenbelt S. Z.; Rubie N. D.; Joshi H. K.; Kirk M. L.; Enemark J. H. Inorg. Chem. 2006, 45, 967. [DOI] [PubMed] [Google Scholar]
- Gruber S.; Kilpatrick L.; Bastian N.; Rajagopalan K.; Spiro T. J. Am. Chem. Soc. 1990, 112, 8179. [Google Scholar]
- Kilpatrick L.; Rajagopalan K. V.; Hilton J.; Bastian N. R.; Stiefel E. I.; Pilato R. S.; Spiro T. G. Biochemistry 1995, 34, 3032. [DOI] [PubMed] [Google Scholar]
- Garton S. D.; Hilton J.; Oku H.; Crouse B. R.; Rajagopalan K. V.; Johnson M. K. J. Am. Chem. Soc. 1997, 119, 12906. [Google Scholar]
- Johnson M. K.; Garton S. D.; Oku H. J. Biol. Inorg. Chem. 1997, 2, 797. [Google Scholar]
- Schindelin H.; Kisker C.; Hilton J.; Rajagopalan K. V.; Rees D. C. Science 1996, 272, 1615. [DOI] [PubMed] [Google Scholar]
- Vibrational Spectra of Dithiolene Complexes; Johnson M. K., Ed.; John Wiley and Sons, Inc.: Hoboken, NJ, 2004; Vol. 52. [Google Scholar]
- Qiu D.; Kilpatrick L. T.; Kitajima N.; Spiro T. G. J. Am. Chem. Soc. 1994, 116, 2585. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.