

Abstract
The loss of β-cell function and β-cell death are key features of diabetes. A range of mechanisms are thought to contribute to β-cell loss, including islet amyloid formation by the neuropancreatic hormone amylin (islet amyloid polypeptide, IAPP). Islet amyloid deposition also contributes to the failure of islet transplants. There are no therapeutic strategies for the treatment or prevention of islet amyloidosis. Aspirin and the nonsteroid anti-inflammatory drug (NSAID) ketoprofen, at clinically relevant doses, have been proposed to inhibit amyloid formation by amylin and thus may hold promise for treatment of islet amyloidosis. These compounds are potentially attractive given the importance of inflammation in islet amyloidosis and given the fact that there are no anti-islet amyloid agents in the clinic. We show that aspirin, even in 20-fold excess, has no effect on the kinetics of amyloid formation by amylin as judged by thioflavin-T binding, right angle light scattering, and transmission electron microscopy, nor does it alter the morphology of resulting amyloid fibrils. Aspirin showed no ability to disaggregate preformed amylin amyloid fibrils under the conditions of these studies, 25 °C and pH 7.4. Ketoprofen is similarly ineffective at inhibiting amylin amyloid formation. The compounds do, however, interfere with circular dichroism- and Congo Red-based assays of amylin amyloid formation. This study highlights the importance of using multiple methods to follow amyloid formation when screening inhibitors.
Type 2 diabetes has reached epidemic proportions, and it is now recognized that β-cell death and β-cell dysfunction play important roles in the disease.1 A range of mechanisms contribute to β-cell loss and dysfunction in vivo, including inflammation and the deposition of amyloid in the islets of Langerhans.2−5 Rapid formation of amyloid also leads to islet graft failure, whereas its prevention has been shown to prolong graft survival and lead to improved glycemic control.6,7 The neuropancreatic hormone amylin is responsible for islet amyloid formation. Amylin plays a role in controlling food intake, gastric emptying, and glucose homeostasis, but it aggregates to form islet amyloid in type 2 diabetes. Amylin is stored in the insulin secretory granule and is thus released in response to insulin secretion.8
Analogues of human amylin that are less aggregation prone than wild-type human amylin have been approved as an adjunct to insulin therapy,9 but there is no treatment for islet amyloidosis, and there are no approved therapeutic strategies to prevent islet amyloid deposition. The search for inhibitors of amyloid aggregation and amyloid formation is an active area of research,10−17 but comparatively few anti-amylin amyloid compounds have been developed, and the vast majority of those are not drug-like. Recently, the intriguing possibility that clinically relevant doses of aspirin and the nonsteroid anti-inflammatory drug (NSAID) ketoprofen may inhibit amylin amyloid formation and might disaggregate preformed amylin amyloid fibrils has been raised.18 This could open very attractive, inexpensive therapeutic approaches if the compounds were indeed effective anti-amylin amyloid agents, particularly because inflammation is believed to play a role in islet amyloidosis toxicity and a central role in type-2 diabetes.19−24 Here, we critically examine the effects of aspirin and ketoprofen on amylin amyloid formation and the effects of aspirin on preformed amylin amyloid fibrils. Aspirin does not inhibit amylin amyloid formation, even when added at 20-fold excess, and is unable to disassemble preformed amylin amyloid. Ketoprofen is similarly ineffective at inhibiting amyloid formation. The reasons for the discrepancy with prior reports are examined, and it is concluded that they are due to interference of the compounds with the analytical assays used and with the difficulty in analyzing small changes in circular dichroism (CD) spectra. The implications for the testing of amyloid inhibitors are discussed. The present study emphasizes the necessity of employing multiple techniques to avoid false positives or false negatives in inhibition assays.
Results and Discussion
Mature, fully processed amylin is 37 residues in length, contains a disulfide bond between residues 2 and 7, and has an amidated C-terminus (Figure 1). We first examined the effects of aspirin on the kinetics of amyloid formation using fluorescence-detected thioflavin-T binding assays. This is the standard assay in this field. Thioflavin-T is a small fluorescent dye that binds to the cross β-structure of amyloid fibrils, presumably in the surface grooves formed by the parallel β-sheets. Binding constrains the conformation of the dye and relieves self-quenching, resulting in an increase in quantum yield.25 Figure 2 displays the results of kinetic experiments conducted in the presence of aspirin. The expected sigmoidal time course is observed in the absence of aspirin, with a T50, defined as the time required to reach half of the total signal change in the thioflavin-T assay, of 20 h. Addition of aspirin, up to even a 20-fold excess, had no detectable effect on the rate of amyloid formation, as judged by the values of T50. The compound also had no detectable effect on the final thioflavin-T intensity. Thioflavin-T binding assays are indirect because they rely on the binding of an extrinsic probe and can sometimes give misleading results,26 but they do have the advantage that they report on the kinetics of amylin amyloid formation in the absence of conflicting factors. We also used transmission electron microscopy (TEM) to monitor the effects of aspirin. Aliquots were removed from each sample at the end of kinetic experiments, blotted onto TEM grids, and imaged. Extensive mats of fibrils were observed in the sample of amylin alone and in all of the samples that contained aspirin.
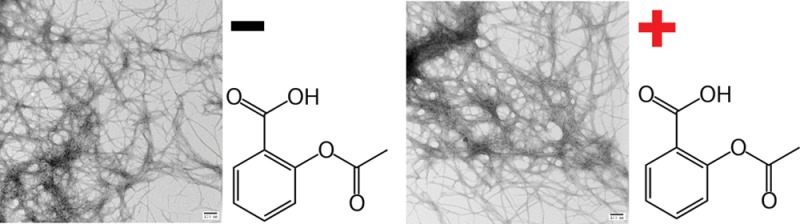
Figure 1.

Primary sequence of human amylin and the structure of aspirin and ketoprofen. Amylin contains a disulfide bridge between residues 2 and 7, and the C-terminus is amidated.
Figure 2.
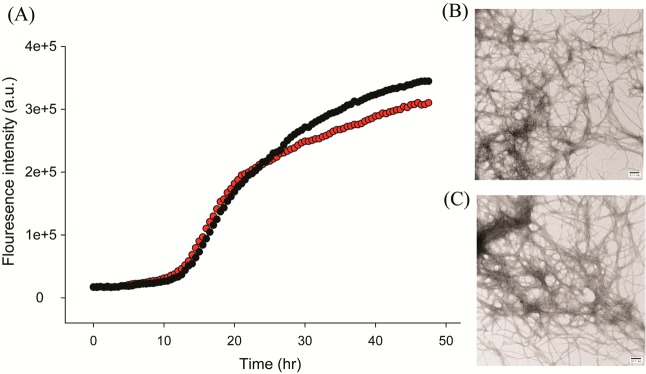
Aspirin does not inhibit amyloid formation by human amylin. (A) Thioflavin-T fluorescence assays of the time course of amyloid formation in the absence (black) and presence (red) of a 20-fold excess of aspirin. (B) TEM image of the sample of amylin without aspirin recorded at the end of the kinetic experiment. (C) TEM image of the sample of amylin with a 20-fold excess aspirin recorded at the end of the kinetic experiment. Scale bars represent 100 nm. Experiments were conducted at pH 7.4 and 25 °C in 20 mM Tris buffer with 0.25% DMSO (v/v) in the absence of any fluorinated alcohol cosolvent. The concentration of amylin was 16 μM, and the concentration of aspirin was 320 μM.
Initial reports of the ability of aspirin to inhibit amylin amyloid formation did not use thioflavin-T assays or TEM, but rather used CD and Congo red binding assays. It is possible, although unlikely, that thioflavin-T could displace aspirin from amylin and interfere with its effects. This does not seem plausible because thioflavin-T has no such effect on a wide range of other inhibitors27,28 and does not bind to preamyloid intermediates. Nonetheless, we also tested the effects of aspirin in the absence of thioflavin-T using right angle light scattering (RALS) and TEM. Similar aggregation curves are observed in the presence and absence of a 20-fold excess of aspirin using RALS (Figure 3). TEM of the samples reveals the presence of dense mats of fibrils in each sample. Note that these samples did not contain thioflavin-T.
Figure 3.
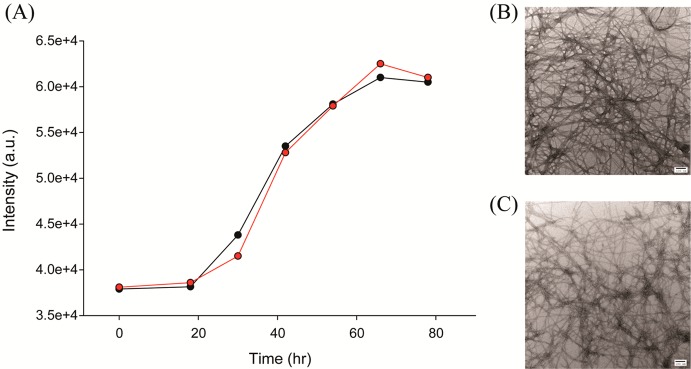
Aspirin does not inhibit amyloid formation by human amylin. (A) Right angle light scattering assays of the time course of amyloid formation in the absence (black) and in the presence (red) of a 20-fold excess of aspirin. No thioflavin-T was added to either sample. (B) TEM image of the sample of amylin without aspirin recorded at the end of the kinetic experiment. (C) TEM image of the sample of amylin with a 20-fold excess aspirin recorded at the end of the kinetic experiment. Scale bars represent 100 nm. Experiments were conducted at pH 7.4 and 25 °C in 20 mM Tris buffer with 0.25% DMSO (v/v) in the absence of any fluorinated alcohol cosolvent. The concentration of amylin was 16 μM, and the concentration of aspirin was 320 μM.
We next examined the ability of aspirin to disaggregate preformed amylin amyloid fibrils. Some, but not all, inhibitors of amyloid formation have this property. We monitored amyloid formation using thioflavin-T assays (Figure 4) and then added a 20-fold excess of aspirin after amyloid formation was complete and the reaction had reached the saturation phase. Aliquots were removed for TEM analysis just before addition of aspirin, immediately afterward, and 48 h later. Addition of the compound did not perturb the thioflavin-T time course; in contrast, compounds that disaggregate amyloid fibrils lead to a decay in the thioflavin-T signal as a function of time.10 The TEM images recorded before and after addition of aspirin are very similar and reveal extensive deposits of amyloid fibrils, confirming that the compound does not disaggregate amylin amyloid (Figure 4B–D). In addition, CD spectra recorded before and after the addition of aspirin have an identical shape, consistent with a high degree of β-structure, although there is a modest decrease in intensity. Furthermore, no changes in the CD spectra are observed upon further incubation of up to 12 h (Supporting Information). The CD studies are fully consistent with thioflavin-T, TEM, and RALS experiments.
Figure 4.

Aspirin does not disaggregate preformed human amylin amyloid fibrils. (A) Thioflavin-T fluorescence assays of the time course of amyloid formation. A 20-fold excess of aspirin was added at the time point indicated by the red arrow. (B) TEM image of the sample just before the addition of a 20-fold excess aspirin (indicated by the black star). (C) TEM image of the sample just after the addition of a 20-fold excess aspirin (indicated by the green star). (D) TEM image of the sample 48 h after the addition of 20-fold excess aspirin (indicated by the blue star). Scale bars represent 100 nm. Experiments were conducted at pH 7.4 and 25 °C in 20 mM Tris buffer with 0.25% DMSO (v/v) in the absence of any fluorinated alcohol cosolvent. The concentration of amylin was 16 μM, and the concentration of aspirin was 320 μM.
The NSAID ketoprofen has also been proposed to be an inhibitor of amylin amyloid formation, again on the basis of Congo red assays and CD spectroscopy. We examined the ability of the compound to inhibit amyloid formation by human amylin using TEM to test its effect on amyloid formation directly (Figure 5). TEM analysis revealed the presence of extensive mats of amyloid fibrils in all samples, showing that the compound is not an amylin amyloid inhibitor.
Figure 5.

Ketoprofen does not inhibit amyloid formation by human amylin. TEM images of samples recorded after incubating amylin with varying amount of ketoprofen for 42 h. Scale bars represent 100 nm. (A) Amylin alone. (B) Mixture of amylin with a 20-fold excess of ketoprofen. (C) Mixture of amylin with a 10-fold excess of ketoprofen. (D) Mixture of amylin with a 5-fold excess of ketoprofen. (E) Mixture of amylin with a 2-fold excess of ketoprofen. (F) Mixture of amylin with an equimolar amount of ketoprofen. Experiments were conducted at pH 7.4 and 25 °C in 20 mM Tris buffer with 0.25% DMSO (v/v) in the absence of any fluorinated alcohol cosolvent. The concentration of amylin was 16 μM.
Conclusions
The data presented here show that aspirin and ketoprofen do not inhibit amylin amyloid formation under the conditions used, pH 7.4 and 25 °C. Why do the conclusions of this study differ from previous work? The earlier studies used trifluoroethanol (TFE) to promote amyloid formation. TFE and hexafluoroisopropanol (HFIP) stabilize secondary structure of peptides, and even a modest amount of HFIP or TFE can accelerate amyloid formation.29 The use of a nonaqueous solvent to induce amyloid formation may contribute to the different conclusions, but the methods used also play a role. The previous study made use of CD and absorbance-detected Congo red binding assays. The reported CD spectra are different in the presence of high concentrations of aspirin; however, the reported spectrum, even at the highest concentration of aspirin, is not that of a random coil and has a shape consistent with significant β-sheet intensity. In addition, the absorbance of aspirin and ketoprofen in the range of 200–250 nm can interfere with CD measurements. We employed different conditions to induce amyloid formation here and observed no change in the shape of the CD spectrum upon the addition of aspirin to preformed amyloid fibrils. Congo red binding was also used to test for the presence of amyloid in the original studies. Congo red staining is a classic method to probe amyloid formation, particularly for ex vivo amyloid deposits, and usually involves monitoring birefringence, but the absorbance-based assays are also employed. In either case, the dye is an extrinsic probe, and it has been shown that it is not amyloid specific.30 In the case of absorbance assays, addition of compounds can interfere by contributing background absorbance or by interfering with the binding of the dye. These considerations and the data presented here highlight the importance of using multiple probes to study amyloid inhibition, particularly methods such as TEM, which directly detect amyloid fibrils.
Methods
Peptide Synthesis and Purification
Human amylin was synthesized on a 0.1 mmol scale using a CEM Liberty microwave peptide synthesizer utilizing Fmoc chemistry. Solvents used were ACS-grade. The methods have been described previously.31,32 In order to afford a peptide with an amidated C-terminus, 5-(4′-fmoc-aminomethyl-3′,5-dimethoxyphenol) valeric acid (Fmoc-PAL-PEG-PS) resin was used and purchased from Life Technologies. Standard Fmoc reaction cycles were used. Fmoc-protected pseudoproline dipeptide derivatives were incorporated at positions 9–10, 19–20, and 27–28 to facilitate the synthesis. The β-branched residues, Arg, and all pseudoproline dipeptide derivatives were double-coupled. A maximum temperature of 50 °C was used for the coupling of His and Cys in order to reduce the possibility of racemization. Peptides were cleaved from the resin by standard trifluoroacetic acid (TFA) methods; ethanedithiol, thioanosole, and anisole were used as scavengers. Crude peptides were partially dissolved in 20% acetic acid (v/v), frozen in liquid nitrogen, and lyophilized to increase their solubility. The dry peptide was redissolved in 100% dimethyl sulfoxide (DMSO) at room temperature to promote the formation of the disulfide bond.33,34 Peptides were purified by reverse-phase HPLC using a Proto 300 C18 preparative column (10 mm × 250 mm). A two-buffer gradient was used: buffer A consisted of 100% H2O and 0.045% HCl (v/v) and buffer B included 80% acetonitrile, 20% H2O, and 0.045% HCl. HCl was used as the counterion instead of TFA because residual TFA can influence amyloid formation. MALDI-TOF mass spectrometry confirmed the correct molecular weight (expected, 3903.3 Da; observed, 3902.8 Da).
Sample Preparation
Human amylin was first dissolved in 100% HFIP at a concentration of 1.6 mM and then filtered to remove any preformed amyloid aggregates. For thioflavin-T fluorescence assays, aliquots were lyophilized and redissolved in 20 mM Tris buffer, pH 7.4, at the desired concentration. Aspirin and ketoprofen were prepared in 100% DMSO.
Thioflavin-T Fluorescence Assays
Solutions were prepared by adding 20 mM Tris buffer, pH 7.4, and thioflavin-T to lyophilized dry peptides for a final peptide concentration of 16 μM. For the studies of aspirin and ketoprofen, 0.25% DMSO was present in the solution. Measurements were made at 25 °C using a Beckman Coulter DTX880 plate reader without stirring. An excitation filter of 430 nm and an emission filter of 485 nm were used. To test the potential disaggregation activity of aspirin, peptide was first incubated in a low-binding 96-well plate and monitored using a plate reader to ensure the formation of amyloid fibrils. Aspirin was added at a 20-fold excess after amyloid formation.
Right Angle Light Scattering Assays (RALS)
Solutions were prepared by adding 20 mM Tris buffer, pH 7.4, without thioflavin-T to lyophilized dry peptides for a final peptide concentration of 16 μM. Experiments were conducted using an Applied Phototechnology fluorescence spectrophotometer. RALS assays used excitation and emission wavelengths of 500 nm.
Transmission Electron Microscopy (TEM)
TEM was performed at the Life Science Microscopy Center at Stony Brook University. Aliquots were removed from the same solutions that were used for the fluorescence measurements. Five microliters of peptide solution was placed on carbon-coated Formvar 300 mesh copper grid for 1 min and then negatively stained by incubation with saturated uranyl acetate for another 1 min.
Circular Dichroism (CD) Experiments
CD experiments were performed using an Applied Photophysics Chirascan circular dichroism spectrometer. The solutions for the CD experiments were prepared by diluting the filtered stock peptide solutions into 20 mM Tris buffer at pH 7.4. The final concentration of peptide was 16 μM in 1% HFIP. Spectra were recorded from 198 to 260 at 1 nm intervals in a quartz cuvette with a 0.1 cm path length at 25 °C. Data were averaged from three scans. A background spectrum was subtracted from the collected data.
Acknowledgments
This work was supported by NIH grant GM078114 (D.P.R). We thank M. Watson and O. Levsh for helpful discussions.
Supporting Information Available
CD spectra of pre-formed amyloid fibrils recorded before the addition of aspirin, immediately after the addition of aspirin, 1 h after the addition of aspirin, and 12 h after the addition of aspirin. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ashcroft F. M.; Rorsman P. (2012) Diabetes mellitus and the beta cell: The last ten years. Cell 148, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath M. Y.; Shoelson S. E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107. [DOI] [PubMed] [Google Scholar]
- Clark A.; Wells C. A.; Buley I. D.; Cruickshank J. K.; Vanhegan R. I.; Matthews D. R.; Cooper G. J. S.; Holman R. R.; Turner R. C. (1988) Islet amyloid, increased alpha-cells, reduced beta-cells and exocrine fibrosis-quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 9, 151–159. [PubMed] [Google Scholar]
- Lorenzo A.; Razzaboni B.; Weir G. C.; Yankner B. A. (1994) Pancreatic-islet cell toxicity of amylin associated with type-2 diabetes-mellitus. Nature 368, 756–760. [DOI] [PubMed] [Google Scholar]
- Konarkowska B.; Aitken J. F.; Kistler J.; Zhang S. P.; Cooper G. J. S. (2006) The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS. J. 273, 3614–3624. [DOI] [PubMed] [Google Scholar]
- Westermark G. T.; Westermark P.; Berne C.; Korsgren O. (2008) Widespread amyloid deposition in transplanted human pancreatic islets. N. Engl. J. Med. 359, 977–979. [DOI] [PubMed] [Google Scholar]
- Potter K. J.; Abedini A.; Marek P.; Klimek A. M.; Butterworth S.; Driscoll M.; Baker R.; Nilsson M. R.; Warnock G. L.; Oberholzer J.; Bertera S.; Trucco M.; Korbutt G. S.; Fraser P. E.; Raleigh D. P.; Verchere C. B. (2010) Islet amyloid deposition limits the viability of human islet grafts but not porcine islet grafts. Proc. Natl. Acad. Sci. U.S.A. 107, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S. E.; Dalessio D. A.; Schwartz M. W.; Fujimoto W. Y.; Ensinck J. W.; Taborsky G. J.; Porte D. (1990) Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 39, 634–638. [DOI] [PubMed] [Google Scholar]
- Hollander P. A.; Levy P.; Fineman M.; Maggs D. G.; Shen L. Z.; Strobel S. A.; Weyer C.; Kolterman O. G. (2003) Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes – A 1-year randomized controlled trial. Diabetes Care 26, 784–790. [DOI] [PubMed] [Google Scholar]
- Cao P.; Raleigh D. P. (2012) Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 51, 2670–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F. L.; Raleigh D. P. (2011) Inhibition of glycosaminoglycan-mediated amyloid formation by islet amyloid polypeptide and proIAPP processing intermediates. J. Mol. Biol. 406, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat Y.; Mazor Y.; Efrat S.; Gazit E. (2004) Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry 43, 14454–14462. [DOI] [PubMed] [Google Scholar]
- Meng F. L.; Raleigh D. P.; Abedini A. (2010) Combination of kinetically selected inhibitors in Trans leads to highly effective inhibition of amyloid formation. J. Am. Chem. Soc. 132, 14340–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R.; Sellin D.; Radovan D.; Gohlke A.; Winter R. (2009) Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. ChemBioChem 10, 445–449. [DOI] [PubMed] [Google Scholar]
- Kapurniotu A.; Schmauder A.; Tenidis K. (2002) Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J. Mol. Biol. 315, 339–350. [DOI] [PubMed] [Google Scholar]
- Porat Y.; Abramowitz A.; Gazit E. (2006) Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 67, 27–37. [DOI] [PubMed] [Google Scholar]
- Noor H.; Cao P.; Raleigh D. P. (2012) Morin hydrate inhibits amyloid formation by islet amyloid polypeptide and disaggregates amyloid fibers. Protien Sci. 21, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T.; Nadackal G. T.; Thomas K. (2003) Aspirin and diabetes: Inhibition of amylin aggregation by nonsteroidal anti-inflammatory drugs. Exp. Clin. Endocrinol. Diabetes 111, 8–11. [DOI] [PubMed] [Google Scholar]
- Ehses J. A.; Perren A.; Eppler E.; Ribaux P.; Pospisilik J. A.; Maor-Cahn R.; Gueripel X.; Ellingsgaard H.; Schneider M. K.; Biollaz G.; Fontana A.; Reinecke M.; Homo-Delarche F.; Donath M. Y. (2007) Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370. [DOI] [PubMed] [Google Scholar]
- Donath M. Y.; Shoelson S. E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107. [DOI] [PubMed] [Google Scholar]
- Masters S. L.; Dunne A.; Subramanian S. L.; Hull R. L.; Tannahill G. M.; Sharp F. A.; Becker C.; Franchi L.; Yoshihara E.; Chen Z.; Mullooly N.; Mielke L. A.; Harris J.; Coll R. C.; Mills K. H.; Mok K. H.; Newsholme P.; Nuñez G.; Yodoi J.; Kahn S. E.; Lavelle E. C.; O’Neill L. A. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 11, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwell-Roper C. Y.; Ehses J. A.; Verchere C. B. (2014) Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes 63, 1698–1711. [DOI] [PubMed] [Google Scholar]
- Goldfine A. B.; Silver R.; Aldhahi W.; Cai D. S.; Tatro E.; Lee J.; Shoelson S. E. (2008) Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. CTS: Clin. Transl. Sci. 1, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y.; Dobrian A. D.; Morris M. A.; Nadler J. L. (2013) Islet inflammation: a unifying target for diabetes treatment?. Trends Endocrinol. Metab. 24, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine H. (1995) Thioflavine-T interaction with amyloid beta-sheet structures. Amyloid 2, 1–6. [Google Scholar]
- Meng F. L.; Marek P.; Potter K. J.; Verchere C. B.; Raleigh D. P. (2008) Rifampicin does not prevent amyloid fibril formation by human islet amyloid polypeptide but does inhibit fibril thioflavin-T interactions: Implications for mechanistic studies beta-cell death. Biochemistry 47, 6016–6024. [DOI] [PubMed] [Google Scholar]
- Marek P.; Gupta R.; Raleigh D. P. (2008) The fluorescent amino acid p-cyanophenylalanine provides an intrinsic probe of amyloid formation. ChemBioChem 9, 1372–1374. [DOI] [PubMed] [Google Scholar]
- Cao P.; Tu L. H.; Abedini A.; Levsh O.; Akter R.; Patsalo V.; Schmidt A. M.; Raleigh D. P. (2012) Sensitivity of amyloid formation by human islet amyloid polypeptide to mutations at residue 20. J. Mol. Biol. 421, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi K.; Ashizaki M.; Yagi H.; Sakurai K.; Lee Y. H.; Goto Y. (2011) Hexafluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J. Biol. Chem. 286, 23959–23966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R.; Uversky V. N.; Nielsen L.; Fink A. L. (2001) Is Congo red an amyloid-specific dye?. J. Biol. Chem. 276, 22715–22721. [DOI] [PubMed] [Google Scholar]
- Abedini A.; Raleigh D. P. (2005) Incorporation of pseudoproline derivatives allows the facile synthesis of human IAPP, a highly amyloidogenic and aggregation-prone polypeptide. Org. Lett. 7, 693–696. [DOI] [PubMed] [Google Scholar]
- Marek P.; Woys A. M.; Sutton K.; Zanni M. T.; Raleigh D. P. (2010) Efficient microwave-assisted synthesis of human islet amyloid polypeptide designed to facilitate the specific incorporation of labeled amino acids. Org. Lett. 12, 4848–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Singh G.; Raleigh D. P. (2006) Recovery and purification of highly aggregation-prone disulfide-containing peptides: Application to islet amyloid polypeptide. Anal. Biochem. 351, 181–186. [DOI] [PubMed] [Google Scholar]
- Tam J. P.; Wu C. R.; Liu W.; Zhang J. W. (1991) Disulfide bond formation in peptides by dimethyl-sulfoxide – Scope and applications. J. Am. Chem. Soc. 113, 6657–6662. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.