

Summary
This study identified a novel pathway consisting of miR-551b/catalase/ROS that results in MUC1 overexpression in lung cancer cells with acquired chemoresistance. MUC1 expression promoted EGFR-mediated cell survival signaling. Intervention against this pathway could be exploited to overcome acquired chemoresistance.
Abstract
Acquired chemoresistance is a major challenge in cancer therapy. While the oncoprotein Mucin-1 (MUC1) performs multiple roles in the development of diverse human tumors, whether MUC1 is involved in acquired chemoresistance has not been determined. Using an acquired chemoresistance lung cancer cell model, we show that MUC1 expression was substantially increased in cells with acquired apoptosis resistance (AR). Knockdown of MUC1 expression effectively increased the sensitivity of these cells to the apoptotic cytotoxicity of anticancer therapeutics, suggesting that MUC1 contributes to acquired chemoresistance. Decreased catalase expression and increased cellular reactive oxygen species (ROS) accumulation were found to be associated with MUC1 overexpression. Scavenging ROS with butylated hydroxyanisole or supplying exogenous catalase dramatically suppressed MUC1 expression through destabilizing MUC1 protein, suggesting that reduced catalase expression mediated ROS accumulation is accounted for MUC1 overexpression. Further, we found that increased miR-551b expression in the AR cells inhibited the expression of catalase and potentiated ROS accumulation and MUC1 expression. Finally, by manipulating MUC1 expression, we found that MUC1 promotes EGFR-mediated activation of the cell survival cascade involving Akt/c-FLIP/COX-2 in order to protect cancer cells from responding to anticancer agents. Thus, our results establish a pathway consisting of miR-551b/catalase/ROS that results in MUC1 overexpression, and intervention against this pathway could be exploited to overcome acquired chemoresistance.
Introduction
While tremendous effort has been devoted to improving cancer chemotherapy, the mortality of lung cancer patients has not been significantly reduced. Chemoresistance is the major hindrance diminishing the anticancer efficacy of chemotherapeutics (1). Although many patients initially respond to chemotherapy, acquired chemoresistance arises rapidly resulting in therapy failure (2,3). Notably, in cancer cells with chemoresistance where chemotherapeutics lose their anticancer activity, they also promote cancer progression, converting anticancer agents into tumor promoters (4). Importantly, lung cancer cells with resistance to one drug may also become resistant to other anticancer therapeutics (4–8). It is believed that chemotherapeutics kill cancer cells mainly through the activation of apoptosis; and apoptosis resistance substantially contributes to chemoresistance (9,10). Thus, elucidating the mechanisms for apoptosis resistance and acquired chemoresistance is highly significant for improving the survival of lung cancer patients.
The O-glycosylated, membrane-bound protein Mucin-1 (MUC1) is expressed on the apical cellular membrane of bronchial epithelium and is induced by airway inflammation. During bacterial respiratory tract infection, MUC1 is important in controlling the extent of inflammation (11,12). The MUC1 gene encodes a single polypeptide precursor that is processed into two subunits to generate the mature MUC1 protein. While the N-terminal subunit, containing highly conserved repeats of 20 amino acids is highly O-glycosylated and secreted during inflammation, the transmembrane C-terminal subunit containing 72 amino acid residues binds to various proteins involved in signal transduction (13,14). Known as a tumor antigen, MUC1 is aberrantly overexpressed in various cancers with loss of its apical polarity (15–17). MUC1 is overexpressed in non-small cell lung cancer and is correlated with poor patient survival (18). Numerous cellular proteins implicated with MUC1 are involved in the malignancy of cancer cells and their resistance to chemotherapy (14). MUC1 also regulates microRNA expression for prostate cancer progression (19). We recently found that chronic cigarette smoke exposure induces persistent MUC1 overexpression in human lung bronchial epithelial cells, which facilitates cigarette smoke-induced cell transformation through EGFR-mediated cell survival signaling (20). In addition, MUC1 mediates cigarette smoke extract-induced TNFα secretion from macrophages to engender a lung cancer-prone microenvironment (21), suggesting that MUC1 functions in both lung macrophages and bronchial epithelial cells for lung cancer development. However, direct evidence for the role and mechanisms of MUC1 in acquired chemoresistance in lung cancer is lacking.
MicroRNAs (miRNAs) are small single-stranded non-coding RNA molecules that regulate gene expression mainly at the post-transcriptional level. By base-pairing to complementary sequences within mRNA, the miRNA silences gene expression through the repression of mRNA translation (22). While miRNAs are widely involved in various malignant properties of cancers and regulation of MUC1 expression with miRNA has been reported (23–25), miRNA regulation of MUC1 function in apoptosis and chemotherapy-response has not been well elucidated.
In this report, with use of an acquired chemoresistance cell model, we obtained evidence showing that MUC1 contributes to acquired chemoresistance in human lung cancer cells. Overexpression of MUC1 is associated with acquired apoptosis- and chemo-resistance involving EGFR-mediated cell survival signaling. We further identify a pathway resulting in MUC1 overexpression, consisting of miR-551b/catalase/ROS, which could be exploited as an intervention target for overcoming acquired chemoresistance.
Materials and methods
Reagents
Glutathione S-transferase (GST) -TRAIL was prepared as previously described (26). Butylated hydroxyanisole (BHA), catalase from bovine liver, Cycloheximide (CHX), Chloroquine (CQ) and MG132 were purchased from Sigma–Aldrich (St Louis, MO). Small interfering RNA (siRNA; SiGenome SMARTpool) for MUC1, miR-551b and negative control siRNA were purchased from Dharmacon. The primary antibody against MUC1 (GP1.4) was from Santa Cruz Biotechnology. MUC1 Ab-5, a hamster monoclonal antibody that recognizes the MUC1 CT domain was purchased from Lab Vision (Fremont, CA). The antibody against β-actin was from Sigma–Aldrich. The antibody against phospho-EGFR (Y1068) was purchased from Abcam. Antibodies against ERK, and phospho-ERK (Y185/187), and phospho-Akt (Ser 473) were from Invitrogen. Antibodies against EGFR and Akt were from Cell Signaling Technology. Antibodies against c-FLIP and Mcl-1 were from Alexis and BioVision, respectively. Antibodies against poly (ADP-ribose) polymerase (PARP), caspase 8 and caspase 3 were from BD Biosciences. 5-(and -6)-chloromethyl-2′, 7′-dichlorodihydro-fluorescein diacetate acetyl ester (CM-H2DCFDA) was purchased from Molecular Probes (Eugene, OR). EGFR inhibitor II (a selective and irreversible inhibitor that blocks EGFR autophosphorylation), PI3 kinase (PI3K)/Akt inhibitor LY294002 (LY), ERK inhibitor U0126, IκB kinase (IKK) inhibitor (SC-514) and COX-2 inhibitor were purchased from Calbiochem. pSG5-GST plasmid was a kind gift of Dr Seishi Murakami from Kanazawa University (27). To construct pSG5-GST-MUC1 CT, an oligonucleotide containing a Hind III site (5′-GGG GGA TCC CTC GAG CTG CAG AAG CTT GAT ATC GCG GCC GCA GAT CTT TT-3′) was first inserted into the Bam HI site of the pSG5-GST plasmid, followed by insertion of the MUC1 CT fragment between the BamH I and Hind III sites. The MUC1 CT fragment was synthesized by PCR amplification with the human MUC1 expressing vector as a template and the hMUC1 CT primers (hMUC1 CT forward: 5′-TGG GGA TCC TGT CAG TGC CGC CGA AAG AAC-3′; hMUC1 CT reverse: 5′-TTT AAG CTT CAA GTT GGC AGA AGT GGC TGC-3′). The construct was verified by DNA sequencing and the expressed protein was verified by Western blot with antibodies against either GST or MUC1 CT (data not shown). The MUC1 shRNA plasmid was constructed by inserting a synthetic oligonucleotide encoding a hairpin sequence with a 19-nucleotide stem that is homologous to the target sequence of human MUC1, CCGGGATACCTACCATCCTAT and a 9-base loop sequence into pSilencer (Oligoengine, Seattle, WA) and verified by DNA sequencing.
Cell culture and establishment of apoptosis-resistant cells
The human lung cancer cell lines A549 and H460 were obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2mM of L-glutamine, 100U/ml of penicillin and 100μg/ml of streptomycin. A549- and H460-AR cells were established by continuous exposure of the cells to gradual increased doses of GST-TRAIL as previously described (7,8,28).
Transfections
For transfection of siRNA, A549- and H460-AR cells were seeded in 12-well plates at about 50% confluence and cultured overnight. The cells were transfected with MUC1, miR-551b, or the control small interfering RNA (siRNA) with INTERFERin™ siRNA transfection reagent (Polyplus-transfection) according to manufacturer’s instructions. MUC1 protein level was determined by Western blot 48h after transfection.
For stable transfection, A549- and H460-AR cells were transfected with control shRNA and MUC1 shRNA with FuGENE HD transfection reagent (Promega). Stable transfected clones were selected and maintained in medium containing hygromycin (25 µg/ml). The cells were co-transfected with a mixture of GST-MUC1 or GST control plasmid and pcDNA His 3.1B plasmid (wt/wt = 10:1) using FuGENE HD transfection reagent. pcDNA His 3.1B plasmid was used to expressing neomycin for selection. Stable transfected clones were selected and maintained in medium containing G418 (200 µg/ml).
Lentiviral vector infections
The V5-catalase expressing lentiviral vector was from Biodesign Institute and viruses were produced and packaged in HEK293T cells following the manufacturer’s instruction (29). A lentiviral vector having no ectopic protein expression was used as a negative control. Twenty-four hours post-infection, the cells were treated and assayed as described in figures.
Cytotoxicity assay
Cytotoxicity was assessed using a lactate dehydrogenase (LDH) release based cytotoxicity detection kit (Promega, Madison, WI). Cells were seeded in 48-well plates at 50–60% confluence, cultured overnight and then treated as indicated in the figures. Cell death based on the release of LDH was calculated as described previously (30).
Detection of ROS
A549- or H460-WT and -AR cells were seeded in 12-well plates at about 80% confluence and cultured overnight. The fluorinated dye CM-H2DCFDA (5 μM) was applied to the culture for 30–60min after which cells were washed twice with cold PBS and harvested. To investigate the effects of exogenous catalase and BHA on ROS levels, A549- and H460-AR cells were treated with catalase or BHA for 1 h and then cells were stained with CM-H2DCFDA. ROS levels were measured using a fluorescence plate reader with excitation wavelength at 485nm and emission wavelength at 535nm. The relative fluorescence intensity was normalized to the total protein concentration of each sample (31). The results were presented as mean ± SD which was plotted for three replicates from each condition.
Reverse transcription-polymerase chain reaction
Total RNA was extracted from each sample using TRIzol® Reagent (Life Technologies). Two micrograms of RNA were used as a template for cDNA synthesis with a reverse transcription kit (Promega). An equal volume of cDNA product was subjected to reverse transcription–polymerase chain reaction (RT–PCR) analysis according to our previous study (21). The following primers were used in the PCR reactions: MUC1, forward primer 5′-ACAATTGACTCTGGCCTTCCG-3′ and reverse primer 5′-TGGG TTTGT GTA AG AGAGGCT-3′; β-actin, forward primer: 5′-CCA GCCTTCCTTCCTGGGCAT-3′ and reverse primer 5′-AGGAGCAATGATCT TGATCTTCATT-3′; Catalase, forward primer 5′- GGGAGAAGGCAAATCT GTGA-3′ and reverse primer 5′- GCACA TCTA GCAC AGGA GAAT-3′. For MUC1 and catalase, the PCR cycles were 32 and 42, respective, while for β-actin the cycles were 23. The amplified PCR products were resolved on 2% agarose gels with 0.5 μg/ml ethidium bromide, visualized and photographed.
Quantitative real-time PCR (RT–qPCR) was carried out with the ABI PRISM 7900HT, using Power SYBR Green PCR Master Mix (Applied Biosystems). Results were generated in triplicate independent experiments. The sequence of miR-551b primer is: 5′-GCGACCCATACTTGGTTTCAG-3′. Gene expression was quantified using the 2−ΔΔCt method (29).
Western blot
Cells were washed twice with cold PBS, collected and lysed with M2 buffer (20mM Tris-HCl, pH 7.6, 0.5 % Nonidet P-40, 250mM NaCl, 3mM EDTA, 3mM EGTA, 2mM dithiothreitol, 0.5mM phenylmethylsulfonyl fluoride, 20mM β-glycerophosphate, 1mM sodium vanadate and 1 μg/ml leupeptin). Concentration of protein was measured by Bradford assay (Bio-Rad). Equal amounts of proteins were separated with 12 % SDS-polyacrylamide gels and then transferred to PVDF membranes. The proteins were detected with enhanced chemiluminescence (Immobilon Western Chemiluminescent HRP Substrate, Millipore).
Reporter assay
A549-AR cells were seeded in 12-well plates overnight and transfected with miR-551b and control siRNA for 24h with INTERFERin siRNA Transfection Reagent (Polyplus-transfection) according to the manufacture’s instructions. After siRNA transfection, the cells were transfected with 0.5 µg luciferase Catalase 3′UTR reporter (Active Motif) together with 0.5 µg pRSV-LacZ to monitor transfection efficiency, using FuGENE™ HD (Roche, Indianapolis, IN) according to the manufacturer’s instructions. Twenty-four hours after plasmid transfection, luciferase reporter assay was performed using a kit from Promega (Madison, WI) and normalized to β-gal.
A549-WT cells were directly transfected with 0.5 µg luciferase Catalase 3′UTR reporter plus 0.5 µg pRSV-LacZ for reporter assay.
Statistics
All data are presented as mean ± SD. Statistical significance was examined by one-way analysis of variance. In all analyses, P < 0.05 was considered statistically significant.
Results
Increased MUC1 expression contributes to chemoresistance in lung cancer cells with acquired apoptosis resistance
A549 and H460 cells with acquired apoptosis resistance (designated as A549- and H460-AR, respectively) were used to investigate the role of MUC1 in chemoresistance (7,8,28). To examine MUC1 expression in AR cells, we compared both MUC1 mRNA and protein expression between AR and WT cells. MUC1 mRNA and protein expression levels were remarkably increased in A549- and H460-AR cells, compared with their respective WT control cells (Figure 1A). MUC1 has been reported to be involved in the regulation of the apoptosis response to genotoxic anticancer agents (32). Previously, we have shown that the response to anticancer agents TRAIL, cisplatin (CDDP) and Adriamycin was reduced in AR cells (7,8,28). Therefore, we investigated if MUC1 is involved in the response to these anticancer agents. Suppression of MUC1 expression by RNA interference significantly sensitized TRAIL-, CDDP- and Adriamycin-induced cell death in both A549- and H460-AR cells (Figure 1B). To substantiate this observation, we used a complementary approach to ectopically express the potent oncogenic MUC1 subunit, MUC1 CT, in A549- and H460 cells by stable transfection of the GST-MUC1 CT plasmid (Figure 1C). The GST-expressing vector was used as a negative control. GST-MUC1 CT significantly attenuated TRAIL-, CDDP- and Adriamycin-induced cell death in both the A549- and H460 cells (Figure 1D). Overexpression of GST-MUC1 CT significantly attenuated TRAIL- CDDP- or Adriamycin-induced apoptosis, which was detected as PARP cleavage and the activation of caspases (caspase 8 and caspase 3 for TRAIL, and caspapse 3 for CDDP and Adriamycin) (Figure 1E–G). Supporting these results, stable knockdown of MUC1 expression in AR cells enhanced PARP cleavage and caspase activation induced by TRAIL, CDDP or Adriamycin (Figure 1C, E–G). Together, these results suggest that increased MUC1 expression in the AR cells is associated with acquired apoptosis- and drug-resistance.
Fig. 1.

Increased MUC1 expression contributes to acquired chemoresistance in lung cancer cells. (A) MUC1 protein and mRNA expression in A549- and H460-WT and -AR cells were detected by Western blot and RT–PCR, respectively. β-Actin was detected as an input control. (B) A549- and H460-AR cells were transfected with MUC1 siRNA and negative control siRNA for 48h. Knockdown of MUC1 was confirmed by Western blot (inserts). β-Actin was detected as an input control. Control and MUC1 knockdown cells were treated with TRAIL (600ng/ml), CDDP (30 µM) and Adriamycin (2 µg/ml) for 36h. Cell death was detected by LDH release assay. Data shown are mean ± SD; *P < 0.05. (C) Upper, A549 and H460 cells were stably transfected with GST-MUC1 CT plasmid and the GST-expressing vector. GST and GST-MUC1 CT expression were detected by Western blot. Lower, MUC1 expression level in both A549 control and MUC1 stable knockdown cells was confirmed by Western blot. β-Actin was detected as an input control. (D) A549 and H460 cells overexpressing GST-MUC1 CT were treated with TRAIL (600ng/ml), CDDP (30 µM) and Adriamycin (2 µg/ml) for 36h. Cell death was detected by LDH release assay. Data shown are mean ± SD; **P < 0.01, *P < 0.05. (E) A549 GST control and GST-MUC1 cells were treated with different concentrations of TRAIL for 4h. A549 control and MUC1 stable knockdown cells were exposed to TRAIL (400ng/ml) at different time points. PARP cleavage and the activation of caspase 8 and caspase 3 were detected by Western blot. β-Actin was detected as an input control. (F) and (G) A549 GST control,GST-MUC1 cells and A549 control and MUC1 stable knockdown cells were exposed to CDDP (20 µM) and Adriamycin (2 µg/ml) at different time points. PARP cleavage and the activation of caspase 3 were detected by Western blot. β-Actin was detected as an input control.
Increased ROS levels are associated with MUC1 overexpression
It has been reported that MUC1 expression is up-regulated by oxidative stress (33,34). In order to explore the mechanism by which the expression of MUC1 is increased in AR cells, we assessed the ROS levels in the AR cells. The ROS levels were remarkably higher in both A549- and H460-AR cells than in their respective WT control cells (Figure 2A). The ROS scavenger BHA effectively suppressed MUC1 mRNA and protein expression in both A549- and H460-AR cells (Figure 2B), suggesting that the up-regulation of MUC1 expression in AR cells was associated with increased ROS accumulation.
Fig. 2.

Increased ROS levels are required for MUC1 overexpression in the AR cells. (A) A549- and H460-WT and -AR cells were incubated with CM-H2DCFDA (5 μM) for about 1h before the cells were collected for ROS detection. Data shown are mean ± SD; **P < 0.01. (B) A549- and H460-AR cells were treated with BHA (100 μM) for 24h and 1h, respectively, before the cells were lifted for Western blot and RT–PCR assays. MUC1 protein and mRNA expression were detected. β-Actin was detected as an input control.
Decreased catalase expression contributes to ROS accumulation and MUC1 overexpression
ROS levels are predominantly regulated by antioxidant enzymes such as superoxide dismutase (SOD) and catalase (35). Since SOD expression was unchanged in the AR cells (data not shown), we assessed the expression of catalase, the key enzyme catalyzing hydrogen peroxide. Compared with that in their respective WT control cells, catalase protein and mRNA expression levels were remarkably suppressed in both A549- and H460-AR cells, which was associated with increased MUC1 expression (Figure 3A). Consistently, knockdown of catalase expression in both parental A549 and H460 cells resulted in an increase of MUC1 expression (Supplementary Figure S1, available at Carcinogenesis Online). Addition of catalase to the culture medium, efficiently suppressed ROS accumulation in AR cells (Figure 3B), and inhibited MUC1 expression and cell death induced by TRAIL (Figure 3C and D, and data not shown). To further validate the role of catalase in MUC1-mediated apoptosis resistance, ectopic expression of catalase was established to effectively suppress ROS accumulation and MUC1 expression, and enhance TRAIL-induced cytotoxicity (Figure 3E–G). These results clearly indicate that catalase suppression contributes to the increased ROS levels and MUC1 expression in AR cells.
Fig. 3.

Decreased catalase expression contributes to ROS increase and MUC1 expression in the AR cells. (A) Catalase protein and mRNA expression in A549- and H460-WT and -AR cells were detected by Western blot and RT–PCR, respectively. β-Actin was detected as an input control. (B) A549- and H460-AR cells were treated with catalase (0.6mg/ml) and BHA (100 μM) for 1h before the cells were subjected to ROS detection. (C) A549- and H460-AR cells were treated with catalase (0.6mg/ml) for 24h. MUC1 expression was detected by Western blot. β-Actin was detected as an input control. (D) A549- and H460-AR cells were pre-treated with catalase (0.6mg/ml) overnight, followed by TRAIL treatment (600ng/ml) for 24h. Cell death was detected by LDH release assay. Data shown are mean. (E) A549- and H460-AR cells were infected with NC or V5-catalase virus for 24h. The expression of V5-catalase and MUC1 were detected by Western blot. β-Actin was detected as an input control. (F) After infection with NC or V5-catalase virus for 24h, A549- and H460-AR cells were incubated with CM-H2DCFDA (5 μM) for 1h before the cells were collected for ROS detection. Data shown are mean ± SD. (G) After infection with NC or V5-catalase virus for 24h, A549- and H460-AR cells were treated with TRAIL (600ng/ml) for 24h. Cell death was detected by LDH release assay. Data shown are mean ± SD. **P < 0.01, *P < 0.05.
miR-551b is responsible for catalase suppression and MUC1 expression in AR cells
Catalase expression is regulated at multiple levels including translation regulation by different microRNAs in different cell types (25,36–38). Thus, we investigated the potential microRNA-mediated regulation of catalase. In a search with miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/), miR-146a, -551b and -943 were found to be candidate microRNAs with the potential to bind to the 3′UTR of catalase mRNA. We first examined the expression of these microRNAs in AR cells using a qPCR assay and found that miR-551b was the only one whose expression was significantly increased in both A549- and H460-AR cells (Figure 4A). Knockdown of miR-551b by RNA interference suppressed ROS accumulation in both A549- and H460-AR cells (Figure 4B and C). Furthermore, suppression of miR-551b rescued catalase expression in a dose-dependent manner, while MUC1 expression was suppressed in A549-AR cells (Figure 4D). Consistently, suppression of miR-551b also rescued catalase expression and inhibited MUC1 expression in H460-AR cells (Figure 4D). To elucidate if miR-551b directly binds to catalase 3′UTR, a reporter assay was performed with A549-AR miR-551b knockdown cells transfected with catalase 3′UTR-luciferase plasmid. Inhibition of miR-551b in A549-AR cells increased catalase 3′UTR-luciferase reporter activity (Figure 4E). Compared with A549-AR cells, the reporter activity was higher in A549 WT cells that express lower levels of miR-551b (Figure 4E). These results suggest that miR-551b directly binds to catalase 3′UTR to suppress catalase expression. Interestingly, treating AR cells with the ROS scavenger BHA resulted in increased miR-551b expression (Figure 4F), suggesting a negative feedback from ROS that controls miR-551b expression. In addition, the PI3K/Akt inhibitor LY294002, but not the inhibitors for EGFR (EGFRin), ERK (U0126) and NF-κB (SC-514), slightly suppressed miR-551b expression (Figure 4G), implying that increased miR-551b expression involves the PI3K/Akt pathway. Altogether, these results suggest that MUC1 overexpression in the AR cells involves miR551b-mediated catalase suppression.
Fig. 4.

Increased miR-551b expression is responsible for catalase suppression and MUC1 expression in AR cells. (A) Expression levels of miR-551b in A549- and H460-WT and -AR cells were detected by real-time PCR. Data shown are mean ± SD; *P < 0.05. (B) Confirmation of miR-551b knockdown in both A549- and H460-AR cells by real-time PCR. **P < 0.01. (C) A549- and H460-AR cells were transfected with miR-551b or negative control siRNA for 24h before the cells were subjected to ROS detection. *P < 0.05, **P < 0.01. (D) A549- and H460-AR cells were transfected with miR-551b siRNA for 48h. The expression of catalase and MUC1 were detected by Western blot. β-Actin was detected as an input control. (E) A549-AR cells were transfected with miR-551b or negative control siRNA. Twenty-four hour post-transfection, the cells were transfected with 0.5 µg luciferase Catalase 3′UTR reporter plus 0.5 µg pRSV-LacZ for another 24h. Luciferase reporter assays were carried out using a kit from Promega (Madison, WI) and normalized to β-gal. A549-WT cells were directly transfected with 0.5 µg luciferase Catalase 3′UTR reporter plus 0.5 µg pRSV-LacZ for reporter assay. **P < 0.01. (F) A549-AR cells were treated with BHA (100 μM) for 4 and 16h, respectively, before the cells were collected for RNA isolation with TRIzol reagent. The expression level of miR-551b was detected by real-time PCR. Data shown are mean ± SD; **P < 0.01 versus control. (G) A549-AR cells were treated overnight with different inhibitors (EGFRin, U0126, LY, SC-514). The expression level of miR-551b was detected by real-time PCR. Data shown are mean ± SD; *P < 0.05 versus control.
Lysosomal degradation of MUC1 was retarded in AR cells, which was associated with increased ROS levels
We also examined if stabilization of the MUC1 protein is involved in MUC1 protein overexpression in AR cells. The WT and AR cells were treated with cycloheximide to prevent protein synthesis, and then MUC1 protein expression was monitored at different time points. The half-life of MUC1 protein in AR cells was about 4h, which is a much longer time interval than that in WT cells (~ 1.8h) (Figure 5A and B). It is noted that in mouse uterine epithelial cells the half-life of Muc1 is about 16 h (39), which is significantly different from our results. It would be interesting to determine if the mechanisms of MUC1 turnover are dramatically different in cancer cells than in normal cells and if cell culture method (polarized versus unpolarized monolayer culture) affects MUC1 turnover. In Addition, MUC1 degradation in AR cells was blocked by the lysosome inhibitor CQ but not the proteasome inhibitor MG132, indicating that MUC1 was degraded at the lysosome (Figure 5C). Actually, MG132 slightly suppressed MUC1 expression, which maybe due to crosstalk occurring between the proteasome and lysosome (40). Collectively, these results suggest that the reduced degradation of MUC1 through the lysosome also contributes to the up-regulation of MUC1 expression in AR cells. Because MUC1 protein expression was associated with elevated ROS levels in AR cells (Figure 2A), we treated the AR cells with BHA to examine if ROS were involved in MUC1 protein stability regulation. BHA treatment shortened the half-life of MUC1 to 1.4h (Figure 5D and E), suggesting that ROS accumulation not only activated MUC1 transcription, but also stabilized MUC1 protein through suppressing the lysosomal degradation of MUC1 in the AR cells.
Fig. 5.

ROS-mediated suppression of lysosomal degradation in AR cells. (A) and (B) A549 TR cells were treated with cycloheximide (CHX, 10 µM) for the indicated time periods. MUC1 expression was detected by Western blot. β-Actin was detected as an input control. The intensity of the individual bands was quantified by densitometry (Image J) and normalized to the corresponding input control bands. MUC1 expression changes were calculated with the control taken as 100%. (C) A549 TR cells were treated with CQ (20 µM) and MG132 (10 µM) for 24h. MUC1 expression was detected by Western blot. β-Actin was detected as a loading control. (D) and (E) H460 TR cells were treated with CHX (10 µM) with or without BHA (100 μM) for the indicated time periods. MUC1 expression was detected by Western blot. β-Actin was detected as an input control. The intensity of the individual bands was quantified by densitometry (Image J) and normalized to the corresponding input control bands. MUC1 expression changes were calculated with the control taken as 100%.
MUC1 potentiates the antiapoptosis cascade consisting of EGFR, Akt, c-FLIP and COX-2 in AR cells
Our previous studies have established a cell survival signaling cascade consisting of EGFR, Akt, c-FLIP, Mcl-1 and COX-2 that contributes to apoptosis resistance induced by anticancer drugs in AR cells (7,8,28). Thus, we examined whether MUC1 regulates this signaling cascade in AR cells. Suppressing MUC1 expression with MUC1 siRNA significantly reduced the expression levels of phospho-EGFR, phospho-Akt, c-FLIP and COX-2 in both A549- and H460-AR cells (Figure 6A and Supplementary Figure S2A, available at Carcinogenesis Online). Consistently, overexpressing MUC1 CT in both wild-type A549 and H460 cells robustly activated expression of these factors (Figure 6B and Supplementary Figure S2B, available at Carcinogenesis Online). However, ERK and Mcl-1 were barely affected by either MUC1 siRNA or MUC1 overexpression (Figure 6A and B, Supplementary Figure S2A and B, available at Carcinogenesis Online).
Fig. 6.

MUC1 potentiates EGFR/Akt/c-FLIP/COX-2 in the AR cells. (A) A549-AR cells were transfected with MUC1 siRNA and negative control siRNA for 48h. The indicated proteins were detected by Western blot. β-Actin was detected as an input control. (B) A549 cells were stably transfected with GST-MUC1 CT and GST control plasmids. The indicated proteins were detected by Western blot. β-Actin was detected as an input control. (C) A549 GST and GST-MUC1 CT cells were pre-treated with the indicated inhibitors (EGFRin (6 µM) for EGFR, LY294002 (LY, 6 µM) for Akt, and COX-2 inhibitor (10 µM) for COX-2 for 30min followed by exposure to TRAIL (600ng/ml), CDDP (30 µM), and Adriamycin (2 µg/ml) for 36h. Cell death was detected by LDH release assay. Data shown are mean ± SD; **P < 0.01, *P < 0.05.
MUC1-mediated chemoresistance involves the EGFR/Akt/COX-2 cascade
We next investigated if the EGFR/Akt/COX-2 cascade is involved in MUC1-mediated resistance to anticancer drugs. A549 and H460 cells overexpressing GST-MUC1 CT were pre-treated with pharmacological inhibitors to block each target in their respective pathway before challenging with TRAIL, CDDP and Adriamycin, respectively. EGFR inhibitor, PI3 kinase (PI3K)/Akt inhibitor LY294002 and COX-2 inhibitor substantially increased TRAIL-, CDDP- and Adriamycin-induced cell death, while these inhibitors alone had marginal toxicity (Figure 6C and Supplementary Figure S2C, available at Carcinogenesis Online). All the inhibitors effectively blocked their respective pathways (data not shown). Together, these results suggest that MUC1 potentiates EGFR activation and the EGFR-mediated cell survival pathway involving Akt and COX-2 to protect cancer cells from anticancer drug-induced cell death.
Discussion
In this report, we investigated the mechanisms for acquired chemoresistance and have obtained evidence substantiating that MUC1 plays an important role in chemoresistance in lung cancer cells. Increased expression of MUC1 is associated with acquired drug resistance and MUC1 knockdown significantly increased apoptotic cytotoxicity induced by cisplatin, Adriamycin and TRAIL in apoptosis-resistant lung cancer cells. ROS levels were increased, which was conversely associated with catalase expression and scavenging ROS suppressed MUC1 expression in AR cells. Further, miR-551b expression was increased in AR cells and knockdown of miR-551b increased catalase while suppressing MUC1 expression. Finally, MUC1 promoted EGFR-mediated activation of the cell survival cascade involving Akt/c-FLIP/COX-2. These results establish a pathway consisting of miR-551b/catalase/ROS that results in MUC1 overexpression, and intervention against this pathway could be exploited for overcoming acquired chemoresistance (Figure 7).
Fig. 7.
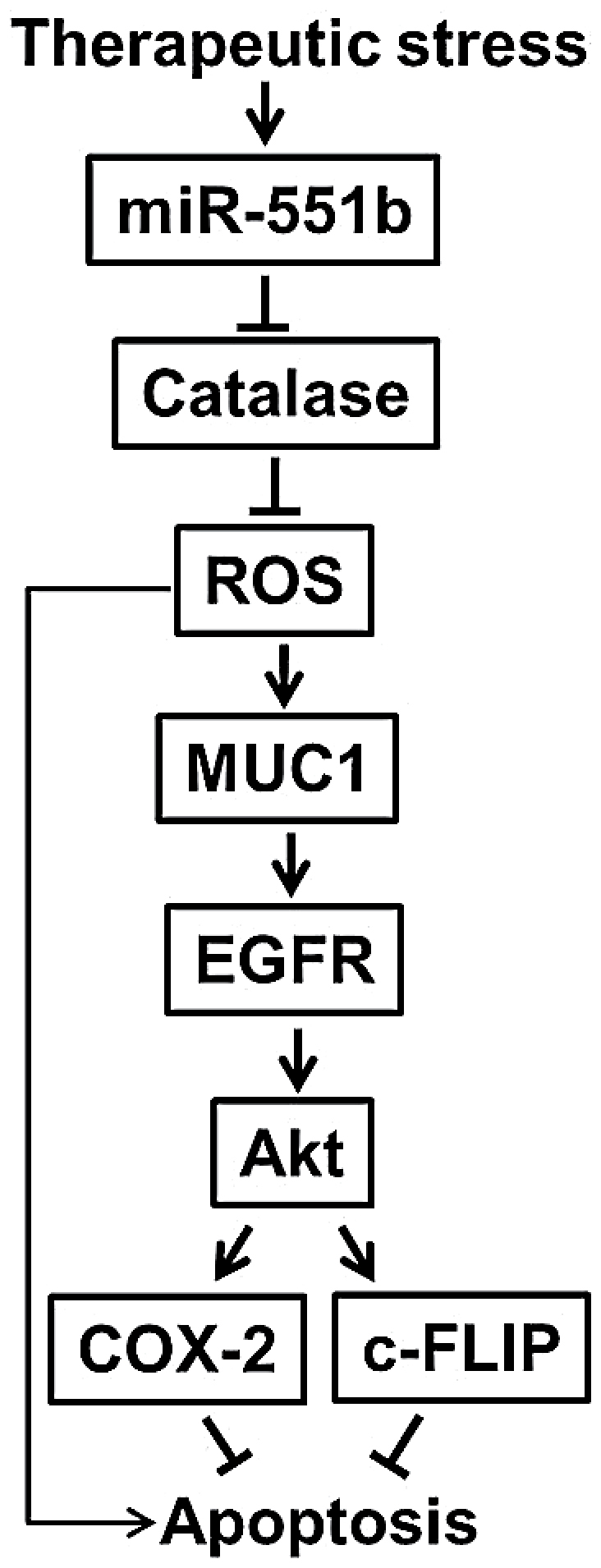
A proposed model for MUC1 expression and function in acquired apoptosis resistance.
Multiple mechanisms, such as enhanced cell survival signals and autophagy and/or suppressed apoptosis, are involved in chemoresistance (9,10,41,42). While MUC1 expression is increased in cancer cells and MUC1 has served as an immunotherapy target in different tumor types, the role of MUC1 in acquired chemoresistance has not been studied. In this report, for the first time, we show clear evidence that MUC1 expression is substantially increased when lung cancer cells acquire drug resistance. Importantly, through manipulation of MUC1 expression in AR cells, we demonstrate that the MUC1 levels are conversely associated with cancer cells’ susceptibility to apoptotic cytotoxicity induced by anticancer drugs. Therefore, we conclude that MUC1 directly contributes to acquired chemoresistance.
Although increased MUC1 expression is common in a wide variety of tumors, the mechanisms of MUC1 expression regulation in cancer are complex and not well elucidated. In normal cells including human lung and gastric epithelial cells, macrophages and mouse placenta, MUC1 transcription is under the control of several transcription factors such as NF-κB, STAT3, HIF-1, SP1 and PPAR-γ (21,34,43–45). In addition, epigenetic regulation including demethylation of the MUC1 promoter, and histone H3-K9 demethylation and acetylation are involved (46). Furthermore, suppressing microRNA-mediated downregulation of MUC1 expression was recently reported (25,36,37). MUC1 expression was associated with ROS increase in the AR cells, which is consistent with a previous report (33). We found MUC1 mRNA was increased in AR cells, which was associated with ROS accumulation resulting from catalase inhibition. ROS activated MAPKs, including ERK, which stimulates MUC1 transcription (38,45). Thus, it is possible that activation of transcription is involved in increased MUC1 expression in AR cells. In addition, we found that lysosomal degradation of MUC1 protein was suppressed in a ROS-dependent manner in AR cells. Therefore, our results suggest a dual mechanism for MUC1 overexpression in AR cells: enhanced MUC1 gene transcription and decreased MUC1 protein degradation. Interestingly, the ROS scavenger BHA strongly suppressed MUC1 mRNA expression (Figure 2B), suggesting that ROS may also stimulate MUC1 gene transcription. While we focus on post-transcriptional regulation of MUC1 expression in this study, how ROS mediates MUC1 transcription deserves future study.
We further found that the miR-551b-mediated suppression of catalase expression is responsible for the increased oxidative stress in the AR cells. Catalase expression is regulated through different mechanisms including proteasomal protein degradation and miRNA-mediated protein synthesis suppression (25,36–38). We found that miR-551b, which is predicted to target the 3′UTR of catalase mRNA, was increased in AR cells and knockdown of miR-551b strongly rescued catalase expression, suggesting an important mechanism in the upregulation of MUC1 through miR-551b-mediated catalase suppression. Interestingly, different siRNAs are reported to suppress catalase expression (25,36,37), suggesting that catalase is controlled by complex mechanisms, and different miRNAs are utilized for regulating catalase expression under different conditions or in different cell contexts. While our results suggest that the PI3K/Akt pathway plays a role in increased miR-551b expression in the AR cells, defining these mechanisms deserves further study.
In addition to promoting MUC1 mRNA expression, ROS also maintain MUC1 protein stability by suppressing lysosomal degradation. While ROS induce lysosomal degradation of some proteins, in certain circumstances, impairing lysosome function with ROS has also been seen (47,48). Thus, it appears that ROS mediate regulation of MUC1 expression across multiple levels. It is possible that therapeutic stress causes ROS-mediated cytotoxicity in cancer cells. In coping with ROS, AR cells acquire MUC1 overexpression to sustain their survival (Figure 7). Through this mechanism, MUC1 functions as a defensive factor to keep cancer cells alive under therapeutic stress. Therefore, to suppress therapeutic-induced MUC1 expression would prevent or attenuate acquired chemoresistance.
There are numerous cellular partners for MUC1 that are involved in cell signaling for cell proliferation and survival (14). However, which pathway plays a major role in acquired chemoresistance is still elusive. Using a lung cancer acquired chemoresistance cell model, we previously identified EGFR-mediated activation of the Akt and ERK pathways that lead to increased expression of c-FLIP, Mcl-1, COX-2 and TGM2 (7,8,28). Using the same cell model, we found MUC1 is involved in acquired chemoresistance. Further, our results show that MUC1 potentiates EGFR activation. Interestingly, MUC1 mainly triggers the Akt but not ERK pathway, and COX-2 and c-FLIP are the main factors affected by MUC1 manipulation. These results suggest that in addition to EGFR activation, MUC1 uses a second level of regulation for the downstream pathways of EGFR. All these results suggest that cancer cells utilize complex mechanisms involving MUC1 as a key mediator for acquired chemoresistance.
Altogether, this study clearly demonstrates that MUC1 is an important factor for acquired chemoresistance in lung cancer cells. Intervention against the pathway consisting of miR551b/catalase/ROS that results in MUC1 overexpression may be exploited for overcoming acquired chemoresistance.
Supplementary material
Supplementary Figures 1 and 2 can be found at http://carcin.oxfordjournals.org/
Funding
National Institute of Environmental Health Sciences (NIEHS)/ National Institutes of Health (NIH) (R01ES017328); Low Dose Radiation Research Program Office of Science, Department of Energy (DE-FG02-09ER64783).
Supplementary Material
Acknowledgements
We thank Dr Seishi Murakami from Kanazawa University, Japan for providing the pSG5-GST plasmid.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- AR
apoptosis resistance
- BHA
butylated hydroxyanisole
- CDDP
cisplatin
- GST
glutathione S-transferase
- LDH
lactate dehydrogenase
- MUC1
Mucin-1
- PARP
poly (ADP-ribose) polymerase
- ROS
reactive oxygen species
- RT–PCR
reverse transcription–polymerase chain reaction.
References
- 1. Chang A. (2011). Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer, 71, 3–10 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., et al. (2011). Hallmarks of cancer: the next generation. Cell, 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3. Lønning P.E. (2010). Molecular basis for therapy resistance. Mol. Oncol., 4, 284–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oliver T.G., et al. (2010). Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer. Genes Dev., 24, 837–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malhi H., et al. (2006). TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene, 25, 7333–7335 [DOI] [PubMed] [Google Scholar]
- 6. Stegehuis J.H., et al. (2010). TRAIL receptor targeting therapies for non-small cell lung cancer: current status and perspectives. Drug Resist. Updat., 13, 2–15 [DOI] [PubMed] [Google Scholar]
- 7. Li Z., et al. (2011). Epidermal growth factor receptor-mediated tissue transglutaminase overexpression couples acquired tumor necrosis factor-related apoptosis-inducing ligand resistance and migration through c-FLIP and MMP-9 proteins in lung cancer cells. J. Biol. Chem., 286, 21164–21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen W., et al. (2010). Acquired activation of the Akt/cyclooxygenase-2/Mcl-1 pathway renders lung cancer cells resistant to apoptosis. Mol. Pharmacol., 77, 416–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghavami S., et al. (2009). Apoptosis and cancer: mutations within caspase genes. J. Med. Genet., 46, 497–510 [DOI] [PubMed] [Google Scholar]
- 10. Ocker M., et al. (2012). Apoptosis-modulating drugs for improved cancer therapy. Eur. Surg. Res., 48, 111–120 [DOI] [PubMed] [Google Scholar]
- 11. Lu W., et al. (2006). Cutting edge: enhanced pulmonary clearance of Pseudomonas aeruginosa by Muc1 knockout mice. J. Immunol., 176, 3890–3894 [DOI] [PubMed] [Google Scholar]
- 12. Kyo Y., et al. (2012). Antiinflammatory role of MUC1 mucin during infection with nontypeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol., 46, 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hattrup C.L., et al. (2008). Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol., 70, 431–457 [DOI] [PubMed] [Google Scholar]
- 14. Kufe D.W. (2013). MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene, 32, 1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baldus S.E., et al. (2004). MUC1 and the MUCs: a family of human mucins with impact in cancer biology. Crit. Rev. Clin. Lab. Sci., 41, 189–231 [DOI] [PubMed] [Google Scholar]
- 16. Woenckhaus M., et al. (2008). Prognostic value of FHIT, CTNNB1, and MUC1 expression in non-small cell lung cancer. Hum. Pathol., 39, 126–136 [DOI] [PubMed] [Google Scholar]
- 17. Schroeder J.A., et al. (2004). MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene, 23, 5739–5747 [DOI] [PubMed] [Google Scholar]
- 18. Guddo F., et al. (1998). MUC1 (episialin) expression in non-small cell lung cancer is independent of EGFR and c-erbB-2 expression and correlates with poor survival in node positive patients. J. Clin. Pathol., 51, 667–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mohr A.M., et al. (2013). MUC1 regulates expression of multiple microRNAs involved in pancreatic tumor progression, including the miR-200c/141 cluster. PLoS One, 8, e73306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu X., et al. (2012). MUC1 contributes to BPDE-induced human bronchial epithelial cell transformation through facilitating EGFR activation. PLoS One, 7, e33846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu X., et al. (2014). MUC1 in macrophage: contributions to cigarette smoke-induced lung cancer. Cancer Res., 74, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartel D.P. (2009). MicroRNAs: target recognition and regulatory functions. Cell, 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ortholan C., et al. (2009). MicroRNAs and lung cancer: new oncogenes and tumor suppressors, new prognostic factors and potential therapeutic targets. Curr. Med. Chem., 16, 1047–1061 [DOI] [PubMed] [Google Scholar]
- 24. Lovat F., et al. (2011). MicroRNAs in the pathogenesis of cancer. Semin. Oncol., 38, 724–733 [DOI] [PubMed] [Google Scholar]
- 25. Jin C., et al. (2010). miR-1226 targets expression of the mucin 1 oncoprotein and induces cell death. Int. J. Oncol., 37, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin Y., et al. (2000). The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol. Cell. Biol., 20, 6638–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yi M., et al. (1997). Delineation of regions important for heteromeric association of hepatitis C virus E1 and E2. Virology, 231, 119–129 [DOI] [PubMed] [Google Scholar]
- 28. Wang X., et al. (2008). Akt-mediated eminent expression of c-FLIP and Mcl-1 confers acquired resistance to TRAIL-induced cytotoxicity to lung cancer cells. Mol. Cancer Ther., 7, 1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Q., et al. (2014). Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway. J. Biol. Chem., 289, 5654–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang X., et al. (2007). Sensitization of TNF-induced cytotoxicity in lung cancer cells by concurrent suppression of the NF-kappaB and Akt pathways. Biochem. Biophys. Res. Commun., 355, 807–812 [DOI] [PubMed] [Google Scholar]
- 31. Bai L., et al. (2012). A superoxide-mediated mitogen-activated protein kinase phosphatase-1 degradation and c-Jun NH(2)-terminal kinase activation pathway for luteolin-induced lung cancer cytotoxicity. Mol. Pharmacol., 81, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ren J., et al. (2004). Human MUC1 carcinoma-associated protein confers resistance to genotoxic anticancer agents. Cancer Cell, 5, 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yin L., et al. (2003). Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J. Biol. Chem., 278, 35458–35464 [DOI] [PubMed] [Google Scholar]
- 34. Kuwahara I., et al. (2007). The signaling pathway involved in neutrophil elastase stimulated MUC1 transcription. Am. J. Respir. Cell Mol. Biol., 37, 691–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu L., et al. (2006). Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. U. S. A., 103, 4952–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sachdeva M., et al. (2010). MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res., 70, 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rajabi H., et al. (2010). Mucin 1 oncoprotein expression is suppressed by the miR-125b oncomir. Genes Cancer, 1, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q., et al. (2013). RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis, 34, 2119–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pimental R.A., et al. (1996). Synthesis and intracellular trafficking of Muc-1 and mucins by polarized mouse uterine epithelial cells. J. Biol. Chem., 271, 28128–28137 [DOI] [PubMed] [Google Scholar]
- 40. Korolchuk V.I., et al. (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell, 33, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He W., et al. (2012). Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy, 8, 1811–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin Y., et al. (2010). The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets, 14, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park Y.S., et al. (2012). PPARγ inhibits airway epithelial cell inflammatory response through a MUC1-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol., 302, L679–L687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shalom-Barak T., et al. (2004). Peroxisome proliferator-activated receptor gamma controls Muc1 transcription in trophoblasts. Mol. Cell. Biol., 24, 10661–10669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jonckheere N., et al. (2010). The membrane-bound mucins: From cell signalling to transcriptional regulation and expression in epithelial cancers. Biochimie, 92, 1–11 [DOI] [PubMed] [Google Scholar]
- 46. Yamada N., et al. (2011). Epigenetic regulation of mucin genes in human cancers. Clin. Epigenetics, 2, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kurz T., et al. (2008). Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta, 1780, 1291–1303 [DOI] [PubMed] [Google Scholar]
- 48. Heid M.E., et al. (2013). Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol., 191, 5230–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.