

Abstract
Prioritization of cancer implicated genes has received growing attention as an effective way to reduce wet lab cost by computational analysis that ranks candidate genes according to the likelihood that experimental verifications will succeed. A multitude of gene prioritization tools have been developed, each integrating different data sources covering gene sequences, differential expressions, function annotations, gene regulations, protein domains, protein interactions, and pathways. This review places existing gene prioritization tools against the backdrop of an integrative Omic hierarchy view toward cancer and focuses on the analysis of their text mining components. We explain the relatively slow progress of text mining in gene prioritization, identify several challenges to current text mining methods, and highlight a few directions where more effective text mining algorithms may improve the overall prioritization task and where prioritizing the pathways may be more desirable than prioritizing only genes.
Keywords: gene prioritization, text mining, cancer omics, pathway prioritization, machine learning
Introduction
Many studies have been dedicated to efficient screening and identification of causative genes and pathways implicated in cancerous phenotypes. Much attention has been drawn to gene prioritization that helps to select from a large number of candidate genes a subset of those that are biologically most relevant and are worth further validation experiments.
To understand the pathophysiological mechanisms, it is insufficient to investigate only at the level of single-nucleotide polymorphisms (SNPs) or copy number variations (CNV) as in genome-wide association study (GWAS). This is partly due to the lack of statistical strength that plagues GWAS,1 which stems from the need to correct for multiple testing, and makes it difficult to implicate causative genes. On the other hand, cancers that involve complex and epistatic genetic traits cannot be adequately explained by additive genetic models. Moreover, cancer phenotypes necessarily involve the full scope of Omics including genomics, transcriptomics, epigenomics, proteomics, and metabolomics, as well as their interaction and correlation with pheno-types. Rather, levels in the Omic hierarchy need to be considered simultaneously and interactively. This is because the genes, RNAs, proteins, and epigenetic factors interplay in the form of signaling networks, metabolic networks, regulatory networks, etc. Moreover, biological networks further interact and exchange information among each other, as shown in Figure 1.
Figure 1.

The Omic hierarchy on the left, biological networks on the right, and their interactions. TF stands for transcription factor, The figure shows some typical network interaction scenarios such as: a signaling network activates transcription factors for a regulatory network; transcription factor complexes that control a regulatory network may be formed through protein interactions (eg, binding); a metabolic network may produce energy (through catabolism) and amino acids (through anabolism) that are necessary for other functional networks; and enzymes that catalyze many metabolic networks are in fact proteins and are produced and regulated by other biological networks. Note that regulatory networks often have participants from multiple levels of the Omic hierarchy.
The importance of the integrated Omic view toward cancer is increasingly realized, and drives collaborative efforts toward development of shared expression, functional annotation, protein–protein interaction (PPI) network, experimental Omic data sources, literature, etc. Most data sources are structured and have predefined schemas for prioritization tools to follow. In fact, gene prioritization tools have been progressively refining their customized components for analyzing structured data sources; see reviews.2–5 In contrast, text mining methods on literature data source and text descriptions in structured data sources have seen slow progress. Thus unlike previous reviews, this review focuses on the text mining methodology used in gene prioritization tools and recent developments on the emerging direction of pathway prioritization.
Heterogeneous Data Sources
With recent advances in the Omic era, large volumes of data are gathered from multiple levels of the Omic hierarchy including genome, transcriptome, epigenome, proteome, and metabolome. These data sources may focus on different subsets of the Omic hierarchy. These data sources also present themselves in various formats such as sequence reads, numerical expression levels, and narrative text in the scientific literature. The increasing availability of high throughput technology such as next-generation sequencing (NGS) not only calls for a changing perspective on the Omic model, but also calls for upgrading hypotheses, algorithms, and software that integrate the evolution of knowledge. In particular, in addition to subsequent analysis on GWAS results to accumulate evidence by statistical procedures,6 gene prioritization methods also need to integrate an increasing number of heterogeneous data sources, in terms of both content and data formats.
There are a multitude of data sources that encode different types of information regarding the Omic hierarchy and information exchange. These data sources have been conventionally categorized based on their purposes, for example, functional annotation, ontologies, and sequence data. Here, we try to summarize the currently active data sources that have been used by gene prioritization tools as well as by cancer biologists and clinicians in their routine research and practice, grouping them according to their primary utility categories. Table 1 lists the summarized data sources. From the table it can be seen that many data sources contain narrative text or literature, suggesting the broad applicability of text mining to gene prioritization. Thus closer understanding and more improvements on text mining methods in gene prioritization is likely to make a general impact.
Table 1.
Data sources for gene and pathway prioritization according to their primary utility.
| UTILITY CATEGORY | DATA SOURCES |
|---|---|
| Literature | PubMed9, MEDLINE, OMIM10 |
| Terminology & Ontology | GO11, UMLS12, DO13, MeSH14, eVOC15, HPO16, MPO17 |
| Pathway | KEGG18, BioCarta19, BioCyc20, Reactome21, GenMAPP22, MSigDB23, Brenda24, CTD, HPRD25, GXD, BIND, MGI, PharmGKB, PID26 |
| Protein sequence & Domain | InterPro27, Aceview28, Pfam29, SMART30, PROSITE31, Gene3D32, ProDom33, Ensembl, Swiss-Prot, TrEMBL, HPRD, RefSeq, GenBank, MGI, CDD34, Entrez Gene35 |
| Regulation | MSigDB, TargetScan36, TRANSFAC37, BioCyc, Reactome, Brenda, TOUCAN |
| Gene expression | Ensembl38, BIOGPS39, GEO40, MBA41, HBA42, Reactome, GenMAPP, GXD, STRING, MGI, SOURCE43 |
| Gene-Protein and Disease | CTD44, HPRD, GXD, COSMIC, TCGA, MGI, ClinVar, NHGRI GWAS, Phar-mGKB, Orphanet45, HuGENavigator46, GHR47, SOURCE, GAD48 |
| Gene & Protein variation | Ensembl, OMIM, HGMD49, Swiss-Prot50, HPRD, GXD51, TrEMBL52, COSMIC53, TCGA54, dbSNP55, GEO, RefSeq, ClinVar56, NHGRI GWAS57,58, PharmGKB59, GeneTests60, MGI, CDD, GHR, ALFRED61, HapMap62 |
| Gene function annotation | PROSITE, GO, BioCyc, Reactome, GenMAPP, Brenda, BIOGPS, Swiss-Prot, TrEMBL, dbSNP, MGI, SOURCE |
| Gene, Protein & Chemical interaction | STRING63, HPRD, BioGrid64, BIND65, IntACT66, DIP67, Gene3D, Drugbank68, Matador69, CTD, Stitch70, Swiss-Prot, TrEMBL, HPRD, PharmGKB, Entrez Gene, PID |
| Gene sequence & Locus | BLAST71, RefSeq72, TOUCAN73, GenBank74, BioCyc, Brenda, MBA, HBA, HPRD, GXD, STRING, BIND, MGI, Entrez Gene |
| Homology analysis | MGI75,76, HomoloGene77 and OMA78, Inparanoid79, BioCyc, Reactome, GXD, Ref-Seq, Entrez Gene |
Notes: Bold font indicates the source has narrative text and is suitable for text mining. This does not include data sources that only points to literature data sources such as PubMed. We also exclude data sources that are built solely by automatic mining of other data sources, eg, GeneCards.7,8
Abbreviations: OMIM, Online Mendelian Inheritance in Man; GO, Gene Ontology; UMLS, Unified Medical Language System; DO, Disease Ontology; MeSH, Medical Subject Heading; HPO, Human Phenotype Ontology; MPO, Mammalian Phenotype Ontology; GEO, Gene Expression Omnibus; CTD, Comparative Toxicogenomics Database; GXD, Gene Expression Databas; MGI, Mouse Genome Informatics; HPRD, Human Protein Reference Database; HGMD, Human Gene Mutation Database; MBA, Mouse Brain Atlas; HBA, Human Brain Atlas; CDD, Conserved Domain Database; GHR, Genetics Home Reference; GAD, Genetic Association Database; OMA, Orthologous Matrix.
Text Mining in Gene Prioritization
This section provides an extensive review of computational tools for gene prioritization. Because we are focusing on text mining in such tools, we categorize them according to the characteristics of their text mining components. In each category, prioritization methods are ordered chronologically.
Prioritization without text mining
Despite the prevalence of literature and narrative text segments in many data sources, earlier gene prioritization methods may leave out the text mining component and simply rely on mining structured data.
POCUS80 relies on statistics composed of term IDs in functional annotation databases that are shared by gene– disease pairs. PROSPECTR81 uses alternating decision trees to classify whether the candidate genes and known disease genes share similar sequence patterns. Gentrepid82 is directly based on biological data including protein domain similarity, protein interaction, and pathway membership. PhenoPred83 is a supervised method based on experimental PPI networks, protein–disease association, protein sequences, and protein function annotations. PhenoPred encodes gene features using shortest-path distances to all disease genes or genes with known function annotation, as well as sequence and function features using physicochemical or predicted structural properties (real-valued) and GO terms and PROSITE patterns. Principal component analysis (PCA)84 was used to reduce dimensionality and support vector machine (SVM)85 was used to predict individual disease–gene correlation.
Prioritization with keyword search text mining
Prioritization methods falling in this category rely on key-word matching when retrieving literature or text segment in response to query terms such as phenotype descriptions or gene names. Indexing is frequently performed to preprocess the narrative text in data sources in order to categorize the literature and reduce the search time.
GeneSeeker86 depends on gene positional data and expression data, as well as phenotype data extracted using keyword search from MEDLINE and OMIM. GeneSeeker uses synonym lists compiled from Swiss-Prot and the now inactive Human Genome Database87 to unify nomenclature across different data sources. Prioritizer88 constructs the gene network using Bayesian integration to account for both manually curated protein interaction and pathway information as well as co-expression data, yeast-two-hybrid (Y2H) interaction data, etc. To initialize the network, known contributing genes are identified by text mining on the OMIM disease entries. CANDID89 asks the users to input keywords related to the traits of interest and uses them to query the literature and protein domain descriptions (keyword matching). CAN-DID also uses other data sources to generate source-specific numeric scores, such as those on phylogenetic conservation, gene expression, protein interactions, linkage, SNP association, and user provided gene scores.
All source-specific scores are merged into a final score by user provided weights. PGMapper90 uses keywords to search phenotype databases such as OMIM and PubMed, and combines linkage, expression, and sequence data sources to search candidate genes for 16 species including human. GeneProspector91 keeps an in-house literature database by first using text mining to screen PubMed, and then having curators review and manually index screened abstracts. Indexing tasks include associating the publications with gene symbols, predefined categories, and study types, in order to calculate a heuristic score for each gene. MaxLink92 relies on clues based on connectivity of candidate genes to the known disease genes in a PPI network to rank genes and uses keyword searches to provide known gene sets.
Prioritization with vector space model based text mining
Vector space model represents text document and segments as vectors of identifiers, eg, index terms. Compared with keyword search, vector space model can take into consideration counts and weights of index terms. Vector space model based text mining is frequently explored in gene prioritization tools.
G2D93–95 performs text mining on MEDLINE to associate MeSH phenotype terms with MeSH chemistry terms and to associate MeSH chemistry terms with GO terms. For both associations, co-occurrence statistics are calculated. Association between MeSH phenotype terms and GO terms is then calculated transitively from the two associations, based on fuzzy set theory. G2D then searches the RefSeq and STRING databases and assigns gene score by averaging across GO terms matching that gene. SNPs3D96 uses text profiling on MEDLINE abstracts to rank candidate genes. More specifically, the authors collected nouns and adjectives as keywords, tallied the raw counts of keywords and gene–keyword pairs within the corpus, devised heuristic scores for gene–gene interaction and disease–gene, and applied a rule-based approach to assign the final rank. MimMiner97 uses the anatomy and the disease sections of MeSH to extract terms from OMIM records and builds a vector space model, weighting the vector element according to the MeSH hierarchy and inverse document frequency. The authors reported correlations between phenotype similarity and gene sequence similarity, correlations between phenotype similarity and protein interaction, correlations between phenotype similarity and gene functions in pathways, and the implication of phenotype grouping on the modular nature of human disease genes, all derived from text mining. Endeavour98,99 trains separate models on different data sources including literature, functional annotation, gene expression, protein domains, protein interactions, pathway membership, cis-regulatory modules, transcriptional motifs, sequence similarity, and user provided data. Endeavour then ranks the target genes against individual models and pools the individual ranks to get an overall rank, using a customized Q-statistic. CAESAR100 accepts as input a text excerpt containing previously implicated genes, performs text mining (vector-space model) against phenotypic, anatomic, and genetic ontologies and extracts ontology terms. CAESAR then maps such terms to candidate genes in expression, interaction, and pathway data sources and generates data-source specific gene scores that are in turn aggregated into a final score. ToppGene101,102 uses co-citation counts in PubMed as an indication of gene relationship. Using data sources covering gene, protein, and pathway; ToppGene implements two gene prioritization systems: one based on function annotation and the other based on interaction network. CIPHER103 is a network based prioritization method integrating PPI networks, disease phenotype similarity, and known gene–phenotype associations. CIPHER uses the text mining component of MimMiner97 to calculate phenotype similarity. GeneDistiller104 uses literature co-occurrence statistics to filter the candidate genes, besides integrating data sources on genes, proteins, and pathways. PRINCE105 is a network based algorithm that not only predicts gene associations but also infers protein complex associations with diseases. The construction of the network begins with scanning causal genes of known diseases that share phenotype similarity to the target disease, where the authors used the MeSH based phenotype similarity metric from MimMiner.97 The network is then initialized with prior knowledge and gene scores are computed using an iterative network propagation algorithm. The web tool PolySearch106 searches PubMed abstracts as well as gene, protein, pathway, and metabolite data sources to integrate their text content. PolySearch manually curates a corpus consisting of terms and synonyms from dictionaries compiled from gene function databases. PolySearch also adopts a heuristic scoring system to rank disease queries and matching gene terms where scoring is based on a discretized sentence relevancy. Gene-Wanderer107 applies random walk and diffusion kernel on PPI networks and compares the PPI domain knowledge collected with and without text mining. The authors pointed out that integrating literature data may lead to better performance on retrospective studies than prospective settings, a phenomenon referred to as “knowledge contamination” as retrospective knowledge is already present in the literature. GPsy108 profiles candidate genes using data sources on gene sequence, expression, function annotation, and gene–disease association, augmented with orthologous genes extracted from Homolo-Gene77 and OMA (Orthologous MAtrix).78 Text mining is used to extract phenotype annotation based on co-occurrence statistics in the biomedical literature.
Prioritization with text mining using ontology structure
In addition to using vector space model and using ontology only as a terminology mapping tool, gene prioritization methods in this category further explore the ontology structure (eg, the is-a relation between parent and child nodes in an ontology), in order to better quantify the semantic similarity between concepts.
Tiffin et al.109 use the eVOC anatomical ontology to combine literature text mining and gene expression data. The eVOC terms serve as a bridge to connect the PubMed literature (via association frequency between eVOC term and disease names) and RefSeq genes (via frequency of RefSeq genes annotated with eVOC terms). A heuristic ranking score is then used to prioritize the bridged disease–gene pair. SUSPECTS110 uses four data sources including gene sequence patterns (obtained from PROSPECTR), gene expression, shared rare protein domains, and semantic similarity of the associated GO terms between the candidate and seed genes. The calculation of semantic similarity is done using the metric of information content proposed by Lord et al.111, which is based on counting how many times a GO term occurs in the Swiss-Prot database. MedSim112 uses GO terms as the function annotation of a gene and experiments with multiple configurations on whether to include GO terms of ortholog genes, interacting genes, or semantically similar GO terms. The semantic similarity is calculated with the simRel score, taking into account the differences, commonalities, and specificities of GO terms.113
Prioritization with statistical text mining
Recently, statistical text mining in gene prioritization has gained traction, where a pre-defined distribution or a Bayesian model is used to fit the data in order to boost the prediction power.
GRAIL114 is a text mining approach that resembles correlating gene pathways to diseases in that it identifies not only the disease genes but also their biological relationships. GRAIL takes seeds from the implicated SNPs identified by GWAS to anchor seed chromosomal regions, so it is less susceptible to knowledge contamination. A gene is represented as a feature vector by using a term frequency-inverse document frequency (tf–idf) weighted vector space model to analyze the PubMed corpus, from which the relatedness between genes can be calculated and connections between candidate genes and seed regions (containing implicated genes) can be made. By fitting a Poisson distribution over the connections between candidate genes and seed regions, a P-significance can be calculated to show how likely a candidate gene is implicated. Besides text similarity-based gene relatedness, GRAIL also calculates annotation-based relatedness and expression-based relatedness to quantify biological relations between genes. Genie115 is a literature-based prioritization method that mines MEDLINE abstracts and explores orthologs to identify more abstracts containing related orthologous genes to complement the genes of under studied organisms. In order to retrieve orthologous genes, a sample set of abstracts with the genes from the target organism is used to train a Bayesian linear classifier that picks discriminative keywords to be used in orthologous gene abstract search. MetaRanker116 applies large-scale text mining on all MEDLINE abstracts available by then using customized statistical models of genes and MeSH terms adjusting for publication bias, similar to GRAIL. In addition, MetaRanker also integrates GWAS SNP–phenotype associations, PPI networks, linkage analysis data, gene expression, CNV, and user provided NGS data.
There are also efforts toward comparing and integrating computational tools for correlating candidate genes, such as gene prioritization portal117, which links to 19 previous computational solutions. In Ref. 118, the authors compared algorithms that use PPI networks including network neighbor analysis, unsupervised clustering, semi-supervised clustering, network flow with prior information, and random walks, and reported superior performance of random walk on network data. Although both works did not have text mining as their focus, they can potentially shed lights on best practice when integrating analysis on structured data with text mining in gene prioritization. They also serve to illustrate the imbalance between the advances in mining structured data and mining text in gene prioritization, which is readily evidenced by the complexity of models employed. We summarize the text mining components in the above gene prioritization methods in Table 2, grouped using the same categorization as we introduced these methods. We also briefly comment the advantages and disadvantages of the text mining components.
Table 2.
Summarization of text mining components in gene prioritization methods.
| CATEGORIZATION | PRIORITIZER | TEXT MINING USAGE | PROS AND CONS OF TEXT MINING METHODS |
|---|---|---|---|
| No text mining | POCUS PROSPECTR Gentrepid PhenoPred |
NA | Lack of textual evidence |
| Keyword search | GeneSeeker | Extract phenotype data | Requires prior knowledge in selecting and hand tuning keyword sets; subject to selection bias when picking keywords |
| Prioritizer | Extract known genes | ||
| CANDID | Match protein domain description | ||
| PGMapper | Extract phenotype data | ||
| GeneProspector | PubMed screening before reviews by curators | ||
| MaxLink | Extract known genes | ||
| Vector space model | G2D | Associate MeSH phenotype terms, MeSH chemistry terms, and GO terms | Possible to calculate semantic similarities automatically; on the other hand, the accuracy of the semantic similarity will be restricted by the co-occurrence counts of words or citations, which are only approximations for real semantics |
| SNPs3D | Score candidate genes by profiling noun and adjective counts in the MEDLINE abstracts | ||
| MimMiner | Extract correlations of pheno-type similarity protein interaction, and gene functions in pathways | ||
| Endeavour | Rank genes separately on literature evidence, before pooling an overall rank | ||
| CAESAR | Match database description of genes to ontology descriptions of phenotype, anatomy and genes | ||
| ToppGene | Use co-citation counts in Pub-Med as indication of gene relationship | ||
| CIPHER | Use the text mining component of MimMiner | ||
| GeneDistiller | Use literature co-occurrence statistics to filter the candidate genes | ||
| PRINCE | Use the text mining component of MimMiner | ||
| PolySearch | Rank gene terms based on a discretized sentence relevancy to disease queries | ||
| GeneWanderer | Use text evidence to augment PPI networks | ||
| GPsy | Extract phenotype annotation based on co-occurrence statistics in the biomedical literature | ||
| With ontology structure | Tiffin et al. | Use the eVOC anatomical ontology to connect the PubMed literature and RefSeq genes | Richer and hierarchical semantics from ontology, but accuracy depend on resolution, noise and irregularities from ontologies |
| SUSPECTS | Calculate semantic similarity between GO terms by exploring GO structure and how many times these GO terms occur in the Swiss-Prot database | ||
| MedSim | Calculate semantic similarity between GO terms by exploring GO structure | ||
| Statistical text mining | GRAIL | Represent genes using a tf-idf weighted feature vector to analyze PubMed abstracts to calculate the gene-gene and gene-SNP correlation | More detailed and advanced modeling of text distributions opens the avenue to even richer semantic analysis. Requires large training corpus, time consuming |
| Genie | Train a Bayesian linear classifier that picks discriminative keywords to be used in orthologous gene abstract search | ||
| MetaRanker | Large-scale text mining on MEDLINE abstracts using customized statistical models of genes and MeSH terms adjusted for publication bias |
Challenges of Text Mining in Gene Prioritization
From the progression of gene prioritization methods over time as summarized in the previous section, it becomes clear that the application of text mining has seen slow methodology advance. In our opinion, the evidence from the literature has been largely treated in an oversimplified fashion. This section lays out some of the challenges encountered by text mining in gene prioritization, and the next section enumerates several promising directions emerged lately.
Noisy, contaminated and biased knowledge
Representation of gene–disease relations extracted by current text mining in gene prioritization is often noisy because of the insufficient level of detail in co-occurrence counts or simple statistics. Mined relations are also biased toward genes and phenotypes that are already present in the literature and may offer only limited insights to new discoveries. This effect of knowledge contamination is said to make the prioritization task easier for retrospective discovery than for novel discovery. In fact, this problem is not specific to text mining only, because after publication in literature, knowledge is also quickly integrated into structured data in sources such as KEGG, STRING, and InterPro. Thus cross-validation based methods for checking generalizability may not guarantee good performance on novel genes. Previous works usually tried to reduce such overestimation by sub-setting data prior to the real discovery of the cross-validated genes. However, this does not eliminate another, more subtle form of knowledge contamination, as literature based text mining may still present bias toward existing concepts about the disease. GRAIL addressed this problem by not using known pathways and genes at all, but increased the risk of reinventing the wheel for known discoveries. A finer statistical approach should be devised to utilize existing knowledge and eliminate or offset the potential bias.
Evaluation of text mining components in gene prioritization methods
Ideally, we need objective evaluation for this ever expanding array of text mining components in gene prioritization methods. In typical text mining settings, a ground truth needs to be established as a gold standard to perform intrinsic evaluation and compare different text mining approaches as isolated systems. For example, in a related task of extracting mentions of genes from documents, experts need to first manually annotate the corpora and mark the gene mentions. With such a ground truth produced, evaluation metrics such as precision, recall, and f-measure can be used to assess the effectiveness of text mining approaches in recognizing gene mentions. Let TP denote the number of true positives in the contingency table, FP denote the number of false positives, and FN denote the number of false negatives, the definition of precision is P = TP/(TP + FP), recall is R = TP/(TP + FN), f-measure is F = 2 × P × R/(P + R). However, the task of text mining for gene prioritization is more involved, and the structure of the ground truth is less obvious. This is further complicated by the fact that text mining is embedded inside the pipeline of gene periodization as one component, which often requires extrinsic evaluation that assesses the performance of the overall task. Because of the explorative nature of gene prioritization tasks, experimental bench-marking and statistical benchmarking by cross-validation are frequent choices of assessing prioritization methods.2 Experimental benchmarking requires lab experiments to determine the proportion of false positives in the top ranked genes. Statistical benchmarking requires known disease-associated genes and evaluates how well the top ranked genes overlap with the known set of genes. Note that both benchmarking evaluations are necessary but not sufficient conditions for a good prioritization.
Problems with guilt-by-association
Network based gene prioritization methods integrate evidence from text mining and similarity profiling, as well as function and interaction annotations. The assumption shared by those methods is the so called Guilt-by-Association (GBA), which states that functionally related genes will give rise to similar mutational phenotypes. GBA has been used to statistically infer previously unknown functions of a gene from prior knowledge about known genes and association data such as PPI and co-expression. However, recent studies have shown that GBA may not in general hold true.119,120 Further, most prioritization tools do not distinguish the types of gene relations, hence they are “black-box” in nature. The black-box nature is partly responsible for the poor performance of GBA. Typically, the construction of functional networks is done by aggregating context independent data such as physical PPI and context specific data such as co-expression121 without distinguishing them. This is especially true for gene and protein interactions extracted from literature using text mining with simple co-occurrence or co-citation statistics. Separating context specific evidence may lend better insights to improve the robustness of the results.122 Thus taking finer semantic information into text mining consideration is likely to improve the accuracy of the mined relations. For example, distinguishing “gene A promotes expression of gene B”, “gene A inhibits gene B,” and “gene A inhibits gene B under the co-expression of gene C” will produce more context specific relations and networks than “gene A and gene B are related”. Further, improving explanatory power can also help elucidate molecular mechanisms of major pathways that underlie cancer development.
Heterogeneous data sources
In gene prioritization, multiple heterogeneous data sources often need to be taken into account, such as expression level, protein interaction, domain interaction, sequence reads, and literature data. Although previous works strive for increasingly comprehensive coverage for those data sources, to date none covers all data sources. Moreover, the inter-operative information exchanges between data sources in previous gene prioritization approaches are largely based on customized ontology and keyword term matching. On the other hand, most gene prioritization methods analyze different types of evidence separately and only pool scores produced by individual types of evidence at a later stage. In keeping with the integrative Omic view in Figure 1, data integration should happen at an earlier stage so that reasoning can be on a more integrative level: “enrichment in expression of gene A” (expression evidence) and “protein A binds protein B” (PPI evidence) together implicate a phenotype C. This level of integration is yet to be achieved and there is still no principled way to construct a unified reasoning platform across data sources.
Cross-species insights
There are genetic knowledge bases describing thoroughly studied model organisms, such as gene–phenotype associations in mouse, worm, fly, and yeast in the STRING database. They can be leveraged to draw insights into human pathways that are not as heavily investigated, through orthologous genes,79 or through cross-species annotation transfer of interolog PPIs.123 In fact, some gene prioritization methods have exploited ortholog and interolog data sources to cover the understudied human cancer phenotypes or genes, using either manually curated lists (eg, GPsy108) or text mining on the literature (eg, MedSim,112 Genie115). However, transferring knowledge across species remains a significant challenge as biological context and environmental factors also contribute to the ways genes and proteins interact. Thus, there is no guaranteed mapping from sequence similarity to functional similarity. For this reason, we also expect that taking finer semantic information into text mining consideration to provide context specificity to ortholog and interolog gene relations is likely to help the overall prioritization task.
Emerging and Future Directions
This section presents several recent advances that may provide partial solutions to some challenges raised in the analysis of the previous section.
Converting literature and clinical records to networks
The task of automatically annotating biomedical text with semantic information is an area of active research of medical Natural Language Processing (NLP). There are existing methods that extract named entities such as genes and proteins, as well as their relations (see related work section of124 for an overview). Previous gene prioritization methods have partially used these methods to identify gene and protein names, but not as much their relations, when constructing gene and protein networks. This is partly due to the fact that most NLP based semantic relation extraction tools identify only binary relations that are too coarse for gene prioritization tasks so that they do not offer much more information compared to simple co-occurrence statistics. Recently, Luo et al.124 proposed an algorithm that translates text into a network representation, where the nodes of the network may be nominal concepts such as genes and proteins or relational concepts such as a verb specifying an interaction. The edges are syntactic dependency links. We give an example sentence–network translation in Figure 2, which shows the network representation for a sentence from the first paragraph in an example paper,125 along with two of its sub-networks.
Figure 2.
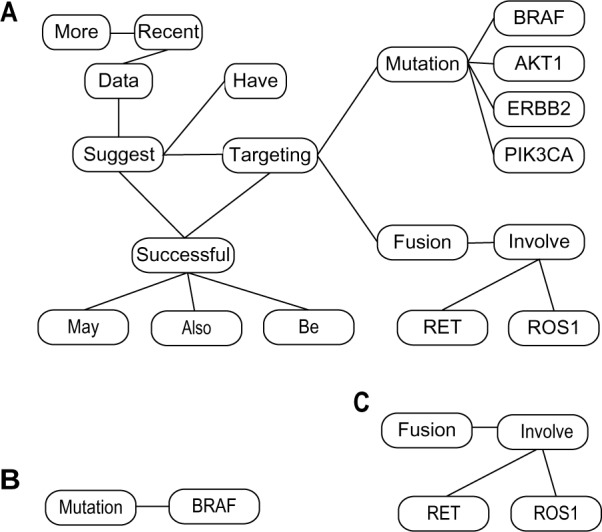
(A) The network representation for the example sentence: “More recent data have suggested that targeting mutations in BRAF, AKT1, ERBB2 and PIK3CA and fusions that involve ROS1 and RET may also be successful”. (B) and (C) are two sub-networks of (A).
Given the text translated networks, useful sub-networks can be harvested under predefined criteria (eg, frequency, P-significance, or other customized criteria). With proper set criteria, sub-networks may be chosen to represent context specific associations between genes and proteins.
Pathway prioritization
Most gene prioritization methods only focus on using network analysis to rank genes’ relatedness to diseases. A particular gene often participates in multiple pathways, yet maybe only some of those pathways are implicated in a cancer phenotype. Moreover, existing pathways may only have part of themselves involved in a disease process. To this end, associating and prioritizing genetic networks or sub-networks instead of genes can be both more discriminative and more informative, to identify new pathways. Finding cancer correlated pathways is mostly carried out on a case-by-case basis, targeting specific cancer types (eg, 126,127). Moreover, identifying such pathways often starts from implicating candidate genes, and known functional pathways are then filtered (sometimes manually) based on prior knowledge. In reality, cancer pathogeneses range from monogenic and oligogenic disorders to complex and epistatic disorders. Direct identification of carcinogenesis pathways may provide more intuition about the underlying molecular mechanisms and hence be more amenable for experimental biologists to start with. On the other hand, gene prioritization methods on direct identification of pathways involved in carcinogenesis have been studied little. GRAIL can be seen as an initial step toward this goal in that it identifies biological relationships among the genes in addition to ranking them. However, GRAIL does not identify what types of relations hold between genes. Hence the predicted genes plus their relationships do not form a pathway as they lack putative mechanism information. The aforementioned text-to-network translation tool may be used for producing context specific gene associations with detailed relation types in order to “build the bridge to the pathway”.
Conclusions
In this review, we focused on text mining in gene prioritization approaches, which is demonstrably an important component because of the large volume and the prevalence of the biomedical literature and narrative text across many heterogeneous data sources. We reviewed and categorized gene prioritization text mining methods according to progressively advanced models employed, and pointed out that they have seen slower advances compared to the other components in gene prioritization that analyze structured data. We discussed several key challenges to text mining in gene prioritization. We also identified promising future directions including text–network translation to provide finer semantic information and pathway prioritization to offer more mechanism information.
Acknowledgments
We thank Dr. Wen Xue for helpful discussion on the utilization of gene and pathway prioritization. We also thank the anonymous reviewers for their comments and feedback.
Footnotes
Author Contributions
YL wrote the first draft of the manuscript. GR contributed to writing the section of data sources for gene and pathway prioritization. PSZ contributed to the analysis and categorization of text mining methods in gene and pathway prioritization. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: The work described was supported in part by Grant Number U54LM008748 from the National Library of Medicine and by the Scullen Center for Cancer Data Analysis. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Library of Medicine or the National Institutes of Health.
COMPETING INTERESTS: PS is an advisor to Health Fidelity, which applies natural language processing methods to clinical text. This paper is unrelated to the company. Other authors disclose no competing interests.
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants. Provenance: the authors were invited to submit this paper.
REFERENCES
- 1.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moreau Y, Tranchevent LC. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat Rev Genet. 2012;13:523–36. doi: 10.1038/nrg3253. [DOI] [PubMed] [Google Scholar]
- 3.Lahti L, Schäfer M, Klein H-U, Bicciato S, Dugas M. Cancer gene prioritization by integrative analysis of mRNA expression and DNA copy number data: a comparative review. Brief Bioinf. 2013;14:27–35. doi: 10.1093/bib/bbs005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bromberg Y. Disease gene prioritization. PLoS Comput Biol. 2013;9:e1002902. doi: 10.1371/journal.pcbi.1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piro RM, Di Cunto F. Computational approaches to disease-gene prediction: rationale, classification and successes. FEBS J. 2012;279:678–96. doi: 10.1111/j.1742-4658.2012.08471.x. [DOI] [PubMed] [Google Scholar]
- 6.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: a novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics. 1998;14:656–64. doi: 10.1093/bioinformatics/14.8.656. [DOI] [PubMed] [Google Scholar]
- 8.Safran M, Solomon I, Shmueli O, et al. GeneCards 2002: towards a complete, object-oriented, human gene compendium. Bioinformatics. 2002;18:1542–3. doi: 10.1093/bioinformatics/18.11.1542. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler DL, Barrett T, Benson DA, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007;35:D5–12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s online Mendelian inheritance in man (OMIM®) Nucleic Acids Res. 2009;37:D793–6. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindberg DA, Humphreys BL, McCray AT, et al. The Unified Medical Language System. Methods Inf Med. 1993;32:281. doi: 10.1055/s-0038-1634945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schriml LM, Arze C, Nadendla S, et al. Disease ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40:D940–6. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Library of Medicine MeSH. [Accessed March 28. 2014]. Available at http://www.ncbi.nlm.nih.gov/mesh.
- 15.Kelso J, Visagie J, Theiler G, et al. eVOC: a controlled vocabulary for unifying gene expression data. Genome Res. 2003;13:1222–30. doi: 10.1101/gr.985203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–5. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith CL, Goldsmith C-AW, Eppig JT. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2004;6:R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.BioCarta BioCarta Pathways. [Accessed March 28, 2014]. Available at http://biocarta.com.
- 20.Karp PD, Ouzounis CA, Moore-Kochlacs C, et al. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005;33:6083–9. doi: 10.1093/nar/gki892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joshi-Tope G, Gillespie M, Vastrik I, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–32. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- 23.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schomburg I, Chang A, Placzek S, et al. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res. 2013;41:D764–72. doi: 10.1093/nar/gks1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peri S, Navarro JD, Amanchy R, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–71. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer CF, Anthony K, Krupa S, et al. PID: the pathway interaction database. Nucleic Acids Res. 2009;37:D674–9. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter S, Jones P, Mitchell A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–12. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts. Genome Biol. 2006;7:S12. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn RD, Tate J, Mistry J, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–8. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34:D257–60. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hulo N, Bairoch A, Bulliard V, et al. The PROSITE database. Nucleic Acids Res. 2006;34:D227–30. doi: 10.1093/nar/gkj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeats C, Maibaum M, Marsden R, et al. Gene3D: modelling protein structure, function and evolution. Nucleic Acids Res. 2006;34:D281–4. doi: 10.1093/nar/gkj057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bru C, Courcelle E, Carrère S, Beausse Y, Dalmar S, Kahn D. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 2005;33:D212–5. doi: 10.1093/nar/gki034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchler-Bauer A, Anderson JB, Cherukuri PF, et al. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–6. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–7. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Matys V, Fricke E, Geffers R, et al. TRANSFAC®: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–8. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flicek P, Ahmed I, Amode MR, et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su AI, Cooke MP, Ching KA, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–70. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett T, Troup DB, Wilhite SE, et al. NCBIGEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–90. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 42.Jones AR, Overly CC, Sunkin SM. The Allen brain atlas: 5 years and beyond. Nat Rev Neurosci. 2009;10:821–8. doi: 10.1038/nrn2722. [DOI] [PubMed] [Google Scholar]
- 43.Diehn M, Sherlock G, Binkley G, et al. SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res. 2003;31:219–23. doi: 10.1093/nar/gkg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis AP, Murphy CG, Saraceni-Richards CA, Rosenstein MC, Wiegers TC, Mattingly CJ. Comparative toxicogenomics database: a knowledge-base and discovery tool for chemical-gene-disease networks. Nucleic Acids Res. 2009;37:D786–92. doi: 10.1093/nar/gkn580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rath A, Olry A, Dhombres F, Brandt MM, Urbero B, Ayme S. Representation of rare diseases in health information systems: the Orphanet approach to serve a wide range of end users. Hum Mutat. 2012;33:803–8. doi: 10.1002/humu.22078. [DOI] [PubMed] [Google Scholar]
- 46.Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A navigator for human genome epidemiology. Nat Genet. 2008;40:124–5. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- 47.National Library of Medicine Genetics Home Reference. [Accessed March 29, 2014]. Available at http://ghr.nlm.nih.gov/
- 48.Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet. 2004;36:431–2. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 49.Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998;26:285–7. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.UniProt Consortium The universal protein resource (UniProt) Nucleic Acids Res. 2008;36:D190–5. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ringwald M, Eppig JT, Begley DA, et al. The mouse gene expression database (GXD) Nucleic Acids Res. 2001;29:98–101. doi: 10.1093/nar/29.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–8. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forbes S, Bhamra G, Bamford S, et al. The catalogue of somatic mutations in cancer (COSMIC) Curr Protoc Hum Genet. 2008;10:11. doi: 10.1002/0471142905.hg1011s57. Unit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sherry ST, Ward M-H, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hewett M, Oliver DE, Rubin DL, et al. PharmGKB: the pharmacogenetics knowledge base. Nucleic Acids Res. 2002;30:163–5. doi: 10.1093/nar/30.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pagon RA, Tarczy-Hornoch P, Baskin PK, et al. GeneTests-GeneClinics: genetic testing information for a growing audience. Hum Mutat. 2002;19:501–9. doi: 10.1002/humu.10069. [DOI] [PubMed] [Google Scholar]
- 61.Rajeevan H, Soundararajan U, Kidd JR, Pakstis AJ, Kidd KK. ALFRED: an allele frequency resource for research and teaching. Nucleic Acids Res. 2012;40:D1010–15. doi: 10.1093/nar/gkr924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibbs RA, Belmont JW, Hardenbol P, et al. The international HapMap project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 63.Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9. 1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breitkreutz B-J, Stark C, Tyers M. The GRID: the general repository for interaction datasets. Genome Biol. 2003;4:R23. doi: 10.1186/gb-2003-4-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alfarano C, Andrade CE, Anthony K, et al. The biomolecular interaction network database and related tools 2005 update. Nucleic Acids Res. 2005;33:D418–24. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kerrien S, Alam-Faruque Y, Aranda B, et al. IntAct-open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–5. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–51. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Günther S, Kuhn M, Dunkel M, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008;36:D919–22. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuhn M, Szklarczyk D, Franceschini A, von Mering C, Jensen LJ, Bork P. STITCH 3: zooming in on protein-chemical interactions. Nucleic Acids Res. 2012;40:D876–80. doi: 10.1093/nar/gkr1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 72.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–4. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aerts S, Van Loo P, Thijs G, et al. TOUCAN 2: the all-inclusive open source workbench for regulatory sequence analysis. Nucleic Acids Res. 2005;33:W393–6. doi: 10.1093/nar/gki354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blake JA, Eppig JT, Richardson JE, Bult CJ, Kadin JA. The Mouse Genome Database (MGD): integration nexus for the laboratory mouse. Nucleic Acids Res. 2001;29:91–4. doi: 10.1093/nar/29.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shaw DR. Searching the Mouse Genome Informatics (MGI) resources for information on mouse biology from genotype to phenotype. Curr Protoc Bioinf. 2009:1–7. doi: 10.1002/0471250953.bi0107s25. [DOI] [PubMed] [Google Scholar]
- 77.HomoloGene HomoloGene. [Accessed March 29, 2014]. Available at http://www.ncbi.nlm.nih.gov/homologene.
- 78.Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–94. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’Brien KP, Remm M, Sonnhammer EL. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:D476–80. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turner FS, Clutterbuck DR, Semple CA. POCUS: mining genomic sequence annotation to predict disease genes. Genome Biol. 2003;4:R75–75. doi: 10.1186/gb-2003-4-11-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. Speeding disease gene discovery by sequence based candidate prioritization. BMC Bioinformatics. 2005;6:55. doi: 10.1186/1471-2105-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.George RA, Liu JY, Feng LL, Bryson-Richardson RJ, Fatkin D, Wouters MA. Analysis of protein sequence and interaction data for candidate disease gene prediction. Nucleic Acids Res. 2006;34:e130–130. doi: 10.1093/nar/gkl707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Radivojac P, Peng K, Clark WT, et al. An integrated approach to inferring gene–disease associations in humans. Proteins Struct Funct Bioinf. 2008;72:1030–7. doi: 10.1002/prot.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jolliffe I. Principal Component Analysis. Wiley Online Library; 2005. [Google Scholar]
- 85.Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–97. [Google Scholar]
- 86.Driel MA, van Cuelenaere K, Kemmeren PP, Leunissen JA, Brunner HG. A new web-based data mining tool for the identification of candidate genes for human genetic disorders. Eur J Hum Genet. 2003;11:57–63. doi: 10.1038/sj.ejhg.5200918. [DOI] [PubMed] [Google Scholar]
- 87.Letovsky SI, Cottingham RW, Porter CJ, Li PW. GDB: the human genome database. Nucleic Acids Res. 1998;26:94–9. doi: 10.1093/nar/26.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Franke L, van Bakel H, Fokkens L, De Jong ED, Egmont-Petersen M, Wijmenga C. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am J Hum Genet. 2006;78:1011–25. doi: 10.1086/504300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hutz JE, Kraja AT, McLeod HL, Province MA. CANDID: a flexible method for prioritizing candidate genes for complex human traits. Genet Epidemiol. 2008;32:779–90. doi: 10.1002/gepi.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiong Q, Qiu Y, Gu W. PGMapper: a web-based tool linking phenotype to genes. Bioinformatics. 2008;24:1011–3. doi: 10.1093/bioinformatics/btn002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu W, Wulf A, Liu T, Khoury MJ, Gwinn M. Gene Prospector: an evidence gateway for evaluating potential susceptibility genes and interacting risk factors for human diseases. BMC Bioinformatics. 2008;9:528. doi: 10.1186/1471-2105-9-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Östlund G, Lindskog M, Sonnhammer EL. Network-based Identification of novel cancer genes. Mol Cell Proteomics. 2010;9:648–55. doi: 10.1074/mcp.M900227-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perez-Iratxeta C, Bork P, Andrade MA. Association of genes to genetically inherited diseases using data mining. Nat Genet. 2002;31:316–9. doi: 10.1038/ng895. [DOI] [PubMed] [Google Scholar]
- 94.Perez-Iratxeta C, Bork P, Andrade-Navarro MA. Update of the G2D tool for prioritization of gene candidates to inherited diseases. Nucleic Acids Res. 2007;35:W212–6. doi: 10.1093/nar/gkm223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perez-Iratxeta C, Wjst M, Bork P, Andrade MA. G2D: a tool for mining genes associated with disease. BMC Genet. 2005;6:45. doi: 10.1186/1471-2156-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA. A text-mining analysis of the human phenome. Eur J Hum Genet. 2006;14:535–42. doi: 10.1038/sj.ejhg.5201585. [DOI] [PubMed] [Google Scholar]
- 98.Aerts S, Lambrechts D, Maity S, et al. Gene prioritization through genomic data fusion. Nat Biotechnol. 2006;24:537–44. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 99.Tranchevent LC, Barriot R, Yu S, et al. ENDEAVOUR update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36:W377–84. doi: 10.1093/nar/gkn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gaulton KJ, Mohlke KL, Vision TJ. A computational system to select candidate genes for complex human traits. Bioinformatics. 2007;23:1132–40. doi: 10.1093/bioinformatics/btm001. [DOI] [PubMed] [Google Scholar]
- 101.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–11. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics. 2007;8:392. doi: 10.1186/1471-2105-8-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu X, Jiang R, Zhang MQ, Li S. Network-based global inference of human disease genes. Mol Syst Biol. 2008;4 doi: 10.1038/msb.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seelow D, Schwarz JM, Schuelke M. GeneDistiller–distilling candidate genes from linkage intervals. PLoS One. 2008;3:e3874. doi: 10.1371/journal.pone.0003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010;6:e1000641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cheng D, Knox C, Young N, Stothard P, Damaraju S, Wishart DS. Poly-Search: a web-based text mining system for extracting relationships between human diseases, genes, mutations, drugs and metabolites. Nucleic Acids Res. 2008;36:W399–405. doi: 10.1093/nar/gkn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Köhler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82:949–58. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Britto R, Sallou O, Collin O, Michaux G, Primig M, Chalmel F. GPSy: a cross-species gene prioritization system for conserved biological processes–application in male gamete development. Nucleic Acids Res. 2012;40:W458–65. doi: 10.1093/nar/gks380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tiffin N, Kelso JF, Powell AR, Pan H, Bajic VB, Hide WA. Integration of text-and data-mining using ontologies successfully selects disease gene candidates. Nucleic Acids Res. 2005;33:1544–52. doi: 10.1093/nar/gki296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics. 2006;22:773–4. doi: 10.1093/bioinformatics/btk031. [DOI] [PubMed] [Google Scholar]
- 111.Lord PW, Stevens RD, Brass A, Goble CA. Investigating semantic similarity measures across the Gene Ontology: the relationship between sequence and annotation. Bioinformatics. 2003;19:1275–83. doi: 10.1093/bioinformatics/btg153. [DOI] [PubMed] [Google Scholar]
- 112.Schlicker A, Lengauer T, Albrecht M. Improving disease gene prioritization using the semantic similarity of Gene Ontology terms. Bioinformatics. 2010;26:i561–7. doi: 10.1093/bioinformatics/btq384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schlicker A, Rahnenführer J, Albrecht M, Lengauer T, Domingues FS. GOTax: investigating biological processes and biochemical activities along the taxonomic tree. Genome Biol. 2007;8:R33. doi: 10.1186/gb-2007-8-3-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raychaudhuri S, Plenge RM, Rossin EJ, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fontaine J-F, Priller F, Barbosa-Silva A, Andrade-Navarro MA. Genie: literature-based gene prioritization at multi genomic scale. Nucleic Acids Res. 2011;39:W455–61. doi: 10.1093/nar/gkr246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pers TH, Dworzyski P, Thomas CE, Lage K, Brunak S. MetaRanker 2.0: a web server for prioritization of genetic variation data. Nucleic Acids Res. 2013;41:W104–8. doi: 10.1093/nar/gkt387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tranchevent L-C, Capdevila FB, Nitsch D, De Moor B, De Causmaecker P, Moreau Y. A guide to web tools to prioritize candidate genes. Brief Bioinf. 2011;12:22–32. doi: 10.1093/bib/bbq007. [DOI] [PubMed] [Google Scholar]
- 118.Navlakha S, Kingsford C. The power of protein interaction networks for associating genes with diseases. Bioinformatics. 2010;26:1057–63. doi: 10.1093/bioinformatics/btq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gillis J, Pavlidis P. Guilt by association is the exception rather than the rule in gene networks. PLoS Comput Biol. 2012;8:e1002444. doi: 10.1371/journal.pcbi.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gillis J, Pavlidis P. The impact of multifunctional genes on. PLoS One. 2011;6:e17258. doi: 10.1371/journal.pone.0017258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21:1109–21. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pavlidis P, Gillis J. Progress and challenges in the computational prediction of gene function using networks. F1000Res. 2012;1:14. doi: 10.12688/f1000research.1-14.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yu H, Luscombe NM, Lu HX, et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–18. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo Y, Szolovits P, Sohani A, Hochberg E. Automatic lymphoma classification with sentence subgraph mining from pathology reports. Am Med Inf Assoc. 2014 doi: 10.1136/amiajnl-2013-002443. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One. 2010;5:e8918. doi: 10.1371/journal.pone.0008918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang Y, Han L, Yuan Y, Li J, Hei N, Liang H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun. 2014;5:3231. doi: 10.1038/ncomms4231. [DOI] [PMC free article] [PubMed] [Google Scholar]